
INTRODUCTION
The gene coding for cellular tumor protein p53, TP53, is the most frequently mutated gene in human cancers, being altered in approximately 50% of malignancies (for a review, see [1]). p53 is a transcriptional factor able to interact both with DNA, regulating the expression of a myriad of target genes, and with numerous proteins, mutually modifying their activity. p53 is usually found at very low levels in the cells, but it accumulates in case of genetic damage. p53 promotes multiple cell functions associated to tumor suppression, as cell cycle arrest, apoptosis and cellular senescence, being fundamental in the prevention of the division of cells that have suffered DNA damage; for all these functions, p53 has been nicknamed as “guardian of the genome” [2]. p53 also precludes tumor formation by regulating additional mechanisms, as genetically modified mice expressing a mutant form of p53 unable to direct cell cycle arrest, apoptosis and senescence do not suffer from early onset tumor formation [3]. Among these additional mechanisms of cancer prevention mediated by p53 are regulation of DNA stability, cellular metabolism, autophagy, stem cell maintenance and metastasis (reviewed in [4]). The cancer preventive activity of p53 could also be mediated by other pathways; for example, it has been shown that rapamycin, a known inhibitor of mTOR pathway, decreases the number of tumors originated in p53-/- or p53+/- mice [5, 6], suggesting a causative role of mTOR inhibition in tumor prevention. As p53 induces the expression of several negative regulators of mTOR pathway [7], part of the antitumoral effect of p53 could be mediated by mTOR downregulation.
Despite the intensive studies performed in the last 35 years, we are far from having a precise knowledge of p53 functions. The complete comprehension of p53 roles is hindered by the multiplicity and complexity of the biological processes it regulates. So, besides its role in cancer, p53 is implicated in clearance of apoptotic cells and prevention of autoimmune diseases [8], as well as in embryonic development, resulting both its inactivation and its hyperactivation in developmental defects [9, 10]. Another source of complexity comes from the fact that some functions of wild type (wt) or mutant p53 are not equally regulated in different tissues or cell types [11]. In addition, there are more than ten isoforms transcribed from the TP53 gene, with important differences in subcellular location and biological activity among them, and scarcely known cross-regulation relationships. Illustrative of this complexity is the recent discovery of a p53 isoform with a surprising role in metastasis promotion [12]. So new studies and experimental models are required for deepening in the understanding of p53 functions in cell physiology and tumor transformation.
Mutation of p53 can result in cancer by the effects associated to loss of its normal functions or by the acquisition of new transforming capabilities, acting as a dominant-negative mutant of the wild type p53 or as a bona fide oncogene with transformation potential [13]. The presence and the type of p53 mutations in tumors are clinically important, as mutated versions of p53 can greatly modify the response to therapies [14].
Many animal models have been developed for the study of p53. Knockout mice lacking p53 die at early age and develop an array of spontaneous tumors, mainly lymphomas and sarcomas, with different frequency depending on the genetic background [15, 16]. More recently, after the generation of p53 floxed (fl) mice [17], a plethora of studies have been performed by using animal models with p53 inactivation restricted to specific cell types. Transgenic (Tg) mouse models bearing different mutant forms of p53, alone or in association with mutations in other genes, have also rendered relevant information about the role of p53 in tumor transformation in different types of cancer [18].
Skin cancer is the most abundant human oncogenic lesion, and TP53 is often mutated in patients with skin cancer [19]. Different spectra of TP53 mutations have been found in different skin tumor types (i.e., basal cell carcinoma (BCC), squamous cell carcinoma (SCC), and melanoma) [20].
Data in the literature indicate that p53 cooperates with a plethora of oncogenic insults, such as Ras activation [21], Rb inactivation [22], loss of αv integrin [23] or Snail overexpression [24], in the development of skin carcinomas. Indeed, a number of experiments performed in different experimental models indicate a tumor suppressor role of p53 in skin cancer. So p53 null mice are prone to the development of skin SCCs after UV light radiation [25], and a requirement for p53 loss for malignant conversion of skin tumors has been described, even in the context of various other genetic oncogenic insults [26]. In addition, mice lacking p53 in epidermis develop spontaneous skin tumors [22, 23, 27]; interestingly, skin and tumors lacking p53 showed higher chromosomal instability than those with p53wt background [28, 29]. In a different cancer model (i.e., mouse keratinocytes grafted onto nude mice), aimed to study the cooperation of p53 loss and v-rasHa-mediated initiation, the tumor originated showed increased growth and malignancy in the absence of one or two p53 alleles [30]. Altogether, these results seem to demonstrate a protective role of p53 in skin carcinogenesis and malignization.
But there are also reports indicating an apparently discrepant protumoral role of p53 in skin carcinogenesis. So two-stage skin carcinogenesis experiments show that p53 null mice develop fewer and smaller skin tumors than p53 wild type mice [31, 32], indicating that the presence of p53 in skin cells provokes the emergence of more tumors, at difference with the results found in epithelial cells of the intestine [33] and other cell types. Other experimental settings have also led to the notion of an oncogenic role of p53 in skin cancer; so p53 loss precludes tumor formation in transgenic mice overexpressing activated oncogenes or growth factors, as v-rasHa, v-fos or human TGFα in epidermis [34]. In short, there exist some discrepancies about the specific role of p53 in skin cancer, which could be related to inaccurate estimation of p53 inactivation in the experiments involving cre-mediated tissue-specific p53 knock-out, to differences among the experiments regarding the genetic background of the animal models or to differences in the p53 status in dermal cells and the possible existence of cell non-autonomous effects of p53.
Given the generally accepted protective role of p53 in carcinogenesis, our hypothesis is that p53 absence in keratinocytes will cause both spontaneous and chemically induced carcinogenesis. In this work, we have generated mice lacking p53 specifically in keratinocytes (p53EKO mice) and have assessed the efficiency of this deletion. Then, we tried to elucidate the role of epidermal p53 in skin cancer by studying both spontaneous and chemically induced two-stage skin carcinogenesis in mice of homogeneous genetic background. We conclude that p53 in epidermal keratinocytes truly protects against tumor promotion, progression and malignancy in skin, both in chemically-induced and in spontaneous carcinogenesis, thus acting as a bona fide tumor suppressor gene, in accordance to the generally admitted role of p53 in other cell types.
RESULTS AND DISCUSSION
Efficient p53 inactivation in the skin of p53fl/fl; K14-Cre mice
All the cells in the epidermis derive from basal epidermal cells expressing keratin K14. So it is expected that the expression of Cre recombinase from a K14-Cre transgene will lead to the recombination of floxed alleles in the cells of stratified epithelia, including epidermis, although many of these cells actually stop expressing K14 as they proceed through the differentiation process. We generated cohorts of p53fl/fl/K14-Cre mice to study the importance of p53 in skin cancer. We did not observe significant deviations between the frequency obtained for each genotype and the expected ratios (not shown). We assessed the efficiency of Trp53 recombination in epidermis of p53fl/fl/K14-Cre mice by PCR analysis using primer combinations able to distinguish between the floxed and the deleted forms of the Trp53 gene (Figure 1A and 1B). Simultaneous PCR analysis for the Cre transgene and for the floxed form of Trp53 in DNA from epidermal and dermal tail skin showed the band characteristic of the fl allele (584 bp) in both dermis and epidermis in mice lacking the K14-Cre transgene (Figure 1B, upper panel); this band is also clearly detectable in dermal DNA, but not in DNA from epidermis of p53fl/fl/K14-Cre mice (Figure 1B, upper panel). Correspondingly, PCR analysis with primers specific for the deleted allele revealed a prevalent 612 bp band in the epidermis of p53fl/fl/K14-Cre mice. This band was also detected, although with less intensity, in the dermis of these mice, probably due to the presence of hair follicles expressing the K14-Cre transgene in the dermis (Figure 1B, lower panel). As expected, the 612 bp deletion-specific band was not detected in mice lacking the K4-Cre transgene (Figure 1B, lower panel).
