
INTRODUCTION
Given the importance of p53 in tumor development, genomic integrity and normal aging process, p53 activities are tightly regulated at multiple levels by a delicate network through various positive/negative regulators, cofactors, and a large number of posttranslational modifications, including phosphorylation, ubiquitination and acetylation [1-5]. Accumulated evidence shows that acetylation is essential for p53 activation and plays an important role in regulating promoter-specific activation of p53 target genes in response to various stress signals [6]. In addition to the six lysine (K) residues clustered at the C-terminal domain, we and others identified additional acetylation sites of p53, K120 and K164, situated within the DNA-binding domain. K120 and K164 are highly conserved throughout evolution and crucial for p53-medited apoptosis and cell cycle arrest respectively [7-9]. To understand the in vivo roles of p53 acetylation, we previously generated the p533KR/3KR knock-in mouse model in which three corresponding acetylation sites (K117, K161 and K162 in mouse p53) were mutated to the non-acetylable arginine [10]. While loss of acetylation at these sites completely abrogated p53-mediated cell cycle arrest, apoptotic cell death and cellular senescence, p533KR/3KR mice do not succumb to spontaneous tumors as documented for previous reported p53-/- mice [11, 12], indicating that loss of p53-mediated acute DNA damage response is not sufficient for tumorigenesis [10]. Studies of other mouse models, including p5325,26 and p21-/-Puma-/-Noxa-/- also suggested that p53-mediated tumor suppression activity cannot be solely attributed to these well known targets of p53 in stress responses [13, 14]. Taken together, these studies imply that other mechanisms are critical for p53 to exert its tumor suppressor function in vivo.
Numerous studies show that p53 plays a critical role in the maintenance of genomic integrity through its role in DNA damage responses [3, 15]. Loss of p53 function promotes chromosomal instability and wild-type p53 functions as a hub of DNA damage activated checkpoints and as a barrier against genomic instability [16, 17]. In this context, p53 deficiency rescues embryonic lethality caused by the inactivation of many DNA double stand break(DSB) repair genes, such as XRCC4, by attenuating cell cycle checkpoint control, apoptosis and senescence to allow cell survival with genomic instabilities [18-21]. The inherent drawbacks of these rescues are often early onset lethal tumors, which preclude long term studies. The p533KR/3KR mutant fail to induce p53-mediated cell cycle arrest, apoptotic cell death and senescence in response to DNA damage, yet still retains tumor suppression capacities, providing a unique tool to dissect the mechanisms of p53-mediated activities in vivo [10]. As such, p533KR/3KRXrcc4-/- mice were generated by crossing p533KR and XRCC4 DNA repair-impaired mutants, and here we show that p533KR/3KR mutant completely rescue the embryonic lethality caused by XRCC4 deficiency, and unlike p53-/- Xrcc4-/- mice, p533KR/3KR Xrcc4-/- mice show a strong resistance to tumor formation, but surprisingly, display the accelerated aging phenotypes. We further showed that p53-mediated ferroptosis is significantly induced in both spleens and testis of p533KR/3KRXRCC4-/- mice. These results redefine the role of p53-mediated cell-cycle arrest, senescence and apoptosis and have significance implications for the roles of p53-mediated ferroptosis in vivo.
RESULTS
p533KR/3KR cells display high levels of aneuploidy in response to DNA damage treatment
Aneuploidy caused by cell division errors is one of the most common types of genomic instabilities found in human cancers [22]. The increased level of aneuploidy was frequently observed in cells from p53-/- mice, which exhibited high levels of genomic instability and early onset thymic lymphomas with aneuploidy [23-25]. So we first examined the aneuploidy level in p533KR/3KR MEFs. DNA content analysis by FACS shows that primary p533KR/3KR MEFs at passage 1 (P1) have a slightly higher basal level of aneuploidy compared with WT MEFs (P1) (Figure 1A and Figure 1B). In response to ionizing radiation (IR), p53-mediated transactivation of Puma and p21 are completely abrogated in p533KR/3KR MEFs as shown in Figure 1C, however, unlike WT MEFs, p533KR/3KR MEFs exhibit an increased level of aneuploidy 24 hours post-radiation, which is comparable to p53-/- MEFs (Figure 1A and 1B), suggesting that the p533KR/3KR MEFs is prone to radiation-induced aneuploidy.
The embryonic lethality caused by the deficiency of XRCC4 can be fully rescued in the p533KR/3KR background
In normal cells, the genome integrity is constantly challenged by inevitable DNA lesions often arising as byproducts of normal cellular processes such as reaction oxygen species or DNA replication stress, leading to DSBs in chromosome; unrepaired DNA DSBs can activate DNA damage responses and induce p53 activation [26, 27]. Homologous recombination (HR) and non-homologous end-joining (NHEJ) are two major DNA DSB repair pathways in mammalian cells [28]. XRCC4 is essential for the protein stability of Ligase 4 - the DNA ligation component of the NHEJ pathway, which is also required for V(D)J recombination in developing lymphocytes. XRCC4-deficient embryos are growth-retarded and die at embryonic day 15.5 with massive p53-mediated neuronal apoptosis [29, 30]. While p53 deficiency full resuced the embryonic lethality of Xrcc4-/- mice, p53-/-Xrcc4-/- mice routinely succumb to pro-B-cell lymphomas and medulloblastomas [19, 21]. To investigate the genomic instability caused by loss of p53-mediated cell cycle arrest, apoptosis, and senescence in vivo, we crossed p533KR mice with XRCC4 mutant mice and eventually obtained p533KR/3KRXrcc4-/- mice from breedings between p533KR/3KRXrcc4+/- mice. p533KR/3KRXrcc4-/- mice were born at the expected Mendelian ratio (44 out of 180), indicating p533KR/3KR fully rescues the embryonic lethality caused by XRCC4 deficiency (Figure 1D). p533KR/3KRXrcc4-/- mice are morphologically normal but slightly smaller than p533KR/3KRXrcc4+/+ mice at birth (Figure 1E). To examine the genomic instability, we first measured aneuploidy in p533KR/3KRXrcc4-/- MEFs together with WT and p53-/-Xrcc4-/- control MEFs. MEFs were either left untreated or exposed to 10 Gy γ-irradiation and analyzed 24 hours post-radiation. FACS analyses of cell cycle distribution using DNA content measurement revealed that the percentage of cells with aneuploidy in p533KR/3KRXrcc4-/- MEFs (10%) is similar to that in p53-/-Xrcc4-/- MEFs (10.5%), but doubled in comparison with WT MEFs (5.1%). In contrast to the WT MEFs that had similar aneuploidy percentage before and after 10 Gy IR treatments, indicative of intact DNA damage responses, p533KR/3KRXrcc4-/- MEFs, similar to p53-/-Xrcc4-/- MEFs, exhibited further augmented aneuploidy levels 24 hours after γ-irradiation (Figure 1F). Taken together, these results demonstrate that the embryonic lethality of Xrcc4-/- mutant mice can be completely rescued in the p533KR/3KR background. A markedly increase in aneuploidy of p533KR/3KRXrcc4-/- cells suggests that the loss of p53-mediatded cell cycle arrest, apoptosis and senescence leads to genomic instability.
