
INTRODUCTION
Thyroid cancer is the most common endocrine malignancy [1]. Clinical outcomes vary depending on the pathologic subtype, of which there are four: papillary (PTC), follicular (FTC), medullary (MTC), and anaplastic (ATC). Differentiated tumors (papillary or follicular) are highly treatable and usually can be cured. Poorly differentiated, medullary or anaplastic tumors are much less common, more aggressive, metastasize early, and have a far poorer prognosis. While the majority of patients respond well to surgical resection, a significant number suffer from recurrence and some patients have an aggressive course and present with locally advanced disease or metastasis [2]. Anaplastic thyroid cancer (ATC) is one of the worst human malignancies, associated with median survival of only 2-3 months [3]. As the mutational landscape of thyroid cancer is being better defined through recent large-scale efforts by the NIH Human Genome Atlas Project, as well as by individual labs, targeted therapies aimed at these mutations are entering the clinical environment [4, 5].
In the current era of personalized medicine, strategies that exploit the mutational profile of individual tumors have come to the fore. Recent work in both preclinical studies and clinical trials has focused on the known driver mutations in thyroid cancer. Altered MAPK signaling pathway in many thyroid cancers leads to ERK phosphorylation, proliferation, and invasion hence BRAF inhibitors (BRAFi) and MEK inhibitors (MEKi) have been the subject of intense investigation. Various inhibitory molecules have been tested, including vemurafenib (PLX4032), which demonstrated a reasonable response in patients with progressive metastatic, radio-iodine refractory BRAF-positive PTC [6], but the response was relatively short-lived in an anecdotal report of patients with ATC [7]. Unfortunately, resistance to BRAFi therapy has proved to be a significant clinical obstacle in treating patients with melanoma, where it appears to lead to reactivation of the MAP kinase pathway within several months [8, 9]. Although few patients with aggressive thyroid cancers, such as ATC, have been treated with BRAFi therapy, resistance is expected to develop in these patients as well.
An alternative approach involving immune therapy, which previously had been rejected by most physician scientists as ineffective and cumbersome, has regained favor as a result of new knowledge about the immune checkpoint receptor, PD-1 and its ligand, PD-L1, which regulate T-cell activity in the tumor microenvironment. A role of the immune checkpoint regulators is to prevent autoimmunity and to reduce collateral tissue damage by modulating the immune response in chronic infection. Cancer cells capable of up regulating the expression of PD-L1 have the potential to become immune resistant [10], thereby evading destruction by T cells. The initial foray into the study of immune checkpoint regulators started with descriptions of CTLA-4 as a co inhibitory receptor. Since then several other immune checkpoint regulators have been identified [11]; the most widely studied has been the interaction between programmed death 1 (PD-1), a T cell co-inhibitory receptor, and its ligand, programmed death ligand 1 (PD-L1) [12].
PD-L1, also known as CD274, is a cell-surface glycoprotein of the B7 family. It is expressed on various tissues including solid tumors and can facilitate immune evasion and T cells exhaustion [13]. The interaction of PD-1 on the surface of T cells with PD-L1 on tumor cells, suppresses TCR-mediated proliferation and activation, and inhibits T cell cytolysis. As a result, increased PD-L1 expression by cancer cells is a fundamental host immune escape mechanism [14]. Many tumor cells increase PD-L1 expression in response to local factors such as Interferon-γ (IFN-γ) released from CD4 helper, activated CD8+ T cells or NK cells in the tumor microenvironment. Indeed, human tumors show a correlation between PD-L1 levels and T cell infiltration reflecting an ongoing but unsuccessful anti-tumor immune response. PD-L1 has been investigated as a potential marker for tumor aggressiveness [15, 16], and anti PD-1 and anti PD-L1 therapies in lung, renal cancer and melanoma are showing promising results [17, 18]. The critical role of PDL1-PD1 interaction has been shown in both preclinical models and clinical trials in many solid cancers but its role in thyroid cancer has been scarcely investigated and mostly in ex vivo experiments. [19–24]
Our study was designed to advance current understanding of the role of PD-L1 in thyroid cancer cells, thereby paving a path for future testing of PD-L1-based therapies in thyroid cancer patients. It is the first study to look at the expression profile of PD-L1 in a panel of nonmedullary thyroid cancer cells at baseline, after IFN-γ stimulation, and after treatment with MAP kinase inhibitors. It also represents the first attempt to determine the impact of PD-L1 antibodies, alone or in combination with BRAF inhibitor, on tumor volume in an in vivo immunocompetent murine model of anaplastic thyroid cancer. In undertaking this study, we hypothesized that PD-L1 expression in non-medullary thyroid cancer would correlate with MAP kinase signaling pathway activity, and as a result, targeted therapies that reduce MAP kinase activity, such as BRAFi and MEKi, would be found to regulate PD-L1 expression in BRAF-mutated tumors. We further proposed that blocking the interaction between PD-L1 and PD-1 with an anti-PD-L1 antibody, would have the added effect of increasing the anti-tumor activity of BRAFi-induced infiltrating T cells. In the first phase of our investigation, we studied 5 human and 4 murine thyroid cancer cell lines to determine baseline expression of PD-L1. Next, we investigated the effect of manipulating MAP kinase activity on PD-L1 expression in vitro and in vivo. Finally, we tested the effect of combining BRAFi and anti-PD-L1 antibody on tumor regression and intra-tumoral immune response in an orthotopic immunocompetent mouse model of ATC.
RESULTS
Thyroid cancer cell lines with the BRAFV600E mutation express higher baseline levels of PD-L1 mRNA compared with BRAFWT
Given the known therapeutic implications of PD-L1 expression in melanoma, PD-L1 mRNA levels in 6 human thyroid cancer cell lines was quantified against 3 BRAFV600E melanoma cell lines (A375,A2058 and UACC903) and one BRAFWT (MelJuso) (Figure 1). BRAFV600E mutant thyroid cancer cell lines showed significantly higher baseline expression of PD-L1 than the BRAFV600E -mutant melanoma cell lines; with 8505c cells showing the highest expression at 93-fold compared with A375 melanoma cells. Thyroid cell lines with the BRAFV600E mutation also showed significantly higher baseline expression of PD-L1 mRNA compared with BRAFWT thyroid cells (P<0.05). In fact, the normal HTORi cell line had the lowest expression of PD-L1. Western blot analysis pointed toward higher PD-L1 protein expression in the mutant BRAF cells compared with wild type across all cell lines investigated.
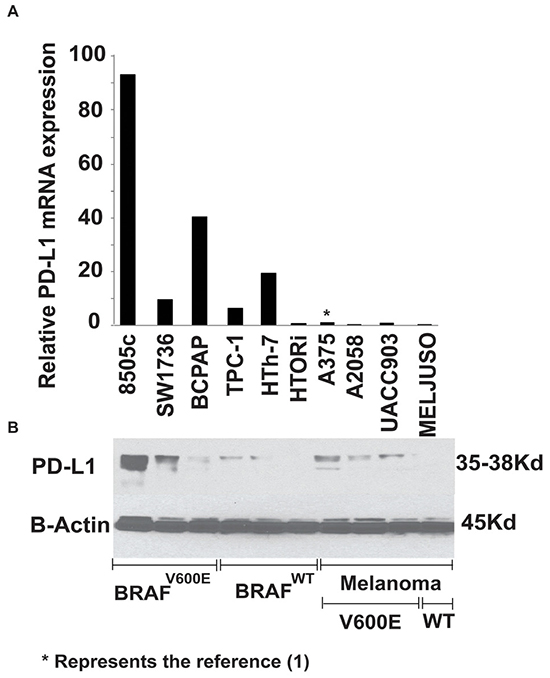
Figure 1: PD-L1 mRNA A. and protein B. expression of different human thyroid and melanoma cell lines. The graph is representative of 3 different experiments using technical triplicate and is plotted as fold change normalized to the expression of A375. PD-L1 mRNA expression of BRAFV600E (8505c, BCPAP, SW1736) thyroid cell lines showed higher base line expression compared to BRAFWT cell lines (TPC-1, HTh-7 and the normal thyroid cells HTORi) (Mann-Whitney U, P≤0.05), thyroid cells as a group showed higher base line expression compared to 4 melanoma cell lines (A375, A2085, MEL-Juso and UACC-903) (Mann-Whitney U, P≤0.05). Western blot (B) analysis showed protein levels corresponded to mRNA levels in thyroid cells.
BRAFV600E mutated PTC tumors from patients showed higher PD-L1 expression compared to BRAFWT tumors
To determine whether tissue from patients with BRAFV600E-mutated tumors also expressed higher levels of PD-L1, PD-L1 mRNA expression levels were analyzed in randomly selected 28 fresh frozen PTC tumors and their matched normal thyroid tissue samples. Fifty-seven percent had BRAFV600E mutations on routine sequencing. None of the demographic or tumor characteristics were significantly different between the BRAFV600E (n=16) and the BRAFWT (n=12) groups (Table 1). PD-L1 mRNA expression levels in the normal thyroid were set to one for analysis purposes, and log2 of fold changes was used for plotting the box graph. The BRAFV600E-mutated tumors showed significantly higher expression of PD-L1 mRNA compared to BRAFWT tumors (log2 of fold changes: median (inter quartile range): 0.51 (-0.05 to 1.04) vs. -0.70 (-2.24 to 0.28) (P = 0.015) (Figure 2).
