

INTRODUCTION
The RecQ family of helicases contains highly conserved and ubiquitously expressed proteins that unwind DNA in the context of replication, repair, transcription, chromatin remodeling and telomere maintenance [1, 2]. Human and most mammalian cells encode five RecQ-like (RECQL) helicases: RECQL1, Bloom (BLM), Werner (WRN), RECQL4 and RECQL5. These helicases display unique as well as overlapping functions in DNA metabolism. They bind specific DNA structures and catalyze unwinding and annealing of DNA strands to resolve DNA replication forks, D-loops, G-quadruplex structures and Holliday junctions [1, 2]. Mutations in BLM, WRN and RECQL4 are associated with autosomal recessive diseases. Loss of function of BLM and WRN is associated with Bloom syndrome (BS) and Werner syndrome (WS) respectively, while RECQL4 is associated with Rothmund-Thomson (RTS), RAPADILINO and Baller-Gerold (BGS) syndromes[1-3].
In general, cells with defects in DNA repair have increased risk of transformation to a pre-cancer or cancer phenotype. WS and BS patients exhibit increased incidence of cancer. The most common neoplasias in WS patients are thyroid cancer, malignant melanoma, meningioma, soft tissue sarcoma, osteosarcoma, breast cancer and leukemias [3, 4]. Increased WRN expression is observed in several cancer cell lines and depletion of WRN induces cell death in these cells [5]. Irinotecan treatment enhanced the survival of colorectal cancer patients who expressed lower WRN [6].
The plant alkaloid camptothecin (CPT) and its derivatives, irinotecan and topotecan, represent an important class of drugs used in chemotherapy. These drugs specifically target DNA topoisomerase I (Top1), an enzyme that transiently creates DNA single-strand breaks to reduce supercoiling during replication and transcription [7, 8]. CPT generates cytotoxic covalent reaction intermediates, CPT-DNA-Top1, by inhibiting the re-ligation step of the Top1 catalytic cycle. The cytotoxic effect of the CPT-DNA-Top1 intermediate is S-phase-specific, and is thought to reflect collision events between the replication machinery and the cytotoxic lesion [7, 8]. When cells accumulate many CPT-DNA-Top1 lesions, the DNA damage response (DDR) and associated pathways are activated [8]. Subsequent to DDR activation, DNA repair factors, including RecQ helicases are recruited to the DNA lesions and/or to stalled DNA replication forks. All human RecQ helicases are important for cell survival after CPT treatment [9-13]. WS and BS patient cells are hypersensitive to inhibitors of Top1 and DNA interstrand crosslinking agents, and a synergistic increase in chromosomal aberrations is observed in BLM-WRN double knockout cells exposed to these agents [11]. RECQL4-deficient RTS patient cells and RECQL1 and RECQL5 knockdown cells are also sensitive to CPT [9, 12, 13]. However, studies identifying the mechanisms by which CPT or its analogs exert their effects on human RecQ helicases are limited. In this study, we tested the effects of CPT on the five RecQ helicases in cellular studies and bioinformatically analyzed the association between CPT sensitivity and WRN gene expression. Further we analyzed the expression profiles of WRN and Top1 in a large cohort of human breast cancers to identify any correlations between gene expression and breast cancer specific survival. This study spans from biochemical and cellular work through bioinformatics to a clinical study.
CPT treatment specifically altered the stability and subcellular localization of WRN, while similar effects on other RecQ helicases were not observed. In CPT-treated cells, a large fraction of WRN re-localized to the cytoplasm and was selectively degraded by the ubiquitin proteasome pathway. CPT-induced WRN degradation was independent of p53 status, and the extent of degradation was associated with the sensitivity of the tumor cells to the anticancer drug. WRN degradation was more extensive in CPT-sensitive breast cancer cells than in CPT-resistant cells. However, CPT-dependent degradation of Top1 was extensive in all cell lines tested. In the METABRIC (Molecular Taxonomy of Breast Cancer International Consortium) cohort comprising 1977 breast cancers, ~20% of tumors were found to express high Top1 mRNA and ~83% were found to express high WRN mRNA. Altered Top1 and WRN expression was not only associated with aggressive breast cancers but also correlated with adverse prognostic outcome in patients. Interestingly, in patients with estrogen receptor (ER)-positive breast cancers, high WRN and high Top1 levels were associated with a bad prognosis. Together these results suggest that WRN, but not the other RecQs, is a target of CPT in mediating chemotherapeutic effects in the tumor cells.
RESULTS
Stability, expression and subcellular distribution of WRN in CPT-treated cells
CPT and its derivatives specifically target Top1, and the five human RecQ helicases play essential roles in cell survival after CPT treatment [9-13], thus we tested the effects of CPT on the expression dynamics of all five human RecQ helicases to identify unique or shared responses to CPT treatment.
U2OS cells were treated with 10 μM CPT for 1, 2, 6, 8 or 10 hours, and lysed with IP lysis buffer [14]. Proteins in the supernatant fraction, obtained after centrifugation, were analyzed by immunoblotting for WRN, BLM, RECQL1, RECQL4, RECQL5, Ku80, CtIP and XLF (Figure 1a). Exposure to CPT caused a decrease in the abundance of WRN protein, but not of the other proteins tested (Figure 1a and 1b). Quantification of the immunoblots indicated that WRN protein decreased significantly in a time-dependent manner from ~1 to 10 h after treatment with CPT (Figure 1a graph). This reduction was not observed for BLM, RECQL1, RECQL4 and RECQL5 or for the DNA repair proteins Ku80, CtIP, and XLF (Figure 1a and 1b). The half-life of WRN protein is reported to be approximately 6 h [15], consistent with its apparent half-life in this study (Figure 1a graph). Interestingly, cells treated with 1 or 10 Gy ionizing radiation (IR) did not show a decrease in WRN abundance at time points up to 10 h after irradiation (Figure 1c). These results indicate that the effect of CPT on WRN protein levels could be specific.
The mRNA transcripts encoding all five RecQ helicases were also quantified in CPT-treated and untreated cells (Figure 1d). For all RecQ helicases, transcript abundance was unaffected up to 24 h with CPT treatment. This indicates that CPT induces (Figure 1a) specific post-translational degradation of WRN protein, or that it destabilizes WRN protein by another mechanism.
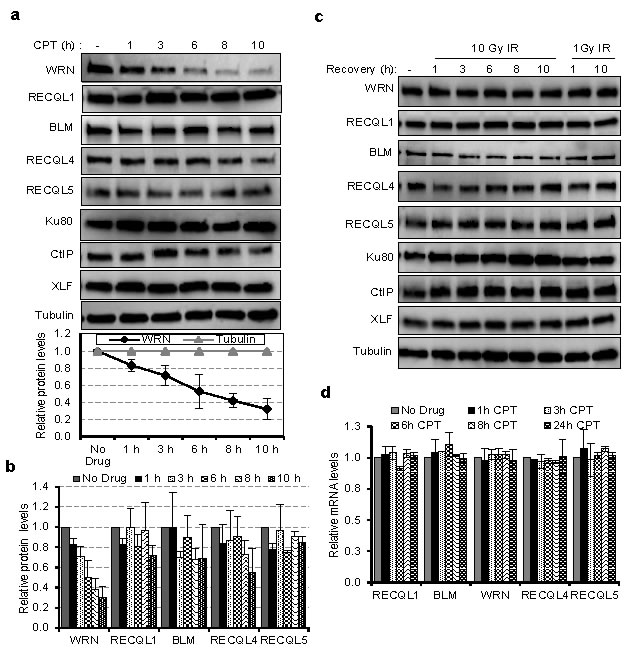
Figure 1: Expression of RecQ helicases in CPT and IR treated cells. a. Half-life of DNA repair proteins in CPT treated cells. Lysates from U2OS cells were treated with 10 µM CPT for the indicated times and immunoblotted for DNA repair proteins viz., Ku70, CtIP, XLF and the RecQ helicases WRN, BLM, RECQL1, RECQL4 and RECQL5. Below; graph showing the relative levels of WRN protein normalized to tubulin from three independent experiments. b. Bar graph showing the protein levels of RecQ helicases in CPT treated cells. c. Immunoblots showing the expression of DNA repair proteins as in panel A from IR treated U2OS cells. d. Quantitative RT-PCR analysis showing mRNA levels of RecQ genes in CPT treated U2OS cells. Expression of GAPDH mRNA was used for normalization. Error bars represent standard deviation from two independent experiments.
