
Introduction
Cancer is a major health issue, constituting the second leading cause of death worldwide and expected to surpass heart diseases as the leading cause of death in the next few years [1, 2]. In 2013, the incidence of cancer cases worldwide was 14.9 million, with 8.2 million cancer-related deaths. Lung cancer was the most common incident form of cancer, with an estimated 1.8 million new cases having deaths that exceeded those from any other type of malignancy worldwide, accounting for nearly one in five deaths (1.6 msillion deaths in total) [2]. Most lung cancers (~85%) are non-small cell lung cancers (NSCLC) which are divided according to two major histologic subtypes: the non-squamous carcinomas (mainly adenocarcinomas) and the squamous-cell carcinomas [3]. The parenchyma and the stroma are the two almost-indistinguishable compartments that compose the NSCLC and build up the tumor microenvironment [4]. The stromal cells contribute to the development and expression of certain cancer hallmark capabilities, defined by Hanahan and Weinberg in 2011 [5]. Among these, angiogenesis assumes major importance, since rate-limiting steps in tumor progressions include gaining access to the host vascular system and the generation of a tumor blood supply to obtain oxygen and nutrients, growth factors and hormones [6].
Angiogenesis and cancer
While the identification of massive vascularization in tumors dates back to 1863 [7] and the importance of tumor angiogenesis has been recognised since 1908 [8], it was only through the work of Folkman in the early 1970s that the scientific community acknowledge angiogenesis as a potential target to inhibit cancer progression [9-12]. The therapeutic potential of anti-angiogenic strategies boosted this field of research, placing angiogenesis as one of the major hubs of current cancer research.
It is now widely accepted that most tumors and metastases originate as small avascular structures which must induce the development of new blood vessels from pre-existing ones in order to grow beyond a minimum size of 2-3 mm3 [6, 13]. To achieve this, tumors undergo an angiogenic switch, disrupting the equilibrium between pro and anti-angiogenic regulators and favouring pro-angiogenic mechanisms. Signalling molecules induce quiescent endothelial cells to continuously sprout from existing blood vessels, thereby forming new vessels that help to sustain expanding neoplastic growth [6, 14, 15], according to the conventional model of angiogenesis known as angiogenic sprout [16].
Decades of research investigating the molecular basis of angiogenesis led to the discovery of a number of angiogenic molecules that promote tumor angiogenesis [15]. Of all the identified angiogenic pathways, the most critical appears to be the one involving the VEGF family and their receptors (VEGFR1-2-3) [17-19], although a number of other important molecules and their receptors have also proven to work in combination with VEGF/VEGFR signalling in tumor angiogenesis [19]. These include the fibroblast growth factor receptors (FGFRs) family and their ligands, particularly FGF1 and FGF2, that induce the proliferation and migration of endothelial cells [20]; as well as the platelet-derived growth factor receptors (PDGFRs) and their ligands (PDGFs) that, either alone or in combination with FGF and VEGF, are associated with tumor vascularization in malignant disease, including NSCLC [21, 22] and the Ang-Tie-2 system [19, 22]. Ever since the identification of VEGF as the first endothelium-acting specific cytokine in 1983 [13, 23, 24], its overexpression has been found in several human tumors, including NSCLC [25-29]. More recently, scientists are gaining a better understanding of the many functions of this molecule in the tumor angiogenic process [29]: it triggers multiple signalling networks that enhance endothelial cell proliferation and survival, increases migration and invasion of endothelial cells, increases vascular permeability of existing vessels, and enhances chemotaxis and mobilization of bone marrow derived endothelial progenitor cells (EPCs) into the peripheral circulation [30, 31].
The growing acknowledgment of VEGF’s key role in tumor angiogenesis has made it an attractive target for therapeutic intervention in cancer. The VEGF pathway is a promising avenue in research that aims to uncover more effective, targeted anti-angiogenic strategies [23, 32]. The extensive investigation in this field has led to the study of several anti-angiogenic agents, including monoclonal antibodies to block VEGF and its receptor VEGFR2 and VEGFR tyrosine kinase inhibitors (TKIs) [33].
Anti-angiogenic therapy and lung cancer
From the multitude of potential therapeutic options that target angiogenesis in NSCLC, [34] (Table 1), there are currently three anti-angiogenic compounds approved by EMA for the treatment of NSCLC. Bevacizumab, an anti-VEGF monoclonal antibody that blocks the binding of VEGF to its high-affinity receptors, was the first angiogenic inhibitor to complete clinical development, showing clinical benefit in patients with metastatic colorectal cancer when combined with chemotherapy [31, 33]. It was approved in 2006 for the treatment of advanced non-squamous NSCLC in the first line setting in combination with chemotherapy [29]. In 2014, ramucirumab, a fully humanized monoclonal antibody that targets angiogenesis by specifically binding to VEGFR-2 with higher affinity than its natural ligand VEGF [35], was approved for the treatment of patients with metastatic NSCLC in second line setting, in combination with docetaxel [36]. In the same year, nintedanib, an oral medication that can simultaneously inhibit triple angiokinase, VEGFR, platelet-derived growth factor receptors (PDGFR), and fibroblast growth factor receptors (FGFR) signalling pathways, was approved to be used in combination with docetaxel in patients with locally advanced, metastatic, or locally recurrent NSCLC adenocarcinoma, after first-line chemotherapy [37]. There are also other potential agents that are under clinical evaluation, whether that be in the clinical trial stage or currently waiting for approval for treatment of metastatic or recurrent NSCLC [38, 39].