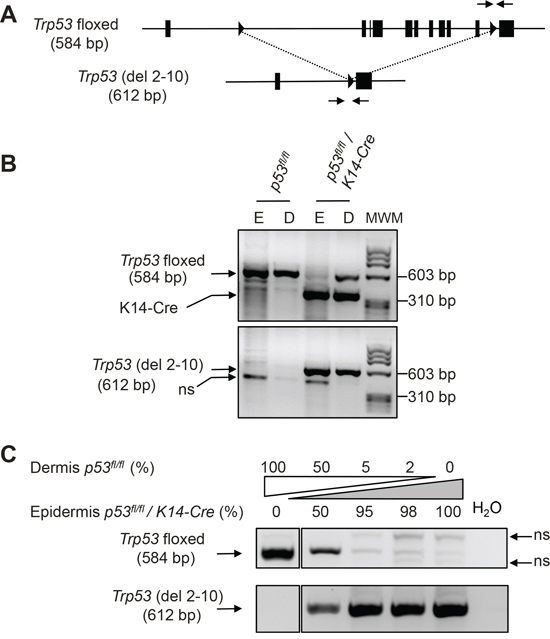
Figure 1: Cre-mediated recombination of floxed Trp53 alleles affects the majority of epidermal cells. A. Schematic structure of floxed Trp53 locus and of the deleted locus lacking exons 2 to 10 (del 2-10) once Cre-mediated recombination has taken place. Triangles represent loxP sites, and the arrows represent the primers used for allele-specific detection. Numbers in parenthesis indicate the length of the amplified fragment in base pairs (bp). B. Representative example of allele-specific analysis of p53 in DNA samples obtained from epidermis (E) and dermis (D) of a transgenic mouse with floxed p53 alleles (lanes marked p53fl/fl) and from double transgenic mice carrying also the K14-Cre transgene. Primers specific of K14-Cre transgene were also included in the amplification reaction of the upper panel. MWM: DNA molecular weight marker. C. Competition PCR of the indicated mixes of DNA form dermis of a non-recombined p53fl/fl mouse and from the epidermis of double p53fl/fl/K14-Cre transgenic mice (that presumably have recombined completely the floxed alleles). ns: non-specific bands.
Competition PCRs using mixed DNAs from epidermis of a p53fl/fl/K14-Cre mouse (presumably containing mainly recombined Trp53 alleles) and dermis of a p53fl/fl mouse (containing only floxed Trp53 alleles) further confirmed that recombination affects to the vast majority of the epidermal cells in mice of the p53fl/fl/K14-Cre genotype, as the addition of only 2% or 5% of dermal non-recombined DNA results in a detectable increase in the band indicative of the floxed allele and a reduction in the intensity of non-specific bands (Figure 1C). We reasoned that if the addition of these small amounts of DNA containing floxed p53 alleles is detectable, the initial amount of floxed alleles in the epidermis of p53fl/fl/K14-Cre mice must be very low and the recombination efficiency in epidermis must be very high.
From these results, we conclude that p53 deletion is efficient, affecting the majority of epidermal cells of p53fl/fl/K14-Cre mice (henceforth, p53EKO mice), and that epidermal p53 deletion does not result in gross deleterious effects over embryonic development or in postnatal life before the age of genotyping and weaning (3 weeks of age).
p53EKO mice are more susceptible to two-stage skin carcinogenesis than p53wt mice
As a first approach for studying the effect of epidermal p53 on cancer, we studied the susceptibility of p53EKO mice to develop skin cancer in a two-stage chemical carcinogenesis protocol; the murine tumors so generated present a mutational landscape highly similar to that found in human SCCs [29]. We performed topical treatment of back skin with the carcinogen DMBA and the hyperplastic agent TPA as indicated in Figure 2A, and registered weekly both the number and size of skin tumors. The experiment finished at week 18 because of the high number and the great size of some of the tumors that arose in p53EKO mice (Figure 2B). Although tumors began to arise on week 6 in both genotypes, and by week 9 all the animals had developed at least one skin tumor, tumors arose faster in p53EKO mice than in p53wt mice, as indicated by the higher percentage of mice bearing tumors in p53EKO compared to p53wt mice in this period of time (Figure 2C). Representative examples of tumor development in p53wt and p53EKO mice at week 14 are shown in Figure 2B. p53wt control mice developed typical exophytic pedunculated cauliflower-like small tumors with gross papillomatous appearance, hard (hyperkeratotic) in consistency, with a dry surface and a poor blood supply (arrowheads in left photograph of Figure 2B). By contrast, tumors developed in p53EKO mice were generally larger, sessile and firmly infiltrated in the dermis (for example, see arrowheads in right photograph of Figure 2B), some of them showing discontinuity of the epidermal barrier, with an ulcerated bleeding surface that did not heal with time; macroscopically, these tumors had a more malignant aspect, resembling skin carcinomas more than papillomas (see arrows in Figure 2B). In addition, although tumors became visible in both genotypes at the same time, p53EKO mice developed more tumors on average than control mice from week 6; these differences were statistically significant between weeks 8 and 11 (Figure 2D). Measurement of tumor size confirmed that lesions from p53EKO mice were larger than those from p53wt mice (Figure 2E). Both the total amount and the percentage of tumors bigger than 2 mm of diameter were greater in p53EKO than in p53wt mice. It is also remarkable the absence of tumors larger than 5 mm of diameter in p53wt mice even at week 14; by contrast, some of the tumors in the population of p53EKO mice reached this size at week 10, and by week 14, 17% of p53EKO tumors (33 out of 196 tumors) had reached this size. In summary, we observed in animals treated with DMBA and TPA that lack of p53 in epidermal cells leads to higher rate of tumor transformation of the skin. These results contrast with the published reduced yield of papillomas in p53 null mice in comparison with p53wt mice [31, 32], and could be explained by differences in the genetic background or perhaps by other differences between the different animal models, such as the different moment of p53 deletion [from the conception in p53 null mice, and around embryonic day 12,5 in our experimental model [35]] or the presence of p53 in dermal cells in p53EKO, that do not occur in p53 null mice. In fact, a non-cell-autonomous effect of dermal p53 over tumor growth in xenografts of human tumoral mammary gland or prostate cell lines has been described, although p53 in stromal cells usually hinders tumor cell growth [36, 37]. In sum, our data indicate that, at difference of the described effect of p53 absence in general knock-out mice [31, 32], p53 in epidermal cells actually protects in the first phases of skin tumor development, as p53EKO mice showed increased rates of tumor appearance and tumor growth than mice bearing non-recombined alleles of p53 in their epidermal cells. So we conclude that p53 is relevant in the initiation process of skin carcinogenesis, at least in the context of chemical insults that activates Ras signaling.