WRN protein distribution was analyzed in U2OS cells stably expressing YFP-WRN and treated with CPT using confocal microscopy (Figure 2a). Live-cell microscopy results indicated that YFP-WRN localized primarily to the nucleolus, re-localized to the nucleoplasm ~60 min after exposure to CPT, after which YFP-WRN fluorescence declined to a low level in the nucleolus (Figure 2a). YFP-WRN foci were also observed in the nucleoplasm of CPT-treated cells, suggesting recruitment of WRN to CPT-induced DNA damage. To examine the subcellular distribution of WRN, proteins from the cytoplasm, cytoplasmic organelles, nuclear-soluble and chromatin fractions were isolated and analyzed by Western blotting (Figure 2b). The results suggest that WRN is enriched in cytoplasmic organelles (Figure 2b, compare lane 5 with 6-8) after CPT treatment. Analysis of subcellular protein fractions from IR treated cells did not show significant changes in the distribution pattern of WRN compared to untreated cells (Supplementary Figure S1a). Further, results from immunostained cells indicated that endogenous WRN resides in the nucleolus of cells (Figure 2c top panel and [16]) and that, upon treatment with CPT, re-localizes to the nucleoplasm and cytoplasm (Figure 2c bottom panels). To test the role of translation in CPT-dependent enrichment of WRN in the cytoplasmic organelle fraction observed in Figure 2b, cells were treated with cycloheximide (CHX) along with CPT. Inhibition of translation did not affect CPT-induced re-localization of WRN (Supplementary Figure S1b), suggesting that the WRN enrichment is not dependent on new protein synthesis. Collectively these results demonstrate that WRN undergoes re-localization in cancer cells treated with CPT.
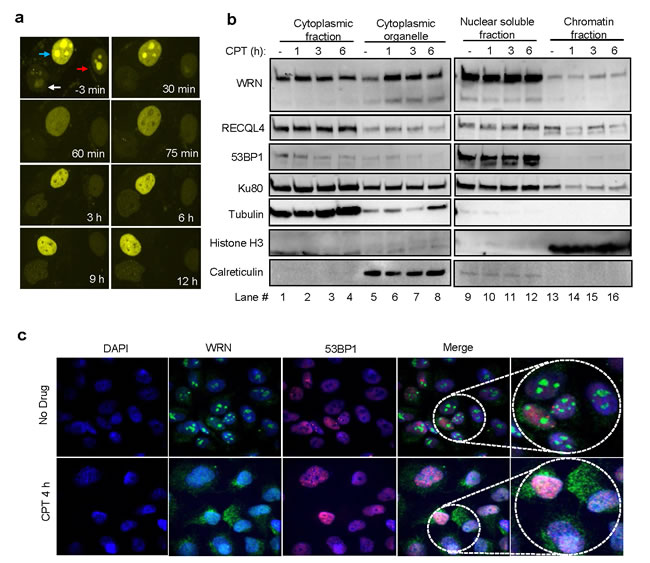
Figure 2: CPT-induced re-localization of WRN. a. Dynamic distribution of YFP-WRN in CPT-treated cells. Live cell time-lapse micrographs from CPT (10 µM) treated U2OS cells stably expressing YFP-WRN. Arrows point the nucleus of YFP-WRN expressing cells. b. WRN distribution in the subcellular compartments. Immunoblots indicating the relative levels of WRN in the cytoplasmic, cytoplasmic organelle, nuclear soluble and chromatin fractions of U2OS cells treated with and without CPT. Time indicate the length of the exposure to CPT. c. Enrichment of endogenous WRN in the cytoplasm. Confocal microphotographs showing WRN and 53BP1 localization in U2OS cells treated with CPT. Representative images from two independent experiments. Microscope images were taken with 40X objective.
CPT-induced ubiquitination and degradation of WRN
To identify the CPT concentrations that induce WRN degradation, U2OS cells were treated with 0.01, 0.05, 0.1, 0.5, 1, 5, 10 and 20 µM CPT for 16h and extracts were prepared by lysing the cells with RIPA buffer followed by brief sonication. Our results showed that the WRN protein is downregulated in cells exposed to 0.5 to 20 µM CPT, conditions that induce degradation of Top1 (Figure 3a and Supplementary Figure S2a). We previously reported that WRN interacts with Top1 [17] and other studies have shown that CPT-induced degradation of Top1 occurs in an ubiquitin- and proteasome-dependent manner [18, 19]. Therefore, it seemed possible that WRN downregulation might be ubiquitin- and proteasome-dependent. Consistent with this possibility, the proteasome inhibitor MG132 suppressed CPT-induced degradation of WRN and Top1 in cells treated with 1 µM CPT (Figure 3b and Supplementary Figure S2b). Further, when 293T cells co-expressing 3xFlag-WRN and HA-ubiquitin were treated with CPT and MG132 (Figure 3c), and cell lysates were analyzed after immunoprecipitation, ubiquitin and WRN co-immunoprecipitated with HA or FLAG antibodies. MG132 stabilized HA-ubiquitinated Flag-WRN, implicating proteasome-mediated degradation of WRN in CPT-treated cells (Figure 3c). We found no evidence that WRN was enriched in lysosomes, using cells in which the lysosome was fluorescence-labeled with lysosome-specific GFP (Supplementary Figure S3). These results suggest that CPT-stimulated degradation of WRN is lysosome-independent, but ubiquitin- and proteasome-dependent.
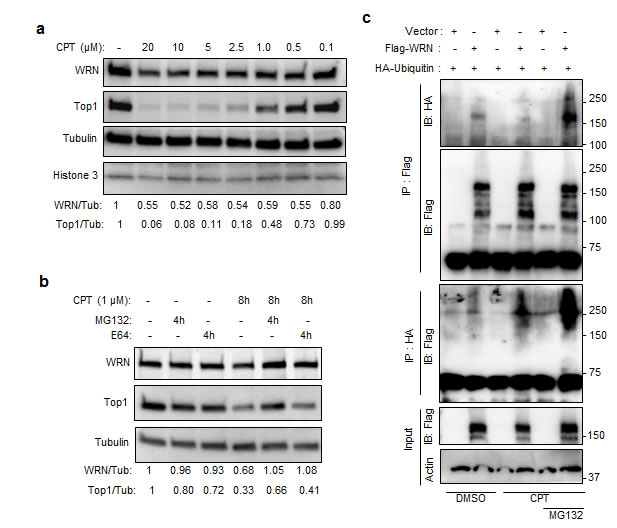
Figure 3: Ubiquitin proteasome-mediated degradation of WRN in CPT treated cells. a. CPT-induced WRN down regulation is associated with Top1 degradation. Immunoblots showing WRN and Top1 protein levels in U2OS cells treated with indicated concentration of CPT for 16 h. b. Proteasome inhibitor MG132 inhibits the effect of CPT on WRN and Top1. U2OS cells were treated with 10 µM MG132 to inhibit proteasome pathway and with 10 µM E64 to inhibit lysosome pathway. c. Ubiquitination of WRN in CPT treated cells. Immunoblots showing ubiquitinated (HA-ubiqutin) 3xFlag-WRN in immunoprecipitated fractions of CPT (1µM) treated 293T cells expressing 3xFlag-WRN and HA-ubiqutin.