Table 1: Angiogenesis inhibitors in non-small cell lung cancer (NSCLC)
Approved |
||
Drug |
Target |
Indication |
Bevacizumab |
VEGF |
First-line treatment of nonsquamous NSCLC with CT |
Nintedanib |
VEGFR 2, FGFR 1-3, PDGFRα and β TKI |
Second-line treatment of adenocarcinoma NSCLC with CT |
Ramucirumab |
VEGFR-2 |
Second-line treatment of NSCLC with CT |
On clinical trials or not approved |
||
Drug |
Target |
|
Vandetanib |
VEGFRs, EGFR, and RET |
|
Sunitinib |
VEGFRs, PDGFRs, KIT, FLT3, CSF-1R, and RET |
|
Aflibercept |
VEGF |
|
Sorafenib |
VEGFR, PDGFRs, FGFR, KIT, and RAF |
|
Motesanib |
VEGFRs, PDGFRs, and KIT |
|
Pazopanib |
VEGFRs, PDGFRs, FGFR, and KIT |
|
Cediranib |
VEGFRs |
|
Cabozantinib |
VEGFR, RET, and MET |
|
Axitinib |
VEGFRs, PDGFRs, and KIT |
|
CT - chemotherapy; VEGF - vascular endothelial growth factor; VEGFR - vascular endothelial growth factor receptor; FGFR - fibroblast growth factor receptor; PDGFR - platelet-derived growth factor receptor; KIT - stem cell factor receptor; FLT3 - Fms-like tyrosine kinase-3; CSF-1R - colony stimulating factor receptor; RET - glial cell-line derived neurotrophic factor receptor; MET - met proto-oncogene;
In spite of the impressive clinical efficacy of bevacizumab, ramucirumab, and nintedanib in various cancer treatment settings, the results were relatively modest and limited [40]. In addition, the clinical use of VEGF/VEGFR blockers as anti- angiogenic therapy for patients with advanced NSCLC has been more challenging than anticipated by the preclinical experiments in which long-term benefit of VEGF/VEGFR inhibition was achieved [41]. Anti-angiogenic agents are usually given to all patients for the approved indications; in a high fraction of these patients, however, the tumor is intrinsically refractory to the anti-angiogenic therapy and the disease progresses ceaselessly [42]. Moreover, when there is no intrinsic resistance, acquired resistance to therapy can rapidly occur and limit the efficacy of the anti-angiogenic treatments [41, 43], and the clinical benefit of prolonging cancer patients survival with advanced disease becomes limited, often in the order of weeks or months [16, 44].
Tumor resistance to the anti-angiogenic therapies (whether intrinsic or acquired), represents a significant problem faced in routine clinical practice. The mechanisms underlying the response to these therapies are far from being clearly understood, further fuelling this active field of research [43]. Preclinical investigations have shed some light on the subject, and although different authors propose escape ways from angiogenic inhibitors that are somewhat distinct, some key features appear to be consensual among most of them; these features are likely to be involved in primary and acquired resistance and deserve consideration [16, 18, 41-43, 45]. One of such features is invasive (or metastatic) co-option of normal quiescent vessels without requisite of angiogenesis.
Vessel co-option and Lung Cancer growth
It is widely accepted that tumor progression is heavily dependent on angiogenesis. Much less understood, is the concept that angiogenesis is necessary for a tumor to become larger than a few millimetres and become clinically detectable, as some research has shown that angiogenesis is not always a pre-requisite for tumor growth [46]. Hence, one possibility for anti-angiogenic therapy resistance is that some primary and metastatic tumors are non-angiogenic, meaning that these tumors do not need angiogenic sprout to obtain an efficient blood supply [47]. Rather, the tumors use alternative vascularization mechanisms. For example, in vessel-dense tissues, the most likely route is hijacking the pre-existing normal blood vessels [42, 44, 48], and more aggressive tumors can undergo vasculogenic mimicry, a process by which tumor cells dedifferentiate to an endothelial phenotype forming structures that provide tumour cells with a secondary circulation system independently of angiogenesis [49].
When tumors arise in well-vascularized organs, their growth will rely on the invasion of host tissue. Enhancement of invasion and metastasis facilitates access to normal tissue vasculature, and cancer cells stay in close contact with the surface of blood vessels [39, 50, 51]. This allows tumor cells to grow and migrate along quiescent normal vessels and take their oxygen and essential nutrients without obligate neovascularization, in a process known as vessel (or vascular) co-option [42, 43, 49]. These non-angiogenic tumors are a separate group of fast-growing malignancies with little apoptosis and very efficient mitochondrial metabolism [52]. This seems to be the case of tumors arising in the lungs, liver, and brain, areas where this form of vascularization appears to assume a major role [31, 50, 51, 53, 54]. This is also true for tumor metastasis that occurs through lymph and blood vessels and outgrow mostly in these vessel-dense organs [55-58].
In recent years, research related to angiogenesis has been massive; but on the contrary, there is a scarcity of research focusing on tumors that escape pathways of classical angiogenesis and use vessel co-option as an alternative blood supply for tumor growth. This has led to a dearth in information regarding the mechanisms and players involved in that process.