Table 1: Patient and tumor characteristics
Total n=28 |
BRAFV600E n=16 |
BRAFWT n=12 |
P value |
|
|---|---|---|---|---|
Female Sex (%) |
16 (57) |
10 (63) |
6 (50) |
0.70 |
Age [year](range) |
53 (29-84) |
53 (35-84) |
51 (29-76) |
0.88 |
Tumor size (%) |
0.45 |
|||
T1-2 |
15 (54) |
10 (63) |
5 (42) |
|
T3-4 |
13 (46) |
6 (37) |
7 (58) |
|
Lymph node status (%) |
1.00 |
|||
Nx/N0 |
18 (64) |
10 (63) |
8 (67) |
|
N1 |
10 (36) |
6 (37) |
4 (33) |
|
TNM stage (%) |
0.53 |
|||
1 |
7 (25) |
4 (25) |
3 (25) |
|
2 |
2 (7) |
1 (6) |
1 (8) |
|
3 |
13 (47) |
9 (56) |
4 (33) |
|
4 |
6 (21) |
2 (13) |
4 (33) |
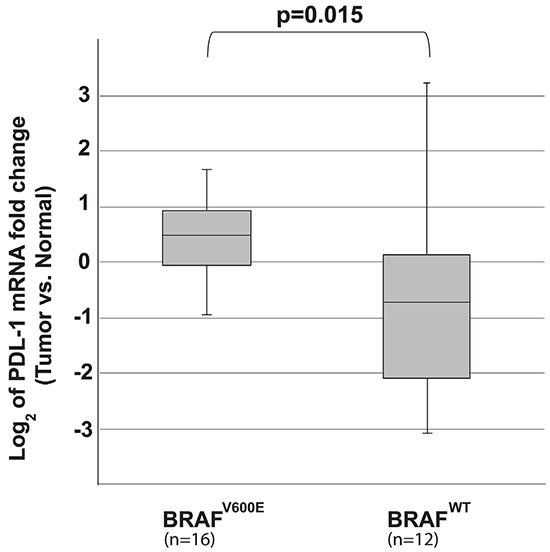
Figure 2: The log2 of the PD-L1 mRNA fold change of tumor vs. adjacent normal tissue in 28 patients. Patients samples carrying BRAFV600E (n=16) showed higher induction of PD-L1 compared to BRAFWT tumors (n=12) (medians: 0.51 vs. -0.70, P = 0.015).
PD-L1 expression in human thyroid cancer cell lines is dependent on MAP kinase pathway activation
Generally, infiltrating T cells in the tumor stroma produce IFN-γ, which is a potent inducer of PD-L1 expression on tumor cells, thus potentially diminishing the anti-tumor immune response [25–27]. In our thyroid cancer cell line panel, IFN-γ appropriately increased both mRNA and protein expression of PD-L1 in all cell lines (Figure 3A).
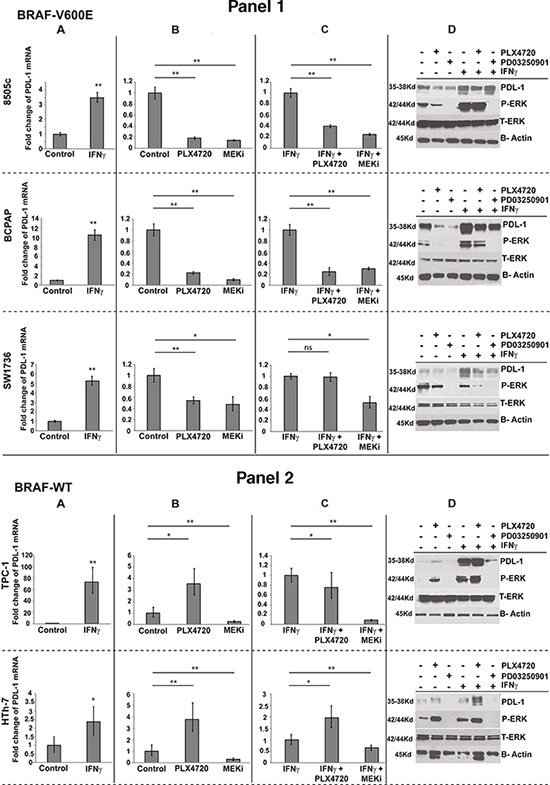
Figure 3: Five human thyroid cancer cell lines were treated for 24 hrs with A. IFNγ. alone B. PLX4720 or PD03250901 alone or in combination with IFNγ C. Quantitative PCR fold change of PD-L1 mRNA was normalized to control (A, B) or to IFNγ treatment state (C). D. Western blot analysis for PD-L1, total ERK, phospho ERK and β-actin were done for all treatment combination mentioned above (* indicate p<0.01, ** indicate p<0.001, ns- not significant).
To assess whether MAP kinase pathway activity alters PD-L1 expression, we measured PD-L1 expression in our thyroid cancer cell line panel after treating with BRAFi PLX4720 or MEKi PD0325901. All cells responded appropriately to MEK inhibition, showing a decrease in pERK. Similarly, BRAF inhibitor decreased pERK in cells with BRAFV600E mutations (Figure 3 Panel 1,D), but as expected, caused a paradoxical increase in pERK in the BRAFWT cancer cells (Figure 3 Panel 2,D) [28]. Both pERK and PD-L1 both mRNA (Figure 3B) and protein (Figure 3D) expression were significantly reduced in all cells treated with MEKi, regardless of mutation status. In contrast, when treated with BRAFi, mutation status of the cell was important in determining the changes in PD-L1 expression. While treatment with BRAFi caused significant decreases in both pERK and PD-L1 expression in BRAFV600E cells (Figure 3 Panel 1B, D), the BRAFWT cells treated with BRAFi showed the expected paradoxical increases in pERK which was accompanied by increases in PD-L1 expression (Figure 3 Panel 2B, D). BCPAP showed the largest reduction when exposed to MEKi in comparison with untreated control, with a 10-fold decrease of PD-L1 expression, and 8505 cells showed a 6.9-fold decrease. Western blot analysis demonstrated tight correlation between the mRNA and protein expression of PD-L1 through the different treatments and tight correlation of PD-L1 protein expression with MAP kinase phosphorylation status (Figure 3D).
When cells were treated in the context of stimulation by IFN-γ, similar trends were seen depending on the mutation status of the cells. Treatment with MEKi in the presence of IFN-γ resulted in similar impressive decreases in PD-L1 mRNA and protein expression in all cell lines regardless of mutation status (Figure 3C, D). Treatment with BRAFi in the presence of IFN-γ resulted in significant decreases in PD-L1 mRNA and protein levels in two of the BRAFV600E-mutated cell lines (8505 and BCPAP), whereas in the SW1736, only decreases in PD-L1 protein expression were observed. In BRAFWT cells treated with PLX4720 in the presence of IFN-γ, consistent increases were only observed in the protein levels of PD-L1. Significant increases in PD-L1 mRNA were observed only in the HTh-7 (ATC) cell line (Figure 3C, D).
MAP kinase pathway inhibition decreases PD-L1 expression in human thyroid cell lines in vivo
To test whether BRAF inhibition also causes a decrease of PD-L1 expression in vivo, SCID mutant mice bearing 8505c and BCPAP orthotropic tumors were treated with BRAF inhibitor for two weeks, and PD-L1 mRNA and protein expression levels were subsequently measured (Figure 4A). Treatment with PLX4720 significantly reduced in vivo expression of PD-L1 mRNA in both tumor models; two-fold for 8505c and 37-fold for BCPAP. PD-L1 protein levels as measured by immunohistochemistry also showed a significant decrease in both tumors (Figure-4B).