WRN attenuates CPT-induced cellular senescence
To explore the functional consequences of WRN downregulation in CPT treated cells, we analyzed cell survival and cellular senescence after CPT treatment. Compared to control cells, WRN depleted cells displayed dose-dependent reduction in cell viability in the presence of CPT (Supplementary Figure S4a). However, knockdown of RECQL4 did not increase the sensitivity of cells to CPT. These results suggest a distinct protective role of WRN against the cytotoxic effects of CPT.
It is well established that DNA damage induces cellular senescence [20], and we and others have shown that WRN plays a role in preventing this process [21, 22]. Because the results presented above showed that WRN is downregulated in CPT-treated cells we hypothesized that decreased abundance of WRN could exacerbate CPT-induced senescence. To test this possibility, we measured senescence-associated β-galactosidase (SA-β-gal) activity, a marker of senescence [21], in fibroblasts and U2OS cells treated with CPT. Knockdown of WRN in human fibroblasts, GM0637, induced cellular senescence in ~60% of the cell population (Figure 4a). Treatment of cells with 5 and 20 µM CPT, levels that downregulated WRN, also induced senescence in ~35 and ~45% of cells, respectively (Figure 4b). To evaluate any protective role(s) that WRN may provide against CPT-induced cellular senescence, GM0637 cells were transfected with 3xFlag-WRN (Supplementary Figure S4b) for 24 hours before treatment with CPT. Consistent with the results obtained in the untransfected cells (Figure 4b), vector-transfected cells exposed to 5 and 20 µM CPT accumulated ~35 and ~40% senescent cells. However, vector-transfected cells treated with DMSO only accumulated ~8% senescent cells (Figure 4c) and the ability of CPT to induce the senescent phenotype was attenuated in cultures expressing 3xFlag-WRN (Figure 4c). The 3xFlag-WRN expressing cultures contained ~14% and ~24% senescent population in the presence of DMSO and CPT, respectively (Figure 4c). Similar results were observed in U2OS cells expressing C-terminal GFP-tagged WRN (Supplementary Figure S4c). However, ectopic expression of 3xFlag-RECQL4 (Supplementary Figure S4b) did not significantly inhibit CPT-induced cellular senescence (Figure 4c). These results demonstrate that WRN provides partial protection against CPT-induced cellular senescence.
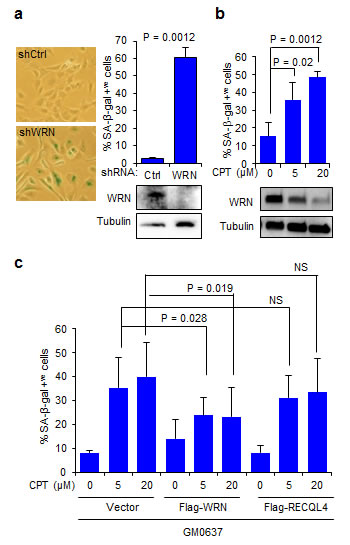
Figure 4: WRN attenuates CPT-induced cellular senescence. a. Knockdown of WRN induces cellular senescence in human fibroblasts. Left, micrographs showing SA-β-gal stained GM637 cells; right, bar graph showing SA-β-gal positive cells (n = three independent experiments). Immunoblots showing knockdown levels of WRN. shCtrl, control shRNA; shWRN, WRN shRNA b. CPT-induced cellular senescence in human fibroblasts. Bar graph showing SA-β-gal positive GM637 cells treated with CPT for 18 h. Error bars represent standard deviation from two independent experiments. Immunoblots indicate WRN degradation after CPT treatment. c. Ectopic expression of WRN attenuates CPT-induced cellular senescence. GM637 cells transfected with vector, Flag-WRN and Flag-RECQL4 were treated with indicated CPT concentrations and stained for SA-β-gal (n = three independent experiments).
WRN degradation and CPT sensitivity in breast cancer cells
A specific mutation (Phe1074Leu) in WRN is found to increase the risk of breast cancer incidence [23, 24], and dysregulation of WRN expression is observed in breast cancer cells lines [5, 6]. Lately there has been an interest in using Top1 inhibitors in the treatment of breast cancer [25, 26], which traditionally has been treated with other approaches. Since Top1 protein expression is tightly correlated with mRNA expression in various cancer cell lines [27] and the above results show that WRN and Top1 are targets for degradation in CPT treated cells, we conducted an in silico search to find correlations between the steady-state expression of Top1 and WRN and the sensitivity to Top1 inhibitors using Cancer Cell Line Encyclopedia (CCLE) and Cancer Genome Atlas (TGCA) databases. The results suggest that, irrespective of cancer types, toptecan sensitivity is generally associated with Top1 (p-value = 7.7e-03) and WRN (p-value = 2.27e-06) mRNA expression (Supplementary Figure S5a, S5b and 5c). Additionally, mining of breast cancer specific RNAseq data from TCGA, indicated a strong correlation between Top1 and WRN expression (Supplementary Figure S5a; r = 0.325, p = 5.83e-26) suggesting the importance of these two genes in breast cancer.
To investigate WRN and Top1 expression and the phenomenon of CPT-induced WRN degradation in breast cancer cells, we used three CPT-sensitive (MCF-7, T47D and ZR-75-1) and three CPT-resistant (BT549, MDA-MB-231 and BT-474) cell lines. The Cell Miner database (http://discover.nci.nih.gov/cellminer/) information on the breast cancer cell lines showed that WRN expression was low in two of the CPT-sensitive cell lines (MCF-7 and T47D), high in one CPT-resistant cell line (BT-549) and low in another CPT-resistant (MDA-MB-231) cell line (Figure 5a). Western blot analysis of WRN in the six breast cancer cell lines showed that these cells expressed WRN at relatively similar levels (within 20% of each other: Figure 5b). However, upon treatment with CPT, a significant difference in WRN degradation was observed between the CPT-sensitive and -resistant cell lines (Figure 5c). The CPT-sensitive cells showed significant WRN degradation after CPT treatment, whereas the CPT-resistant cells showed minimal WRN degradation (Figure 5c and 5d). In contrast, CPT-induced Top1 degradation was extensive in both (Figure 5c and 5e) demonstrating that Top1 has no predictive power and further that dose was sufficient to induce degradation in all cell lines.
A seminal study reported a high degree of heterogeneity in Top1 degradation after CPT treatment [28]. Our results confirm this observation in MCF-7, T47D, HCT116, and WI38 cell lines treated with CPT for 8 h (Supplementary Figure S6a); however Top1 was extensively degraded in all tested cell lines, including ZR-75-1, after 16 h of treatment (Figure 5c and Supplementary Figure S6a and S6b). Supporting the importance of WRN in cell proliferation after DNA damage, CPT-sensitive breast cancer cells, which displayed drug-induced WRN degradation, showed compromised cell proliferation after CPT treatment (Figure 5f). Cell survival and proliferation of CPT-resistant cells was largely unaffected by CPT (Figure 5f). Together these results demonstrate that CPT-sensitivity correlated with WRN degradation and not Top1 degradation.