The first insights into the relationship between vessel co-option and lung cancer were made by Pezzella and co-workers, who described NSCLC that grew without morphological evidence of neoangiogenesis but with signs of normal tissue vessel exploitation [59]. They characterized these tumors as having an alveolar pattern, with tumor cell nests filling the alveolar spaces without destruction of the lung parenchyma. The only vessels evident in these tumors appeared to belong to the trapped alveolar septa [59]. Moreover, patients with alveolar pattern tumors presented a worse survival rate than their angiogenic counterparts. Later, when investigating the possible role of microvessel count in NSCLC as a potential marker of disease prognosis, Offersen and colleagues [60] identified the same special vascular pattern in 17 out of 35 NSCLC samples, thus confirming the description of Pezzella’s group. Their observations led them to the hypothesis that these alveolar tumors are nonangiogenic and invasive and exploited the pre-existing vascular beds. They also noted that some tumors exhibited only the alveolar pattern while other tumors presented a mixed alveolar pattern consisting of both alveolar and angiogenic features [60]. There was no correlation, however, between angiogenic or vessel co-option status and disease aggressiveness.
Taking into account the NSCLC growth patterns, Nia Sardari et al. suggested a modification of Pezzella’s classification according to morphological features, based on the biological properties of the tumor-lung interface, which is the region where the tumor expands and the tumor-stroma interactions are more active and homogeneous [61]. According to them, NSCLCs can be classified as having a destructive growth pattern (angiogenic growth pattern), papillary growth pattern (with preservation of the alveolar structure of the lung parenchyma at the interface with co-option of alveolar blood vessels with formation of stromal stalks and subsequent angiogenesis), and alveolar growth pattern (preservation of the alveolar structure of lung parenchyma with co-option of septal blood vessels and without evidence of new stroma formation at the interface). Moreover, they suggested that, in NSCLC, a low degree of ongoing angiogenesis is predictive of poorer prognosis [61, 62].
The hypothesis of co-option by lung metastases, which are often the main cause of death in many solid malignancies, was also proposed by Pezzella’s group back in the 1990’s. They observed that, regardless of the angiogenic status of the primary breast carcinomas, they could relapse as nonangiogenic tumors in the lungs. This was also true for lung metastases of human renal and colorectal carcinomas [56, 63, 64]. In a very recent study, Szabo and co-workers used cell lines from six different solid tumors, and showed that lung metastases vascularize by co-opting the pulmonary microvasculature. The investigated cell lines incorporated the pre-existing host tissue capillaries within the alveolar walls, striping the epithelium from these co-opted alveolar walls [57]. Once there, the metastases expand as the malignant cells spread from one alveolar space to another. Their work not only shed some light on the mechanisms underlying this phenomenon, but it also raised some questions surrounding the biology of the nonangiogenic tumors, further advocating the need for additional exploration in this subject.
Vessel co-option and Angiopoietin-2
In the lungs, the normal co-opted vessels trapped in the tumor can be very effective because they allow for more efficient tumor growth by exploiting the highly regular vascular network of the lungs and progressively filling the empty alveolar spaces [46]. Regardless of the efficacy of vessel co-option in sustaining tumor growth, the quiescent blood vessels co-opted by tumors suffer extreme changes over time [65]. While there is still debate if this due to a host defence mechanism against tumor development [47] or whether dependence on the survival of endothelial cells (ECs) [50], there is little doubt on the subsequent alterations observed. First, in the centre of the tumor, there is widespread regression of the co-opted vessels associated with the regression of the EC, turning it progressively hypoxic, with subsequent massive tumor cell loss [13, 32], followed by a robust de novo angiogenesis at the outer rim of the tumor, that rescues the remaining tumor cells in a later stage [13, 31].
The key regulator in the regression of the initially co-opted blood vessels appears to be Angiopoietin-2 (Ang-2) [49, 53, 66], a cytokine that belongs to the Angiopoietins family, an important class of angiogenic molecules. It is a natural ligand of the endothelial tyrosine kinase-receptor, Tie-2, primarily synthetized and secreted by ECs at sites of vascular remodelling, like tumors, in a tightly regulated fashion [66-68]. Ang-2 is overexpressed in a number of tumors including NSCLC [69, 70], and there is also evidence that it is deeply involved in lung metastases homing and progression [71, 72]. Experimental evidence supports the notion that, soon after vessel co-option, host vessels start to express high levels of Ang-2 that acts through an endogenous autocrine loop mechanism that is context dependent [73, 74]. When it binds to its Tie-2 receptor, it functions as a vessel-destabilizing molecule that converts mature vessels to a tenuous and plastic state by inducing loosening of endothelial cell interactions with pericytes and smooth muscle cells, leading to the loss of vascular integrity and increased vascular permeability. The ECs of such destabilized vessels can be prone to two fates, depending on the local cytokine milieu [74, 75]. In the presence of VEGF, these cells will respond to the proliferating signals induced by the pro-angiogenic molecule and will migrate or proliferate, triggering a sprouting angiogenesis [13, 66, 70, 73, 76, 77]. In the absence of VEGF, however, the expression of Ang-2 causes irreversible loss of vascular structures [76,78] with marked regression of the co-opted vessels, as is the case when tumors co-opt pre-existing vessels [77]. This is due to the fact that, without the pericytes coverage, the ECs of the Ang-2-unstable vessels will die [79] in a very similar fashion to what happens with primitive vessels during development [74]. This generates the hypoxic core and the apoptotic tumor cell loss observed in nonangiogenic tumors [47, 76], that presumably act as the initial stimulus for the molecular changes that culminate in VEGF expression by the remaining tumor cells and in neoangiogenesis [69], mediated both by VEGF and Ang-2 [47] (Figure 1).