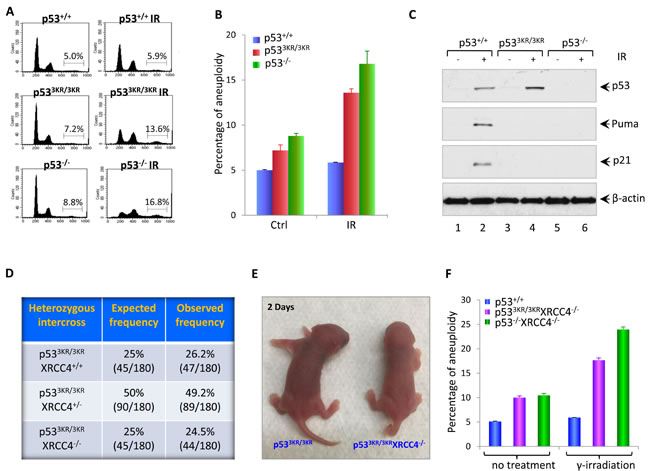
Figure 1: Loss of p53-mediated acute DNA damage response causes genomic instability. A. Flow cytometric analysis of cell cycle distribution in p53+/+, p533KR/3KR, and p53-/- MEFs. MEFs were either left untreated or exposed to 10 Gy of γ-irradiation; 24 hours later, MEFs were collected and fixed with 70% ethanol for 1hour at 4oC, then subjected to FACS after propidium iodide (PI) staining. B. Quantification of the percentage of MEFs with aneuploidy. Error bars represent averages ± SD from at least three independent MEF lines for each genotype. C. Western blot analysis of p53+/+, p533KR/3KR and p53-/- MEFs. Cells were either untreated or exposed to 10 Gy of γ-irradiation, then lyzed and analyzed for the expression of p53, p21, and Puma. β-actin was used as a loading control. D. Table showing the expected and observed frequency from the intercross of p533KR/3KRXrcc4+/- mice. E. Representative pictures of p533KR/3KRXRCC4-/- mice and p533KR/3KR littermates at 2 days of age. F. The percentage of aneuploidy by FACS analysis of cell cycle distribution in p53+/+, p533KR/3KRXRCC4-/-, and p53-/-XRCC4-/- MEFs. Cells were either left untreated or exposed to 10 Gy of γ-irradiation. 24 hours post-radiation, MEFs were collected, fixed with 70% ethanol for 1hour at 4oC, and then subjected to FACS after propidium iodide (PI) staining. Data are shown as averages ± SD from three independent MEF lines for indicated genotypes.
Spontaneous genomic instability in p533KR/3KRXrcc4-/- mice
To further characterize the spontaneous genomic instability observed in p533KR/3KRXrcc4-/- MEFs, we obtained metaphase spreads from early passage (P1) p533KR/3KRXrcc4-/- MEFs as well as WT, p533KR/3KR, p53-/- and p53-/-Xrcc4-/- control MEFs and quantified chromosomal breaks with telomere-fluorescence in situ hybridization (T-FISH) analysis [31, 32]. As shown in Figure 2A and Figure S1, overall percentage of abnormal metaphases in p533KR/3KR MEFs (18.2%) is similar to that in p53-/- MEFs (17.8%), in contrast to only 6.3% of p53+/+ MEFs. XRCC4 deficiency dramatically increased the frequency of abnormal metaphases in both p53-/- and p533KR/3KR background to 48.7% and 46.7%, respectively. T-FISH can identify three types of cytogenetic aberrations, namely chromatid breaks - breaks at one of two sister chromatids, chromosome breaks - breaks at both sister chromatids and chromosomal fusion - joining of two chromosome ends in the absence of telomere signals (Figure 2C). Chromosomal translocations without altering telomere signal cannot be unequivocally identified in T-FISH analyses, leading to underestimation of chromosomal fusion by T-FISH assay. Chromosome breaks, chromatid breaks and chromosome fusion were increased significantly in p533KR/3KRXrcc4-/- MEFs as seen in p53-/-Xrcc4-/- MEFs (Figure 2B and Figure S2), validating the genomic instability in those cells. Moreover, Western blot analyses revealed that several DNA damage markers such as phospho-ATM, phospho-p53, phospho-Kap1 and phospho-H2AX (γ-H2AX) were readily detected in p533KR/3KRXrcc4-/- MEFs (lane 5, Figure S3).
To assess the levels of spontaneous genomic instability observed in vivo, we isolated bone marrow from 4 weeks and 22 weeks old p533KR/3KRXrcc4-/- mice using age-matched p53+/+ mice as controls. Metaphase spreads were prepared from total bone marrows after incubation with 0.1 µg/ml colcemid for 6 hours and subjected to T-FISH analyses. As shown in Figure 2D, the percentage of abnormal metaphases in p533KR/3KRXrcc4-/- bone marrow cells increased significantly from 10.9% at 4 weeks to 19.8% at 22 weeks, whereas abnormal metaphase counts remain steady in p53+/+ mice at the same age period (also see Figure S4). We also examined the levels of proteins involved in the maintenance of genome stability in the spleens of p533KR/3KRXrcc4-/- mice at 4 weeks and 22 weeks of age by Western blot analyses. As shown in Figure 2E, phospho-p53, phospho-Kap1 and γ-H2AX were readily detected in splenocytes from p533KR/3KRXrcc4-/- mice at 4 weeks of age whereas none of them was detected in the splenocytes of p53+/+, p533KR/3KR and p53-/- mice at the same age. Moreover, the levels of these DNA damage markers were further increased in p533KR/3KRXrcc4-/- mice at 22 weeks of age compared with age-matched control mice. Immunohistochemical analyses also confirmed a marked increase of the percentage of γ-H2AX positive cells in the spleens of p533KR/3KRXrcc4-/- mice at 22 weeks of age (Figure 2F). Taken together, these data demonstrate spontaneous genomic instability in p533KR/3KRXrcc4-/- mice.