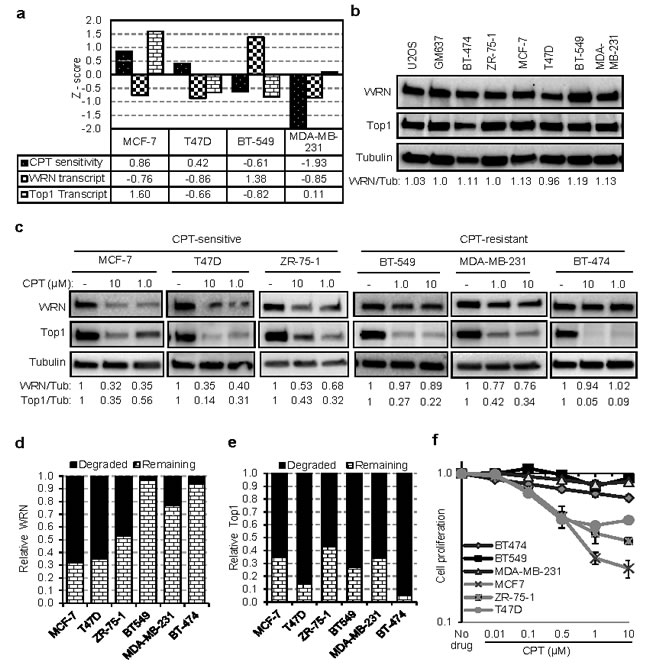
Figure 5: WRN degradation is associated with sensitivity of tumor cells to CPT. a. WRN expression and CPT sensitivity of breast cancer cells lines. Graph showing Z scores of WRN transcript expression and CPT sensitivity as documented in NCI-60 Cell Miner database. Z score for WRN transcript intensity indicates relative mRNA expression compared to average expression of NCI-60 panel of cell lines. Negative Z score for CPT sensitivity indicates the resistance of the cell line to CPT. b. WRN protein expression in tumor cell lines. c. CPT-dependent WRN degradation in CPT-sensitive and CPT-resistant breast cancer cell lines. Immunoblots represents protein levels in cells treated with CPT for 16 h. Graphs showing relative degradation of WRN d. and Top1 e. in breast cancer cell lines treated with 10 µM CPT. f. CPT-dependent cell viability and proliferation in sensitive and resistant breast cancer cells. Graph showing relative cell proliferation after 24 h of CPT (0.01 to 10 µM) treatment. Error bars represent SEM from three independent experiments.
Top1 and WRN mRNA expression in human breast cancers
Given the essential role of WRN and Top1 in DNA repair and replication we studied their role in breast cancer pathogenesis and prognosis. Inhibition of WRN promoter methylation increased the WRN mRNA levels which in turn increased the protein levels in several cancer cells [6], and a tight correlation between Top1 mRNA and protein expression was observed in the NCI-60 panel of cell lines [27] implying that high mRNA leads to high protein expression. Therefore we investigated the mRNA levels of WRN and Top1 in the METABRIC (Molecular Taxonomy of Breast Cancer International Consortium) cohort. Primary tumors were expression profiled [29] to investigate associations with clinic-pathological parameters and survival in patients. Nearly 16.5% (326/1977) of tumors had low WRN mRNA expression and ~85.5% (1651/1977) tumors had high WRN mRNA expression (Figure 6a). Low WRN mRNA expression was significantly associated with aggressive clinicopathological features including high histological grade, lymph node stage, high risk Nottingham prognostic index (NPI) > 3.4 and Her-2 over expression (ps≤0.01) (Table 1). Interestingly, ER negative and triple negative phenotypes were more common in tumors with high WRN mRNA expression (ps≤0.01). Low WRN mRNA expression was significantly associated with molecular phenotypes: PAM50.Her2, PAM50.LumB, Genufu subtype (ER+/Her2-/High proliferation) and Genufu subtype (Her2 positive) breast tumors (ps≤0.01). On the other hand, PAM50.LumA tumors, PAM50.basal and Genufu subtype (ER+/Her2-/low proliferation) express high levels of WRN mRNA (ps≤0.01). Similarly, the WRN mRNA level was significantly associated with the various integrative clusters (Table 1) described in the METABRIC study which was based on gene copy number changes and gene expression data [29]. Low WRN mRNA expression was significantly associated with clusters, which had poor clinical outcome in the METABRIC study [29]. Low WRN mRNA expression in tumors was associated with adverse Breast Cancer Specific Survival (BCSS) in the whole cohort (p = 0.002) (Figure 6a). The data provides evidence that low WRN mRNA expression is associated with aggressive phenotypes and poor survival in patients.
In the METABRIC cohort, ~20% (390/1977) of tumors had high Top1 mRNA expression and 80% (1586/1977) tumors had low Top1 mRNA expression (Figure 6b). High Top1 mRNA expression was significantly associated with aggressive clinicopathological features including high histological grade, larger tumor size, high risk Nottingham prognostic index (NPI) > 3.4, Her2 over expression, and ER- and PR- tumors (ps < 0.05) (Table 1). High Top1 mRNA expression in tumors was associated with adverse BCSS in the whole cohort (p = 0.002) (Figure 6b). Of note, patients in the METABRIC cohort did not receive Top1 inhibitor based therapy. Nevertheless, taken together, the data provides evidence that high Top1 mRNA expression is associated with aggressive breast cancers.
Table 1: Clinico-pathological characteristics of METABRIC cohort based on WRN and Top1 expression
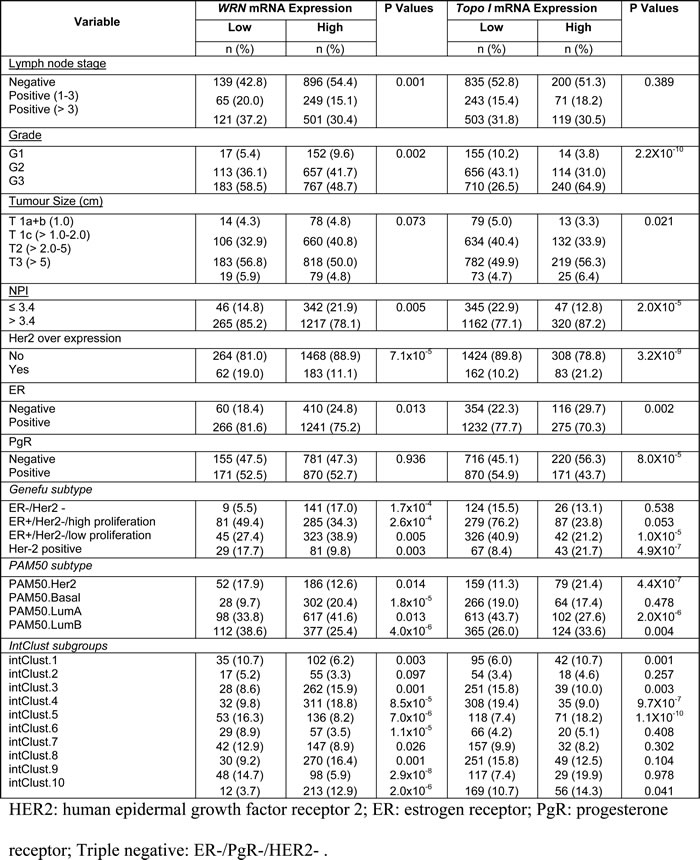
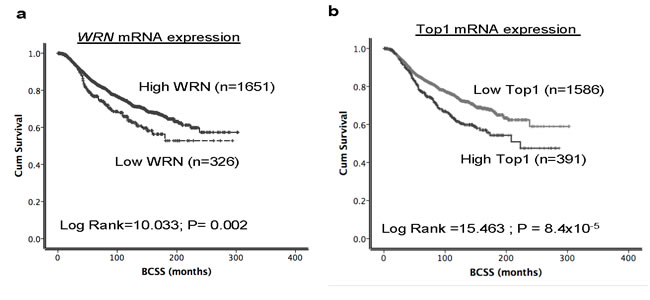
Figure 6: Top1 and WRN mRNA expression in METABRIC cohort. Kaplan Meier curves showing BCSS (Breast cancer specific survival) based on Top1 expression a. and WRN expression b.
