
INTRODUCTION
Renal cell carcinoma (RCC) is a common cancer which accounts for ~90% of kidney cancer cases in adults, with over 200,000 cases worldwide per year [1] and its incidence steadily rising in most areas of the world [2, 3]. Despite of recent advances in RCC diagnosis and therapy, many patients are still present with metastasis at initial diagnosis and poor treatment response rates [4]. Therefore, elucidation of the molecular mechanisms underlying RCC development and progression is urgently needed [5].
Cancer is caused by cumulative genetic and epigenetic alterations [6]. Epigenetic silencing of tumor suppressor genes (TSGs) through promoter CpG methylation confers selective advantages of clonal expansion, leading to tumor initiation and progression [7]. Promoter methylation of TSGs can also be used as epigenetic biomarkers for tumor diagnosis [8]. A series of genes have been identified to be methylated in RCC with some of them linked to tumor prognosis [9]. For example, VHL is one of TSGs early identified to be epigenetically inactivated by promoter CpG methylation in RCC [10]. RASSF1A, which maps to the 3p21 region of frequent allele loss, was methylated in 30~50% of sporadic clear cell (cc) RCC and papillary RCC [11, 12]. SLC16A3 promoter methylation is a predictive marker for the prognosis and clinical outcome of ccRCC [13]. Our group has also identified several novel TSGs silenced by promoter methylation in RCC, including DLC1, DLEC1 and IRF8. The methylation of these TSGs is associated with patient poor prognosis, thus as potential biomarkers for RCC [14–16].
Chromodomain helicase DNA binding (CHD) genes encode a class of ATPase-dependent DNA-binding proteins interacting with histones to modulate chromatin structure and transcription [17]. CHD5, located at 1p36.3, is the fifth member of a nine-member family of CHD chromatin remodeling proteins (CHD1-CHD9) [18]. CHD5 consists of two tandem plant homeodomains (PHDs), dual chromodomains, SNF2 family N-terminal domain (SNF2N), ATP-dependent helicase conserved C-terminal domain (HELIC) followed by other domains with unknown functions [19–21]. CHD5 has been reported frequently methylated in multiple human cancers, including glioma, breast, lung, gastric, colon, ovarian and prostate cancers [18, 22]. Reduced CHD5 expression is associated with unfavorable clinical features and outcome of cancer patients [23–26]. In mice, Chd5 functions as a dose-dependent TSG through regulating cell proliferation, apoptosis and senescence, due to upregulation of p19Arf and TP53 [27]. CHD5 significantly inhibits clonogenic growth and tumor xenograft growth, thus as a functional tumor suppressor in multiple common cancers, including breast, colon, lung, ovary and prostate cancers [18], although no report about the expression and function of CHD5 has been reported in RCC yet.
We have identified CHD5 as a methylated target in RCC cell lines. Here, we further studied the epigenetic alteration of CHD5 in RCC cells, and characterized its tumor suppressive functions and the underlying molecular mechanisms during RCC pathogenesis.
RESULTS
Identification of CHD5 as a methylated TSG candidate for urological cancers
We have previously identified CHD5 as a methylated target through epigenome study [28]. Meanwhile, through analyzing microarray data from GENT dataset [29], we found that CHD5 was underexpressed in 366 kidney cancer tissues, 87 bladder cancer tissues and 244 prostate cancer tissues, compared with the corresponding normal tissues (Figure 1A). Data from Oncomine database [30] also indicated that CHD5 mRNA expression was frequently decreased in kidney [31–33], bladder [34] and prostate cancers [35] compare with their adjacent control tissues (Supplementary Table 1). Our semi-quantitative RT-PCR data showed that CHD5 was silenced or downregulated in 7/9 RCC, 2/3 prostate and 1/3 bladder tumor cell lines, but readily detected in most human normal adult tissues including kidney and prostate, as well as immortalized normal cell lines (HEK293 and RHEK-1) (Figure 1B and 1C, Supplementary Figure 1A). Then we analyzed the promoter region of CHD5, and found that there was a typical CpG island spanning the transcription start site (Figure 1D). These results indicated that CHD5 is a downregulated candidate TSG for urological cancers and possibly subjected to methylation-mediated silencing.
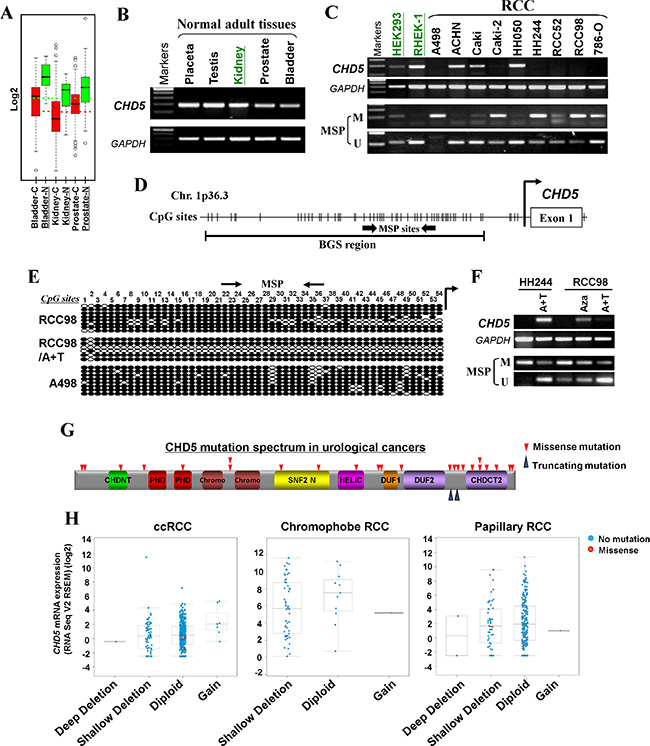
Figure 1: CHD5 is predominantly inactivated by promoter CpG methylation in urological cancers. (A) Expression profiles of CHD5 in urological tumors and normal tissues. C, cancer; N, normal (GENT datasets, http://medical-genome.kribb.re.kr/GENT/search/search.php). (B) CHD5 expression in a panel of human normal adult tissues detected by RT-PCR, with GAPDH as an internal control. (C) CHD5 was downregulated or silenced by promoter methylation in RCC cell lines as determined by RT-PCR and MSP, but expressed and unmethylated in HEK293 and RHEK-1 cell line. M, methylated; U, unmethylated. (D) Schematic structure of the CHD5 promoter region. Exon 1, CpG sites (short vertical lines), MSP sites and BGS region analyzed are shown. (E) BGS analysis of the CHD5 promoter in representative RCC cells. Each row of circles represented an individual promoter allele. Filled circle, methylated CpG site; open circle, unmethylated CpG site. (F) Pharmacologic demethylation with Aza alone or combined with TSA (A + T) restored CHD5 expression in methylated/silenced tumor cell lines. (G) Schematic representation of CHD5 somatic mutations identified in urological cancers. CHDNT, CHD N-terminal domain; PHD, plant homeodomain; Chromo, CHRromatin Organisation MOdifier domain; SNF2 N, SNF2 family N-terminal domain; HELIC, Helicase conserved C-terminal domain; DUF, Domain of Unknown Function; CHDCT2, CHD C-terminal domain. (H) CHD5 putative copy number alterations from GISTIC (Genomic Identification of Significant Targets in Cancer): loss of an allele of CHD5 correlated with decreased mRNA expression in ccRCC (left), chromophobe RCC (middle) and papillary RCC (right).