Not surprisingly, the discovery of the role of Ang-2 in tumor progression led to the suggestion that its inhibition could translate into clinically meaningful responses, opening the door to multiple approaches that have been used to experimentally inhibit Ang-2 as well as explore its effects on angiogenesis and tumor growth [80-82]. Pre-clinical models revealed that Ang-2 inhibition reduces the growth of a broad range of tumors. Although some of the results were modest, some revealed to be very promising and there is now a robust pipeline of drugs targeting the Ang/Tie-2 system in different clinical trials phases (Table 2) [67, 78, 83]. Furthermore, with Ang-2 being required to render endothelium responsive to VEGF and with both molecules contributing to tumor angiogenesis and metastases [84, 85], there seems to be a more encouraging response to the straightforward question of whether co-targeting of both ligands in a bispecific manner would improve the outcomes of current anti-angiogenic therapies [80, 83, 86-88].
Table 2: Ang-1/Ang-2 and Tie inhibitors in development for NSCLC
Drug |
Target |
Studies |
Regorafenib |
VEGFRs, PDGFRs, FGFR, RET, Kit, B-Raf and Tie-2 |
NCT01187615 |
Trebananib Fc fusion peptibody |
Ang-1 and Ang-2 |
NCT01666977, EudraCT 2011-001111-31 |
Foretinib |
VEGFRs, PDGFR β, FLT3, MET, and Tie-2 |
NCT01068587 |
MGCD265 |
VEGFRs, MET, and Tie-2 |
NCT02544633, EudraCT 2015-002070-21 |
AMG 780 Fully human anti Ang-1/2 mAb |
Ang-1 and Ang-2 |
NCT01137552 |
VEGFR - vascular endothelial growth factor receptor; PDGFR - platelet-derived growth factor receptor; FGFR - fibroblast growth factor receptor; RET - glial cell-line derived neurotrophic factor receptor; KIT - stem cell factor receptor; Tie - endothelial tyrosine kinase-receptor; FLT3 - Fms-like tyrosine kinase-3; B-Raf - serine/threonine-protein kinase;
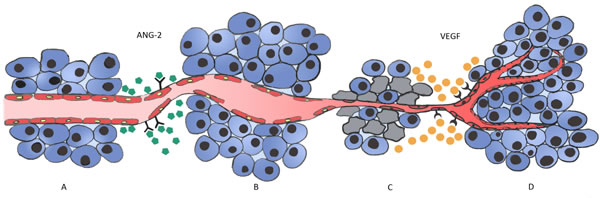
Figure 1: Vessel co-option and Ang-2 regulation in cancer development in vessel dense tissues. A. In well vascularized organs, such as the lung, tumor cells grow and migrate along quiescent normal vessels (vessel co-option). B. Over time, tumor cells induce extreme changes in the co-opted vessels and ECs start to express Ang-2, leading to vascular disruption and vessel regression. C. Regression of the co-opted vessel associated with regression of the ECs generates a hypoxic core in the tumor centre, with massive tumor cell loss. This triggers the angiogenic switch, with the remaining tumor cells expressing high amounts of VEGF. D. VEGF expression induces a robust angiogenic response that ultimately rescues the tumor and allows its growth and progression.
Vessel co-option and clinical implications
The ability to identify tumors that make vascular co-option their primary source of blood supply does not envisage an easy task, hence why few strategies have been used to achieve this goal [48]. Research in the field has been scarce, especially when compared to the angiogenic field that has largely overshadowed alternative blood sources for tumor development. Moreover, much of the research has been performed in cell lines or murine models and only a few in human tissues [48]. While the findings are limited so far, what has been discovered highly advocates for unravelling the vessel co-option mechanisms and involved players. The precise identification of tumors that preferentially use this route to support growth and the factors driving them to switch from this to an angiogenic pattern may be crucial to delineate future cancer treatments for two main reasons. The first is that vascular co-option may represent a clever strategy by which tumors partly evade and resist conventional anti-angiogenic treatments [89]. Even if a treatment like bevacizumab is effective against one angiogenic factor such as VEGF, the therapy can still fail if this factor is not important for the endothelium in that given tumor, as appears to be the case in tumors that co-opt pre-existing vessels in NSCLC [45]. In these cases, vessel co-option may serve as a pathologic biomarker for selecting potentially nonresponsive patients [43]. There is also evidence that in some nonangiogenic tumors, cancer cells adapt by migrating more aggressively into normal tissue [42]; and when anti-angiogenic treatments are used indiscriminately, they may contribute to the selection of clones of nonangiogenic cells that will progress with a more aggressive behaviour [89, 90]. These features should be carefully considered when planning anti-angiogenic therapeutic interventions, suggesting the need for tailor-made treatments against such tumors.
Secondly, anti-angiogenic compounds do not affect incorporated pre-existent vasculature or matured tumor vasculature, making targeting existing vessels on which the tumor growth relies, an attractive approach to accomplish tumor regression [91]. This is also of primordial importance in cases of metastases that establish in well-vascularized organs, since vessel co-option may constitute their primary feeding option [57]. Moreover, it can be speculated that in earlier stages of the tumor, the interval that mediates Ang-2 overexpression, co-opted vessels regression, and de novo angiogenesis seems to be the perfect therapeutic window for intervention using a dual-pronged approach with Ang-2 and VEGF blockers rather than in more advanced stages of the disease. This issue should be addressed by investigators developing pre-clinical/clinical trials of drugs that target angiogenesis or envisage tumor arrest by anti-angiogenic strategies.
Conclusions
Anti-angiogenic strategies focusing on VEGF/VEGFR in combination with chemotherapy marked a milestone in the field of cancer treatment, including NSCLC. However, a relevant number of patients are unresponsive or refractory to anti-angiogenic treatments. Some tumors obviate the need to generate angiogenesis by co-opting host mature vessels and growing along them, using them as blood sources. Vessel co-option is a mechanism that may help explain the limited success of anti-angiogenic therapy in these patients in an adjuvant setting.