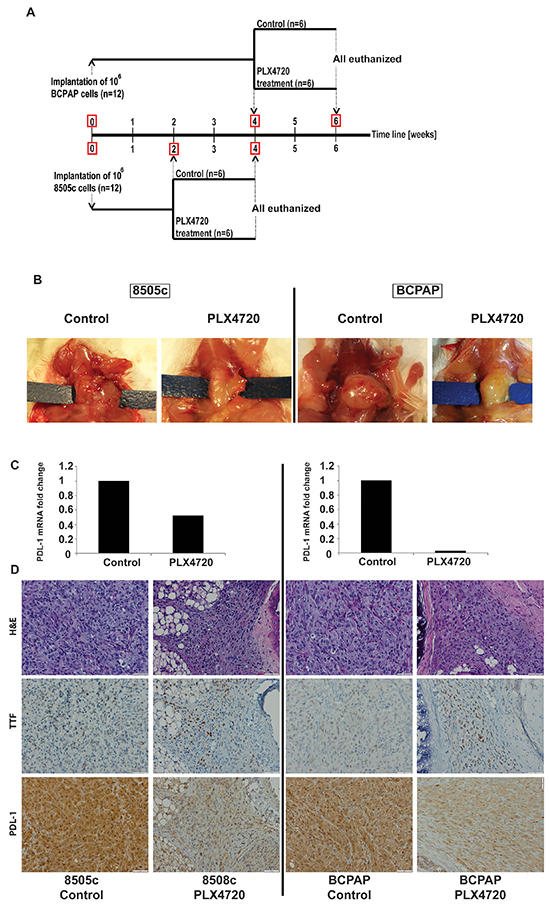
Figure 4: Schedule for the experiment using Orthotropic SCID mouse model for 2 human thyroid cancer cell lines (8505c; ATC derived, and BCPAP; PTC derived cells) is outlined in A. In vivo results of PLX4720 treatment for both cell lines showing obvious tumor volume reduction B. PD-L1 mRNA expression of tumors pooled for each group showing significant reduction of PD-L1 (2 and 37 fold for 8505c and BCPAP respectively) when treated with PXL4720. C. IHC analysis for human PD-L1 showed a marked reduction of staining with PLX4720 treatment for both cell lines D.
BRAF inhibition does not regulate PD-L1 expression in BRAFV600E murine thyroid cancer cell lines
We checked the PD-L1 mRNA expression of genetically engineered murine thyroid cancer lines (3601R, 3868, 3743 and 3403) at baseline and in response to PLX720, IFN-γ, or both. Although IFN-γ caused a dramatic increase of PD-L1 expression; 3601R (93.7-fold), 3868 (13.9- fold), 3743 (74.8-fold), 3403 (20.2-fold) as compared to untreated control, none of the four cell lines showed significant (>2-fold) change in PD-L1 expression in the presence of BRAFi (Figure 5A), regardless of the presence of IFN-γ. Cell surface analysis of PD-L1 protein expression using FACS was consistent with this observation (Figure 5B).
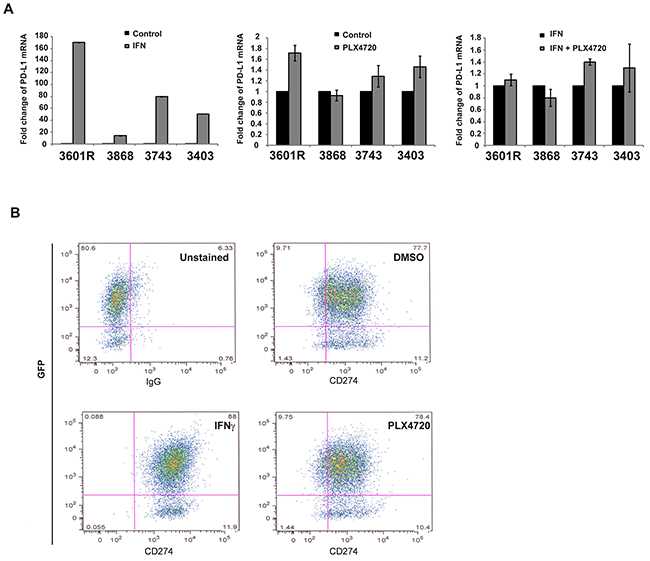
Figure 5: Murine cell lines were treated with PLX4720, IFNγ and the combination of the two. PD-L1 mRNA expression was measured against GAPDH expression and compared to non treated cells. While IFNγ robustly increased PD-L1 mRNA expression in all cell lines, none of the four cell lines exhibited a significant change in PD-L1 expression (<2 fold change) when treated with PLX4720 (compared to control) or with the combination of IFNγ + PLX4720 (compared to IFNγ alone) A. Cell surface analysis of PD-L1 using FACS showed a saturation of PD-L1+ cells while PLX4720 treatment did not differ from DMSO alone. The same result was seen for all four murine cell lines. A representative of one cell line (3743) is shown B.
Combination of anti-PD-L1 antibody with PLX4720 was highly effective in reducing tumor volume in an immunocompetent orthotopic mouse model of thyroid cancer
We used our previously described immunocompetent mouse model of ATC in which 105 genetically engineered murine ATC; TBP 3743 BRAFV600E/WT P53−/− cells are implanted into an immunocompetent mouse thyroid, resulting in a very rapidly enlarging tumor with marked infiltration of CD8+ T lymphocytes when treated with PLX4720 [29]. Given the rapid nature of this model (life expectancy of control animals is two weeks), one week after tumor implantation, the mice were randomized to treatment with either anti-PD-L1 Ab, PLX4720, or a combination of both agents (see schema in Figure 6A). All animals were euthanized and tumor volume was measured after 8 days of treatment (i.e., 14 days post-implantation). Control animals had a median tumor volume of 782±175 mm3. Treatment with anti-PD-L1 Ab alone did not significantly reduce tumor volume (717±62mm3, p=1.00), whereas PLX4720 treatment resulted in significant tumor volume reduction (439±188 mm3, 44% tumor reduction, P = 0.007). Using the Wald chi-square for statistical analysis showed that the combination of PLX4720 and anti-PD-L1 antibody acted synergistically, and was the most effective in reducing tumor volume compared to all other treatment groups (147.3±60.8 mm3, showing a dramatic 81% tumor volume reduction compared to control (P < 0.001) and impressively over 66% better than those treated with PLX4720 alone (P=0.023) (Figure 6B, 6C).
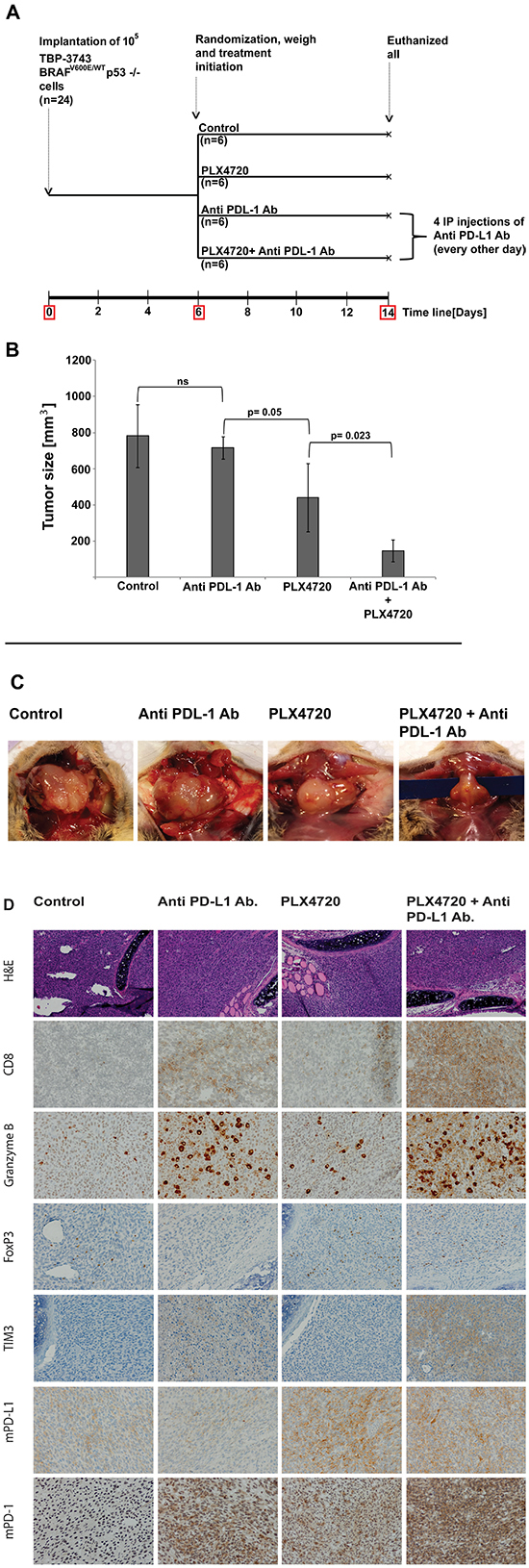
Figure 6: Schedule for the experiment using immunocompetent orthotropic mouse model implanted with murine cells 3473 A. TBP: TPOCreER; Braf tm1Mmcm/WT; Trp53tm1Brn/tm1Brn (TBP). IP: intraperitoneal. Tumor volume represents in cubic millimeters as mean±SD. The combination of PLX4720 and Anti PD-L1 monoclonal antibody had a dramatic effect on tumor volume reduction compared to control and to either treatment alone (147±61g, P<0.05) B, C. The presence of CD8+-CTL, the cytotoxicity marker; Granzyme B, Treg marker; FoxP3, T cell exhaustion marker; TIM3 and the expression of mouse PD-L1 and mouse PD-1 were analyzed using IHC. CD8+-CTL infiltration and cytoxicity were evident with anti PD-L1 treatment alone and were intensively stained when combined with PLX4720. The exhaustion markers TIM3 and PD-1 were evident with anti PD-L1 treatment but were highly induced when combining with PLX4720 D.