Silencing of CHD5 due to its promoter CpG methylation in urological cancers
We next studied whether promoter CpG methylation was involved in silencing CHD5 in urological cancers. Methylation-specific PCR (MSP) analysis showed that CHD5 was frequently methylated in RCC, prostate and bladder tumor cell lines, which was negatively correlated with the corresponding expression levels (Figure 1C, Supplementary Figure 1A). To examine the methylation status of CHD5 promoter in more detail, bisulfite genomic sequencing (BGS) analysis was performed for a 539-bp region with 54 CpG sites spanning the CHD5 core promoter and exon 1. High density of methylated CpG sites were detected in two representative RCC cell lines (RCC98 and A498) and one prostate tumor cell line (PC3), which confirmed the MSP data (Figure 1E, Supplementary Figure 1B).
To determine whether methylation directly contributes to CHD5 silencing, we treated two RCC cell lines (HH244 and RCC98) with DNA methyltransferase inhibitor Aza, alone or in combination with histone deacetylase inhibitor TSA. After pharmological treatment, CHD5 expression was restored in HH244 and RCC98 cells, accompanied by significant increase of unmethylated alleles and decrease of methylated alleles (Figure 1F). Demethylation of CHD5 promoter in RCC98 cells was confirmed by BGS analysis (Figure 1E).
Moreover, the data retrieved from cBio database [36, 37] revealed the presence of copy number loss of CHD5 in ccRCC [31], papillary RCC (TCGA) and prostate cancer [35, 38, 39] (Supplementary Figure 2). Meanwhile, the data also indicated that CHD5 was rarely mutated in urological cancers [40–42] (Figure 1G and Supplementary Table 2). Loss or downregulation of CHD5 expression correlated with copy number loss using data from the TCGA RCC cohorts (Figure 1H). Together, these findings suggested CHD5 as a candidate TSG that was epigenetically silenced in urological cancers.
Frequent methylation of CHD5 in primary RCC tumors
Through MSP analysis, we found that normal urological tissues exhibited unmethylated CHD5 promoter (Figure 2A). Meanwhile, we examined CHD5 methylation in our RCC samples. MSP analysis showed that CHD5 was methylated in 44% (24/55) of RCC samples (Figure 2B). High-resolution BGS analysis of two representative cases further confirmed the methylation (Figure 2C). The frequency of CHD5 methylation in our patients group was comparable to the data obtained from The Cancer Genome Altas (TCGA) RCC cohorts. The overall frequency of CHD5 methylation in TCGA RCC cohorts was 49% (334/688), with highest in ccRCC (100%, 320/320) [31], followed by papillary RCC (4.1%,12/292) and chromophobe RCC (3%, 2/66) [43] (Figure 2D).
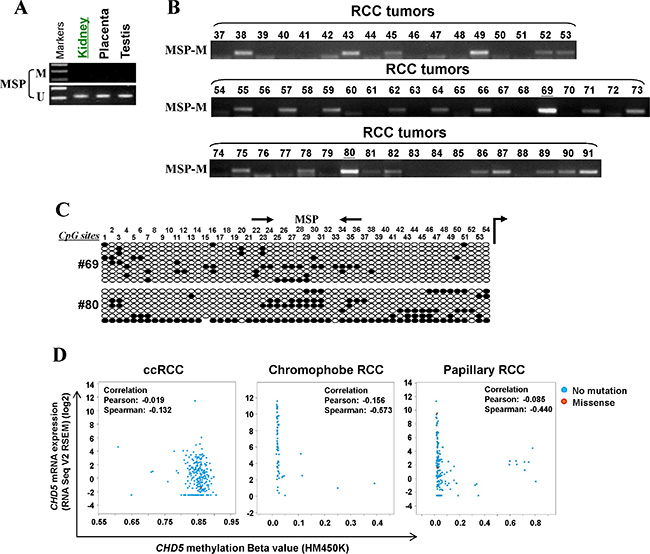
Figure 2: CHD5 is methylated in primary RCC tissues. (A) MSP analysis of CHD5 methylation in normal urological tissues. (B) Representative MSP analysis of CHD5 methylation in RCC tissues. All these samples have been shown to have sufficient bisulfite converted DNA by unmethylation detection for TSGs. (C) BGS analysis confirmed CHD5 methylation in two representative primary RCC tissues. (D) CHD5 methylation was observed in all of ccRCC (320/320), 3.0% (2/66) chromophobe RCC and 4.1% (12/292) in papillary RCC primary tumors from TCGA RCC cohorts.