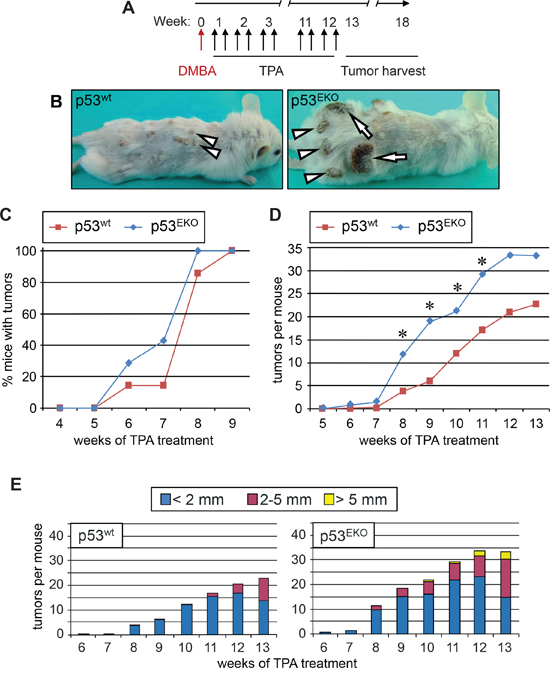
Figure 2: Absence of p53 in epidermis predispose to tumor development in a two-stage chemical carcinogenesis protocol. A. Timeline of the skin carcinogenesis experiment. 8 to 9 week-old mice were shaved, and three days later treated topically with the carcinogen DMBA. TPA was administered twice a week for 12 weeks. Tumor sampling extended until week 18 in p53EKO mice. B. Representative example of a p53wt (left) or p53EKO (right) mouse at week 14. Note the small pedunculated skin tumors with papillomatous aspect in the p53wt mouse and the larger skin tumors in p53EKO mice (arrowheads); in addition, some of the tumors in p53EKO mice are sessile and ulcerated, resembling malignant carcinomas (arrows). C. Tumors emerge earlier in p53EKO mice than in p53wt mice when subjected to a DMBA/TPA chemical carcinogenesis protocol. D. Time evolution of mean tumor number by phenotypes. p53EKO mice developed more tumors than p53wt mice. Asterisks indicate p value < 0.05 in the t-Student test. E. Size distribution of skin tumors in p53EKO and p53wt mice. Note the earlier emergence and the higher number of medium (diameter > 2 mm) and large (diameter > 5 mm) tumors in p53EKO mice in comparison to p53wt mice.
Different skin tumor types emerge in p53EKO mice
As indicated, p53EKO tumors were frequently larger than those obtained in p53wt mice, indicating a faster growth. Macroscopically, tumors arising in wild type mice were typically exophytic and papillomatous. By contrast, several p53EKO skin tumors had an external appearance suggestive of a more malignant status: they were sessile, with a tendency to infiltrate deeply in the subcutaneous tissue, frequently showing persistent ulceration of the surface. We assessed histologically 18 and 51 tumors from p53wt and p53EKO mice, respectively, collected at weeks 14-18 of the experiment. Microscopically, tumors from p53wt mice were homogeneous, with high level of differentiation and keratinization of the stratified epithelium covering a branched conjunctive tissue stroma, poorly vascularized, featuring the classical histological appearance of squamous papillomas (Figure 3A). Only two of the papillomas showed early signs of malignancy, in the form of small foci of microcarcinoma, composed by nests of epidermal cells with a lack of differentiation of a basal layer invading the stroma (Figure 3B and Table 1). However, p53EKO mice produced more varied tumor histotypes: we found squamous papillomas (Figure 3C) and papillomas that showed higher basal atypia than p53wt papillomas with multiple foci of microcarcinoma (Figure 3D, arrows). Some tumors found in p53EKO mice were tricoepitheliomas, benign lesions from hair follicles composed of cornified cysts, unable to form normal hair (arrow in Figure 3E); others were classified as basosquamous tumors (Figure 3F), lesions derived from hair follicles resembling basosquamous carcinomas, but benign due to the integrity and continuity of the basal membrane. Finally, we also found ulcerative lesions, that correspond to highly malignant skin tumors: infiltrating poorly-differentiated SCCs (18 out of 51 tumors in p53EKO mice, none in p53wt mice), which showed solid areas of increased cellularity infiltrating from the epidermis to the dermis and subcutaneous tissue, composed by keratinocytes with marked cellular pleomorphism, most of them with fibroblastoid (spindle) cell shape, being common the presence of multinucleated cells, cells with aberrant giant nuclei (e.g., arrow in Figure 3H) and mitotic figures (arrowheads in Figure 3H); these features resemble those found in human keratinocytes with p53 loss by means of shRNAs against p53 [38]. These poorly-differentiated SCCs showed a very homogenous solid pattern with little evidence of squamous differentiation and/or keratin deposition (Figure 3G and 3H). Strikingly, all the p53EKO mice included in this study developed at least one poorly-differentiated SCC, precluding the prolongation of the study for the analysis of the evolution of the other tumor types. The incidence of each tumor type is indicated in Table 1. From the data included in Figure 3 and Table 1, we conclude that p53 absence in epidermal cells leads to a marked acceleration of the malignant transformation process of the tumors arisen after treatment with DMBA and TPA. Furthermore, the more varied tumor types observed in p53EKO mice suggests that p53 acts as a tumor suppressor gene in different types of epidermal cells; so lack of p53 would result in different tumor types depending on the degree of commitment or differentiation of the transformed cell type.
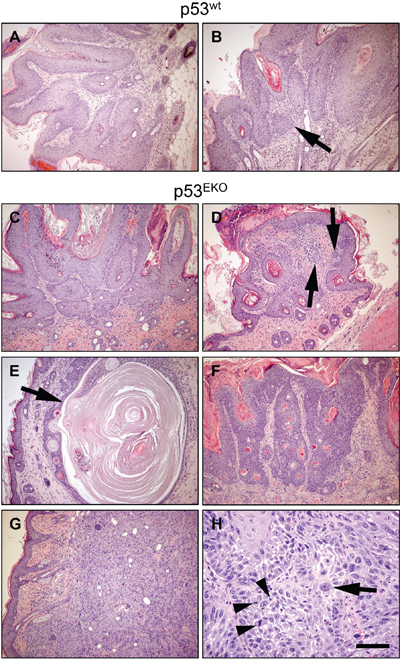
Figure 3: Histological aspect of chemically induced skin tumors. Tumors developed in p53wt mice were typical well-differentiated papillomas A. that sometimes showed foci of microcarcinoma as groups of epidermal cells deepening in the dermis (B., arrow). C-H. Tumors developed in p53EKO mice were histologically more diverse. They ranged from classical papillomas (C) and papillomas with abundant foci of microcarcinoma (D), to tricoepithelioma (E), basosquamous tumors (F) and poorly-differentiated carcinomas (G and H). Scale bars equal 200 microns in A-G and 50 microns in H.