Blockade of the PD-L1 – PD-1 axis resulted in increased CD8+ T-cell infiltration and improved the cytotoxic profile of the T cells
To investigate the influence of anti-PD-L1 treatment alone and in combination with BRAFi on T-cell infiltration, the treated tumors from the immunocompetent mice were immunostained for CD8; TIM3, a marker of CD8+ cell exhaustion; and Granzyme B, a marker of T-cell cytotoxicity. Tumors from animals treated with PD-L1 blockade or PLX4720 alone had visibly higher infiltration of CD8+ T cells compared to controls; in the tumors treated with the BRAF inhibitor most of the T cells appeared to have infiltrated into the peripheral aspects of the tumor. Combining both drugs, however, dramatically increased CD8+ T-cell infiltration throughout the tumor (Figure 6D). Granzyme B staining is thought to show T cells involved in acute cytotoxicity, and again, the Granzyme B staining was most dramatically increased in the combinatorial treatment (Figure 6D). Patterns of T cells showing expression of TIM3 exhaustion markers were similar, with the highest levels of exhausted T cells seen in the combinatorial treatments.
T-regulatory cells (Tregs) have been shown to selectively interfere with the release of cytolytic granules by cytotoxic T lymphocytes (CTLs) in a reversible and TGFβ-dependent manner, thereby attenuating CTL-mediated cytotoxicity. Previous studies have shown that the CD8:Treg ratio is an important indicator of therapeutic benefit in melanomas, where potent synergistic effects of combination therapy using BRAFi and PD1 blockade cause increased CD8+:Treg ratio and enhanced cytokine production, compared with BRAFi alone in both mice and humans [30]. Indeed, in our model as well, we found that while PLX4720 alone appears to increase the infiltration of CD8+ T cells into the periphery of the tumor, unfortunately there was also a moderate increase in FoxP3 T regulatory cells. However, the combination of BRAFi and PD-L1 antibody yielded an obvious increased CD8+:Treg ratio in the tumor, which should be beneficial (Figure 6D).
The baseline expression of mouse PD-L1 (mPD-L1), as seen in the IHC staining of the control group, was observed to increase dramatically upon treatment with PLX4720 alone; while treatment with anti-PD-L1 antibody alone or in combination with BRAFi reduced the IHC staining expression of mPD-L1 as compared with control and BRAFi alone, respectively.
DISCUSSION
During the last decade, most thyroid cancer therapeutics have targeted known oncogenic mutations, such as BRAFV600E, in an effort to alter thyroid cancer progression. Unfortunately, the response to these efforts to inhibit the MAP kinase pathway has been relatively short-lived as a consequence of the development of resistance to treatment. Coincident with this work, immune therapy, has regained favor as a result of new knowledge about the immune checkpoint receptor, PD-1 and its ligand, PD-L1, which regulate T-cell activity in the tumor microenvironment. In particular, studies involving the use of monoclonal antibodies directed against the PD-1 receptor and its ligand PD-L1 have shown promising response rates, leading to FDA approval of PD-1 drugs for advanced melanoma, lung and renal cancer [31, 32]. One exciting application of this approach is being tested clinically to determine which combinations of BRAF or MEK inhibitors, coupled with immunotherapy, will generate the more sustained tumor response.
In melanoma patients, the initiation of BRAFi therapy causes rapid T-cell infiltration into tumor, but the T cells are rendered ineffective after two weeks, probably resulting from a surge of expression of PD-L1 on the tumor cells [33]. This finding suggests that PD-1 pathway blockade might prolong the immune response. Moreover, by combining BRAFi and PD-1 pathway blockade, it might be possible to significantly augment and prolong the therapeutic response to BRAF inhibitors.
This approach might also be the logical next step in thyroid cancer, given the deregulation of the MAP kinase pathway in many aggressive thyroid cancers, but little is known about the baseline characteristics of these immune regulators in thyroid malignancy. Furthermore, as clinical trials in other malignancies mature, it is becoming increasingly clear that toxicity is a major barrier to treatment. Additional preclinical data providing mechanistic insight into the possible beneficial synergistic and toxic effects of these combinations in the various tumor components is essential.
The presence of PD-L1 in thyroid tumors was first described in 2003 by Brown et al. [24]. Subsequently, the role of PDL1-PD1 interaction in thyroid cancer has only been investigated in a handful of in vitro experiments and through staining of patient tissue samples; Cunha et al demonstrated higher levels of PD-L1 in papillary thyroid cancer tissue samples compared to normal and benign lesions and showed a positive correlation between PD-L1 levels and infiltration of CD8+, CD4+, FoxP3+ lymphocytes and tumor associated macrophages [19, 20]. French and colleagues found PD-1+ T lymphocytes to be enriched in metastatic lymph nodes especially in recurrent disease and were markedly associated with extra nodal invasion [21]. Interestingly, this same group recently suggested that tumor associated lymphocytes, taken from grossly involved lymph nodes, exhibit incomplete exhaustion which may thus be reversible [22]. Given that in patients, the immune system itself does not appear capable of actively engaging and destroying metastatic deposits in lymph nodes, testing combinations of therapeutics that inhibit the suppressive activity of Tregs may be valuable. The MAP kinase pathway-dependent regulation of PD-L1 in thyroid cancer was implied when a positive correlation between BRAFV600E and high level of PD-L1 in 33 PTC samples was recently reported [23]. Both BRAF and PD-L1 expression was tested in 13 patients with ATC, 3 patients were found to have BRAFV600E mutations, and 3 had high expression of PD-L1, there was one patient with both [34].
In this study, we have established that thyroid cancer cell lines and tumor samples from patients with BRAFV600E -mutated tumors have higher levels of PD-L1 compared with either BRAFWT tumors or matched normal tissue. Additionally, we have validated the combination of PD-L1 blockade and BRAF-targeted therapy in a novel immunocompetent model of ATC using rapidly growing genetically engineered BRAFV600E-mutated tumors. Our model is clinically relevant on multiple fronts. First, the genetically engineered cell lines have both BRAF and p53 mutations allowing for treatment to be initiated when the tumors are large and still rapidly growing. Second, the immune competent nature of our model permits us to study the immune milieu inside the tumor. Our studies did show that PD-L1 blockade has very minimal effects on the tumor when given alone, despite fairly impressive infiltration of CD8+ T cells and high activity of Granzyme B. This finding is not entirely unexpected, however, given the aggressiveness of this specific ATC model in which more conventional agents such as BRAF or Src inhibitors were less effective [35]
In contrast, the combination of PD-L1 antibody and BRAF inhibitor produced a powerful synergistic response. The synergistic improvement in tumor shrinkage in those animals treated with a combination of BRAFi and PD-L1 antibody was also associated with an increase in tumor-infiltrating lymphocytes (TIL) and TIL function. The mechanisms underlying TIL infiltration were not studied here. It could be the result of clonal expansion or increased trafficking of T cells into the tumor. Previous studies in melanoma animal models and patient trials indicate that both mechanisms are likely at play [33]. Clearly, combined therapy with BRAFi and PD1 blockade caused an increase of the CD8+:Treg ratio, which favors cytokine production and destruction of tumor cells. In fact, the observed increase of Granzyme B activity, signaling increased T-cell cytotoxicity, in tumors treated with combination therapy also supports this view. Our results are consistent with several other published studies showing synergy of BRAF inhibitors with other immunotherapies in murine models [36, 37].
Little is known about the effects of immune checkpoint regulators in thyroid cancer. The therapeutic effect of blocking either PD-1 or PD-L1 previously has not been tested. PD-L1 is expressed by multiple cell types in tumors, thus underscoring the importance of these in vivo studies. PD-L1 expression in lymphoid organs may suppress the initial activation of T cells or perhaps even induce Tregs. PD-L1, expressed on both tumor and non-tumor cells has the potential to suppress antitumor immunity by contributing to T-cell exhaustion.
Treatment with MAP kinase inhibitors, such as BRAFi and MEKi, can result in complex interactions given the varied effects of these inhibitors on immune cells and tumor cells (Figure 7). In this study we show that treatment with MEK inhibitor consistently decreased PD-L1 mRNA and protein expression levels in vitro. Both thyroid cancer cell lines and tumor samples from patients with BRAFV600E-mutated tumors had higher levels of PD-L1 compared with either BRAFWT tumors or matched normal tissue.