CHD5 suppresses RCC cell clonogenicity and induces apoptosis
Next, we assessed the impact of CHD5 on clonogenicity with monolayer cell colony formation assay in three RCC cell lines (A498, RCC98 and HH244). We transfected A498, RCC98 and HH244 cell line with empty vector or pcDNA3.1/CHD5, and the restored expression of CHD5 was validated by semi-quantitative RT-PCR. Results showed that ectopic expression of CHD5 significantly decreased the numbers of RCC cell colonies, compared with controls (Figure 3A, 3B and 3C).
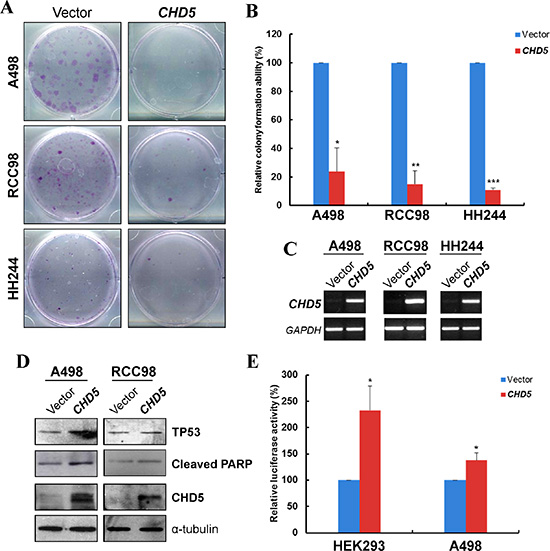
Figure 3: CHD5 suppresses RCC cell growth and induced TP53-related apoptosis. (A) Representative monolayer culture colony formation assay of A498, RCC98 and HH244 cells. (B) Quantitative analysis of colony numbers. Data are presented as mean ± SD of three independent experiments. *P < 0.05, **P < 0.01 ***P < 0.001. (C) Restored expression of CHD5 in transfected cell lines as confirmed by RT-PCR. (D) Western blot showed upregulation of cleaved PARP and TP53 in CHD5-expressing RCC cells. α-tubulin was used as a loading control. (E) CHD5 expression can increase the transcription activity of TP53-binding site promoter construct as determined by luciferase reporter assay in HEK293 and A498 cell lines. *P < 0.05.
As the growth inhibition could be attributed to the induction of apoptosis, we then examined the effect of CHD5 on the apoptosis of RCC cells. We observed the cleavage of poly (ADP-ribose) polymerase (PARP), a classic apoptotic marker in cells with ectopic expression of CHD5 (Figure 3D). Additionally, CHD5 has been reported to regulate TP53-induced apoptosis [27]. Hence we assessed the expression and activation of TP53 in CHD5-expressing RCC cells. As shown in Figure 3D, CHD5 could upregulate TP53 expression in RCC cells. Moreover, through dual luciferase reporter assay of TP53 binding-site promoter construct, CHD5-expressing HEK293 and A498 cells showed significantly elevated TP53 transcriptional activities when compared with controls (Figure 3E). Collectively, the data indicated that CHD5 could suppress the clonogenicity of RCC cells, which might be linked to the enhancement of TP53-induced apoptosis.
CHD5 inhibits the migration and invasion of RCC cells
We also assessed the effects of CHD5 on RCC cell migration and invasion. Scratch wound-healing assay showed that CHD5-expressing A498 and HH244 cells were less proficient in healing an artificial wound than the vector-transfected cells on a confluent monolayer (Figure 4A, 4C). Moreover, CHD5-expressing cells displayed significantly decreased invasiveness compared with controls in Matrigel invasion assay (Figure 4B, 4C).
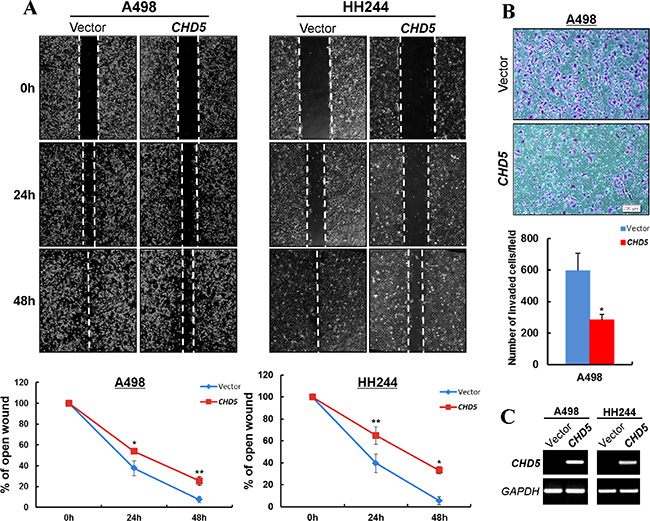
Figure 4: CHD5 inhibits the migration and invasion of RCC cells. (A) Wound healing assay demonstrated a slower wound closure of CHD5-expressing A498 and HH244 cells compared with controls. *P < 0.05; **P < 0.01. (B) Transwell migration assay of CHD5-expressing tumor cells. Migrated cells at the lower surface of the transwell filter were stained (up panel) and counted (bottom panel). *P < 0.05. (C) Expression of CHD5 in transfected cell lines was confirmed by RT-PCR.