WRN and Top1 co-expression in ER positive and ER negative breast cancers
Our in vitro results suggest that WRN degradation could be a useful marker in personalized chemotherapy. To identify potential patient populations that may benefit from CPT-based regimens, we proceeded to sub-group analysis of patients in the METABRIC cohort where ER positive tumors comprised of 1507/1977 (76.2%) of tumors and 470/1977 (23.8%) were ER negative tumors.
When Top1 and WRN were combined together, as expected, patients with high WRN/high Top1 expressing tumors had worst survival compared to tumors with low WRN/low Top1 expression in the whole cohort (p < 0.0001) (Figure 7a and Supplementary Table 1) as well as in the ER positive cohort (p < 0.0001) (Figure 7b). In addition, in high risk ER positive tumors that received adjuvant endocrine therapy, high WRN/high Top1 expressing tumors was associated with poor survival (p < 0.001) (Supplementary Figure S7a). In ER positive tumors that received no endocrine therapy, high WRN/high Top1 expressing tumors remains associated with poor survival compared to tumors with low WRN/low Top1 expressing (p = 0.006) (Supplementary Figure S7b).
Interestingly, when WRN and Top1 examined were combined in ER negative tumors, low WRN/low Top1 expressing tumors had the worst survival compared to patients with high WRN/high Top1 expressing tumors (p = 0.021) (Figure 7c). Collectively, the data suggest that WRN/Top1 expression may have predictive significance in the ER negative sub-group. In patients who had no adjuvant chemotherapy, compared to tumors with low WRN/low Top1 expression, tumors with high WRN/high Top1 expression have a poorer prognosis (p = 0.043) (Supplementary Figure S7d).
Taken together, in ER positive tumors WRN/Top1 co-expression may have predictive significance in patients who received endocrine therapy and prognostic significance in patients who received no endocrine therapy. Importantly, the data also suggest that an alternative form of therapy such as those targeting Top1 could be a promising strategy in high Top1/high WRN expressing tumors. Overall these results identify that ~14% of ER positive and ~34% of ER negative breast cancer patients express high WRN and high Top1 in their tumors and may benefit from CPT-based personalized chemotherapy.
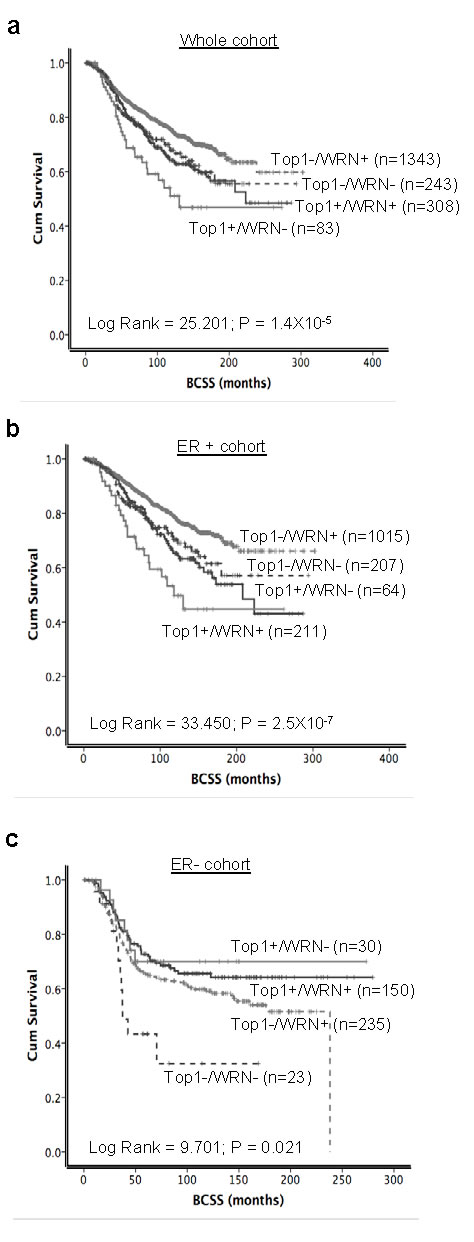
Figure 7: Kaplan Meier curves showing BCSS with WRN and Top1 expression. a. WRN and Top1 expression in whole cohort. b. and c. combined expression of WRN and Top1 in ER positive and ER negative METABRIC cohorts.
DISCUSSION
Cellular studies show that RecQ helicases protect against the cytotoxic effects of CPT [9-13]. To characterize the mechanism, we analyzed the expression and stability of the human RecQ helicases in cells exposed to CPT. CPT affects the degradation of WRN protein without altering expression of WRN mRNA or the abundance of the other RecQ proteins. CPT treatment resulted in re-localization of WRN from the nucleolus to the nucleoplasm and cytoplasmic organelles, and resulted in downregulation of the protein in a time- and CPT concentration-dependent manner. CPT-induced WRN downregulation was mediated by the ubiquitin proteasome pathway, and ectopic expression of WRN attenuated the CPT-induced senescent phenotype. Drug-induced WRN degradation in breast cancer cell lines was associated with the sensitivity of cells to CPT. Breast cancer patients with ER-positive tumors expressing high WRN and Top1 had poor survival. Overall findings presented in this study, based on cellular studies, in silico studies and clinical studies, suggest that WRN helicase along with Top1 can be targeted with CPT at the protein level and could be a used as a potential marker for predicting the efficiency of CPT-based chemotherapy for breast cancer.
WRN, the largest human RecQ protein, contains a nuclease domain, which is not found in other RecQ proteins, and catalyzes four DNA-dependent reactions: 3’-5’-exonuclease, ATPase, DNA strand annealing and 3’-5’-helicase activities. Through its enzymatic functions, WRN acts on various DNA structures to facilitate DNA repair. Mutations in WRN leads to defects in DNA repair, premature aging and to cancer susceptibility [23, 30, 31]. Small molecules targeting DNA repair proteins have profound effects in inhibiting tumor survival. A recent study identified NSC 19630 [1-(propoxymethyl)-maleimide] as a specific inhibitor of WRN, which synergistically inhibited cell proliferation and induced DNA damage with topotecan [32].
CPT and its analogs are used against a broad spectrum of tumors [33]. It covalently binds to the interface between Top1 and DNA, thereby blocking the cleavage/re-ligation activities of the topoisomerase [7]. As a result high loads of protein-linked DNA breaks block replication and transcription events and initiate senescence and cell death [8, 34]. The novel observation reported here is that WRN is specifically degraded in CPT-treated cells exacerbating the adverse consequences of CPT-induced DNA damage and loss of functional Top1. CPT treatment leads to a reduction of Top1 protein, trapped as the CPT-DNA-Top1 complex, by small ubiquitin-like modifier (SUMO) protein conjugation and by the ubiquitin proteasome pathway [18, 35, 36]. As shown here, CPT also causes the specific degradation of WRN, but not of the other RecQ helicases, likely because WRN physically and functionally interacts with Top1 [17]. Thus, the physical association between WRN and Top1 could lead to a shared ‘fate’ of degradation in CPT-treated cells. However, as shown in Figure 5c, Top1 degradation can be uncoupled from WRN degradation in certain cell types.