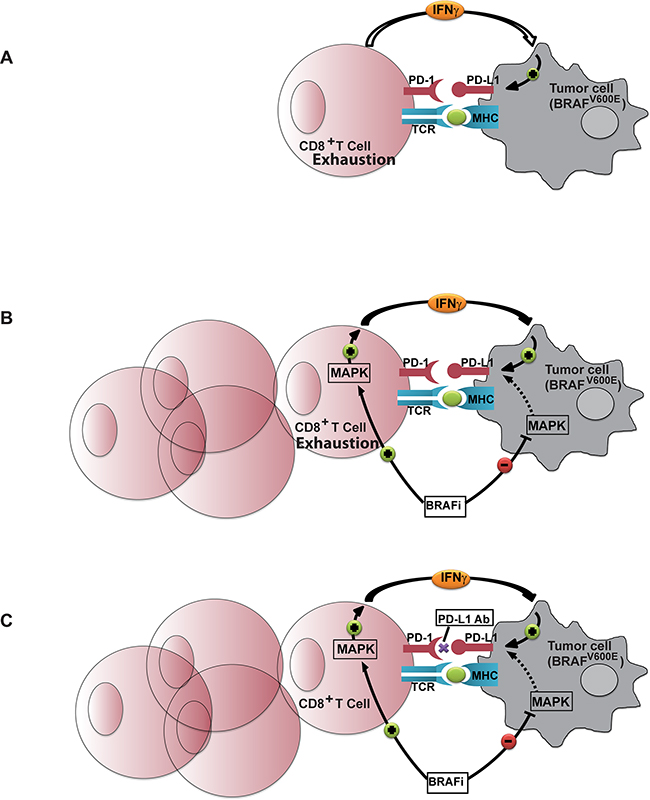
Figure 7: The rational for adding Anti PD-L1 monoclonal antibody to BRAFi treatment. A. In the naïve environment the lack of migration of T-cells into the tumor site and the mild reactivity that drives T-cells to produce cytokine (among them IFNγ) has substantial impact on the expression of PD-L1 which tempers the already weak anti tumor reaction. B. BRAFi treatment results in increased T cell infiltration into the tumor microenvironment. Although BRAFi potentially down-regulates the expression of PD-L1 on BRAFV600E tumor cells by down regulating MAP kinase activity in these cells, it paradoxically up-regulates MAP kinase in the BRAFWT T-cells which results in IFNγ production and intensive up-regulation of PD-L1 on tumor cells. Immune response with BRAFi alone therefore does not come to its full potential. C. Adding anti PD-L1 antibody is an additional step necessary to allow the immune response facilitated by BRAFi to reach its full potential.
Accumulating evidence points to a relationship between MAP kinase signaling activation and the induction of PD-L1 expression [33, 38, 39]. Our study exploits the opposing effect of BRAFi on cells to prove this relationship. It has been reported that BRAF inhibitors block the MAP kinase signaling pathway in BRAFV600E cells, whereas in BRAFWT tumors, BRAFi activates the RAF-MEK-ERK pathway and could enhance tumor growth in xenograft models [40, 41]. Moreover, our lab previously showed persistently higher levels of phospho-ERK-1/ ERK-2 protein when TPC-1 (BRAFWT) cells were treated with PLX4720 [28]. We blocked the hyperactive pathway in BRAFV600E cells with BRAFi and MEKi and we activated and blocked the same pathway in BRAFWT cells with BRAFi and MEKi, respectively. We demonstrated tight correlation between MAP kinase activity (as measured by ERK phosphorylation) and the expression of PD-L1 both in the mRNA and protein levels. IFNγ, a known and potent upregulator of PD-L1, abrogated the suppression that BRAFi imposed on PD-L1, but it was MAP kinase activity that consistently correlated with fine differences of expression as exhibited in our Western blot results.
The combination of BRAFi and immunotherapy is gaining acceptance as a rational approach to cancer treatment, and is undergoing clinical investigation in melanoma [42], where BRAFi has been shown to evoke tumor CD8+ T cell infiltration in melanoma patient samples [33, 43] and in thyroid mouse model [35]. Atefi et al. did point to the effect that BRAFi has on T cells when exposed simultaneously to PD-L1 [44]. These investigators showed that exposure of lymphocytes (which do not carry the mutant BRAF) to PD-L1 decreased activity of the MAP kinase pathway and lowered the production of cytokines. With the addition of vemurafenib (BRAFi), T cell-MAP kinase activity and cytokine production were restored. This surge of cytokine release, including IFNγ, has a potent effect on PD-L1 expression which in turn dampens the full potential of BRAFi on anti-tumor immunity.
Our previously established mouse model served as an ideal platform to explore the effect of PD-L1 blockade in the presence of BRAFi. On the basis of the above rationale, we hypothesized that a synergistic clinical and immunologic effect would be observed by combining both approaches. Indeed, our results showed dramatic tumor shrinkage when anti-PD-L1 Ab and BRAFi were combined compared to BRAFi alone. IHC analyses demonstrated an intense and dense infiltration of CD8+ T cell staining for cytotoxic marker. Moreover, the blockade of PD-L1 mitigated the effect of CD8+ T cells on the number of FoxP3 T regulatory cells in the tumor microenvironment. Multiple previous studies have shown that tumor-specific CD4+FoxP3 Tregs express multiple inhibitory receptors, including PD-1 and TIM3, which likely reduce the capacity of T cells in the tumor microenvironment for proliferation and cytokine production [45].
It is noteworthy that PD-L1 blockade and not BRAFi resulted in higher expression of the exhaustion marker, TIM3. This observation was not consistent with Fredrick et al., who reported increased expression of TIM3 with BRAFi treatment [33]. Ascribing the intensity of TIM3 solely to the intensity of the immune infiltrate would be far too simplistic. The intracellular signal that drives T helpers or CD8+ cells to over express TIM3 is not clear, but its co-expression with PD-1 on tumor specific CD8+ T cells makes it an important future candidate for combined immunotherapy [46–48]. Nevertheless, in the environment of PD-L1 blockade, increased TIM-3 and PD-1 expression may reflect the higher level of T cell activation rather than be an exhaustion marker. Given that the immune system, itself, does not appear capable of actively engaging and destroying metastatic deposits in lymph nodes in patients, testing combinations of therapeutics that inhibit the suppressive activity of Tregs may also be a fruitful line of investigation.
Our study does have a number of important limitations. Although the toxicity and tolerability profile of anti-PD-1/PD-L1 in clinical trials is promising [17, 49], our study was limited by the short duration of treatments as enforced by the rapid tumor growth in our model making it difficult to determine possible toxicity.
Adverse events (AE) related to anti PD-1/PD-L1 antibody as a single therapy and to a greater extent when combined with anti CTLA-4 or BRAFi are well documented in patients with melanoma and lung cancer [50, 51]. Most of the documented AE are low grade and include elevated liver function tests, pneumonitis and other immune-related adverse events that mainly involve the gut, skin, endocrine glands, liver, and lung. This combination of BRAFi and anti PD-1/ PD-L1 was investigated in melanoma murine model with no documented treatment-related toxicity [30].
Since previous studies have not established whether tumors that have been growing slowly over time versus those that grow more aggressively, such as in our model, alter the native immune response to the tumor, it is not entirely clear where our model fits in the spectrum. In addition, genetically engineered tumor cell lines (such as ours) may have a smaller mutation burden compared to the equivalent native tumor. It is not entirely clear whether the immune response in the genetically engineered models is a true representation of the immune response in human tumors. Since thyroid cancers generally lie on the low end of the mutation burden, this might be less of an issue in thyroid cancer than other genetically engineered models used for preclinical studies [52]. Future murine tumor models with a longer pre-established therapeutic window for anti-PD-L1 will give better insight into survival patterns and perhaps a more accurate picture of its effect on tumor volume. In addition, failure of native T cells to clear tumors as they slowly grow is likely not entirely due to the PD-1/PD-L1 interaction. Here, we did not investigate the role of PD-L2, though in general it appears to have more restricted expression, is less prevalent across tumor types and thus does not appear as important in tumor’s immune evasion [13]. However, exceptions do exist and in esophageal cancer, it appears that changes in PD-L2 levels driven by Interleukin-4 feedback loops, is a major part of the suppression of the immune response [53]. In our study, we also did not address possible significant impact of macrophages. Macrophage function is known to be a significant component of human ATC [54], and yet our previous analysis of our immunocompetent ATC model did not show a similarly large influx of macrophages; thus we did not further analyze the macrophage component in this study.
The fact that the study of tumor immunity has gone from a fringe effort to a viable and creative option in a wide range of tumors led us to take the first exploration in aggressive thyroid cancers. Our study is unique for its utilization of an immunocompetent in vivo model that permits exploration of the various players of antitumor immunity in the growing tumor. More than the exciting and robust immune infiltration that occurs when BRAFi and anti-PD-L1 work synergistically; it is obvious from the IHC results of our control group that aggressive thyroid tumors potentially grow without propagating anti tumor immune response. This observation, although hardly new, is striking given the robust immune infiltration when treatment with BRAFi and combinatorial therapies are implemented. Evidence from other malignancies, along with this preclinical study, suggests promise for strategies that combine immunotherapy with targeted therapy in thyroid cancer patients. The scarce number of aggressive thyroid cancer patients treated with BRAFi and the lack of ex vivo samples from these patients is a missing piece in the puzzle. We hope that in the near future, as clinical trials with targeted therapies and immune checkpoint regulators are developed for thyroid cancer, that pre and on treatment biopsy samples will be obtained from these patients to allow detailed analysis of the immune environment inside the tumors. This will allow a better understanding of the immune landscape of those who respond versus those who do not.