CHD5 represses the expression of multiple cancer genes through direct binding to their promoters
We further investigated the possible mechanism of CHD5 functioning as a tumor suppressor in RCC cells. We firstly examined the expression levels of multiple cancer genes in RCC cells with ectopic CHD5 expression, including oncogenes, epigenetic master genes, epithelial-mesenchymal transition (EMT) and stem cell-related genes. Semi-quantitative and qRT-PCR showed that the expression levels of multiple oncogenes (MYC, MDM2, STAT3, CCND1, YAP1 etc.), epigenetic master genes (Bmi-1, EZH2, JMJD2C etc.), epithelial-mesenchymal transition and stem cell markers (FN1, SNAI1, OCT4 and NANOG) and hypoxia-inducible factors (HIF2A and HIG2) were significantly decreased in CHD5-transfected A498 and RCC98 cells (Figure 5A and 5B). Furthermore, we examined the protein expression of some cancer genes by Western blot. We found that the protein levels of EZH2, STAT3, MYC, MDM2 and p-AKT were decreased in CHD5-expressing RCC cells (Figure 5C). We further performed ChIP assay to detect the recruitments of CHD5 to MYC, MDM2, EZH2, Bmi-1 and MCL1 promoters. It was found that CHD5 indeed bound to these promoters with varied binding activities (Figure 5D).
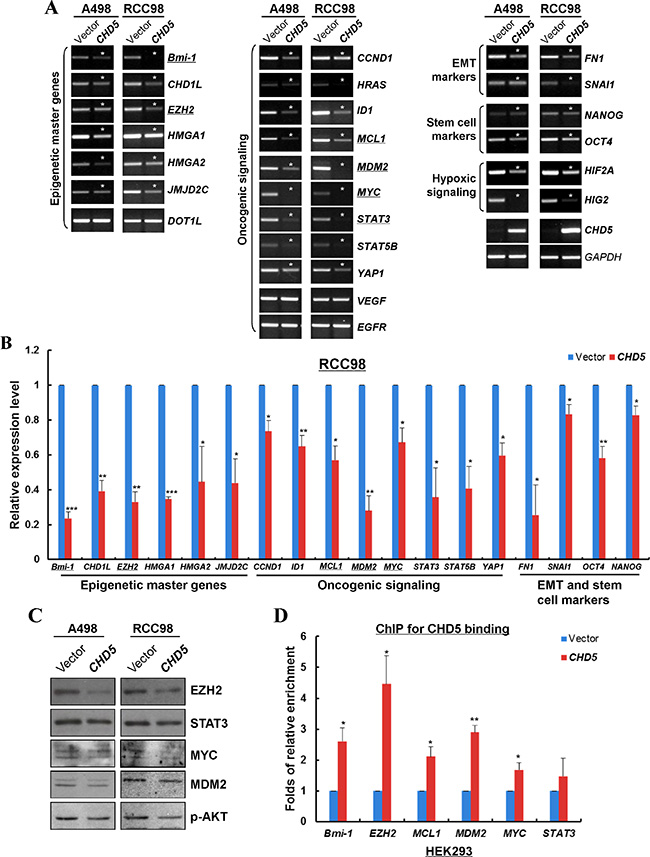
Figure 5: CHD5 represses the expression of multiple cancer genes through direct interaction with their promoters. (A) Semi-quantitative RT-PCR showed that CHD5 repressed the mRNA expression of multiple oncogenic genes in A498 and RCC98 cells. Asterisk (*) indicates significantly downregulation effects. (B) qRT-PCR showed that CHD5 inhibited the expression levels of multiple cancer genes in RCC98 cells, including epigenetic master genes, hypoxia-inducible factors, oncogenic genes, EMT and stem cell markers. *P < 0.05; **P < 0.01; ***P < 0.001. (C) Western blot showed decreased levels of EZH2, STAT3, MYC, MDM2 and p-AKT by CHD5. Gel loading control is the same as in Figure 3D. (D) ChIP-qPCR showed enrichment of CHD5 binding to the promoter regions of EZH2, MYC, MDM2, BMI1 and MCL1 in HEK293 cell lines. *P < 0.05; **P < 0.01.
DISCUSSION
In this study, we demonstrated that CHD5 acted as a functional tumor suppressor and was frequently silenced by promoter CpG methylation in RCC. Recent findings suggest that genetic and epigenetic alterations are both involved in RCC development. Identification of epigenetic alterations in RCC could be helpful to unravel the mechanisms underlying RCC carcinogenesis and develop potential biomarkers for cancer screening and prognosis prediction [31]. CHD5 is located at 1p36, together with other tumor suppressors including p73, CAMTA1, miR-34a, KIF1β, and CASZ1 [18]. CHD5 is reported to be rarely mutated, but with frequent allelic loss in cancers. CHD5 methylation has been identified in several cancer types including glioma, breast, colon, lung, ovary and prostate cancers [18, 22], suggesting the contribution of epigenetic regulation to its biallelic inactivation [18]. Our present data demonstrated that CHD5 could inhibit cell proliferation, migration and invasion, as well as induce apoptosis in RCC cells, supporting the involvement of epigenetic inactivation of CHD5 in RCC tumorigenesis (Figure 6).
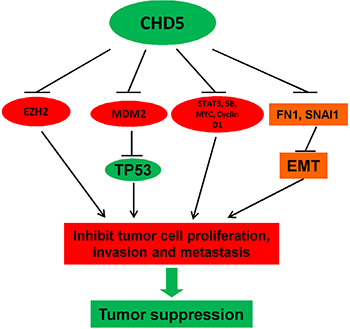
Figure 6: Proposed model of the tumor suppressive functions of CHD5 in RCC. CHD5 promotes TP53-related apoptosis and suppresses multiple cancer gene expression (EZH2, MYC and MDM2, etc), leading further to tumor suppression.