Table 1: Incidence of tumors and tumor histotype obtained in p53wt and p53EKO mice treated with DMBA and TPA
Type of tumor |
p53wt (%) |
p53EKO (%) |
|---|---|---|
Squamous papilloma |
16 (88,9) |
10 (19,6) |
Tricoepithelioma |
0 (0) |
3 (5,9) |
Basosquamous tumor |
0 (0) |
17 (33,3) |
Adenosquamous tumor |
0 (0) |
1 (2,0) |
Papillomas with microcarcinomas |
2 (11,1) |
2 (3,9) |
Poorly-differentiated SCC |
0 (0) |
18 (35,3) |
TOTAL: |
18 |
51 |
Immunohistochemical analysis indicates increased malignancy of p53EKO tumors
In order to define at the molecular level the consequences of p53 absence, we performed an immunohistochemical study of epidermal differentiation markers and of several molecules important in processes related to tumorigenesis, as differentiation, apoptosis or cell cycle regulation (Figures 4 and 5).
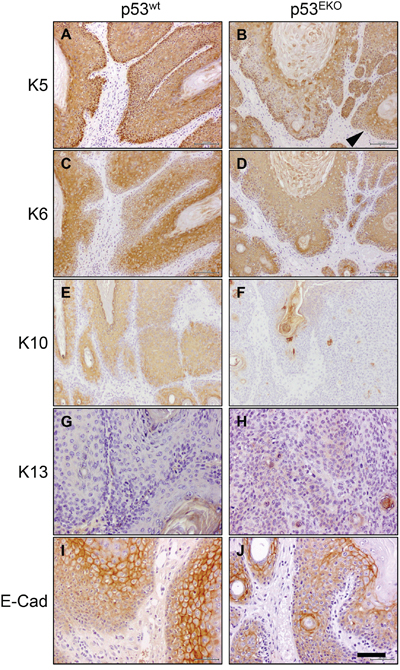
Figure 4: Altered keratin and E-cadherin expression in p53EKO tumors. Immunohistochemical analysis of the expression of keratins K5, K6, K10 and K13 and of adhesion molecule E-cadherin in p53wt and p53EKO papillomas. Left row correspond to p53wt tumors, and right row to p53EKO tumors. Scale bars equal 200 microns in A-F. and 100 microns in G-J.
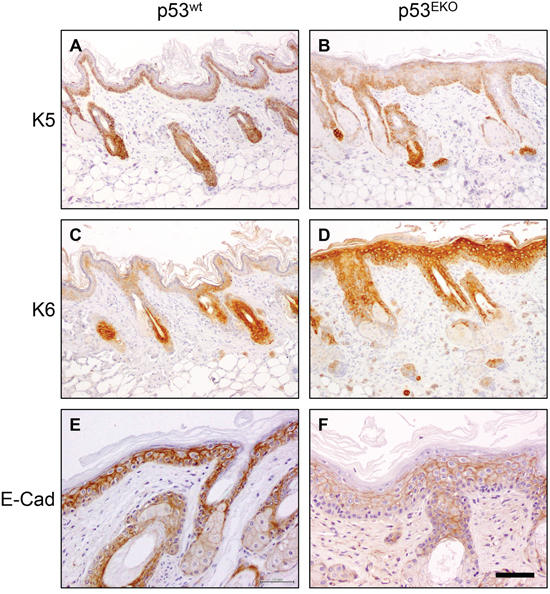
Figure 5: Altered keratin and E-cadherin expression in non-tumoral p53EKO skin. Immunochemical analysis of the expression of keratins K5, K6 and E-Cadherin in hyperplastic p53wt and p53EKO back skin. Scale bars equal 200 microns in A-D. and 100 microns in E and F.
p53wt tumors consistently showed stronger staining than p53EKO tumors when assayed with an antibody specific for keratin K5 (Figure 4A and 4B). Moreover, staining indicative of K5 expression was lost in part of the basal epithelium in p53EKO tumors (arrowhead in Figure 4B). By contrast, K5 staining in basal cells of p53wt tumors was stronger and continuous. When using an antibody specific for the epidermal hyperproliferation marker keratin K6, we observed in p53EKO tumors a tendency to express K6 more widely and in more layers than p53wt tumors (Figure 4C and 4D). The expression of the normal epidermal differentiation marker K10 was higher in p53wt than in p53EKO tumors, indicating a lower differentiation degree in p53EKO tumors (Figure 4E and 4F). Conversely, p53wt tumors did not express keratin K13, a protein associated to epidermal carcinoma progression, but this protein was actually detected, although faintly, in some p53EKO tumors (Figure 4G and 4H). Interestingly, E-cadherin, an adhesion protein downregulated in cancer progression and metastasis, is expressed at higher level in p53wt papillomas than in p53EKO papillomas (Figure 4I and 4J), indicating a stronger tendency to malignancy in papillomas lacking p53 than in papillomas grown in p53wt mice. Taken together, the changes in expression of keratins and E-cadherin in skin tumors indicate molecularly that p53 absence leads to the development of papillomas prone to malignancy, as they express higher amounts of keratins associated to malignant transformation, and less amounts of E-cadherin. We next studied if these alterations in differentiation and adhesion markers were already present in non-tumoral skin. With this aim, we treated p53EKO and p53wt mice twice every other day with TPA, in order to induce a hyperplastic state in the skin that facilitates the detection of differences in these proteins. We could distinguish differences in the expression of K5, K6, and E-cadherin in both genotypes, independent of tumoral transformation: so hyperplastic p53EKO skin showed fainter and irregular K5 staining in the basal layer, more intense and wider staining for K6 and milder E-cadherin staining than hyperplastic p53wt skin (Figures 5A–5F). All these molecular changes found in tumors and in non-tumoral skin are known to be important in the process of malignant transformation and in the development of invasion and metastasis capabilities by epithelial tumor cells [39, 40].