MATERIALS AND METHODS
Cell culture, reagents and antibodiess
Six human thyroid cancer cell lines; 8505c (Deutsche Sammlung von Mikroorganismen und Zellkulturen), BCPAP (previously provided by Dr. G. Damante of the University of Udine, Italy), SW1736, TPC-1 (provided by Dr. F. Frasca, University of Catania, Catania, Italy), HTh-7 and HTORi; four murine thyroid cancer cell lines; 3601R, 3868, 3743, 3403 [55], and four human melanoma cell lines; A375,A2058, UACC903 and MelJuso, were used in the study. All cell lines were cultured in Dulbecco’s Modified Eagle’s Medium (DMEM) supplemented with 10% fetal bovine serum and penicillin/streptomycin and incubated at 37°C in a 5% CO2 incubator. Cells were plated at 70-80% confluence and treated for 24 hours with either 10μM PLX4720 (Plexxikon, Berkeley, CA), 10μM PD03250901 (LC Biolabs, Woburn, MA), or 500U/ml hIFNγ for human cells and 200ng/ml mIFNγ for murine cells (BD Biosciences, San Jose, CA) either as single agents or in combination as specified in the results section. At the end of the treatment, cells were collected for RNA or protein isolation. Cell characteristics and mutational status are presented in Table 2.
Table 2: Cell line characteristics and their mutational status
Cell line |
Type |
Important mutations and deletions |
|---|---|---|
8505c |
ATC |
BRAFV600E/-, P53 mutation |
BCPAP |
PTC |
BRAFV600E/WT, P53 mutation |
SW1736 |
ATC |
BRAFV600E/WT, P53 mutation |
TPC-1 |
PTC |
BRAFWT, RET/PTC, PIK3A |
HTh-7 |
ATC |
BRAFWT, PTEN−/−, NRAS |
HTORi |
NT |
BRAFWT |
A375 |
MEL |
BRAFV600E/WT, P53 mutation |
A2058 |
MEL |
BRAFV600E/WT, PTEN−/−, P53 mutation |
MEL-JUSO |
MEL |
BRAFWT, RET/PTC, NRAS |
UACC-903 |
MEL |
BRAFV600E/WT, PTEN mutation |
NT - Normal thyroid, MEL - Melanoma, ATC – Anaplastic Thyroid Cancer, PTC - Papillary Thyroid Cancer.
In preparation for these studies, a dose-response curve for hIFN-γ treatment was generated and a working concentration of 500U/ml, the minimum dose required to reach a saturated response for the 24-hour treatment, consistent with previously validated concentrations (data not shown), was selected [56].
phospho-p44/42 MAPK (ERK1/2) (Thr202/Tyr204) (phospho-ERK), p44/42 MAPK (ERK1/2) (Total ERK) and β-actin were purchased from Cell Signaling Technology.
The anti-PD-L1 antibody (10F.9G2) has been described previously [57], had less than 2EU Endotoxin per mg protein and was injected intraperitonealy (I.P) as described later.
RNA isolation and real-time polymerase chain reaction
RNA isolation was performed using Trizol (Invitrogen, Carlsbad, CA), and the cDNA was synthesized using the Superscript VILO cDNA synthesis kit (Invitrogen) according to the manufacturer’s instructions. PD-L1 gene expression was measured using TaqMan Gene Expression Assays (Invitogen, Primer i.d Hs-01125301_m1, Mm00452054_m1) by real-time reverse transcriptase polymerase chain reaction (RT-PCR) with technical triplicates and repeated at least twice. Gene expression levels were normalized to GAPDH expression.
Protein extraction and western blot
At the end of treatment, cells were washed twice with PBS and lysed rapidly in RIPA lysis buffer containing protease and phosphatase inhibitor cocktails (Thermo Scientific, Rockford, IL) by incubating on ice for 10 minutes. Total lysates were centrifuged at 15,000 G for 15 minutes at 4°C, and supernatants collected and quantified using Bradford protein assay kit (Pierce, Rockford, IL). Western blot analysis was performed using standard procedure, and the proteins were identified using specific antibodies; total ERK (tERK), phosphorylated p42/44 ERK (pERK), β-actin and PD-L1.
Orthotropic thyroid cancer mouse models of human ATC and PTC cell lines
All animal experiments were conducted at the Massachusetts General Hospital in accordance with federal, local, and institutional guidelines. To test PD-L1 expression and response to BRAFi therapy using human thyroid cancer cells: 24 ten-week-old female SCID mutant mice were injected with human thyroid cancer cells (12 with BCPAP and 12 with 8505c) as previously described [58]. Briefly, mice were anesthetized, the thyroid gland exposed, and 106 cells injected into the left thyroid gland using a 27-gauge needle. The right side of the thyroid gland was not manipulated and was used as an internal control. Mice were randomized and started treatment at two weeks for the 8505 and 4 weeks for the BCPAP-implanted groups (see schema in Figure 4A). Six mice in each group received PLX4720-impregnated chow (Research Diets, Inc.) ad libitum while the other six were fed control diet (Research Diets, Inc.). After 14 days of treatment, mice were euthanized and both fresh and formalin-fixed tumors were collected for RNA isolation and IHC analysis, respectively.
Immunocompetent mouse model of murine ATC using Anti-PD-L1 Ab
To test tumor growth and anti-tumoral immune indices in response to PD-L1 – PD1 blockade, with or without BRAFi therapy, we used the genetically engineered murine ATC cell line in an immunocompetent setting. First we implanted 105 of the TBP (TPOCreER; Braf tm1Mmcm/WT; Trp53tm1Brn/tm1Brn) 3747 BRAFV600E/WT P53−/− murine tumor cell line in 24 immunocompetent syngeneic B6129SF1/J mice as previously described [29, 35]. In this immunocompetent model, the tumor progresses quite rapidly, and by 2 weeks the tumors have reached lethal size. Thus, one week after tumor implantation, the mice were randomized into 4 treatment groups. The control group received control chow (n=6), the PLX-treated group received PLX4720-impregnated chow (n=6), the anti-PD-L1 antibody group (n=6) received control diet and was injected with anti-PD-L1 antibody (10F.9G2) at 200ug/mouse/I.P dose every other day (4 total doses), and the combined treatment group received both PLX4720 chow and anti-PD-L1 antibody (4 total doses) (n=6). On day 14, all groups were euthanized and tumors collected. Tumor volume was calculated using the formula: (π/6) x length x width x height. tissue samples were collected and processed for IHC analysis [59].
Immunohistochemistry
4-μm sections of the formalin-fixed orthotropic thyroid tumors were stained with Hematoxylin and Eosin stain and underwent IHC analysis for CD8 (Leica Microsystems), FoxP3 (eBioscience), TIM3 (Millipore), Granzyme B (abcam), mouse PD-L1 (clone 10F.9G2) (Provided by GF), and human PD-L1 (ProSci-Iinc Poway CA) primary antibodies. The primary antibody was detected using the respective biotin-free secondary antibody.
Patient samples
Using the IRB-approved MGH Endocrine Surgery Tissue Bank and the Endocrine Surgery Data Registry, we obtained clinical data, fresh frozen tumors, and matched normal tissues from randomly selected patients with PTCs >1 cm. A total of 16 BRAFV600E and 12 BRAFWT tumors with their matched normal thyroid tissue samples were analyzed for PD-L1 mRNA expression.
Statistical analysis
Statistical analysis was carried out using Microsoft Excel (Microsoft, Redmond, VA, USA) and IBM SPSS Statistics (version 23.0, IBM, Armonk, NY). BRAF mutant and wild type groups were compared using the Student t-test for age, Fisher’s exact test for sex, tumor size (T1-2 vs. T3-4) and lymph node status, and Pearson chi-square for TNM stage. All experiments were performed in technical triplicate and repeated twice unless specified otherwise. A fold change in mRNA expression of 2 or more was considered significant. A log power of 2 for fold changes in expression was used to generate box-and-whisker plots based on the median and inter quartile range, and the nonparametric Mann-Whitney U- test was used to compare independent groups. Two-way factorial analysis of variance (ANOVA) was applied to test the effects of treatments on reduction in tumor volume with the Wald chi-square test to assess the additive vs. supra-additive (synergistic) effect of two-drug combinations. Two-tailed values of P < 0.05 were considered significant for all analysis.
ACKNOWLEDGMENTS
We thank Patricia Della Pelle of the Department of Pathology at MGH for histology and immunohistochemistry. We also extend gratitude to Gideon Bollag and Paul Lin (Plexxikon) for providing PLX4720.
FUNDING
This work was supported by the National Institution of Health Grant R01 1R01CA149738-01A1 and the ENDOCRINE TISSUE BANK Ruane Fund (SP). NCI P50CA101942 and NIH P01AI054456 (GJF). The Clair and Emanuel G. Rosenblatt Fund and American Healthcare Professionals and Friends for Medicine in Israel (EB).