Nucleosome remodeling and deacetylation (NuRD) complex plays a critical role in the regulation of transcriptional events during normal physiology and cancer pathogenesis including RCC [17, 31]. CHD5, being a component of NuRD complex, has been identified to be capable of suppressing the transcription of cell-cycle regulator WEE1. CHD5 mutant lacking the ATPase activity due to mutation within HELIC domain maintained its interaction with other NuRD subunits, but the ability to repress WEE1 transcription was decreased [44]. The dual PHDs of Chd5 specifically interacts with unmodified N-terminus of histone H3, which is ciritical for Chd5 to exert the regulatory role on gene expression and the potential tumor suppressive function [21, 45]. Therefore, both PHDs and HELIC are required for the tumor suppressive function of CHD5. Recently, Potts RC et al. demonstrated that CHD5 protein can regulate target genes through direct interaction in the form of a NuRD-like multi-protein complex [46]. Here, we demonstrated that CHD5 could repress the transcription of multiple oncogenes, including MYC and EZH2, through direct binding to their promoters.
In conclusion, CHD5 is frequently silenced by promoter methylation in urological cancers including RCC, and functions as a TSG through direct repression of multiple oncogenes in RCC cells. Our present study emphasizes the contribution of epigenetic regulation to RCC carcinogenesis and silencing of CHD5 could be a potential tumor biomarker for RCC diagnosis.
MATERIALS AND METHODS
Database analysis
Databases including GENT [29], Oncomine [30], cBio (MSKCC) [36, 37] and Catalogue of Somatic Mutations in Cancer (COSMIC) (Wellcome-Sanger) [47] database were screened for information specifying genomic alterations and mRNA expression in TCGA cohorts and other published papers. Functionality of CHD5 mutation was evaluated by PolyPhen-2 software (Version 2.2.2) [48].
Cell lines and tissue samples
RCC cell lines (A498, ACHN, Caki, Caki-2, HH050, HH244, RCC52, RCC98 and 786-O) were routinely maintained in RPMI1640 (Invitrogen, Grand Island, NY) with 10% fetal bovine serum. Immortalized, non-transformed normal epithelial cell line HEK293 and RHEK-1 (kindly gifted by Prof. John Rhim, US Naval Medical Research Center, Bethesda, MD) were used as controls. Cell lines were obtained from either the American Type Culture Collection or our collaborators. Human normal tissues RNA were purchased from Stratagene (Santa Clara, CA), Biochain (Newark, CA) or Chemicon (Billerica, MA). DNA samples of RCC cases were obtained from our collaborators as described previously [14].
Semi-quantitative and quantitative RT-PCR
Total RNA was extracted using TRI reagent. Reverse transcription (RT) using random hexamer, and RT-PCR using Go-Taq (Promega, Madison, WI) were performed as previously described [49]. Primers used for RT-PCR were listed in Table 1 and Supplementary Table 3.
Table 1: Primers used for the detection of CHD5 expression and promoter methylation
Type |
Primers |
Sequence (5′–3′) |
Length |
|---|---|---|---|
RT-PCR |
CHD5F |
CCAGTGGGCACCGAGGAG |
|
CHD5R |
CTTCTTCCGCTTCCCTTTAC |
192 bp |
|
MSP |
CHD5m1 |
GTTCGGGGTTTAGCGTTTTC |
|
CHD5m2 |
GAAACTTAACGAACCCGAACG |
108 bp |
|
CHD5u1 |
GGGTTTGGGGTTTAGTGTTTTT |
|
|
CHD5u2 |
TCAAAACTTAACAAACCCAAACA |
112 bp |
|
BGS |
CHD5BGS1 |
GGGTTTTAGTTGTATTTAGTTTG |
|
CHD5BGS2 |
TAACAACAAAAAACAAATTAAAAAAC |
539 bp |
Quantitative real-time PCR (qRT-PCR) was performed as previously described [50]. SYBR Green master mix (Applied Biosystems, Grand Island, NY) was used. The expression levels of target genes in CHD5-transfected cells were normalized to those transfected with vector control. GAPDH was used as an internal control. The sequences of primers used in qRT-PCR were listed in Table 1 and Supplementary Table 3.
Bisulfite treatment and promoter methylation analysis
Bisulfite modification of DNA, MSP and BGS analysis was conducted as previously described [49]. 38 cycles of PCR reaction were performed in MSP using primers amplifying methylated gene allele, with 40 cycles for reaction using primers amplifying unmethylated gene allele. For BGS, PCR products amplified using BGS primers were cloned into pCR4-TOPO vector (Invitrogen, Carlsbad, CA, USA), with 6–10 colonies randomly chosen and sequenced. Primers used for MSP and BGS were listed in Table 1.
Demethylation treatment using 5-Aza-2′-deoxycytidine and trichostatin A
Treatment of RCC cell line using 5-Aza-2′-deoxycytidine (Aza) and trichostatin A (TSA) was carried out as previously described [51]. Briefly, cells were treated with 10 μM Aza (Sigma, Ronkonkoma, NY) for 72 hours and harvested for DNA and RNA extraction. Alternatively, after 72 hours of Aza treatment, cells were incubated with 100 ng/ml TSA for additional 24 hours.
Construction of CHD5 expression vector
The full-length Open Reading Frame (ORF) of CHD5 was amplified and cloned to pcDNA3.1 (+) vector, with a Flag tag added to its N terminus. The sequence and orientation of CHD5 ORF were confirmed by Sanger sequencing.
Colony formation assays
A498, HH244 and RCC98 cells were seeded in a 12-well plate at 1~2 × 105/well. The cells were then transfected with CHD5 plasmid or empty vector using Lipofectamine 2000 (Invitrogen). After 48 hours post-transfection, the transfectants were sub-cultured into 6-well plates for selection with 200 μg/ml G418 (Calbiochem, Darmstadt, Germany). After 1~2 weeks of selection, surviving colonies (> 50 cells/colony) were fixed with methanol and stained with Gentian Violet and counted.
Dual-luciferase reporter assay
TP53 transcriptional activity was determined by luciferase reporter assay. After 48 hours of transfection, luciferase activities were determined using a dual-luciferase reporter assay kit (Promega, Madison, WI, USA). Relative luciferase activities were determined and normalized using Renilla reniformis luciferase activity as an internal control.