Thus far, the only growth factors proven to be associated with vessel co-option are VEGF and Ang-2. This lack of information is likely due to the limited number of studies examining this subject. Ang-2 seems to have a particularly critical role in the process, but is also an extremely laborious study topic due to the complexity of its functions and regulation, which are both highly cell context dependent.
Tumors that grow in non-angiogenic fashions through exploitation of pre-existing vessels are non-responsive to anti-angiogenic molecules and raise a number of concerns in terms of treatment. First, little is known about the modifications a neoplastic cell must go through in order to co-opt a blood vessel, which is a huge obstacle for strategies that aim to interfere with this step in tumor progression. Second, once the tumor is committed to vascular co-option pathway, an effective way of blocking tumor progression would be to target existing tumor vasculature; this would require the availability of tumor-vessel specific targeting agents, however, and the few candidates that have been identified so far have failed to prove their clinical efficacy.
All of these concerns reinforce the need for better understanding of the mechanisms and molecular players underlying vessel co-option during tumor development within the proper biologic context. This would not only explore more assertive cancer treatments and help with the identification of tumors where vessel co-option is the growth support (instead of angiogenesis), but could also help identify patients who may be nonresponsive to current anti-angiogenic treatments. Additionally, it could open doors to novel areas of NSCLC research at both the molecular and microanatomical level.
Acknowledgments
The authors would like to thank the Liga Portuguesa Contra o Cancro - Núcleo Regional do Norte, for the Educational Grant conceded to Ana Luísa Coelho.
The authors would like to thank Dr. Rachel Zsido for her valuable contribution in the critical review of redaction.
Conflicts of Interest
The authors declare they have no potential conflict of interests with this research.
References
1. Siegel RL, Miller KD, Jemal A. Cancer statistics, 2015. CA Cancer J Clin. 2015; 65: 5-29.
2. Fitzmaurice C, Dicker D, Pain A, Hamavid H, Moradi-Lakeh M, MacIntyre MF, Allen C, Hansen G, Woodbrook R, Wolfe C, Hamadeh RR, Moore A, Werdecker A, et al. The Global Burden of Cancer 2013. JAMA Oncol. 2015; 1: 505-27.
3. Reck M, Heigener DF, Mok T, Soria JC, Rabe KF. Management of non-small-cell lung cancer: recent developments. Lancet. 2013; 382: 709-19.
4. Schiavoni G, Gabriele L, Mattei F. The tumor microenvironment: a pitch for multiple players. Front Oncol. 2013; 3: 90.
5. Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011; 144: 646-74.
6. Bergers G, Benjamin LE. Tumorigenesis and the angiogenic switch. Nat Rev Cancer. 2003; 3: 401-10.
7. Fan F, Schimming A, Jaeger D, Podar K. Targeting the tumor microenvironment: focus on angiogenesis. J Oncol. 2012; 2012: 281261.
8. Goldmann E. The Growth of Malignant Disease in Man and the Lower Animals, with special reference to the Vascular System. Proc R Soc Med. 1908; 1: 1-13.
9. Folkman J. Tumor angiogenesis: therapeutic implications. N Engl J Med. 1971; 285: 1182-6.
10. Folkman J, Merler E, Abernathy C, Williams G. Isolation of a tumor factor responsible for angiogenesis. J Exp Med. 1971; 133: 275-88.
11. Holleb AI, Folkman J. Tumor angiogenesis. CA Cancer J Clin. 1972; 22: 226-9.
12. Folkman J. Anti-angiogenesis: new concept for therapy of solid tumors. Ann Surg. 1972; 175: 409-16.
13. Holash J, Wiegand SJ, Yancopoulos GD. New model of tumor angiogenesis: dynamic balance between vessel regression and growth mediated by angiopoietins and VEGF. Oncogene. 1999; 18: 5356-62.
14. Hanahan D, Folkman J. Patterns and emerging mechanisms of the angiogenic switch during tumorigenesis. Cell. 1996; 86: 353-64.
15. Baeriswyl V, Christofori G. The angiogenic switch in carcinogenesis. Semin Cancer Biol. 2009; 19: 329-37.
16. Potente M, Gerhardt H, Carmeliet P. Basic and therapeutic aspects of angiogenesis. Cell. 2011; 146: 873-87.
17. Jain RK. Normalization of tumor vasculature: an emerging concept in antiangiogenic therapy. Science. 2005; 307: 58-62.
18. Dvorak HF. Vascular permeability factor/vascular endothelial growth factor: a critical cytokine in tumor angiogenesis and a potential target for diagnosis and therapy. J Clin Oncol. 2002; 20: 4368-80.
19. Zhao Y, Adjei AA. Targeting Angiogenesis in Cancer Therapy: Moving Beyond Vascular Endothelial Growth Factor. Oncologist. 2015; 20: 660-73.
20. Presta M, Dell’Era P, Mitola S, Moroni E, Ronca R, Rusnati M. Fibroblast growth factor/fibroblast growth factor receptor system in angiogenesis. Cytokine Growth Factor Rev. 2005; 16: 159-78.
21. Cao Y. Multifarious functions of PDGFs and PDGFRs in tumor growth and metastasis. Trends Mol Med. 2013; 19: 460-73.
22. Ferrara N, Kerbel RS. Angiogenesis as a therapeutic target. Nature. 2005; 438: 967-74.
23. Ferrara N. VEGF and the quest for tumour angiogenesis factors. Nat Rev Cancer. 2002; 2: 795-803.
24. Senger DR, Galli SJ, Dvorak AM, Perruzzi CA, Harvey VS, Dvorak HF. Tumor cells secrete a vascular permeability factor that promotes accumulation of ascites fluid. Science. 1983; 219: 983-5.