CONFLICTS OF INTERESTS
GF has patents/pending royalties on the PD-1 pathway from Bristol-Myers-Squibb, Roche, Merck, EMD-Serono, Boehringer-Ingelheim, AstraZeneca, and Novartis. GF has served on advisory boards for CoStim, Novartis, Roche, Eli Lilly and Bristol-Myers-Squibb.
All other authors certify that they have no competing financial interests pertaining to any of the data or statements given in this article.
This work was presented in abstract format at the International thyroid congress 2015 Orlando FL.
REFERENCES
1. Siegel R, Naishadham D and Jemal A. Cancer statistics, 2013. CA. 2013; 63:11-30.
2. Grebe SK and Hay ID. Thyroid cancer nodal metastases: biologic significance and therapeutic considerations. Surgical oncology clinics of North America. 1996; 5:43-63.
3. Siironen P, Hagstrom J, Maenpaa HO, Louhimo J, Heikkila A, Heiskanen I, Arola J and Haglund C. Anaplastic and poorly differentiated thyroid carcinoma: therapeutic strategies and treatment outcome of 52 consecutive patients. Oncology. 2010; 79:400-408.
4. Xing M, Alzahrani AS, Carson KA, Viola D, Elisei R, Bendlova B, Yip L, Mian C, Vianello F, Tuttle RM, Robenshtok E, Fagin JA, Puxeddu E, Fugazzola L, Czarniecka A, Jarzab B, et al. Association between BRAF V600E mutation and mortality in patients with papillary thyroid cancer. Jama. 2013; 309:1493-1501.
5. Cancer Genome Atlas Research Network. Integrated genomic characterization of papillary thyroid carcinoma. Cell. 2014; 159:676-690.
6. Kim KB, Cabanillas ME, Lazar AJ, Williams MD, Sanders DL, Ilagan JL, Nolop K, Lee RJ and Sherman SI. Clinical responses to vemurafenib in patients with metastatic papillary thyroid cancer harboring BRAF(V600E) mutation. Thyroid. 2013; 23:1277-1283.
7. Rosove MH, Peddi PF and Glaspy JA. BRAF V600E inhibition in anaplastic thyroid cancer. The New England journal of medicine. 2013; 368:684-685.
8. Salama AK and Flaherty KT. BRAF in melanoma: current strategies and future directions. Clinical cancer research. 2013; 19:4326-4334.
9. Bollag G, Tsai J, Zhang J, Zhang C, Ibrahim P, Nolop K and Hirth P. Vemurafenib: the first drug approved for BRAF-mutant cancer. Nature reviews Drug discovery. 2012; 11:873-886.
10. Pardoll DM. The blockade of immune checkpoints in cancer immunotherapy. Nature reviews Cancer. 2012; 12:252-264.
11. Shin DS and Ribas A. The evolution of checkpoint blockade as a cancer therapy: what's here, what's next? Current opinion in immunology. 2015; 33:23-35.
12. Mahoney KM, Rennert PD and Freeman GJ. Combination cancer immunotherapy and new immunomodulatory targets. Nature reviews Drug discovery. 2015; 14:561-584.
13. Chen DS, Irving BA and Hodi FS. Molecular pathways: next-generation immunotherapy--inhibiting programmed death-ligand 1 and programmed death-1. Clinical cancer research. 2012; 18:6580-6587.
14. Zou W and Chen L. Inhibitory B7-family molecules in the tumour microenvironment. Nature reviews Immunology. 2008; 8:467-477.
15. Thompson RH, Gillett MD, Cheville JC, Lohse CM, Dong H, Webster WS, Krejci KG, Lobo JR, Sengupta S, Chen L, Zincke H, Blute ML, Strome SE, Leibovich BC and Kwon ED. Costimulatory B7-H1 in renal cell carcinoma patients: Indicator of tumor aggressiveness and potential therapeutic target. Proceedings of the National Academy of Sciences of the United States of America. 2004; 101:17174-17179.
16. Hino R, Kabashima K, Kato Y, Yagi H, Nakamura M, Honjo T, Okazaki T and Tokura Y. Tumor cell expression of programmed cell death-1 ligand 1 is a prognostic factor for malignant melanoma. Cancer. 2010; 116:1757-1766.
17. Brahmer JR, Tykodi SS, Chow LQ, Hwu WJ, Topalian SL, Hwu P, Drake CG, Camacho LH, Kauh J, Odunsi K, Pitot HC, Hamid O, Bhatia S, Martins R, Eaton K, Chen S, et al. Safety and activity of anti-PD-L1 antibody in patients with advanced cancer. The New England journal of medicine. 2012; 366:2455-2465.
18. Topalian SL, Hodi FS, Brahmer JR, Gettinger SN, Smith DC, McDermott DF, Powderly JD, Carvajal RD, Sosman JA, Atkins MB, Leming PD, Spigel DR, Antonia SJ, Horn L, Drake CG, Pardoll DM, et al. Safety, activity, and immune correlates of anti-PD-1 antibody in cancer. The New England journal of medicine. 2012; 366:2443-2454.
19. Cunha LL, Marcello MA, Vassallo J and Ward LS. Differentiated thyroid carcinomas and their B7H1 shield. Future Oncol. 2013; 9:1417-1419.
20. Cunha LL, Marcello MA, Morari EC, Nonogaki S, Conte FF, Gerhard R, Soares FA, Vassallo J and Ward LS. Differentiated thyroid carcinomas may elude the immune system by B7H1 upregulation. Endocrine-related cancer. 2013; 20:103-110.
21. French JD, Kotnis GR, Said S, Raeburn CD, McIntyre RC, Jr., Klopper JP and Haugen BR. Programmed death-1+ T cells and regulatory T cells are enriched in tumor-involved lymph nodes and associated with aggressive features in papillary thyroid cancer. The Journal of clinical endocrinology and metabolism. 2012; 97:E934-943.
22. Severson JJ, Serracino HS, Mateescu V, Raeburn CD, McIntyre RC, Jr., Sams SB, Haugen BR and French JD. PD-1+Tim-3+ CD8+ T Lymphocytes Display Varied Degrees of Functional Exhaustion in Patients with Regionally Metastatic Differentiated Thyroid Cancer. Cancer immunology research. 2015; 3:620-630.
23. Angell TE, Lechner MG, Jang JK, Correa AJ, LoPresti JS and Epstein AL. BRAF V600E in papillary thyroid carcinoma is associated with increased programmed death ligand 1 expression and suppressive immune cell infiltration. Thyroid. 2014; 24:1385-1393.
24. Brown JA, Dorfman DM, Ma FR, Sullivan EL, Munoz O, Wood CR, Greenfield EA and Freeman GJ. Blockade of programmed death-1 ligands on dendritic cells enhances T cell activation and cytokine production. J Immunol. 2003; 170:1257-1266.
25. Eppihimer MJ, Gunn J, Freeman GJ, Greenfield EA, Chernova T, Erickson J and Leonard JP. Expression and regulation of the PD-L1 immunoinhibitory molecule on microvascular endothelial cells. Microcirculation. 2002; 9:133-145.
26. Schreiner B, Mitsdoerffer M, Kieseier BC, Chen L, Hartung HP, Weller M and Wiendl H. Interferon-beta enhances monocyte and dendritic cell expression of B7-H1 (PD-L1), a strong inhibitor of autologous T-cell activation: relevance for the immune modulatory effect in multiple sclerosis. Journal of neuroimmunology. 2004; 155:172-182.
27. Keir ME, Butte MJ, Freeman GJ and Sharpe AH. PD-1 and its ligands in tolerance and immunity. Annual review of immunology. 2008; 26:677-704.
28. Nucera C, Lawler J and Parangi S. BRAF(V600E) and microenvironment in thyroid cancer: a functional link to drive cancer progression. Cancer research. 2011; 71:2417-2422.
29. Vanden Borre P, McFadden DG, Gunda V, Sadow PM, Varmeh S, Bernasconi M, Jacks T and Parangi S. The next generation of orthotopic thyroid cancer models: immunocompetent orthotopic mouse models of BRAF V600E-positive papillary and anaplastic thyroid carcinoma. Thyroid. 2014; 24:705-714.
30. Cooper ZA, Juneja VR, Sage PT, Frederick DT, Piris A, Mitra D, Lo JA, Hodi FS, Freeman GJ, Bosenberg MW, McMahon M, Flaherty KT, Fisher DE, Sharpe AH and Wargo JA. Response to BRAF inhibition in melanoma is enhanced when combined with immune checkpoint blockade. Cancer immunology research. 2014; 2:643-654.
31. Hamid O, Robert C, Daud A, Hodi FS, Hwu WJ, Kefford R, Wolchok JD, Hersey P, Joseph RW, Weber JS, Dronca R, Gangadhar TC, Patnaik A, Zarour H, Joshua AM, Gergich K, et al. Safety and tumor responses with lambrolizumab (anti-PD-1) in melanoma. The New England journal of medicine. 2013; 369:134-144.