Wound healing assay and matrigel invasion assays
Wound healing assay to evaluate cell migration ability was performed as previously described [51]. Briefly, cells transfected with empty vector or CHD5 construct were allowed to growth until confluent (> 95%). Cell scratches were then created in A498 and HH244 cell lines using 200 μl sterile tips and washed twice with culturing medium. After indicated time points of incubation, cells were imaged under a phase-contrast microscope. The experiments were performed in triplicate. In-vitro invasion assays were carried out in Corning BioCoat Matrigel chambers (Corning, NY) as described previously [51].
Western blot
Western blot was performed as described previously [51]. Briefly, membranes were incubated with primary antibody at 4°C overnight, followed by incubation with secondary antibody at room temperature for 1 hour. Immunoreactive bands were detected using Western blot Luminol reagent (GE Healthcare, Waukesha, WI). Antibodies used were CHD5 (23320002, Novus), EZH2 (18-7395, Invitrogen), MDM2 (sc-813, Santa Cruz, CA), MYC (sc-764, Santa Curz, CA), cleaved poly (ADP-ribose) polymerase (9541, Cell Signal), phospho-AKT (4060, Cell Signal), α-tubulin (MS-581, Thermo Lab Vision, MI); p53 (M7001), anti-mouse IgG-HRP (P0161), anti-rabbit IgG-HRP (P0448) (Dako, Glostrup, Denmark).
Chromatin immunoprecipitation
Chromatin immunoprecipitation (ChIP) was carried out as previously described using ChIP-IT Express Kit from Active Motif (53008; Carlsbad, CA) [50]. For each ChIP reaction, 20 μg of total chromatin was incubated with 20 μl of Protein G magnetic beads and 1 μg of FLAG-M2 antibody (F3165; Sigma) at 4°C overnight. Both input and precipitated DNA were purified with QIAamp DNA Mini Kit (Qiagen, Valencia, CA) for subsequent quantitative real-time PCR (ChIP-qPCR). The relative enrichment of precipitated DNA was normalized to normal mouse IgG. Primers used for ChIP assay were listed in Table 2 and the locations of primers were shown in Supplementary Figure 3
Table 2: Primers for ChIP-qPCR used in this study
Gene |
Primers |
Sequence (5′–3′) |
Length (bp) |
|---|---|---|---|
Bmi-1 |
BMI1ChIPF4 |
TCTCTGCAATTTGAGCCCTG |
205 |
BMI1ChIPR4 |
GAAAATGCAAACCGCACTCC |
||
EZH2 |
EZH2ChIPF1 |
AAATTAGTCGGGTGTGGTGG |
152 |
EZH2ChIPR1 |
AAACGGAGTCTCACACTGTC |
||
MDM2 |
MDM2ChIPF1 |
CATTTGGGTACAACTCCAGC |
115 |
MDM2ChIPR1 |
TGGAAACTGCGACAAATGCG |
||
MYC |
MYCChIPF2 |
AAAGGGAGAGGGTTTGAGAG |
226 |
MYCChIPR2 |
GAGATTAGCGAGAGAGGATC |
||
MCL1 |
MCL1ChIPF2 |
CAACAGAGCTAGACTGTCTC |
204 |
MCL1ChIPR2 |
CACGTGCTACCCTAAAGAAC |
||
STAT3 |
STAT3ChIPF2 |
GCTGCTCTCCTCATTGGTC |
258 |
STAT3ChIPR2 |
CCTGTCCAGGATCCGGTTG |
Statistical analysis
Results were presented as mean ± SD. Statistical analysis was carried out with Student’s t-test, P < 0.05 was considered as statistically significant.
ACKNOWLEDGMENTS AND FUNDING
This study was supported by grants from Hong Kong RGC, National Natural Science Foundation (No. 81572327 and 813728898) and The Chinese University of Hong Kong.
CONFLICTS OF INTEREST
The authors declare no conflicts of interest.
REFERENCES
1. Siegel R, Naishadham D, Jemal A. Cancer statistics, 2013. CA Cancer J Clin. 2013; 63:11–30.
2. Mathew A, Devesa SS, Fraumeni JF, Jr., Chow WH. Global increases in kidney cancer incidence, 1973–1992. Eur J Cancer Prev. 2002; 11:171–178.
3. Sun M, Thuret R, Abdollah F, Lughezzani G, Schmitges J, Tian Z, Shariat SF, Montorsi F, Patard JJ, Perrotte P, Karakiewicz PI. Age-adjusted incidence, mortality, and survival rates of stage-specific renal cell carcinoma in North America: a trend analysis. Eur Urol. 2011; 59:135–141.
4. Uchida K, Miyao N, Masumori N, Takahashi A, Oda T, Yanase M, Kitamura H, Itoh N, Sato M, Tsukamoto T. Recurrence of renal cell carcinoma more than 5 years after nephrectomy. International journal of urology. 2002; 9:19–23.
5. Jones J, Libermann TA. Genomics of renal cell cancer: the biology behind and the therapy ahead. Clin Cancer Res. 2007; 13:685s–692s.
6. Shen H, Laird PW. Interplay between the cancer genome and epigenome. Cell. 2013; 153:38–55.
7. Jones PA, Baylin SB. The fundamental role of epigenetic events in cancer. Nat Rev Genet. 2002; 3:415–428.
8. Belinsky SA. Gene-promoter hypermethylation as a biomarker in lung cancer. Nat Rev Cancer. 2004; 4:707–717.
9. Maher ER. Genomics and epigenomics of renal cell carcinoma. Semin Cancer Biol. 2013; 23:10–17.
10. Herman JG, Latif F, Weng Y, Lerman MI, Zbar B, Liu S, Samid D, Duan DS, Gnarra JR, Linehan WM, et al. Silencing of the VHL tumor-suppressor gene by DNA methylation in renal carcinoma. Proc Natl Acad Sci U S A. 1994; 91:9700–9704.