25. Yuan A, Yu CJ, Chen WJ, Lin FY, Kuo SH, Luh KT, Yang PC. Correlation of total VEGF mRNA and protein expression with histologic type, tumor angiogenesis, patient survival and timing of relapse in non-small-cell lung cancer. Int J Cancer. 2000; 89: 475-83.
26. Imoto H, Osaki T, Taga S, Ohgami A, Ichiyoshi Y, Yasumoto K. Vascular endothelial growth factor expression in non-small-cell lung cancer: prognostic significance in squamous cell carcinoma. J Thorac Cardiovasc Surg. 1998; 115: 1007-14.
27. Koukourakis MI, Giatromanolaki A, Thorpe PE, Brekken RA, Sivridis E, Kakolyris S, Georgoulias V, Gatter KC, Harris AL. Vascular endothelial growth factor/KDR activated microvessel density versus CD31 standard microvessel density in non-small cell lung cancer. Cancer Res. 2000; 60: 3088-95.
28. Stefanou D, Batistatou A, Arkoumani E, Ntzani E, Agnantis NJ. Expression of vascular endothelial growth factor (VEGF) and association with microvessel density in small-cell and non-small-cell lung carcinomas. Histol Histopathol. 2004; 19: 37-42.
29. Villaruz LC, Socinski MA. The role of anti-angiogenesis in non-small-cell lung cancer: an update. Curr Oncol Rep. 2015; 17: 26.
30. Hicklin DJ, Ellis LM. Role of the vascular endothelial growth factor pathway in tumor growth and angiogenesis. J Clin Oncol. 2005; 23: 1011-27.
31. Ellis LM, Hicklin DJ. VEGF-targeted therapy: mechanisms of anti-tumour activity. Nat Rev Cancer. 2008; 8: 579-91.
32. Korn C, Augustin HG. Mechanisms of Vessel Pruning and Regression. Dev Cell. 2015; 34: 5-17.
33. Ferrara N, Hillan KJ, Gerber HP, Novotny W. Discovery and development of bevacizumab, an anti-VEGF antibody for treating cancer. Nat Rev Drug Discov. 2004; 3: 391-400.
34. Crino L, Metro G. Therapeutic options targeting angiogenesis in nonsmall cell lung cancer. Eur Respir Rev. 2014; 23: 79-91.
35. Reinmuth N, Heigener D, Reck M. Novel angiogenesis inhibitors in nonsmall cell lung cancer. Curr Opin Oncol. 2015; 27: 79-86.
36. Garon EB, Ciuleanu TE, Arrieta O, Prabhash K, Syrigos KN, Goksel T, Park K, Gorbunova V, Kowalyszyn RD, Pikiel J, Czyzewicz G, Orlov SV, Lewanski CR, et al. Ramucirumab plus docetaxel versus placebo plus docetaxel for second-line treatment of stage IV non-small-cell lung cancer after disease progression on platinum-based therapy (REVEL): a multicentre, double-blind, randomised phase 3 trial. Lancet. 2014; 384: 665-73.
37. Reck M, Kaiser R, Mellemgaard A, Douillard JY, Orlov S, Krzakowski M, von Pawel J, Gottfried M, Bondarenko I, Liao M, Gann CN, Barrueco J, Gaschler-Markefski B, et al. Docetaxel plus nintedanib versus docetaxel plus placebo in patients with previously treated non-small-cell lung cancer (LUME-Lung 1): a phase 3, double-blind, randomised controlled trial. Lancet Oncol. 2014; 15: 143-55.
38. Aggarwal C, Somaiah N, Simon G. Antiangiogenic agents in the management of non-small cell lung cancer: where do we stand now and where are we headed? Cancer Biol Ther. 2012; 13: 247-63.
39. Pallis AG, Syrigos KN. Targeting tumor neovasculature in non-small-cell lung cancer. Crit Rev Oncol Hematol. 2013; 86: 130-42.
40. Jain RK. Normalizing tumor microenvironment to treat cancer: bench to bedside to biomarkers. J Clin Oncol. 2013; 31: 2205-18.
41. Carmeliet P, Jain RK. Molecular mechanisms and clinical applications of angiogenesis. Nature. 2011; 473: 298-307.
42. Bergers G, Hanahan D. Modes of resistance to anti-angiogenic therapy. Nat Rev Cancer. 2008; 8: 592-603.
43. Huang Y, Carbone DP. Mechanisms of and strategies for overcoming resistance to anti-vascular endothelial growth factor therapy in non-small cell lung cancer. Biochim Biophys Acta. 2015; 1855: 193-201.
44. Bertolini F, Marighetti P, Martin-Padura I, Mancuso P, Hu-Lowe DD, Shaked Y, D’Onofrio A. Anti-VEGF and beyond: shaping a new generation of anti-angiogenic therapies for cancer. Drug Discov Today. 2011; 16: 1052-60.
45. Eikesdal HP, Kalluri R. Drug resistance associated with antiangiogenesis therapy. Semin Cancer Biol. 2009; 19: 310-7.
46. Passalidou E, Trivella M, Singh N, Ferguson M, Hu J, Cesario A, Granone P, Nicholson AG, Goldstraw P, Ratcliffe C, Tetlow M, Leigh I, Harris AL, et al. Vascular phenotype in angiogenic and non-angiogenic lung non-small cell carcinomas. Br J Cancer. 2002; 86: 244-9.