32. Motzer RJ, Rini BI, McDermott DF, Redman BG, Kuzel TM, Harrison MR, Vaishampayan UN, Drabkin HA, George S, Logan TF, Margolin KA, Plimack ER, Lambert AM, Waxman IM and Hammers HJ. Nivolumab for Metastatic Renal Cell Carcinoma: Results of a Randomized Phase II Trial. Journal of clinical oncology. 2015; 33:1430-1437.
33. Frederick DT, Piris A, Cogdill AP, Cooper ZA, Lezcano C, Ferrone CR, Mitra D, Boni A, Newton LP, Liu C, Peng W, Sullivan RJ, Lawrence DP, Hodi FS, Overwijk WW, Lizee G, et al. BRAF inhibition is associated with enhanced melanoma antigen expression and a more favorable tumor microenvironment in patients with metastatic melanoma. Clinical cancer research. 2013; 19:1225-1231.
34. Wu H, Sun Y, Ye H, Yang S, Lee SL and de las Morenas A. Anaplastic thyroid cancer: outcome and the mutation/expression profiles of potential targets. Pathology oncology research. 2015; 21:695-701.
35. Vanden Borre P, Gunda V, McFadden DG, Sadow PM, Varmeh S, Bernasconi M and Parangi S. Combined BRAF(V600E)- and SRC-inhibition induces apoptosis, evokes an immune response and reduces tumor growth in an immunocompetent orthotopic mouse model of anaplastic thyroid cancer. Oncotarget. 2014; 5:3996-4010. doi: 10.18632/oncotarget.2130.
36. Koya RC, Mok S, Otte N, Blacketor KJ, Comin-Anduix B, Tumeh PC, Minasyan A, Graham NA, Graeber TG, Chodon T and Ribas A. BRAF inhibitor vemurafenib improves the antitumor activity of adoptive cell immunotherapy. Cancer research. 2012; 72:3928-3937.
37. Knight DA, Ngiow SF, Li M, Parmenter T, Mok S, Cass A, Haynes NM, Kinross K, Yagita H, Koya RC, Graeber TG, Ribas A, McArthur GA and Smyth MJ. Host immunity contributes to the anti-melanoma activity of BRAF inhibitors. The Journal of clinical investigation. 2013; 123:1371-1381.
38. Jiang X, Zhou J, Giobbie-Hurder A, Wargo J and Hodi FS. The activation of MAPK in melanoma cells resistant to BRAF inhibition promotes PD-L1 expression that is reversible by MEK and PI3K inhibition. Clinical cancer research. 2013; 19:598-609.
39. Yamamoto R, Nishikori M, Tashima M, Sakai T, Ichinohe T, Takaori-Kondo A, Ohmori K and Uchiyama T. B7-H1 expression is regulated by MEK/ERK signaling pathway in anaplastic large cell lymphoma and Hodgkin lymphoma. Cancer science. 2009; 100:2093-2100.
40. Heidorn SJ, Milagre C, Whittaker S, Nourry A, Niculescu-Duvas I, Dhomen N, Hussain J, Reis-Filho JS, Springer CJ, Pritchard C and Marais R. Kinase-dead BRAF and oncogenic RAS cooperate to drive tumor progression through CRAF. Cell. 2010; 140:209-221.
41. Hatzivassiliou G, Song K, Yen I, Brandhuber BJ, Anderson DJ, Alvarado R, Ludlam MJ, Stokoe D, Gloor SL, Vigers G, Morales T, Aliagas I, Liu B, Sideris S, Hoeflich KP, Jaiswal BS, et al. RAF inhibitors prime wild-type RAF to activate the MAPK pathway and enhance growth. Nature. 2010; 464:431-435.
42. Kim T, Amaria RN, Spencer C, Reuben A, Cooper ZA and Wargo JA. Combining targeted therapy and immune checkpoint inhibitors in the treatment of metastatic melanoma. Cancer biology & medicine. 2014; 11:237-246.
43. Wilmott JS, Long GV, Howle JR, Haydu LE, Sharma RN, Thompson JF, Kefford RF, Hersey P and Scolyer RA. Selective BRAF inhibitors induce marked T-cell infiltration into human metastatic melanoma. Clinical cancer research. 2012; 18:1386-1394.
44. Atefi M, Avramis E, Lassen A, Wong DJ, Robert L, Foulad D, Cerniglia M, Titz B, Chodon T, Graeber TG, Comin-Anduix B and Ribas A. Effects of MAPK and PI3K pathways on PD-L1 expression in melanoma. Clinical cancer research. 2014; 20:3446-3457.
45. Goding SR, Wilson KA, Xie Y, Harris KM, Baxi A, Akpinarli A, Fulton A, Tamada K, Strome SE and Antony PA. Restoring immune function of tumor-specific CD4+ T cells during recurrence of melanoma. J Immunol. 2013; 190:4899-4909.
46. Jin HT, Anderson AC, Tan WG, West EE, Ha SJ, Araki K, Freeman GJ, Kuchroo VK and Ahmed R. Cooperation of Tim-3 and PD-1 in CD8 T-cell exhaustion during chronic viral infection. Proceedings of the National Academy of Sciences of the United States of America. 2010; 107:14733-14738.
47. Ngiow SF, von Scheidt B, Akiba H, Yagita H, Teng MW and Smyth MJ. Anti-TIM3 antibody promotes T cell IFN-gamma-mediated antitumor immunity and suppresses established tumors. Cancer research. 2011; 71:3540-3551.
48. Sakuishi K, Apetoh L, Sullivan JM, Blazar BR, Kuchroo VK and Anderson AC. Targeting Tim-3 and PD-1 pathways to reverse T cell exhaustion and restore anti-tumor immunity. The Journal of experimental medicine. 2010; 207:2187-2194.
49. Brahmer JR, Drake CG, Wollner I, Powderly JD, Picus J, Sharfman WH, Stankevich E, Pons A, Salay TM, McMiller TL, Gilson MM, Wang C, Selby M, Taube JM, Anders R, Chen L, et al. Phase I study of single-agent anti-programmed death-1 (MDX-1106) in refractory solid tumors: safety, clinical activity, pharmacodynamics, and immunologic correlates. Journal of clinical oncology. 2010; 28:3167-3175.
50. Ascierto PA. Immunotherapies and novel combinations: the focus of advances in the treatment of melanoma. Cancer immunology, immunotherapy. 2015; 64:271-274.
51. Viteri S, Gonzalez-Cao M, Barron F, Riso A and Rosell R. Results of clinical trials with anti-programmed death 1/programmed death ligand 1 inhibitors in lung cancer. Translational lung cancer research. 2015; 4:756-762.
52. Lawrence MS, Stojanov P, Polak P, Kryukov GV, Cibulskis K, Sivachenko A, Carter SL, Stewart C, Mermel CH, Roberts SA, Kiezun A, Hammerman PS, McKenna A, Drier Y, Zou L, Ramos AH, et al. Mutational heterogeneity in cancer and the search for new cancer-associated genes. Nature. 2013; 499:214-218.
53. Derks S, Nason KS, Liao X, Stachler MD, Liu KX, Liu JB, Sicinska E, Goldberg MS, Freeman GJ, Rodig SJ, Davison JM and Bass AJ. Epithelial PD-L2 Expression Marks Barrett's Esophagus and Esophageal Adenocarcinoma. Cancer immunology research. 2015; 3:1123-1129.
54. Ryder M, Ghossein RA, Ricarte-Filho JC, Knauf JA and Fagin JA. Increased density of tumor-associated macrophages is associated with decreased survival in advanced thyroid cancer. Endocrine-related cancer. 2008; 15:1069-1074.
55. McFadden DG, Vernon A, Santiago PM, Martinez-McFaline R, Bhutkar A, Crowley DM, McMahon M, Sadow PM and Jacks T. p53 constrains progression to anaplastic thyroid carcinoma in a Braf-mutant mouse model of papillary thyroid cancer. Proceedings of the National Academy of Sciences of the United States of America. 2014; 111:E1600-1609.
56. Muhlbauer M, Fleck M, Schutz C, Weiss T, Froh M, Blank C, Scholmerich J and Hellerbrand C. PD-L1 is induced in hepatocytes by viral infection and by interferon-alpha and -gamma and mediates T cell apoptosis. Journal of hepatology. 2006; 45:520-528.
57. Rodig N, Ryan T, Allen JA, Pang H, Grabie N, Chernova T, Greenfield EA, Liang SC, Sharpe AH, Lichtman AH and Freeman GJ. Endothelial expression of PD-L1 and PD-L2 down-regulates CD8+ T cell activation and cytolysis. European journal of immunology. 2003; 33:3117-3126.
58. Nucera C, Nehs MA, Mekel M, Zhang X, Hodin R, Lawler J, Nose V and Parangi S. A novel orthotopic mouse model of human anaplastic thyroid carcinoma. Thyroid. 2009; 19:1077-1084.
59. Tomayko MM and Reynolds CP. Determination of subcutaneous tumor size in athymic (nude) mice. Cancer chemotherapy and pharmacology. 1989; 24:148-154.