We also analysed the expression of proteins implicated in cell proliferation, DNA damage and angiogenesis in p53wt papillomas and p53EKO SCCs (Figure 6). Akt signaling pathway is important in the regulation of cell survival, cell cycle progression, metabolism, angiogenesis and in epithelial carcinogenesis [41]. Interestingly, Akt seems to be activated in p53EKO tumors, as indicated by the widespread staining observed for activated P-Akt in these tumors (compare Figure 6A and 6B). We also found more intense staining for Stat3, a mediator of proliferation and antiapoptotic signals, in p53EKO tumors than in p53wt tumors (compare Figure 6C and 6D); interestingly, Stat3 is detected in nuclei in p53EKO samples (arrows in Figure 6D), indicating activation of this transcription factor, a feature widely found in numerous malignancies [42, 43]. It is interesting to note that a regulation of Stat3 by p53 has been described, so that wt p53 (but not mutant forms of p53) reduces Stat3 phosphorylation at least in breast, ovarian and prostate cells [44]. Cyclin D1, a regulator of cell cycle entry, is found in most of the nuclei of the basal layer in p53wt tumors (Figure 6E); interestingly, p53EKO tumors showed Cyclin D1 staining also in other cell layers (Figure 6F), indicating higher proliferative potential. Consistent with the role of p53 as a pivotal molecule in the maintaining of genetic stability, p53EKO tumors showed increased staining for γH2AX, a marker of DNA double stranded breaks [45] (Figure 6G and 6H); this result indicate that p53EKO tumors accumulate more DNA damage, a feature associated to malignant transformation. We also analyzed the expression of α-smooth-muscle-actin (Sma), a marker of perivascular cells of the blood vessels and of tumoral angiogenesis; its expression is increased in p53EKO tumors (Figure 6I and 6J), indicating an increased blood supply and consequently growth ability in these tumors. p19ARF is a protein that mediates the accumulation of p53 in cells subjected to oncogenic signals, and its level is increased in cells lacking p53 [46]. When the expression of p19ARF was analyzed, we found consistently an increase in the p19 nuclear staining in p53EKO tumors (Figure 6K and 6L). Finally, tumor cells in p53EKO mice did not show immunoreactivity against an antibody specific for p53 protein (Figure 6M and 6N), indicating that these lesions are actually formed from epidermal cells lacking p53.
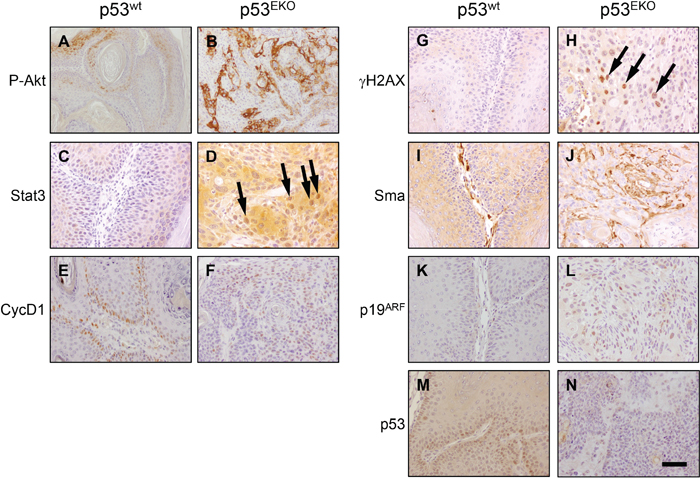
Figure 6: Immunohistochemical analysis of signaling proteins in p53wt and p53EKO tumors. Photographs are representative fields showing the expression of proteins relevant in carcinogenesis: P-Akt, Stat3, CycD1, γH2AX, Sma, p19ARF and p53. Scale bars equal 200 microns in A and B., and 100 microns in C-N.
Western blot analysis of p53wt and p53EKO tumors further confirmed the results obtained in the immunohistochemical analysis (Figure 7). Tumors originated in p53EKO mice showed a marked decrease in the band specific for p53, as expected. Our western blot analysis also showed over activation of Stat3 and Akt pathways and higher levels of CyclinD1 in p53EKO tumors, in concordance with the results shown in Figure 6. The tumor suppressor gene p19ARF is also consistently overexpressed in p53EKO tumors, probably as an attempt of inhibiting tumor growth in epidermal cells lacking p53 by p53-independent mechanisms [32]. Interestingly, these changes in expression are not seen for p16INK4a, indicating the differential roles of these two tumor suppressor proteins in skin cancer. We have not detected differences in mTOR signaling (measured as phosphorylation of the ribosomal protein S6) in tumors or in non-tumoral skin of p53wt and p53EKO genotypes (not shown).
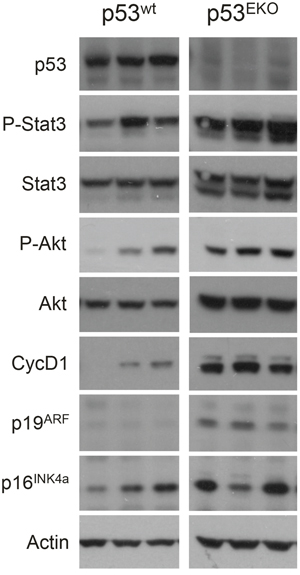
Figure 7: Expression of signaling proteins and cell cycle regulators in p53wt and p53EKO tumors. Western blot analyses of the indicated proteins in three different tumors from p53wt and p53EKO mice are shown.
In summary, the expression analysis performed in tumors from p53wt and p53EKO mice highlight the protective role of p53 in skin cancer, as lack of p53 in epidermal cells results in tumors with activated pro-tumoral signaling pathways and with changes in other molecules that result in more malignant tumors.
Lack of p53 in epidermal cells leads to spontaneous tumor formation
In an attempt to determine the importance of p53 in spontaneous carcinogenesis, independent of the Ras mutations induced by DMBA/TPA treatments, we studied tumor formation in aging mice.
Absence of p53 is able to cause spontaneous tumor formation in different cell types [15], but not in all of them, as enterocytes lacking p53 do not give rise to spontaneous intestinal tumors [33]. We generated a cohort of 21 male- and 16 female-p53EKO mice, and analyzed both tumor emergence and the histological type of the tumors arisen. Mice were maintained in the study until they were 14 months old or had visible tumors larger than 5 mm in diameter. A significant number of the p53EKO mice (11 out of 37) succumbed by unknown pathologies before the end of the experiment; from the rest of the animals, the majority of them (20 out of 26) developed epithelial tumors before the age of 14 months. The histological classification of these tumors is indicated in Table 2, and representative examples are shown in Figure 8. Spontaneous carcinogenesis in wild-type mice is a very rare event in the time period analyzed. We observed 38 different tumors in these 20 p53EKO animals, that arose mainly in skin (32 tumors), but also in other epithelial tissues expressing K14 that presumably suffered also Cre-mediated p53 inactivation, as mammary gland and oral epithelia (3 tumors in each one; Figure 8A and 8B). It is remarkable that, besides some benign tumors (papillomas, keratoacanthomas and basosquamous tumors; e.g., Figure 8C) the majority of the tumors analyzed were malignant, ranging from relatively well differentiated SCCs (Figure 8D) to poorly-differentiated SCCs (Figure 8E) and highly aggressive carcinomas formed by undifferentiated spindle cells (Figure 8F). In summary, mice lacking p53 in stratified epithelia have a shortened lifespan, with a tendency to develop tumoral lesions in epidermis but also in other epithelial tissues, even without the existence of chemically induced Ras mutations; furthermore, these tumors easily become malignant, being relatively frequent the emergence of poorly-differentiated SCCs and spindle SCCs.