47. Ribatti D, Vacca A, Dammacco F. New non-angiogenesis dependent pathways for tumour growth. Eur J Cancer. 2003; 39: 1835-41.
48. Donnem T, Hu J, Ferguson M, Adighibe O, Snell C, Harris AL, Gatter KC, Pezzella F. Vessel co-option in primary human tumors and metastases: an obstacle to effective anti-angiogenic treatment? Cancer Med. 2013; 2: 427-36.
49. Hillen F, Griffioen AW. Tumour vascularization: sprouting angiogenesis and beyond. Cancer Metastasis Rev. 2007; 26: 489-502.
50. Dome B, Hendrix MJ, Paku S, Tovari J, Timar J. Alternative vascularization mechanisms in cancer: Pathology and therapeutic implications. Am J Pathol. 2007; 170: 1-15.
51. Carbonell WS, Ansorge O, Sibson N, Muschel R. The vascular basement membrane as “soil” in brain metastasis. PLoS One. 2009; 4: e5857.
52. Hu J, Bianchi F, Ferguson M, Cesario A, Margaritora S, Granone P, Goldstraw P, Tetlow M, Ratcliffe C, Nicholson AG, Harris A, Gatter K, Pezzella F. Gene expression signature for angiogenic and nonangiogenic non-small-cell lung cancer. Oncogene. 2005; 24: 1212-9.
53. Dvorak HF. Tumor Stroma, Tumor Blood Vessels, and Antiangiogenesis Therapy. Cancer J. 2015; 21: 237-43.
54. Adighibe O, Micklem K, Campo L, Ferguson M, Harris A, Pozos R, Gatter K, Pezzella F. Is nonangiogenesis a novel pathway for cancer progression? A study using 3-dimensional tumour reconstructions. Br J Cancer. 2006; 94: 1176-9.
55. Leenders WP, Kusters B, Verrijp K, Maass C, Wesseling P, Heerschap A, Ruiter D, Ryan A, de Waal R. Antiangiogenic therapy of cerebral melanoma metastases results in sustained tumor progression via vessel co-option. Clin Cancer Res. 2004; 10: 6222-30.
56. Evidence for novel non-angiogenic pathway in breast-cancer metastasis. Breast Cancer Progression Working Party. Lancet. 2000; 355: 1787-8.
57. Szabo V, Bugyik E, Dezso K, Ecker N, Nagy P, Timar J, Tovari J, Laszlo V, Bridgeman VL, Wan E, Frentzas S, Vermeulen PB, Reynolds AR, et al. Mechanism of tumour vascularization in experimental lung metastases. J Pathol. 2015; 235: 384-96.
58. Kusters B, Leenders WP, Wesseling P, Smits D, Verrijp K, Ruiter DJ, Peters JP, van Der Kogel AJ, de Waal RM. Vascular endothelial growth factor-A(165) induces progression of melanoma brain metastases without induction of sprouting angiogenesis. Cancer Res. 2002; 62: 341-5.
59. Pezzella F, Pastorino U, Tagliabue E, Andreola S, Sozzi G, Gasparini G, Menard S, Gatter KC, Harris AL, Fox S, Buyse M, Pilotti S, Pierotti M, et al. Non-small-cell lung carcinoma tumor growth without morphological evidence of neo-angiogenesis. Am J Pathol. 1997; 151: 1417-23.
60. Offersen BV, Pfeiffer P, Hamilton-Dutoit S, Overgaard J. Patterns of angiogenesis in nonsmall-cell lung carcinoma. Cancer. 2001; 91: 1500-9.
61. Sardari Nia P, Colpaert C, Blyweert B, Kui B, Vermeulen P, Ferguson M, Hendriks J, Weyler J, Pezzella F, Van Marck E, Van Schil P. Prognostic value of nonangiogenic and angiogenic growth patterns in non-small-cell lung cancer. Br J Cancer. 2004; 91: 1293-300.
62. Sardari Nia P, Colpaert C, Vermeulen P, Weyler J, Pezzella F, Van Schil P, Van Marck E. Different growth patterns of non-small cell lung cancer represent distinct biologic subtypes. Ann Thorac Surg. 2008; 85: 395-405.
63. Pezzella F, Di Bacco A, Andreola S, Nicholson AG, Pastorino U, Harris AL. Angiogenesis in primary lung cancer and lung secondaries. Eur J Cancer. 1996; 32A: 2494-500.
64. Sardari Nia P, Hendriks J, Friedel G, Van Schil P, Van Marck E. Distinct angiogenic and non-angiogenic growth patterns of lung metastases from renal cell carcinoma. Histopathology. 2007; 51: 354-61.
65. Ziyad S, Iruela-Arispe ML. Molecular mechanisms of tumor angiogenesis. GenesCancer. 2011; 2: 1085-96. doi:10.1177/1947601911432334.
66. Mazzieri R, Pucci F, Moi D, Zonari E, Ranghetti A, Berti A, Politi LS, Gentner B, Brown JL, Naldini L, De Palma M. Targeting the ANG2/TIE2 axis inhibits tumor growth and metastasis by impairing angiogenesis and disabling rebounds of proangiogenic myeloid cells. Cancer Cell. 2011; 19: 512-26.
67. Cascone T, Heymach JV. Targeting the angiopoietin/Tie2 pathway: cutting tumor vessels with a double-edged sword? J Clin Oncol. 2012; 30: 441-4.
68. Augustin HG, Koh GY, Thurston G, Alitalo K. Control of vascular morphogenesis and homeostasis through the angiopoietin-Tie system. Nat Rev Mol Cell Biol. 2009; 10: 165-77.