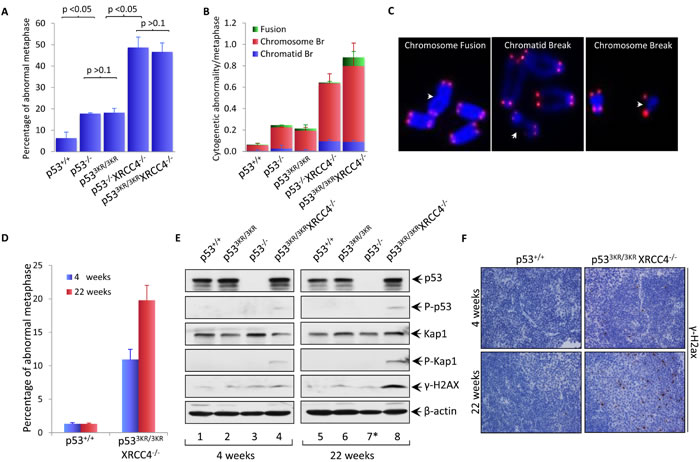
Figure 2: Spontaneous genomic instability in p533KR/3KRXRCC4-/- mice was augmented with age. A. Percentage of abnormal metaphase spreads obtained from early passage p53+/+, p533KR/3KR, p53-/-, p533KR/3KRXRCC4-/-, and p53-/-XRCC4-/- MEFs. Metaphases were prepared from MEFs for the indicated phenotypes after 3 hours treatment with 0.1µg/ml colcemid and analyzed using telomere-FISH assay. At least 100 metaphases were counted. Results are shown as averages ± SD from three different MEF lines of each genotype. B. Frequency of cytogenetic abnormalities in three categories: chromosomal breaks, chromatid breaks and chromosomal fusions, in MEFs with the indicated genotypes. Values shown are the averages ± SD from three independent MEF lines. C. Representative images of abnormalities observed in p533KR/3KRXRCC4-/- MEF metaphases. Chromosomes were stained with telomere specific probes (red) and counterstained with DAPI (blue).D. Percentage of metaphases with abnormalities in bone marrows from 4-week-old and 22-week-old p53+/+ and p533KR/3KRXRCC4-/- mice. Metaphase spreads were obtained from mice bone marrows with the indicated genotypes after with incubation with 0.1µg/ml colcemid for 6 hours and a minimum of 100 metaphases was analyzed for each sample using telomere-FISH. Results are reported as averages ± SD from three mice for each genotype. E. Immunoblot assays of p53, phopho-p53, Kap1, phopho-Kap1, γ-H2ax and β-actin proteins in the lysates prepared from the spleens of p53+/+, p533KR/3KR, and p53-/- p533KR/3KRXRCC4-/- mice at the indicated ages. *; The 3-month-old p53-/- mice (lane 7) was used for control. β-actin was used as a loading control. F. Representative immunohistochemical staining of spleens from 4 week- and 22 week-old p53+/+ and p533KR/3KRXRCC4-/- mice for γ-H2ax.
The p533KR/3KRXrcc4-/- mice are not predisposed to cancer but nevertheless have a short life-span
The above data indicate that p533KR/3KRXrcc4-/- mice show high levels of spontaneous genomic instability, but unlike p53-/-Xrcc4-/- mice that uniformlly succumb to pro-B-cell lymphomas by ~10 weeks [19], p533KR/3KRXrcc4-/- mice lived up to 30 weeks (Figure 3A). Since p53-/-Xrcc4-/- mice mainly succumb to pro-B-cell lymphomas, we performed the further analyses of the spleens. The spleens of p533KR/3KRXrcc4-/- mice appear normal but smaller compared to the age-matched control mice (Figure 3B), likely due to the absence of mature B and T cells caused by Xrcc4 deficiency. Histological examinations of spleens of p533KR/3KRXrcc4-/- mice showed no evidence of lymphomas, indicated by infiltration of clonal enlarged B/T cells (Figure 3C). Further analysis also failed to identify any tumors in other organs of p533KR/3KRXrcc4-/- mice. p533KR/3KRXrcc4-/- newborn mice exhibit slight differences in overall body weight compared to their littermates (Figure 3D). By 4 weeks of age, their body weight was more than 70% lower than control littermates. p533KR/3KRXrcc4-/- mice reach their maximum body weight at 8 weeks of age, which is only about 50% normal control mice, and then started weight loss (Figure 3D). Interestingly, the whole body X-ray analysis showed that p533KR/3KRXrcc4-/- mice displayed kyphosis (also known as hunchback), an abnormal curvature of the spine, at the age of 22 weeks (Figures 3E and 3F), one of the main premature aging associated phenotypes that were previously reported in p53 mutant mice [33, 34].
We next sought to examine the possibility that the shortened lifespans in p533KR/3KRXrcc4-/- mice could be accompanied by more defined premature aging phenotypes in addition to aging-associated kyphosis. Aging related alterations in the male reproductive system occur primarily in the testis. The mammalian testis is a complex organ composed of germ cells and supporting cells such as Leydig cells and Sertoli cells that together produce functional spermatozoa and hormones respectively through a series of orchestrated developmental transitions in the seminiferous tubules. It has been reported that the production of mature sperms and their quality gradually decline with age in humans and studies using mouse models have also shown convincing evidence that the proliferation and maturation of germ cells can become compromised with aging, leading to testicular atrophy [35]. p533KR/3KRXrcc4-/- mice can live up to about 30 weeks but they are all sterile. Histological examinations of testes of 22-week-old p533KR/3KRXrcc4-/- mice revealed a marked reduction of testicular mass, extensive seminiferous epithelium degeneration and severe tubule atrophy in comparison with their control littermates (Figure 3G). In the meantime, spermatogonia were not found in the tubular lumina and significant fractions of the spermatogonia is missing from the basal layer, consistent with the fact that spermatogenesis was completely compromised in p533KR/3KRXrcc4-/- mice (Figure 3G). Taken together, these data indicate that that p533KR/3KRXrcc4-/- mice, unlike p53-/-Xrcc4-/- mice, do not succumb to pro-B-cell lymphomas but have a short life span and exhibit aging-associated kyphosis and testicular atrophy at the age of 22 weeks.