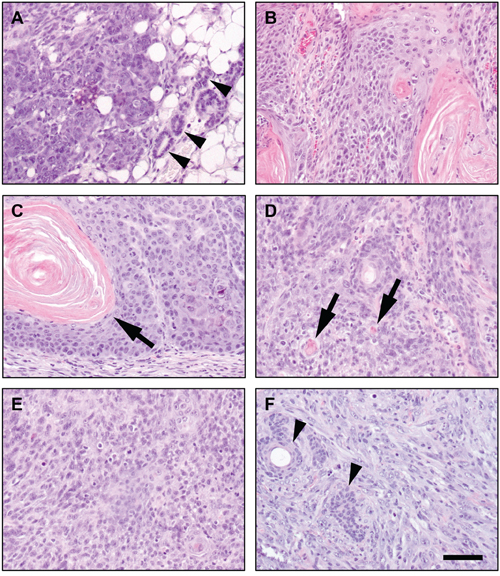
Figure 8: Spontaneous epithelial tumors in mice lacking p53 in stratified epithelial cells. Representative examples of spontaneous tumoral lesions arising in aging p53EKO mice. A. Mammary gland carcinoma. Arrowheads indicate non-tumoral mammary ducts. B. SCC of oral epithelia. C. Benign papilloma in skin. Arrow marks a keratin pearl. Note the well distributed basal cell layer, characteristic of benign skin tumors. D. Well-differentiated SCC of skin. Small squamous differentiation foci around keratin deposits (pearls) are formed in several parts of the tumor (arrows). E. Poorly-differentiated skin SCC, formed by solid masses of keratinocytes showing scattered foci of squamous differentiation. F. Spindle undifferentiated SCC, containing abundant fibroblastoid spindle-shaped cells that have lost the epithelial morphology. Remnants of hair follicles are still visible (arrowheads). Scale bars equal 100 microns.
Table 2: Classification of spontaneous epithelial tumors in p53EKO mice
Tissue |
Type of tumor |
Number of tumors |
|---|---|---|
Oral epithelia |
Carcinoma |
3 |
Mammary gland |
Carcinoma |
3 |
Benign tumor |
9 |
|
Differentiated SCC |
12 |
|
Skin |
Poorly-differentiated SCC |
5 |
Spindle undifferentiated SCC |
6 |
The skin tumors were classified histologically as benign (squamous papillomas, keratoacanthomas and basosquamous tumors), differentiated and poorly-differentiated SCCs, and spindle cell carcinomas, containing highly transformed fusiform cells.
Taken together, the results presented in this report indicates that epidermal loss of p53 function facilitates both papilloma emergence and their growth in two-stage skin chemical carcinogenesis, and also increases the rate of malignant transformation of these tumoral lesions. The increased diversity of epidermal tumor types obtained in p53EKO mice indicates a tumor suppressive role of p53 in a variety of epidermal cell types. p53 absence in keratinocytes also leads to the spontaneous development of highly malignant poorly-differentiated SCCs in skin and other stratified epithelia, without the need of induced mutations in Ras genes that occurs after DMBA treatment. In summary, our results show that p53 absence increases initiation, growth and malignancy of skin tumors, and that its function is truly and totally protective in skin carcinogenesis, confirming a role for p53 as a genuine tumor suppressor gene in skin carcinogenesis.
MATERIALS AND METHODS
Mice and treatments
Animal work was conducted following protocols approved by our institutional Ethical Committee for Animal Experiments and according European, Spanish and local regulations; the experiments included in this publication are under the permit number BME 02/10 of the Ethical Committee for Animal Experiments at CIEMAT. All efforts were made to minimize suffering of the animals employed. Mice were maintained on autoclaved standard rodent chow and water ad libitum and kept under a 12 h light -12 h dark cycle. Floxed Trp53 mice and K14-Cre mice have been described previously [17, 47]. For obtaining mice lacking p53 in epidermis, we crossed p53wt/fl; K14-Cre female mice with p53wt/fl males. In mice inheriting both the K14-Cre transgene and floxed alleles of Trp53, Cre activity would delete exons 2 to 10 of Trp53 in epidermis and other cell types expressing the K14-Cre transgene [47] (Figure 1A). This recombined allele is unable to produce a functional protein, as it lacks part of the transcriptional activation domain, the DNA-binding domain and the tetramerization domain of p53. We selected for the carcinogenesis studies p53fl/fl; K14-Cre mice (referred in this work as p53EKO) and p53wt/wt littermates as control mice (referred in this work as p53wt). All the mice used in the carcinogenesis studies were of the same mixed genetic background (50% hybrid C57BL6JxDBA2J and 50% FVB). Cohorts of seven mice of each genotype were subjected to a cutaneous two-stage chemical carcinogenesis protocol. In this protocol, mice were shaved using a hair clipper and treated three days later with a single topical application of 100 μg of 7,12-dimethylbenz[a]anthracene (DMBA, Sigma-Aldrich, reference D3254) dissolved in 200 μl acetone (week 0). Seven days after DMBA application, 5 μg of 12-O-tetradecanoylphorbol 13-acetate (TPA, Sigma-Aldrich, reference P1585) in 200 μl acetone was applied topically twice a week for 12 weeks. The number and size of tumors per mouse was recorded weekly. p53EKO mice were euthanized when the tumors had a diameter greater than 1 cm or at week 18, due to humanitarian reasons. Four p53wt mice were time-paired sacrificed between weeks 13 and 18, and the rest were sacrificed between weeks 35 and 40, in order to get lesions of enough size to permit the analysis by western blot. Samples were processed for histological and western blot analyses, and the mice genotypes were confirmed by reanalyzing new DNA samples taken postmortem.
For spontaneous carcinogenesis, mice were monitored for tumor development for 14 months. Mice showing tumors or with obvious signs of disease were euthanized for necropsy and histological analysis.
For the study of non-tumoral hyperplastic skin, three p53EKO mice and the same number of p53wt mice were shaved and topically treated twice with 5 μg of TPA or vehicle at days 3 and 5 after shaving.
Genotyping and PCR analysis
DNA was obtained from tail biopsies of 2 week old-mice or from samples taken postmortem. Mice were genotyped by PCR. Primers used for determination of the Trp53 status (i.e.: floxed, deleted or wild type) and for detecting the presence of the K14-Cre transgene are indicated in [47].
Histology and immunohistochemistry
Mouse tumors were dissected and immediately fixed in 10% buffered formalin or 70% ethanol and embedded in paraffin. 5 μm-thick sections were used for H&E staining or immunohistochemical preparations. Most of the tumors were fixed and classified by morphology after sectioning and staining with H&E. The antibodies used in the immunohistochemical analysis were against p53 (NCL-p53-CM5p, Novocastra, Leica Biosystems, NewCastle, UK); keratin K5 (PRB-160P), K10 (PRB-159P) (Covance, San Diego, CA); keratin K13 (ab6112), p19ARF (ab80) (Abcam, Cambridge, UK); smooth muscle actin (C6198, Sigma-Aldrich, St Louis, MO); Cyclin D1(RM-9104-RQ, Thermo Fisher Scientific, MA, USA); Phospho-Akt1(ser 473) (9277), Stat3 (4904) (Cell Signaling Technology, Danvers, MA, USA) and BrdU (11170376001, Roche, Mannheim, Germany). Immunoreactivity was revealed using an ABC avidin-biotin-peroxidase system and ABC substrate (Vector Laboratories), and the sections counterstained slightly with haematoxylin. Control experiments omitting the primary antibody gave no signals.