Protein degradation is orchestrated mainly by the proteasome and lysosome proteolytic systems. Lack of KFERQ motifs in WRN suggests that it may not be targeted for degradation by lysosomes. Our results showed that CPT treatment induced re-localization of WRN from the nucleolus to the cytoplasm where ubiquitin-dependent protein turnover occurs. Ubiquitinylation of proteins occur by the addition of ubiquitin molecules to lysine residues on target proteins. In the G1 phase of the cell cycle, BLM is ubiquitinated and degraded to promote non-homologous end joining [37]. Such degradation pathways might be operating on the other RecQ helicases depending on the cell cycle, as well as the functional contexts. Acetyltransferases CBP and p300 increased the stability of WRN in the presence of mitomycin C, and WRN stability and degradation was respectively associated with acetylation and ubiquitination of six lysine residues of the protein [15]. We observed ubiquitination of WRN after CPT treatment, suggesting decreased WRN stability due to protein degradation by the ubiquitin-proteasome pathway. However, the ubiquitin ligases that mediate CPT-dependent ubiquitination of WRN are yet to be identified. Interestingly, CPT-induced WRN degradation was confined to CPT-sensitive cancer cells suggesting a role for WRN in rendering resistance against CPT. In addition to enhancing the enzymatic activity of Top1 [17], WRN’s nuclease and helicase activities might play critical roles in removing CPT-Top1-DNA cytotoxic lesions to reduce cytotoxic effects of the drug. Therefore it is possible that the WRN contributes to resistance to CPT by enhancing DNA repair capabilities at various levels.
Senescence is enhanced when RecQ helicases are deficient [21], or if there is persistent DNA damage [38] and after treatment with anticancer drugs like CPT [39]. In our recent report we demonstrated that knockdown of RecQ helicases (with the exception of RECQL1) resulted in the accumulation of DNA-SCARS (DNA segments with chromatin alternations reinforcing senescence), increased p21 and p16 along with increased SA-β-gal activity [21]. WRN knockdown and CPT treatment both induce DNA damage and cause increased p21 expression and SA-β-gal activity [39]. Here, we demonstrate that WRN degradation and increased senescence after CPT treatment can be suppressed by ectopically expressing WRN. Rescue of WRN in CPT-treated cells might increase the efficiency of DDR to remove cytotoxic DNA lesions.
Top1 gene amplification has been reported in about 30% of breast cancers [40] and a tight correlation between mRNA and protein expression was observed in the NCI-60 panel of cell lines [27]. Irinotecan, a semisynthetic derivative of CPT is routinely used in colorectal cancer therapy [41]. Although phase II trials have demonstrated a response rate of about 5% to 23%, irinitotecan is not routinely used as monotherapy in breast cancer [26]. However, etirinotecan, a long acting derivative of irinotecan has shown a response rate of 29% and is currently being evaluated in a large phase III clinical trial in metastatic breast cancer [25]. Given the potential promise of Top1 inhibitors in breast cancer, the development of predictive biomarkers to personalize therapy is highly desirable. In the current study, we suggest that CPT-induced WRN degradation in breast cancer cells could be a biomarker for CPT sensitivity. Taken together, the pre-clinical data show that WRN and/or Top1 expression could have prognostic and/or predictive significance in breast cancers. To explore this hypothesis, we conducted the first large study of WRN and Top1 expression in human breast cancers. We observed that Top1 mRNA overexpression was associated with aggressive highly proliferative breast cancers. In contrast, for WRN, low mRNA expression was associated with adverse clinicopathological features and was linked to poor breast cancer specific survival. Taken together, these novel observations suggest that low WRN expression in human tumors may promote a ‘mutator phenotype’ leading to aggressive breast cancers. Although the mechanism of regulation of WRN mRNA expression is not understood, Agrelo et al. have previously shown that epigenetic inactivation of WRN is frequent in solid tumors with the highest prevalence in colorectal tumors [37.9% (69/182 tumors)] [6]. In a small cohort of breast tumors (n = 58), 17.2% (10/58) showed WRN inactivation although the authors did not describe any clinicopathological associations in that study [6]. In our study, we have found that WRN mRNA expression level was low in 326/1977 of breast tumors (16.5%) which concur strikingly with the study by Agrelo et al. In colorectal cancers, WRN hypermethylation and depletion is associated with good response to irinotecan therapy [6]. We speculate that low WRN expression could result in a genomic instability phenotype with an aggressive behavior. To support this hypothesis, we observed that low WRN was associated with lymph node positivity, grade 3 and HER-2 overexpression. As the clinical study presented here provides prognostic information, future studies, particularly in patients who receive neoadjuvant chemotherapy (including Top1 inhibitor) could provide predictive information.
When WRN and Top1 were combined, surprisingly we found that tumors with high Top1/high WRN expression have poor survival, particularly in the ER positive sub-group. Top1 has essential roles during replication and proliferation. Highly proliferative ER positive breast tumors (PAM50. Lum B phenotype) manifest endocrine resistance. As high Top1 expression is significantly associated with PAM50. Lum B breast tumors (Table 1), we speculate that endocrine resistance may be contributing to the poor survival seen in patients. The data for WRN is intriguing. Whether WRN-mediated DNA repair in ER positive tumors would influence therapy outcome remains to be established. Interestingly, high expression of the multifunctional WRN helicase is observed in several cancer cell lines (Supplementary Figure S5 and [5]). As WRN participates in various DNA repair pathways, it is possible that it enhances the DNA repair capabilities of established tumor cells to withstand DNA damage induced by endogenous and exogenous agents.
In conclusion, we provide the first pre-clinical evidence that WRN degradation is a biomarker of CPT sensitivity in breast cancer cells, where it can distinguish CPT- sensitive and resistant breast cancer cells. Therefore, WRN expression and degradation could be used to identify tumors which may be sensitive to CPT and its derivatives. However, a prospective clinical trial of Top1 inhibitor therapy in ER positive breast cancers would be required to confirm our in vitro results. Additionally we show that in human breast cancers, Top1 and/or WRN expression has prognostic and predictive significance. Top1/ WRN expression based stratification could be a promising approach to personalize Top1 inhibitor therapy in breast cancer patients.
MATERIALS AND METHODS
Cell culture
U2OS, 293T, GM637, BT-474, BT-549, MCF-7, MDA-MB-231, T47D, ZR-75-1, HCT116, HCT116 p53-/- and WI-38 were cultured as described [14, 21, 28]. For knockdown experiments, cells were infected with lentivirus carrying either control or WRN or RECQL4 small hairpin RNA (shRNA) as described before [21] for at least 72 hours. For overexpression, transfections were performed with 3 x 105 cells using 1 μg of pCMV-tag4 plasmids with 3xFlag or 3xFlag-WRN or 3xFlag-RECQL4, or pEGFP, pEGFP-WRN, or pcDNA3.1-HA-ubiquitin using jetPRIME (Polyplus transfection) [21]. CPT (Sigma) was dissolved in DMSO, and 10 mM aliquots were stored at -20 0C, and cells were treated at 10 μM unless otherwise specified. For proteasome inhibition, cells were treated with 10 μM MG132 (EMD Millipore) and for inhibiting protein synthesis, cells were pretreated with 10 μg/ml cycloheximide (CHX; Sigma). For detecting lysosomes, cells were infected with CellLight Lysosome-GFP baculovirus (Life technologies). For IR treatments, cells were grown in 6 cm plates and irradiated with 1 or 10 gy of 137Cs gamma rays.