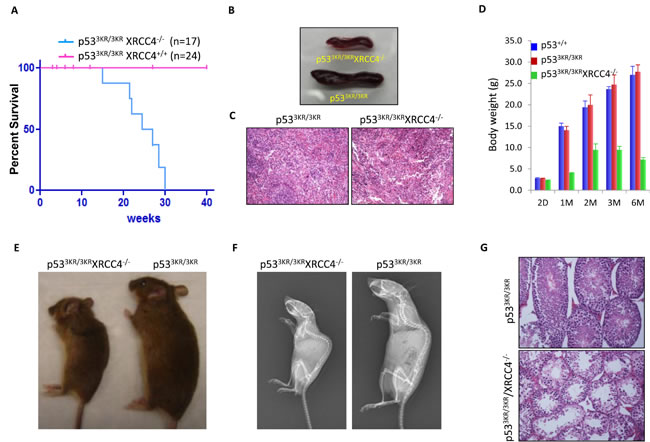
Figure 3: p533KR/3KRXRCC4-/- mice bearing high levels of genomic instability are resistant to cancer but display premature aging phenotypes. A. Kaplan-Meier overall survival curves of p533KR/3KR and p533KR/3KRXRCC4-/- mice. B. Images of spleens of p533KR/3KRXRCC4-/- mouse and p533KR/3KR littermate at the age of 22 weeks.C. Hematoxylin and eosin (H&E) staining of spleens from 22-week-old p533KR/3KR and p533KR/3KRXRCC4-/- mice.D. Average body weight of p53+/+, p533KR/3KR, and p533KR/3KRXRCC4-/- mice at different ages. Error bars means averages ± SD. E. p533KR/3KRXRCC4-/- mouse and p533KR/3KR littermate at 22 weeks of age.F. Representative whole-body radiographs of 22-week-old p533KR/3KRXRCC4-/- mice with p533KR/3KR littermates. G. H&E staining of testis from 22-week-old p533KR/3KR and p533KR/3KRXRCC4-/- mice.
Mechanistic insights into the phenotypes observed in p533KR/3KRXrcc4-/- mice
Although p53-mediated cell cycle arrest, senescence and apoptosis are well accepted as major tumor suppression mechanisms, accumulating evidence indicates that the loss of these canonical functions does not directly lead to tumorigenesis. Our recent studies show that p53-mediated regulation of ferroptosis is a new mechanism of tumor suppression that acts independently of the canonical p53 functions in cell-cycle arrest, senescence and apoptosis through suppressing SLC7A11 expression [36]. Thus, although p533KR is defective in cell-cycle arrest, senescence and apoptosis, it is very likely that p533KR/3KRXrcc4-/- mice do not succumb to pro-B-cell lymphomas by activating p533KR mediated ferroptosis. To this end, we first examined whether SLC7A11 is downregulated in p533KR/3KRXrcc4-/- MEF cells. As shown in Figure 4A, q-PCR analysis revealed a significant downregulation of SLC7A11 in p533KR/3KRXrcc4-/- MEFs compared with the control p53-/-Xrcc4-/- MEFs (also see Figure S5). Consistent with these results, p533KR/3KRXrcc4-/- MEF cells are very sensitive to ferroptosis upon ROS treatment whereas no obvious cell death were detected in the p53-/-Xrcc4-/- MEFs under same conditions (Figure 4B). As expected, ROS-induced cell death was completely rescued in the presence of ferrostatin-1, a specific inhibitor for ferroptosis [36] (Figure 4B and Figure 4C).
To provide the in vivo evidence to support the role of p53-mediated ferroptosis to prevent the development of pro-B-cell lymphomas, we performed the analysis of p53-mediated activities in spleens. Western blot analysis showed that p53 is stabilized in the spleens of p533KR/3KRXrcc4-/- mice, the levels of SLC7A11 are significantly downregulated (lane 2, Figure 4D). Of note, since most p53-/-Xrcc4-/- mice become moribund by 10 weeks due to pro-B-cell lymphomas [19, 37] , we used the tumor-free p53-/- mice at the age of 3 months as a control. Since recent studies identified up-regulation of PTGS2 as a potential molecular marker of ferroptosis [38], we examined the levels of PTGS2 expression in the spleen of p533KR/3KRXrcc4-/- mice. As shown in Figure 4E, Ptgs2 was indeed significantly upregulated in p533KR/3KRXrcc4-/- spleens (vs. p533KR/3KR spleens). To examine whether p53-mediated ferroptosis plays a role in testicular atrophy, we performed the similar analysis of p53-mediated activities in testis. Indeed, p53 was activated and SLC7A11 expression was significantly reduced (Figure 4F); the ferroptosis marker PTGS2 was also dramatically upregulated in p533KR/3KRXrcc4-/- testis (Figure 4G). In contrast, no obvious ferroptotic cell death was detected in the testis from either p533KR/3KRXrcc4+/+ or p53-/- mice. Moreover, downregulation of SLC7A11 was also obesreved in lives, brain and bone morrows of p533KR/3KRXrcc4-/- mice although the levels of PTGS2 expression were not highly induced in those tissues, suggesting that additional factors might be required for ferroptosis induction (Figure S6). Taken together, these data demonstrate that p53-mediated downregulation of SLC7A11 is induced in the cells and tissues of p533KR/3KRXrcc4-/- mice and also suggest that p53-mediated ferroptosis may play a role in both preventing tumor development and testicular atrophy observed in the p533KR/3KRXrcc4-/- mice.
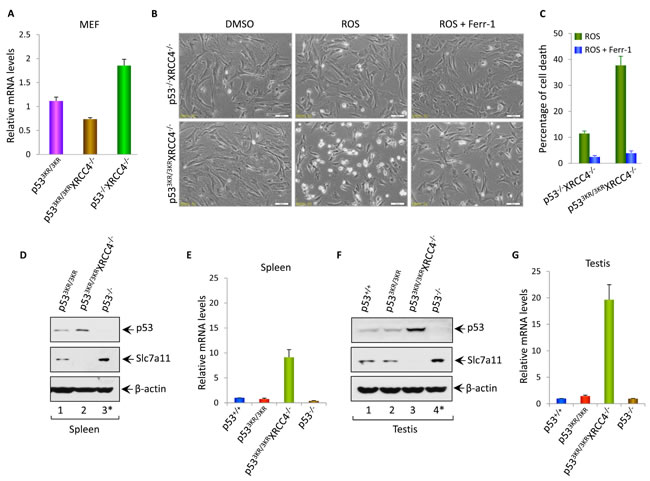
Figure 4: The roles of p53-mediated effects on Slc7a11 and ferroptosis in tumor suppression and aging associated testicular atrophy in p533KR/3KRXRCC4-/- mice. A. qRT-PCR analysis of Slc7a11 mRNA levels in p533KR/3KR, p533KR/3KRXRCC4-/- and p53-/-XRCC4-/- MEFs. Data were shown as average ± SEM from three independent MEF lines.B. p533KR/3KRXRCC4-/- and p53-/-XRCC4-/- MEFs were either left untreated or exposed to ROS (Tert-butyl-hydroperoxide; 400 µM) and ferroptosis inhibitor, ferrostatin-1 (ferr-1, 2 µM) for 8 hours and then representative phase-contrast images were taken.C. Quantification of cell death of MEFs after the treatment with ROS and ferr-1. Data were shown as average ±SD from three independent MEF lines.D. and F. Western blot analysis of p53, Slc7a11 and β-actin proteins in the lysates prepared from the spleens (D) or testis (F) of p53+/+, p533KR/3KR, p533KR/3KRXRCC4-/-, and p53-/- mice at the age of 22 weeks. *; The 3-month-old tumor-free p53-/- mice were used for control.E. and G. qRT-PCR analysis of Ptgs2 mRNA levels in spleens (E) or in testis (G) of 22-week-old p53+/+, p533KR/3KR, p533KR/3KRXRCC4-/- and 3-month-old p53-/- mice. Error bars represent averages ± SEM of three mice for each genotype.