Western blot
In those tumors that were large enough, we froze part of the tumor in liquid nitrogen at the moment of the sacrifice for western blot analysis. Whole-cell protein extracts were subjected to SDS/PAGE using standard techniques. Protein content was determined by the Bradford colorimetric protein assay (BioRad Laboratories; Hercules, CA; USA). The antibodies used in western blots were against p53 (NCL-p53-CM5p, Novocastra, Leica Biosystems, NewCastle, UK); Stat3 (phospho tyr705; 9131), Stat3 (4904); Akt (phospho ser473; 4058) (Cell Signaling Technology, Danvers, MA, USA); p19ARF (ab80) (Abcam, Cambridge, UK); Akt1/2 (sc-1619), p16ink4a (sc-1207), Cyclin D1(sc-753), and actin (sc-1616) as a loading control (Santa Cruz Biotechnology, Santa Cruz, CA, USA).
Statistical analysis
Data are expressed as mean ± SEM. P values were determined by using the unpaired, two-tailed Student t test. P values < 0.05 were considered significant.
ACKNOWLEDGMENTS
We would like to thank Berta Hernanz and Rebeca Sanz for her excellent technical help, Federico Sanchez-Sierra and Pilar Hernández Lorenzo for their assistance with the histological processing of the samples, and Edilia de Almeida, Jesús Martínez and the personnel of the CIEMAT Animal Unit for their help in the treatments and care of the mice used in this work.
CONFLICTS OF INTEREST
The authors declare that no conflicting financial interests exit.
GRANT SUPPORT
This research was supported by grants from the Spanish government (CIEM13-4E-1944 and PI14/01403 to AR; PI13/02580 to MLC and SAF2012-34378 and SAF-2015-66015-R to JMP from the Ministerio de Economía y Competitividad; RD12/0036/0009 –to JMP- from the Instituto de Salud Carlos III and S2011/BMD2470 –to JMP- from the Comunidad Autónoma de Madrid).
Abbreviations
bp, base pairs; H&E, haematoxylin and eosin; Tg, transgenic; fl, floxed and flanked by loxP sites; SCC, Squamous cell carcinoma; SpCC, Spindle cell carcinoma; wt, wild type.
REFERENCES
1. Brosh R, Rotter V. When mutants gain new powers: news from the mutant p53 field. Nat Rev Cancer. 2009; 9:701-713.
2. Lane DP. Cancer. p53, guardian of the genome. Nature. 1992; 358:15-16.
3. Li T, Kon N, Jiang L, Tan M, Ludwig T, Zhao Y, Baer R, Gu W. Tumor suppression in the absence of p53-mediated cell-cycle arrest, apoptosis, and senescence. Cell. 2012; 149:1269-1283.
4. Bieging KT, Mello SS, Attardi LD. Unravelling mechanisms of p53-mediated tumour suppression. Nat Rev Cancer. 2014; 14:359-370.
5. Komarova EA, Antoch MP, Novototskaya LR, Chernova OB, Paszkiewicz G, Leontieva OV, Blagosklonny MV, Gudkov AV. Rapamycin extends lifespan and delays tumorigenesis in heterozygous p53+/- mice. Aging (Albany NY). 2012; 4:709-714.
6. Comas M, Toshkov I, Kuropatwinski KK, Chernova OB, Polinsky A, Blagosklonny MV, Gudkov AV, Antoch MP. New nanoformulation of rapamycin Rapatar extends lifespan in homozygous p53-/- mice by delaying carcinogenesis. Aging (Albany NY). 2012; 4:715-722.
7. Feng Z, Levine AJ. The regulation of energy metabolism and the IGF-1/mTOR pathways by the p53 protein. Trends in cell biology. 2010; 20:427-434.
8. Yoon KW, Byun S, Kwon E, Hwang SY, Chu K, Hiraki M, Jo SH, Weins A, Hakroush S, Cebulla A, Sykes DB, Greka A, Mundel P, Fisher DE, Mandinova A, Lee SW. CELL DEATH. Control of signaling-mediated clearance of apoptotic cells by the tumor suppressor p53. Science. 2015; 349:1261669.
9. Shin MH, He Y, Huang J. Embryonic stem cells shed new light on the developmental roles of p53. Cell & bioscience. 2013; 3:42.
10. Van Nostrand JL, Brady CA, Jung H, Fuentes DR, Kozak MM, Johnson TM, Lin CY, Lin CJ, Swiderski DL, Vogel H, Bernstein JA, Attie-Bitach T, Chang CP, Wysocka J, Martin DM, Attardi LD. Inappropriate p53 activation during development induces features of CHARGE syndrome. Nature. 2014; 514:228-232.
11. Lee MK, Teoh WW, Phang BH, Tong WM, Wang ZQ, Sabapathy K. Cell-type, dose, and mutation-type specificity dictate mutant p53 functions in vivo. Cancer Cell. 2012; 22:751-764.
12. Senturk S, Yao Z, Camiolo M, Stiles B, Rathod T, Walsh AM, Nemajerova A, Lazzara MJ, Altorki NK, Krainer A, Moll UM, Lowe SW, Cartegni L, Sordella R. p53Psi is a transcriptionally inactive p53 isoform able to reprogram cells toward a metastatic-like state. Proceedings of the National Academy of Sciences of the United States of America. 2014; 111:E3287-3296.
13. Caulin C, Nguyen T, Lang GA, Goepfert TM, Brinkley BR, Cai WW, Lozano G, Roop DR. An inducible mouse model for skin cancer reveals distinct roles for gain- and loss-of-function p53 mutations. J Clin Invest. 2007; 117:1893-1901.
14. Muller PA, Vousden KH. Mutant p53 in cancer: new functions and therapeutic opportunities. Cancer Cell. 2014; 25:304-317.
15. Donehower LA, Harvey M, Slagle BL, McArthur MJ, Montgomery CA, Jr., Butel JS, Bradley A. Mice deficient for p53 are developmentally normal but susceptible to spontaneous tumours. Nature. 1992; 356:215-221.
16. Harvey M, McArthur MJ, Montgomery CA, Jr., Bradley A, Donehower LA. Genetic background alters the spectrum of tumors that develop in p53-deficient mice. FASEB. 1993; 7:938-943.
17. Marino S, Vooijs M, van Der Gulden H, Jonkers J, Berns A. Induction of medulloblastomas in p53-null mutant mice by somatic inactivation of Rb in the external granular layer cells of the cerebellum. Genes Dev. 2000; 14:994-1004.
18. Jackson JG, Lozano G. The mutant p53 mouse as a pre-clinical model. Oncogene. 2013; 32:4325-4330.
19. Benjamin CL, Ananthaswamy HN. p53 and the pathogenesis of skin cancer. Toxicol Appl Pharmacol. 2007; 224:241-248.
20. Giglia-Mari G, Sarasin A. TP53 mutations in human skin cancers. Human mutation. 2003; 21:217-228.
21. Lapouge G, Youssef KK, Vokaer B, Achouri Y, Michaux C, Sotiropoulou PA, Blanpain C. Identifying the cellular origin of squamous skin tumors. Proceedings of the National Academy of Sciences of the United States of America. 2011; 108:7431-7436.