Immunoblotting
Cells were lysed in either IP lysis buffer [14] or RIPA buffer (Thermo Scientific) and briefly sonicated. The Subcellular Protein Fractionation kit for Cultured Cells (Thermo Scientific) was used to isolate proteins from specific cellular compartments i.e. cytoplasm, cytoplasmic organelles, nuclear soluble and chromatin. Proteins were detected by Western blotting [14] using in house (anti-WRN, -RECQL4, -RECQL5), Abcam (anti-CtIP, -XLF), Millipore (histone 3), BD Pharmingen (anti-53BP1, -calretuculin, -Top1) and Santa Cruz (anti-RECQL1, -Ku80, -actin, -tubulin) antibodies and quantitated by using ImageJ 1.46r software (National Institutes of Health, USA).
Quantitative real-time polymerase chain reaction (qRT-PCR)
Total cellular RNA was isolated using RNeasy RNA isolation kit (Qiagen) from 500,000 cells treated with or without CPT. Complementary DNA (cDNA) synthesis and qRT-PCR were performed in a single reaction tube with 15 ng of RNA using gene-specific primers and SuperScript III Platinum SYBR Green One-Step qRT-PCR kit (Life Technologies). Reverse transcription and qRT-PCR were performed according to manufacturer’s protocol in a 7900 Fast Real-Time PCR system (Applied Biosystems). Hs00262956_m1 (RECQL1), Hs00172060-m1 (BLM), Hs01087915_m1 (WRN), Hs01548660_g1 (RECQL4), Hs00188633_m1 (RECQL5) and Hs02758991_g1 (GAPDH) primer sets (Applied Biosystems) were used for quantitating mRNA.
Immunostaining and microscopy
Cells grown in chambered glass slides (Labtek) for 24 hours in the presence or absence of bacullovirus expressing lysosome specific GFP were treated with CPT. Post treatment, cells were fixed and immunostained for WRN and 53BP1 as described before [21] and imaged with a Zeiss LSM510 (40X objective) microscope and Volocity 3D image analysis software (Perkinelmer). Time-lapse microscopy was performed with U2OS cells stably expressing YFP-WRN using the above mentioned microscope settings supported with temperature and CO2-regulated incubation chamber.
Cell survival and proliferation assays
Cells (5000/well) were seeded in triplicates in a 96-well plate and treated with CPT for 24h. Cell viability and proliferation were assayed according to the manufacturer’s protocol using WST-1 reagent (Roche). Cell viability was calculated from three independent experiments and normalized to no treatment controls.
SA-β-gal assay
Three biological repeats of SA-β-gal staining was performed with control, WRN and RECQL4 expressing cells treated with CPT for 18 hours as previously described [21].
WRN and Top1 mRNA expression in patient tumors
The METABRIC study protocol, molecular profiling, Tumor Marker Prognostic Studies (REMARK) criteria were followed [29, 42] to study WRN and Top1 mRNA expression. Gene copy number was assayed on the Affymetrix SNP 6.0 platform (data available through the European Genotype Archive, http://www.ebi.ac.uk/ega/page.php under accession Number: EGAS00000000082). The mRNA expression was analyzed as before [29, 43] by using WRN (ILMN_1679881) and Top1 (ILMN_2192316) specific probes.
Breast cancer specific survival (BCSS) was defined as the number of months from diagnosis to the occurrence of BC related-death and the data was analyzed using SPSS (SPSS, version 17 Chicago, IL) as described before [43]. Cumulative survival probabilities were estimated using the Kaplan-Meier method, and differences between survival rates were tested for significance using the log-rank test and multivariate analysis for survival was performed using the Cox proportional hazard model as described before [43].
ACKNOWLEDGMENTS
We thank Drs. Sanjay K Bharti and Beverly Baptiste for critical reading of this manuscript.
Financial support
Intramural Program of the National Institute on Aging and the National Cancer Institute, National Institutes of Health, USA.
ConflictS of Interest
There is no conflict of interest.
REFERENCES
1. Bohr VA. Rising from the RecQ-age: the role of human RecQ helicases in genome maintenance. Trends in biochemical sciences. 2008; 33:609-620.
2. Croteau DL, Popuri V, Opresko PL and Bohr VA. Human RecQ helicases in DNA repair, recombination, and replication. Annual review of biochemistry. 2014; 83:519-552.
3. Lauper JM, Krause A, Vaughan TL and Monnat RJ, Jr. Spectrum and risk of neoplasia in Werner syndrome: a systematic review. PloS one. 2013; 8:e59709.
4. Sugimoto M, Furuichi Y, Ide T and Goto M. Involvement of WRN helicase in immortalization and tumorigenesis by the telomeric crisis pathway (Review). Oncology letters. 2011; 2:609-611.
5. Opresko PL, Calvo JP and von Kobbe C. Role for the Werner syndrome protein in the promotion of tumor cell growth. Mechanisms of ageing and development. 2007; 128:423-436.
6. Agrelo R, Cheng WH, Setien F, Ropero S, Espada J, Fraga MF, Herranz M, Paz MF, Sanchez-Cespedes M, Artiga MJ, Guerrero D, Castells A, von Kobbe C, Bohr VA and Esteller M. Epigenetic inactivation of the premature aging Werner syndrome gene in human cancer. Proceedings of the National Academy of Sciences of the United States of America. 2006; 103:8822-8827.
7. Hsiang YH, Hertzberg R, Hecht S and Liu LF. Camptothecin induces protein-linked DNA breaks via mammalian DNA topoisomerase I. The Journal of biological chemistry. 1985; 260:14873-14878.
8. Tomicic MT and Kaina B. Topoisomerase degradation, DSB repair, p53 and IAPs in cancer cell resistance to camptothecin-like topoisomerase I inhibitors. Biochimica et biophysica acta. 2013; 1835:11-27.
9. Sharma S and Brosh RM, Jr. Human RECQ1 is a DNA damage responsive protein required for genotoxic stress resistance and suppression of sister chromatid exchanges. PloS one. 2007; 2:e1297.
10. Mao FJ, Sidorova JM, Lauper JM, Emond MJ and Monnat RJ. The human WRN and BLM RecQ helicases differentially regulate cell proliferation and survival after chemotherapeutic DNA damage. Cancer research. 2010; 70:6548-6555.
11. Imamura O, Fujita K, Itoh C, Takeda S, Furuichi Y and Matsumoto T. Werner and Bloom helicases are involved in DNA repair in a complementary fashion. Oncogene. 2002; 21:954-963.
12. Jin W, Liu H, Zhang Y, Otta SK, Plon SE and Wang LL. Sensitivity of RECQL4-deficient fibroblasts from Rothmund-Thomson syndrome patients to genotoxic agents. Human genetics. 2008; 123:643-653.
13. Hu Y, Lu X, Zhou G, Barnes EL and Luo G. Recql5 plays an important role in DNA replication and cell survival after camptothecin treatment. Molecular biology of the cell. 2009; 20:114-123.
14. Shamanna RA, Singh DK, Lu H, Mirey G, Keijzers G, Salles B, Croteau DL and Bohr VA. RECQ helicase RECQL4 participates in non-homologous end joining and interacts with the Ku complex. Carcinogenesis. 2014; 35:2415-2424.
15. Li K, Wang R, Lozada E, Fan W, Orren DK and Luo J. Acetylation of WRN protein regulates its stability by inhibiting ubiquitination. PloS one. 2010; 5:e10341.
16. Marciniak RA, Lombard DB, Johnson FB and Guarente L. Nucleolar localization of the Werner syndrome protein in human cells. Proceedings of the National Academy of Sciences of the United States of America. 1998; 95:6887-6892.