DISCUSSION
Normal proliferating cells constantly cope with a variety of stress signals from both outside and inside such as replication stress, telomere shortening and ROS damage, and their genome integrity are inevitably damaged during proliferation, which can induce a p53-mediated temporary arrest at cell cycle checkpoints to allow cells to correct possible defects, thereby avoiding the transmission of genetic lesions to daughter cells [39]. Cell cycle arrest is released and cell proliferation resumes once the damage is repaired. Extensive and irreparable damage can trigger cellular p53-mediated senescence and apoptotic cell death. In contrast, the cell cycle progression proceeds with accumulated unrepaired DNA damage in p53-null cells, leading to mutations, chromosomal aberrations, and aneuploidy. Since p53-mediated cell cycle arrest, apoptosis and senescence are abrogated in p533KR/3KR cells, it is not surprising that DNA damage is accumulated in these cells, leading to aneuploidy. Indeed, we showed that p533KR/3KR MEFs exhibit higher levels of aneuploidy upon DNA damage. To corroborate this finding, we generated p533KR/3KRXrcc4-/- mice by ablating the NHEJ repair pathway and found that tissues and cells derived from p533KR/3KRXrcc4-/- mice have higher basal levels of genomic instability determined by chromosome breaks and even fusions. Our results demonstrate that p53-mediated cell cycle arrest, apoptosis and senescence are absolutely essential for its ability to control genomic integrity in vivo. Consistent with our earlier study [10, 36], p533KR/3KRXrcc4-/- mice do not develop spontaneous tumors, suggesting that the acetylation defective mutant retains its tumor suppression activity through activating its metabolic targets. Notably, p53 is activated in both MEFs and tissues of p533KR/3KRXrcc4-/- mice associated downregulation of Slc7a11 and p53-mediated ferroptosis is induced in vitro and in vivo. Thus, our results suggest that p53-mediated ferroptosis plays a critical role in suppressing tumorigenesis despite high levels of genomic instability in p533KR/3KRXrcc4-/- mice.
The molecular basis for the aging-like phenotypes observed in p533KR/3KRXrcc4-/- mice needs further elucidation. One common denominator of aging is the accumulation of genetic damage throughout life [40] and genomic instability has been well accepted as one of the major hallmarks of aging [41]. Consistent with this notion, several DNA repair defiency mouse models have been reported associated with accelerated aging phenotypes [42-44]. Although it is possible that the role of p53 in those aging mice is simply preventing tumor formation, a direct role of p53 in aging has been supported by several animal models. For example, by generating mice with a p53 truncation mutant encoding a carboxyl-terminal fragment called M protein, Tyner et al., showed that p53+/m mice exhibited an enhanced resistance to cancer that was accompanied by reduced longevity and early onset of a number of aging phenotypes [33]. Similar results was also obtained in a transgenic mouse model overexpressing a modestly truncated, naturally occurring isoform of p53 called p44 [34], indicating that p53 has pro-aging effects regardless of the status of genomic instability. Nevertheless, the mice with extra copies of p53 gene (super-p53), or with extra copies of the Arf gene (super-Arf), or with Mdm2 hypomorphic alleles (Mdm2puro/Δ7-12) have a normal longevity [45-48]. Taken together, these studies indicate that the role of p53 in aging is more complex than originally anticipated. Consistent with this notion, paradoxical roles of p53 in cellular senescence has recently been reported; several studies indicate that p53 is capable of converting senescence into quiescence by inhibiting the mTOR pathway, suggesting that p53 may also have the anti-aging activities in vivo [49-52]. It is very likely that only some aspects of p53 activity are activated in the mice expressing truncated p53 mutants and promote aging whereas increasing the overall function of p53 (e.g. the mice with extra copies of p53 gene or the mice with Mdm2 hypomorphic alleles) may neutralize these pro-aging effects because anti-aging activities of p53 (e.g. through inhibiting the mTOR pathway) are also enhanced in those mice.
Aging is a dynamic and complex process defined as the time-dependent functional decline. With age, homeostasis declines and damage accumulates. One of prime candidates that induce macromolecular damage is oxidative stress from reactive oxygen species (ROS) generated from normal physiological activities. Indeed, many long-lived mutants are resistant to oxidative stress [53]. Ferroptosis involves metabolic dysfunction that results in the production of both cytosolic and lipid ROS [36, 38]. Repression of SLC7A11 transcription by p53 results in reduction of cystine uptake. Because of less cystine uptake, the levels of intracellular glutathione (GSH) will be reduced and the cellular system for defending oxidative stress is abrogated. Thus, the sensitivity of ROS-induced ferroptosis is significantly increased in p53-activating cells. We showed that SLC7A11 is downregulated by p53 and that p53-mediated ferroptosis is dramatically induced in the testis of p533KR/3KRXrcc4-/- mice. Thus, it is very likely that the combination of genomic instability and p53-mediated ferroptosis contributes significantly to the aging associated phenotypes observed in p533KR/3KRXrcc4-/- mice.
MATERIALS AND METHODS
Generation of p533KR/3KRXRCC4-/- mice
The p533KR/3KR and XRCC4+/- mice described previously (Yan et al., 2006) were bred to generate p53+/3KRXRCC4+/- mice. The p533KR/3KR XRCC4-/- double knockout mice were obtained by intercrossing p53+/3KRXRCC4+/- double heterozygous mice. The p533KR/3KR XRCC4-/- offspring were PCR genotyped using primer sets (For p533KR, forward: 5’-CTTCCTGCAGTCTGG GACAGC C-3’, reverse: 5’-GCAGCTGGGCCTACAG CACAC G-3’) and (For XRCC4, p1, 5’-TAAGCTATTACTCCTGCATGGAGCATTATCACC, p2, 5’-GCACCTTTGCCTACTAAGCCATCTCAC and p3, 5’-TTCAGCTAACCAGCATCAAT AG). All mice work was performed in compliance with the IACUC (Institutional Animal Care and Use Committee) of Columbia University.