22. Martinez-Cruz AB, Santos M, Lara MF, Segrelles C, Ruiz S, Moral M, Lorz C, Garcia-Escudero R, Paramio JM. Spontaneous squamous cell carcinoma induced by the somatic inactivation of retinoblastoma and Trp53 tumor suppressors. Cancer Res. 2008; 68:683-692.
23. Savar A, Acin S, Gonzalez CL, El-Sawy T, Mejia O, Li Z, Esmaeli B, Lacy-Hulbert A, El-Naggar AK, McCarty JH, Caulin C. Loss of epithelial p53 and alphav integrin cooperate through Akt to induce squamous cell carcinoma yet prevent remodeling of the tumor microenvironment. Oncogene. 2014.
24. De Craene B, Denecker G, Vermassen P, Taminau J, Mauch C, Derore A, Jonkers J, Fuchs E, Berx G. Epidermal Snail expression drives skin cancer initiation and progression through enhanced cytoprotection, epidermal stem/progenitor cell expansion and enhanced metastatic potential. Cell death and differentiation. 2014; 21:310-320.
25. Li G, Tron V, Ho V. Induction of squamous cell carcinoma in p53-deficient mice after ultraviolet irradiation. The Journal of investigative dermatology. 1998; 110:72-75.
26. Macdonald FH, Yao D, Quinn JA, Greenhalgh DA. PTEN ablation in Ras(Ha)/Fos skin carcinogenesis invokes p53-dependent p21 to delay conversion while p53-independent p21 limits progression via cyclin D1/E2 inhibition. Oncogene. 2014; 33:4132-4143.
27. Lyle S, Hoover K, Colpan C, Zhu Z, Matijasevic Z, Jones SN. Dicer Cooperates with p53 to Suppress DNA Damage and Skin Carcinogenesis in Mice. PloS one. 2014; 9:e100920.
28. Martinez-Cruz AB, Santos M, Garcia-Escudero R, Moral M, Segrelles C, Lorz C, Saiz C, Buitrago-Perez A, Costa C, Paramio JM. Spontaneous tumor formation in Trp53-deficient epidermis mediated by chromosomal instability and inflammation. Anticancer Res. 2009; 29:3035-3042.
29. Nassar D, Latil M, Boeckx B, Lambrechts D, Blanpain C. Genomic landscape of carcinogen-induced and genetically induced mouse skin squamous cell carcinoma. Nat Med. 2015; 21:946-954.
30. Weinberg WC, Azzoli CG, Kadiwar N, Yuspa SH. p53 gene dosage modifies growth and malignant progression of keratinocytes expressing the v-rasHa oncogene. Cancer Res. 1994; 54:5584-5592.
31. Kemp CJ, Donehower LA, Bradley A, Balmain A. Reduction of p53 gene dosage does not increase initiation or promotion but enhances malignant progression of chemically induced skin tumors. Cell. 1993; 74:813-822.
32. Kelly-Spratt KS, Gurley KE, Yasui Y, Kemp CJ. p19Arf suppresses growth, progression, and metastasis of Hras-driven carcinomas through p53-dependent and -independent pathways. PLoS biology. 2004; 2:E242.
33. Schwitalla S, Ziegler PK, Horst D, Becker V, Kerle I, Begus-Nahrmann Y, Lechel A, Rudolph KL, Langer R, Slotta-Huspenina J, Bader FG, Prazeres da Costa O, Neurath MF, Meining A, Kirchner T, Greten FR. Loss of p53 in enterocytes generates an inflammatory microenvironment enabling invasion and lymph node metastasis of carcinogen-induced colorectal tumors. Cancer Cell. 2013; 23:93-106.
34. Greenhalgh DA, Wang XJ, Donehower LA, Roop DR. Paradoxical tumor inhibitory effect of p53 loss in transgenic mice expressing epidermal-targeted v-rasHa, v-fos, or human transforming growth factor alpha. Cancer Res. 1996; 56:4413-4423.
35. Ramirez A, Bravo A, Jorcano JL, Vidal M. Sequences 5’ of the bovine keratin 5 gene direct tissue- and cell-type-specific expression of a lacZ gene in the adult and during development. Differentiation. 1994; 58:53-64.
36. Addadi Y, Moskovits N, Granot D, Lozano G, Carmi Y, Apte RN, Neeman M, Oren M. p53 status in stromal fibroblasts modulates tumor growth in an SDF1-dependent manner. Cancer Res. 2010; 70:9650-9658.
37. Kiaris H, Chatzistamou I, Trimis G, Frangou-Plemmenou M, Pafiti-Kondi A, Kalofoutis A. Evidence for nonautonomous effect of p53 tumor suppressor in carcinogenesis. Cancer Res. 2005; 65:1627-1630.
38. Freije A, Molinuevo R, Ceballos L, Cagigas M, Alonso-Lecue P, Rodriguez R, Menendez P, Aberdam D, De Diego E, Gandarillas A. Inactivation of p53 in Human Keratinocytes Leads to Squamous Differentiation and Shedding via Replication Stress and Mitotic Slippage. Cell reports. 2014; 9:1349-1360.
39. Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011; 144:646-674.
40. Yuspa SH. The pathogenesis of squamous cell cancer: lessons learned from studies of skin carcinogenesis—thirty-third G. H. A. Clowes Memorial Award Lecture. Cancer Res. 1994; 54:1178-1189.
41. Segrelles C, Lu J, Hammann B, Santos M, Moral M, Cascallana JL, Lara MF, Rho O, Carbajal S, Traag J, Beltran L, Martinez-Cruz AB, Garcia-Escudero R, Lorz C, Ruiz S, Bravo A, et al. Deregulated activity of Akt in epithelial basal cells induces spontaneous tumors and heightened sensitivity to skin carcinogenesis. Cancer Res. 2007; 67:10879-10888.
42. Macias E, Rao D, Digiovanni J. Role of stat3 in skin carcinogenesis: insights gained from relevant mouse models. J Skin Cancer. 2013; 2013:684050.
43. Siveen KS, Sikka S, Surana R, Dai X, Zhang J, Kumar AP, Tan BK, Sethi G, Bishayee A. Targeting the STAT3 signaling pathway in cancer: role of synthetic and natural inhibitors. Biochim Biophys Acta. 2014; 1845:136-154.
44. Lin J, Tang H, Jin X, Jia G, Hsieh JT. p53 regulates Stat3 phosphorylation and DNA binding activity in human prostate cancer cells expressing constitutively active Stat3. Oncogene. 2002; 21:3082-3088.
45. Bonner WM, Redon CE, Dickey JS, Nakamura AJ, Sedelnikova OA, Solier S, Pommier Y. GammaH2AX and cancer. Nat Rev Cancer. 2008; 8:957-967.
46. Sherr CJ. Tumor surveillance via the ARF-p53 pathway. Genes Dev. 1998; 12:2984-2991.
47. Jonkers J, Meuwissen R, van der Gulden H, Peterse H, van der Valk M, Berns A. Synergistic tumor suppressor activity of BRCA2 and p53 in a conditional mouse model for breast cancer. Nat Genet. 2001; 29:418-425.