17. Laine JP, Opresko PL, Indig FE, Harrigan JA, von Kobbe C and Bohr VA. Werner protein stimulates topoisomerase I DNA relaxation activity. Cancer research. 2003; 63:7136-7146.
18. Desai SD, Liu LF, Vazquez-Abad D and D’Arpa P. Ubiquitin-dependent destruction of topoisomerase I is stimulated by the antitumor drug camptothecin. The Journal of biological chemistry. 1997; 272:24159-24164.
19. Lin CP, Ban Y, Lyu YL, Desai SD and Liu LF. A ubiquitin-proteasome pathway for the repair of topoisomerase I-DNA covalent complexes. The Journal of biological chemistry. 2008; 283:21074-21083.
20. Chen JH, Hales CN and Ozanne SE. DNA damage, cellular senescence and organismal ageing: causal or correlative? Nucleic acids research. 2007; 35:7417-7428.
21. Lu H, Fang EF, Sykora P, Kulikowicz T, Zhang Y, Becker KG, Croteau DL and Bohr VA. Senescence induced by RECQL4 dysfunction contributes to Rothmund-Thomson syndrome features in mice. Cell death & disease. 2014; 5:e1226.
22. Grandori C, Wu KJ, Fernandez P, Ngouenet C, Grim J, Clurman BE, Moser MJ, Oshima J, Russell DW, Swisshelm K, Frank S, Amati B, Dalla-Favera R and Monnat RJ, Jr. Werner syndrome protein limits MYC-induced cellular senescence. Genes & development. 2003; 17:1569-1574.
23. Ding SL, Yu JC, Chen ST, Hsu GC and Shen CY. Genetic variation in the premature aging gene WRN: a case-control study on breast cancer susceptibility. Cancer epidemiology, biomarkers & prevention. 2007; 16:263-269.
24. Wang Z, Xu Y, Tang J, Ma H, Qin J, Lu C, Wang X, Hu Z, Wang X and Shen H. A polymorphism in Werner syndrome gene is associated with breast cancer susceptibility in Chinese women. Breast cancer research and treatment. 2009; 118:169-175.
25. Awada A, Garcia AA, Chan S, Jerusalem GH, Coleman RE, Huizing MT, Mehdi A, O’Reilly SM, Hamm JT, Barrett-Lee PJ, Cocquyt V, Sideras K, Young DE, Zhao C, Chia YL, Hoch U, et al. Two schedules of etirinotecan pegol (NKTR-102) in patients with previously treated metastatic breast cancer: a randomised phase 2 study. Lancet Oncol. 2013; 14:1216-1225.
26. Kumler I, Brunner N, Stenvang J, Balslev E and Nielsen DL. A systematic review on topoisomerase 1 inhibition in the treatment of metastatic breast cancer. Breast cancer research and treatment. 2013; 138:347-358.
27. Pfister TD, Reinhold WC, Agama K, Gupta S, Khin SA, Kinders RJ, Parchment RE, Tomaszewski JE, Doroshow JH and Pommier Y. Topoisomerase I levels in the NCI-60 cancer cell line panel determined by validated ELISA and microarray analysis and correlation with indenoisoquinoline sensitivity. Molecular cancer therapeutics. 2009; 8:1878-1884.
28. Desai SD, Li TK, Rodriguez-Bauman A, Rubin EH and Liu LF. Ubiquitin/26S proteasome-mediated degradation of topoisomerase I as a resistance mechanism to camptothecin in tumor cells. Cancer research. 2001; 61:5926-5932.
29. Curtis C, Shah SP, Chin SF, Turashvili G, Rueda OM, Dunning MJ, Speed D, Lynch AG, Samarajiwa S, Yuan Y, Graf S, Ha G, Haffari G, Bashashati A, Russell R, McKinney S, et al. The genomic and transcriptomic architecture of 2,000 breast tumours reveals novel subgroups. Nature. 2012; 486:346-352.
30. Chun SG and Yee NS. Werner syndrome as a hereditary risk factor for exocrine pancreatic cancer: potential role of WRN in pancreatic tumorigenesis and patient-tailored therapy. Cancer biology & therapy. 2010; 10:430-437.
31. Chun SG, Shaeffer DS and Bryant-Greenwood PK. The Werner’s Syndrome RecQ helicase/exonuclease at the nexus of cancer and aging. Hawaii medical journal. 2011; 70:52-55.
32. Aggarwal M, Sommers JA, Shoemaker RH and Brosh RM, Jr. Inhibition of helicase activity by a small molecule impairs Werner syndrome helicase (WRN) function in the cellular response to DNA damage or replication stress. Proceedings of the National Academy of Sciences of the United States of America. 2011; 108:1525-1530.
33. Kehrer DF, Soepenberg O, Loos WJ, Verweij J and Sparreboom A. Modulation of camptothecin analogs in the treatment of cancer: a review. Anticancer Drugs. 2001; 12:89-105.
34. Zhang H, Wang JC and Liu LF. Involvement of DNA topoisomerase I in transcription of human ribosomal RNA genes. Proceedings of the National Academy of Sciences of the United States of America. 1988; 85:1060-1064.
35. Mao Y, Sun M, Desai SD and Liu LF. SUMO-1 conjugation to topoisomerase I: A possible repair response to topoisomerase-mediated DNA damage. Proceedings of the National Academy of Sciences of the United States of America. 2000; 97:4046-4051.
36. Beidler DR and Cheng YC. Camptothecin induction of a time- and concentration-dependent decrease of topoisomerase I and its implication in camptothecin activity. Molecular pharmacology. 1995; 47:907-914.
37. Wang J, Chen J and Gong Z. TopBP1 controls BLM protein level to maintain genome stability. Molecular cell. 2013; 52:667-678.
38. Rodier F, Coppe JP, Patil CK, Hoeijmakers WA, Munoz DP, Raza SR, Freund A, Campeau E, Davalos AR and Campisi J. Persistent DNA damage signalling triggers senescence-associated inflammatory cytokine secretion. Nature cell biology. 2009; 11:973-979.
39. Han Z, Wei W, Dunaway S, Darnowski JW, Calabresi P, Sedivy J, Hendrickson EA, Balan KV, Pantazis P and Wyche JH. Role of p21 in apoptosis and senescence of human colon cancer cells treated with camptothecin. The Journal of biological chemistry. 2002; 277:17154-17160.
40. Kumler I, Balslev E, Poulsen TS, Nielsen SL, Nygard SB, Romer MU, Christensen IJ, Hogdall E, Moreira J, Nielsen DL, Brunner N and Stenvang J. Topoisomerase-1 gene copy aberrations are frequent in patients with breast cancer. Int J Cancer. 2015.
41. Gustavsson B, Carlsson G, Machover D, Petrelli N, Roth A, Schmoll HJ, Tveit KM and Gibson F. A review of the evolution of systemic chemotherapy in the management of colorectal cancer. Clin Colorectal Cancer. 2015; 14:1-10.
42. McShane LM, Altman DG, Sauerbrei W, Taube SE, Gion M, Clark GM and Statistics Subcommittee of the NCIEWGoCD. Reporting recommendations for tumor marker prognostic studies (REMARK). J Natl Cancer Inst. 2005; 97:1180-1184.
43. Arora A, Abdel-Fatah TM, Agarwal D, Doherty R, Moseley PM, Aleskandarany MA, Green AR, Ball G, Alshareeda AT, Rakha EA, Chan SY, Ellis IO and Madhusudan S. Transcriptomic and Protein Expression Analysis Reveals Clinicopathological Significance of Bloom Syndrome Helicase (BLM) in Breast Cancer. Molecular cancer therapeutics. 2015; 14:1057-1065.