Cell culture
Mouse embryonic fibroblasts (MEFs) were isolated from 13.5 day postcoital embryos obtained by heterozygous intercross according to the standard procedures. MEFs were cultured in DMEM medium supplemented with heat-inactivated FBS (MEF only) and 1% non-essential amino acids plus 200 unit/ml penicillin/streptomycin. Cells were maintained at 37oC in a 5% CO2 incubator.
Tissue staining
immunohistochemical staining was performed using standard protocols. Tissues from mice were collected and fixed with 10% formalin overnight, then processed, paraffin-embedded, sectioned and stained with anti-mouse γ-H2ax (#2577, Cell Signaling Technologies) antibodies according to the standard procedures.
Metaphase preparation and telomere FISH staining
The metaphase preparation and telomere FISH staining were performed as described as before with minor modifications [54]. Briefly the cells were cultured for 3 (MEFs) or 6 (bone marrow cells) hours with colcemid at the final concentration of 0.1ug/ml (GIBCO). The treated cells were then collected and swollen in 0.57% KCl hypotonic solution for 20 minutes at room temperature. After incubation, cells were fixed by adding ice-cold freshly prepared fixative (3:1 v/v methanol: acetic acid) for > 4 times. Metaphase spreads were obtained by dropping the fixed cells onto slides and steamed over the 85 oC water bath for 10 sec, air-dried for > 1 hour and then checked under a microscope. For telomere FISH staining, slides were fixed in 4% (w/v) formaldehyde/PBS for 15 min at room temperature, followed by three washes with 1xPBS and digestion with pepsin (0.1%, pH was adjusted to 1.2 with HCl) (Sigma) for 10 min at 37 oC. The slides were then dehydrated in ethanol after three washes with PBS. After air dry, a probe mix (70% (v/v) formamide, 2% (w/v) BSA, 100 μg/ml tRNA, 10 mM Tris, 0.5 ug/ml custom Telomere G-strand PNA probe from PNA Bio Inc) was added to each slide, and the slides were denatured by heating for 3 min at 80 °C on a heat block. After 2 hours incubation in the dark wet chamber at 37 oC, slides were washed twice with 70% (v/v) formamide, 0.1% (w/v) BSA, 10 mM Tris-HCl, pH 7.5, followed by three washes in 50 mM Tris-HCl, pH 7.5, 150 mM NaCl, 0.1% (w/v) BSA, and 0.1% (v/v) Tween-20. The slides were then dehydrated in ethanol serial and air dry. DNA was counterstained with DAPI. A minimum of 100 metaphases were captured and analyzed using Metafer MSearch Metaphase Finder developed by MetaSystems, MA.
RNA isolation and qRT-PCR
Total RNA was isolated from MEFs or mouse tissues using Trizol (Invitrogen) and treated with DNase I (Ambion). 1 µg total RNA was reverse-transcribed using SuperScript III First-Strand Synthesis SuperMix (Invitrogen) following manufacturer’s protocol. PCR was performed in triplicate using SYBR green mix (Applied Biosystems), and a 7500 Fast Real-Time PCR System (Applied Biosystems) under the following conditions: 15min at 95oC followed by 40 cycles of 95oC for 15 sec and 60oC for 1 min. qRT-PCR data analysis were performed as described before (Bookout and Mangelsdorf, 2003) and Primers used for RT-PCR and qRT-PCR were shown in qRT-PCR Primers.
Flow cytometry
To analyze cell cycle by DNA content, 2X106 MEFs were suspended in 200ul of PBS/0.1%FBS by vortexing after wash once with ice-cold PBS. 4mls of ice cold 70%ETOH was added to the cells one drop at a time. Cells were pelleted and resuspended in 1ml of PI solution (40ug/ml PI and 100ug/ml RNaseA) after at least 1 hr to overnight fixation at 4oC. Then FACS analysis was performed after 1 hr incubation at 37oC followed by filtering cells through 40-70um mesh. Cells were sorted using a Becton Dickinson FACScalibur machine and data were analyzed using CellQuest.
Western blotting
Cell or tissue lysates were prepared in RIPA buffer (50 mM Tris-HCl [pH 8], 150 mM NaCl, 0.1% SDS, and 0.5% Na deoxycholate, 1% NP40 and fresh proteinase inhibitor cocktail). Protein extracts were analyzed by Western blotting according to standard protocols using primary antibodies specific for p53 (CM5, Leica Microsystems), P-S15p53 (9284, Cell Signaling Technologies), p21 (SX118, Santa Cruz), PUMA (p4743, Sigma), ATM (ab2631, Abcam), phosphor-ATM (ab81292, Abcam), Kap1 (ab10484, Abcam), phosphor-Kap1 (ab70369, Abcam), Slc7a11 (ab37185, Abcam) and β-actin (A3853, Sigma). HRP-conjugated anti-rabbit,-mouse and -rat secondary antibodies (GE Healthcare) were used and signal was detected using an ECL Western blotting detection system (GE Healthcare).
ROS and ferroptosis inhibitor treatment
2 x 105 MEF cells were seeded into 6-well plates and cultured overnight. Then cells were treated with 400 µM tert-butyl hydroperoxide (TBH, Sigma-Aldrich) to generate ROS and at the same time ferroptosis inhibitor, Ferrostatin-1 (Ferr-1, Xcess Biosciences) was added into the culture medium at the concentration of 2 µM. Cells were cultured for 8 hours, then trypsinized and stained with trypan blue followed by counting witha haemocytometer using standard protocol. Cells stained blue were considered as dead cells and cell death quantification was further confirmed by propidium iodide (PI) staining followed by FACS analysis.
qRT-PCR primers
The following primers were used for the quantitative real-time PCR (qRT-PCR) analysis of the indicated mouse mRNAs: β-Actin forward 5’-GGCTGTATTCCCCTCCATCG-3’, β-Actin reverse 5’-CCAGTTGGTAACAATGCCATGT-3’. Slc7a11 forward, TGGGTGGA ACTGCTCGTAAT, reverse, AGGATGTAGCGTCCAAATGC; mouse Ptgs2 forward, GG GAGTCTGGAACATTGTGAA, reverse, GTGCACATTGTAAGTAGGTGGACT.
ACKNOWLEDGMENTS
The authors thank the members of Gu laboratory for insightful commnets, Columbia Cancer Center facility for excellent services.
Conflicts of interests
The authors declare no competing financial interests.