11. Morrissey C, Martinez A, Zatyka M, Agathanggelou A, Honorio S, Astuti D, Morgan NV, Moch H, Richards FM, Kishida T, Yao M, Schraml P, Latif F, et al. Epigenetic inactivation of the RASSF1A 3p21.3 tumor suppressor gene in both clear cell and papillary renal cell carcinoma. Cancer Res. 2001; 61:7277–7281.
12. Dreijerink K, Braga E, Kuzmin I, Geil L, Duh FM, Angeloni D, Zbar B, Lerman MI, Stanbridge EJ, Minna JD, Protopopov A, Li J, Kashuba V, et al. The candidate tumor suppressor gene, RASSF1A, from human chromosome 3p21.3 is involved in kidney tumorigenesis. Proc Natl Acad Sci U S A. 2001; 98:7504–7509.
13. Fisel P, Kruck S, Winter S, Bedke J, Hennenlotter J, Nies AT, Scharpf M, Fend F, Stenzl A, Schwab M, Schaeffeler E. DNA methylation of the SLC16A3 promoter regulates expression of the human lactate transporter MCT4 in renal cancer with consequences for clinical outcome. Clin Cancer Res. 2013; 19:5170–5181.
14. Zhang Q, Ying J, Li J, Fan Y, Poon FF, Ng KM, Tao Q, Jin J. Aberrant promoter methylation of DLEC1, a critical 3p22 tumor suppressor for renal cell carcinoma, is associated with more advanced tumor stage. J Urol. 2010; 184:731–737.
15. Zhang Q, Ying J, Zhang K, Li H, Ng KM, Zhao Y, He Q, Yang X, Xin D, Liao SK, Tao Q, Jin J. Aberrant methylation of the 8p22 tumor suppressor gene DLC1 in renal cell carcinoma. Cancer Lett. 2007; 249:220–226.
16. Zhang Q, Zhang L, Li L, Wang Z, Ying J, Fan Y, Xu B, Wang L, Liu Q, Chen G, Tao Q, Jin J. Interferon regulatory factor 8 functions as a tumor suppressor in renal cell carcinoma and its promoter methylation is associated with patient poor prognosis. Cancer Lett. 2014; 354:227–234.
17. Lai AY, Wade PA. Cancer biology and NuRD: a multifaceted chromatin remodelling complex. Nat Rev Cancer. 2011; 11:588–596.
18. Bagchi A, Mills AA. The quest for the 1p36 tumor suppressor. Cancer Res. 2008; 68:2551–2556.
19. Marfella CG, Imbalzano AN. The Chd family of chromatin remodelers. Mutation research. 2007; 618:30–40.
20. Sims JK, Wade PA. SnapShot: Chromatin remodeling: CHD. Cell. 2011; 144:626–626 e621.
21. Paul S, Kuo A, Schalch T, Vogel H, Joshua-Tor L, McCombie WR, Gozani O, Hammell M, Mills AA. Chd5 requires PHD-mediated histone 3 binding for tumor suppression. Cell Rep. 2013; 3:92–102.
22. Kolla V, Zhuang T, Higashi M, Naraparaju K, Brodeur GM. Role of CHD5 in Human Cancers: 10 Years Later. Cancer Res. 2013; 74:652–658.
23. Wong RR, Chan LK, Tsang TP, Lee CW, Cheung TH, Yim SF, Siu NS, Lee SN, Yu MY, Chim SS, Wong YF, Chung TK. CHD5 downregulation associated with poor prognosis in epithelial ovarian cancer. Gynecologic and obstetric investigation. 2011; 72:203–207.
24. Wang L, He S, Tu Y, Ji P, Zong J, Zhang J, Feng F, Zhao J, Gao G, Zhang Y. Downregulation of chromatin remodeling factor CHD5 is associated with a poor prognosis in human glioma. J Clin Neurosci. 2013; 20:958–963.
25. Fujita T, Igarashi J, Okawa ER, Gotoh T, Manne J, Kolla V, Kim J, Zhao H, Pawel BR, London WB, Maris JM, White PS, Brodeur GM. CHD5, a tumor suppressor gene deleted from 1p36.31 in neuroblastomas. J Natl Cancer Inst. 2008; 100:940–949.
26. Hall WA, Petrova AV, Colbert LE, Hardy CW, Fisher SB, Saka B, Shelton JW, Warren MD, Pantazides BG, Gandhi K, Kowalski J, Kooby DA, El-Rayes BF, et al. Low CHD5 expression activates the DNA damage response and predicts poor outcome in patients undergoing adjuvant therapy for resected pancreatic cancer. Oncogene. 2014; 33:5450–5456.
27. Bagchi A, Papazoglu C, Wu Y, Capurso D, Brodt M, Francis D, Bredel M, Vogel H, Mills AA. CHD5 is a tumor suppressor at human 1p36. Cell. 2007; 128:459–475.
28. Wang J, Xia Y, Li L, Gong D, Yao Y, Luo H, Lu H, Yi N, Wu H, Zhang X, Tao Q, Gao F. Double restriction-enzyme digestion improves the coverage and accuracy of genome-wide CpG methylation profiling by reduced representation bisulfite sequencing. BMC genomics. 2013; 14:11.
29. Shin G, Kang TW, Yang S, Baek SJ, Jeong YS, Kim SY. GENT: gene expression database of normal and tumor tissues. Cancer informatics. 2011; 10:149–157.
30. Rhodes DR, Yu J, Shanker K, Deshpande N, Varambally R, Ghosh D, Barrette T, Pandey A, Chinnaiyan AM. ONCOMINE: a cancer microarray database and integrated data-mining platform. Neoplasia. 2004; 6:1–6.
31. Cancer Genome Atlas Research N. Comprehensive molecular characterization of clear cell renal cell carcinoma. Nature. 2013; 499:43–49.