Grant Support
This work was supported by the National Cancer Institute of the National Institutes of Health under Award 5R01CA172023, 5RO1CA169246, and 5RO1CA085533 to W.G. 2P01CA080058 to W.G. and J. M., and R01CA184187 to S.Z. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health. X.L and S.Z are fellow and scholar of Leukemia Lymphoma Society respecitively.
REFERENCES
1. Feng Z, Lin M and Wu R. The Regulation of Aging and Longevity: A New and Complex Role of p53. Genes Cancer. 2011; 2:443-452. doi: 10.1177/1947601911410223.
2. Gannon HS and Jones SN. Using Mouse Models to Explore MDM-p53 Signaling in Development, Cell Growth, and Tumorigenesis. Genes Cancer. 2012; 3:209-218. doi: 10.1177/1947601912455324.
3. Eischen CM and Lozano G. The Mdm network and its regulation of p53 activities: a rheostat of cancer risk. Hum Mutat. 2014; 35:728-737.
4. Vousden KH and Prives C. Blinded by the Light: The Growing Complexity of p53. Cell. 2009; 137:413-431.
5. Berkers CR, Maddocks OD, Cheung EC, Mor I and Vousden KH. Metabolic regulation by p53 family members. Cell Metab. 2013; 18:617-633.
6. Kruse JP and Gu W. Modes of p53 regulation. Cell. 2009; 137:609-622.
7. Sykes SM, Mellert HS, Holbert MA, Li K, Marmorstein R, Lane WS and McMahon SB. Acetylation of the p53 DNA-binding domain regulates apoptosis induction. Mol Cell. 2006; 24:841-851.
8. Tang Y, Luo J, Zhang W and Gu W. Tip60-dependent acetylation of p53 modulates the decision between cell-cycle arrest and apoptosis. Mol Cell. 2006; 24:827-839.
9. Tang Y, Zhao W, Chen Y, Zhao Y and Gu W. Acetylation is indispensable for p53 activation. Cell. 2008; 133:612-626.
10. Li T, Kon N, Jiang L, Tan M, Ludwig T, Zhao Y, Baer R and Gu W. Tumor suppression in the absence of p53-mediated cell-cycle arrest, apoptosis, and senescence. Cell. 2012; 149:1269-1283.
11. Donehower LA, Harvey M, Slagle BL, McArthur MJ, Montgomery CA, Jr., Butel JS and Bradley A. Mice deficient for p53 are developmentally normal but susceptible to spontaneous tumours. Nature. 1992; 356:215-221.
12. Jacks T, Remington L, Williams BO, Schmitt EM, Halachmi S, Bronson RT and Weinberg RA. Tumor spectrum analysis in p53-mutant mice. Curr Biol. 1994; 4:1-7.
13. Brady CA, Jiang D, Mello SS, Johnson TM, Jarvis LA, Kozak MM, Kenzelmann Broz D, Basak S, Park EJ, McLaughlin ME, Karnezis AN and Attardi LD. Distinct p53 transcriptional programs dictate acute DNA-damage responses and tumor suppression. Cell. 2011; 145:571-583.
14. Valente LJ, Gray DH, Michalak EM, Pinon-Hofbauer J, Egle A, Scott CL, Janic A and Strasser A. p53 efficiently suppresses tumor development in the complete absence of its cell-cycle inhibitory and proapoptotic effectors p21, Puma, and Noxa. Cell Rep. 2013; 3:1339-1345.
15. Batchelor E, Loewer A and Lahav G. The ups and downs of p53: understanding protein dynamics in single cells. Nat Rev Cancer. 2009; 9:371-377.
16. Sengupta S and Harris CC. p53: traffic cop at the crossroads of DNA repair and recombination. Nat Rev Mol Cell Biol. 2005; 6:44-55.
17. Halazonetis TD, Gorgoulis VG and Bartek J. An oncogene-induced DNA damage model for cancer development. Science. 2008; 319:1352-1355.
18. Bogue MA, Zhu C, Aguilar-Cordova E, Donehower LA and Roth DB. p53 is required for both radiation-induced differentiation and rescue of V(D)J rearrangement in scid mouse thymocytes. Genes Dev. 1996; 10:553-565.
19. Gao Y, Ferguson DO, Xie W, Manis JP, Sekiguchi J, Frank KM, Chaudhuri J, Horner J, DePinho RA and Alt FW. Interplay of p53 and DNA-repair protein XRCC4 in tumorigenesis, genomic stability and development. Nature. 2000; 404:897-900.
20. Lim DS, Vogel H, Willerford DM, Sands AT, Platt KA and Hasty P. Analysis of ku80-mutant mice and cells with deficient levels of p53. Mol Cell Biol. 2000; 20:3772-3780.
21. Yan CT, Kaushal D, Murphy M, Zhang Y, Datta A, Chen C, Monroe B, Mostoslavsky G, Coakley K, Gao Y, Mills KD, Fazeli AP, Tepsuporn S, Hall G, Mulligan R, Fox E, et al. XRCC4 suppresses medulloblastomas with recurrent translocations in p53-deficient mice. Proc Natl Acad Sci U S A. 2006; 103:7378-7383.
22. Storchova Z and Kuffer C. The consequences of tetraploidy and aneuploidy. J Cell Sci. 2008; 121:3859-3866.
23. Hainaut P and Hollstein M. p53 and human cancer: the first ten thousand mutations. Adv Cancer Res. 2000; 77:81-137.
24. Donehower LA, Godley LA, Aldaz CM, Pyle R, Shi YP, Pinkel D, Gray J, Bradley A, Medina D and Varmus HE. Deficiency of p53 accelerates mammary tumorigenesis in Wnt-1 transgenic mice and promotes chromosomal instability. Genes Dev. 1995; 9:882-895.
25. Fukasawa K and Vande Woude GF. Synergy between the Mos/mitogen-activated protein kinase pathway and loss of p53 function in transformation and chromosome instability. Mol Cell Biol. 1997; 17:506-518.
26. Kinzler KW and Vogelstein B. Cancer-susceptibility genes. Gatekeepers and caretakers. Nature. 1997; 386:761, 763.
27. Ferguson DO, Sekiguchi JM, Chang S, Frank KM, Gao Y, DePinho RA and Alt FW. The nonhomologous end-joining pathway of DNA repair is required for genomic stability and the suppression of translocations. Proc Natl Acad Sci U S A. 2000; 97:6630-6633.
28. Polo SE and Jackson SP. Dynamics of DNA damage response proteins at DNA breaks: a focus on protein modifications. Genes Dev. 2011; 25:409-433.
29. Barnes DE, Stamp G, Rosewell I, Denzel A and Lindahl T. Targeted disruption of the gene encoding DNA ligase IV leads to lethality in embryonic mice. Curr Biol. 1998; 8:1395-1398.
30. Frank KM, Sekiguchi JM, Seidl KJ, Swat W, Rathbun GA, Cheng HL, Davidson L, Kangaloo L and Alt FW. Late embryonic lethality and impaired V(D)J recombination in mice lacking DNA ligase IV. Nature. 1998; 396:173-177.
31. Franco S, Gostissa M, Zha S, Lombard DB, Murphy MM, Zarrin AA, Yan C, Tepsuporn S, Morales JC, Adams MM, Lou Z, Bassing CH, Manis JP, Chen J, Carpenter PB and Alt FW. H2AX prevents DNA breaks from progressing to chromosome breaks and translocations. Mol Cell. 2006; 21:201-214.
32. Zha S, Sekiguchi J, Brush JW, Bassing CH and Alt FW. Complementary functions of ATM and H2AX in development and suppression of genomic instability. Proc Natl Acad Sci U S A. 2008; 105:9302-9306.
33. Tyner SD, Venkatachalam S, Choi J, Jones S, Ghebranious N, Igelmann H, Lu X, Soron G, Cooper B, Brayton C, Park SH, Thompson T, Karsenty G, Bradley A and Donehower LA. p53 mutant mice that display early ageing-associated phenotypes. Nature. 2002; 415:45-53.
34. Maier B, Gluba W, Bernier B, Turner T, Mohammad K, Guise T, Sutherland A, Thorner M and Scrable H. Modulation of mammalian life span by the short isoform of p53. Genes Dev. 2004; 18:306-319.
35. Martin GM and Oshima J. Lessons from human progeroid syndromes. Nature. 2000; 408:263-266.
36. Jiang L, Kon N, Li T, Wang SJ, Su T, Hibshoosh H, Baer R and Gu W. Ferroptosis as a p53-mediated activity during tumour suppression. Nature. 2015; 520:57-62.
37. Zhu C, Mills KD, Ferguson DO, Lee C, Manis J, Fleming J, Gao Y, Morton CC and Alt FW. Unrepaired DNA breaks in p53-deficient cells lead to oncogenic gene amplification subsequent to translocations. Cell. 2002; 109:811-821.
38. Yang WS, SriRamaratnam R, Welsch ME, Shimada K, Skouta R, Viswanathan VS, Cheah JH, Clemons PA, Shamji AF, Clish CB, Brown LM, Girotti AW, Cornish VW, Schreiber SL and Stockwell BR. Regulation of ferroptotic cancer cell death by GPX4. Cell. 2014; 156:317-331.
39. Serrano M and Blasco MA. Cancer and ageing: convergent and divergent mechanisms. Nat Rev Mol Cell Biol. 2007; 8:715-722.
40. Moskalev AA, Smit-McBride Z, Shaposhnikov MV, Plyusnina EN, Zhavoronkov A, Budovsky A, Tacutu R and Fraifeld VE. Gadd45 proteins: relevance to aging, longevity and age-related pathologies. Ageing Res Rev. 2012; 11:51-66.
41. Lopez-Otin C, Blasco MA, Partridge L, Serrano M and Kroemer G. The hallmarks of aging. Cell. 2013; 153:1194-1217.
42. Li H, Vogel H, Holcomb VB, Gu Y and Hasty P. Deletion of Ku70, Ku80, or both causes early aging without substantially increased cancer. Mol Cell Biol. 2007; 27:8205-8214.
43. Niedernhofer LJ, Garinis GA, Raams A, Lalai AS, Robinson AR, Appeldoorn E, Odijk H, Oostendorp R, Ahmad A, van Leeuwen W, Theil AF, Vermeulen W, van der Horst GT, Meinecke P, Kleijer WJ, Vijg J, et al. A new progeroid syndrome reveals that genotoxic stress suppresses the somatotroph axis. Nature. 2006; 444:1038-1043.
44. Crossan GP, van der Weyden L, Rosado IV, Langevin F, Gaillard PH, McIntyre RE, Sanger Mouse Genetics P, Gallagher F, Kettunen MI, Lewis DY, Brindle K, Arends MJ, Adams DJ and Patel KJ. Disruption of mouse Slx4, a regulator of structure-specific nucleases, phenocopies Fanconi anemia. Nat Genet. 2011; 43:147-152.
45. Garcia-Cao I, Garcia-Cao M, Martin-Caballero J, Criado LM, Klatt P, Flores JM, Weill JC, Blasco MA and Serrano M. “Super p53” mice exhibit enhanced DNA damage response, are tumor resistant and age normally. EMBO J. 2002; 21:6225-6235.
46. Matheu A, Maraver A, Klatt P, Flores I, Garcia-Cao I, Borras C, Flores JM, Vina J, Blasco MA and Serrano M. Delayed ageing through damage protection by the Arf/p53 pathway. Nature. 2007; 448:375-379.
47. Matheu A, Pantoja C, Efeyan A, Criado LM, Martin-Caballero J, Flores JM, Klatt P and Serrano M. Increased gene dosage of Ink4a/Arf results in cancer resistance and normal aging. Genes Dev. 2004; 18:2736-2746.
48. Mendrysa SM, O’Leary KA, McElwee MK, Michalowski J, Eisenman RN, Powell DA and Perry ME. Tumor suppression and normal aging in mice with constitutively high p53 activity. Genes Dev. 2006; 20:16-21.
49. Korotchkina LG, Leontieva OV, Bukreeva EI, Demidenko ZN, Gudkov AV and Blagosklonny MV. The choice between p53-induced senescence and quiescence is determined in part by the mTOR pathway. Aging (Albany NY). 2010; 2:344-352. doi: 10.18632/aging.100160.
50. Blagosklonny MV. Cell cycle arrest is not yet senescence, which is not just cell cycle arrest: terminology for TOR-driven aging. Aging (Albany NY). 2012; 4:159-165. doi: 10.18632/aging.100443.
51. Blagosklonny MV. Tumor suppression by p53 without apoptosis and senescence: conundrum or rapalog-like gerosuppression? Aging (Albany NY). 2012; 4:450-455. doi: 10.18632/aging.100475.
52. Demidenko ZN, Korotchkina LG, Gudkov AV and Blagosklonny MV. Paradoxical suppression of cellular senescence by p53. Proc Natl Acad Sci U S A. 2010; 107:9660-9664.
53. Kenyon CJ. The genetics of ageing. Nature. 2010; 464:504-512.
54. Liu X, Jiang W, Dubois RL, Yamamoto K, Wolner Z and Zha S. Overlapping functions between XLF repair protein and 53BP1 DNA damage response factor in end joining and lymphocyte development. Proc Natl Acad Sci U S A. 2012; 109:3903-3908.