32. Beroukhim R, Brunet JP, Di Napoli A, Mertz KD, Seeley A, Pires MM, Linhart D, Worrell RA, Moch H, Rubin MA, Sellers WR, Meyerson M, Linehan WM, et al. Patterns of gene expression and copy-number alterations in von-hippel lindau disease-associated and sporadic clear cell carcinoma of the kidney. Cancer Res. 2009; 69:4674–4681.
33. Lenburg ME, Liou LS, Gerry NP, Frampton GM, Cohen HT, Christman MF. Previously unidentified changes in renal cell carcinoma gene expression identified by parametric analysis of microarray data. BMC Cancer. 2003; 3:31.
34. Dyrskjot L, Kruhoffer M, Thykjaer T, Marcussen N, Jensen JL, Moller K, Orntoft TF. Gene expression in the urinary bladder: a common carcinoma in situ gene expression signature exists disregarding histopathological classification. Cancer Res. 2004; 64:4040–4048.
35. Grasso CS, Wu YM, Robinson DR, Cao X, Dhanasekaran SM, Khan AP, Quist MJ, Jing X, Lonigro RJ, Brenner JC, Asangani IA, Ateeq B, Chun SY, et al. The mutational landscape of lethal castration-resistant prostate cancer. Nature. 2012; 487:239–243.
36. Cerami E, Gao J, Dogrusoz U, Gross BE, Sumer SO, Aksoy BA, Jacobsen A, Byrne CJ, Heuer ML, Larsson E, Antipin Y, Reva B, Goldberg AP, et al. The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer discovery. 2012; 2:401–404.
37. Gao J, Aksoy BA, Dogrusoz U, Dresdner G, Gross B, Sumer SO, Sun Y, Jacobsen A, Sinha R, Larsson E, Cerami E, Sander C, Schultz N. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Science signaling. 2013; 6:pl1.
38. Cancer Genome Atlas Research N. The Molecular Taxonomy of Primary Prostate Cancer. Cell. 2015; 163:1011–1025.
39. Robinson D, Van Allen EM, Wu YM, Schultz N, Lonigro RJ, Mosquera JM, Montgomery B, Taplin ME, Pritchard CC, Attard G, Beltran H, Abida W, Bradley RK, et al. Integrative clinical genomics of advanced prostate cancer. Cell. 2015; 161:1215–1228.
40. Cancer Genome Atlas Research N. Comprehensive molecular characterization of urothelial bladder carcinoma. Nature. 2014; 507:315–322.
41. Guo G, Gui Y, Gao S, Tang A, Hu X, Huang Y, Jia W, Li Z, He M, Sun L, Song P, Sun X, Zhao X, et al. Frequent mutations of genes encoding ubiquitin-mediated proteolysis pathway components in clear cell renal cell carcinoma. Nat Genet. 2012; 44:17–19.
42. Barbieri CE, Baca SC, Lawrence MS, Demichelis F, Blattner M, Theurillat JP, White TA, Stojanov P, Van Allen E, Stransky N, Nickerson E, Chae SS, Boysen G, et al. Exome sequencing identifies recurrent SPOP, FOXA1 and MED12 mutations in prostate cancer. Nat Genet. 2012; 44:685–689.
43. Davis CF, Ricketts CJ, Wang M, Yang L, Cherniack AD, Shen H, Buhay C, Kang H, Kim SC, Fahey CC, Hacker KE, Bhanot G, Gordenin DA, et al. The somatic genomic landscape of chromophobe renal cell carcinoma. Cancer cell. 2014; 26:319–330.
44. Quan J, Adelmant G, Marto JA, Look AT, Yusufzai T. The chromatin remodeling factor CHD5 is a transcriptional repressor of WEE1. PLoS One. 2014; 9:e108066.
45. Oliver SS, Musselman CA, Srinivasan R, Svaren JP, Kutateladze TG, Denu JM. Multivalent recognition of histone tails by the PHD fingers of CHD5. Biochemistry. 2012; 51:6534–6544.
46. Potts RC, Zhang P, Wurster AL, Precht P, Mughal MR, Wood WH, 3rd, Zhang Y, Becker KG, Mattson MP, Pazin MJ. CHD5, a brain-specific paralog of Mi2 chromatin remodeling enzymes, regulates expression of neuronal genes. PLoS One. 2011; 6:e24515.
47. Forbes SA, Beare D, Gunasekaran P, Leung K, Bindal N, Boutselakis H, Ding M, Bamford S, Cole C, Ward S, Kok CY, Jia M, De T, et al. COSMIC: exploring the world’s knowledge of somatic mutations in human cancer. Nucleic acids research. 2015; 43:D805–811.
48. Adzhubei IA, Schmidt S, Peshkin L, Ramensky VE, Gerasimova A, Bork P, Kondrashov AS, Sunyaev SR. A method and server for predicting damaging missense mutations. Nature methods. 2010; 7:248–249.
49. Li L, Ying J, Tong X, Zhong L, Su X, Xiang T, Shu X, Rong R, Xiong L, Li H, Chan AT, Ambinder RF, Guo Y, et al. Epigenetic identification of receptor tyrosine kinase-like orphan receptor 2 as a functional tumor suppressor inhibiting beta-catenin and AKT signaling but frequently methylated in common carcinomas. Cell Mol Life Sci. 2014; 71:2179–2192.
50. Shu XS, Li L, Ji M, Cheng Y, Ying J, Fan Y, Zhong L, Liu X, Tsao SW, Chan AT, Tao Q. FEZF2, a novel 3p14 tumor suppressor gene, represses oncogene EZH2 and MDM2 expression and is frequently methylated in nasopharyngeal carcinoma. Carcinogenesis. 2013; 34:1984–1993.
51. Li L, Ying J, Li H, Zhang Y, Shu X, Fan Y, Tan J, Cao Y, Tsao SW, Srivastava G, Chan AT, Tao Q. The human cadherin 11 is a pro-apoptotic tumor suppressor modulating cell stemness through Wnt/beta-catenin signaling and silenced in common carcinomas. Oncogene. 2011; 31:3901–3912.