69. Wong MP, Chan SY, Fu KH, Leung SY, Cheung N, Yuen ST, Chung LP. The angiopoietins, tie2 and vascular endothelial growth factor are differentially expressed in the transformation of normal lung to non-small cell lung carcinomas. Lung Cancer. 2000; 29: 11-22.
70. Daly C, Eichten A, Castanaro C, Pasnikowski E, Adler A, Lalani AS, Papadopoulos N, Kyle AH, Minchinton AI, Yancopoulos GD, Thurston G. Angiopoietin-2 functions as a Tie2 agonist in tumor models, where it limits the effects of VEGF inhibition. Cancer Res. 2013; 73: 108-18.
71. Holopainen T, Saharinen P, D’Amico G, Lampinen A, Eklund L, Sormunen R, Anisimov A, Zarkada G, Lohela M, Helotera H, Tammela T, Benjamin LE, Yla-Herttuala S, et al. Effects of angiopoietin-2-blocking antibody on endothelial cell-cell junctions and lung metastasis. J Natl Cancer Inst. 2012; 104: 461-75.
72. Minami T, Jiang S, Schadler K, Suehiro J, Osawa T, Oike Y, Miura M, Naito M, Kodama T, Ryeom S. The calcineurin-NFAT-angiopoietin-2 signaling axis in lung endothelium is critical for the establishment of lung metastases. Cell Rep. 2013; 4: 709-23.
73. Scharpfenecker M, Fiedler U, Reiss Y, Augustin HG. The Tie-2 ligand angiopoietin-2 destabilizes quiescent endothelium through an internal autocrine loop mechanism. J Cell Sci. 2005; 118: 771-80.
74. Yancopoulos GD, Davis S, Gale NW, Rudge JS, Wiegand SJ, Holash J. Vascular-specific growth factors and blood vessel formation. Nature. 2000; 407: 242-8.
75. Nasarre P, Thomas M, Kruse K, Helfrich I, Wolter V, Deppermann C, Schadendorf D, Thurston G, Fiedler U, Augustin HG. Host-derived angiopoietin-2 affects early stages of tumor development and vessel maturation but is dispensable for later stages of tumor growth. Cancer Res. 2009; 69: 1324-33.
76. Yu Q, Stamenkovic I. Angiopoietin-2 is implicated in the regulation of tumor angiogenesis. Am J Pathol. 2001; 158: 563-70.
77. Holash J, Maisonpierre PC, Compton D, Boland P, Alexander CR, Zagzag D, Yancopoulos GD, Wiegand SJ. Vessel cooption, regression, and growth in tumors mediated by angiopoietins and VEGF. Science. 1999; 284: 1994-8.
78. Huang H, Lai JY, Do J, Liu D, Li L, Del Rosario J, Doppalapudi VR, Pirie-Shepherd S, Levin N, Bradshaw C, Woodnutt G, Lappe R, Bhat A. Specifically targeting angiopoietin-2 inhibits angiogenesis, Tie2-expressing monocyte infiltration, and tumor growth. Clin Cancer Res. 2011; 17: 1001-11.
79. Huang H, Bhat A, Woodnutt G, Lappe R. Targeting the ANGPT-TIE2 pathway in malignancy. Nat Rev Cancer. 2010; 10: 575-85.
80. Hashizume H, Falcon BL, Kuroda T, Baluk P, Coxon A, Yu D, Bready JV, Oliner JD, McDonald DM. Complementary actions of inhibitors of angiopoietin-2 and VEGF on tumor angiogenesis and growth. Cancer Res. 2010; 70: 2213-23.
81. Brown JL, Cao ZA, Pinzon-Ortiz M, Kendrew J, Reimer C, Wen S, Zhou JQ, Tabrizi M, Emery S, McDermott B, Pablo L, McCoon P, Bedian V, et al. A human monoclonal anti-ANG2 antibody leads to broad antitumor activity in combination with VEGF inhibitors and chemotherapy agents in preclinical models. Mol Cancer Ther. 2010; 9: 145-56.
82. Oliner J, Min H, Leal J, Yu D, Rao S, You E, Tang X, Kim H, Meyer S, Han SJ, Hawkins N, Rosenfeld R, Davy E, et al. Suppression of angiogenesis and tumor growth by selective inhibition of angiopoietin-2. Cancer Cell. 2004; 6: 507-16.
83. Chae SS, Kamoun WS, Farrar CT, Kirkpatrick ND, Niemeyer E, de Graaf AM, Sorensen AG, Munn LL, Jain RK, Fukumura D. Angiopoietin-2 interferes with anti-VEGFR2-induced vessel normalization and survival benefit in mice bearing gliomas. Clin Cancer Res. 2010; 16: 3618-27.
84. Sheridan C. Amgen’s angiopoietin blocker fails in ovarian cancer. Nat Biotechnol. 2015; 33: 5-6.
85. Eklund L, Saharinen P. Angiopoietin signaling in the vasculature. Exp Cell Res. 2013; 319: 1271-80.
86. Bessho H, Wong B, Huang D, Tan J, Ong CK, Iwamura M, Hart S, Dangl M, Thomas M, Teh BT. Effect of Ang-2-VEGF-A Bispecific Antibody in Renal Cell Carcinoma. Cancer Invest. 2015; 33: 378-86.
87. Kienast Y, Klein C, Scheuer W, Raemsch R, Lorenzon E, Bernicke D, Herting F, Yu S, The HH, Martarello L, Gassner C, Stubenrauch KG, Munro K, et al. Ang-2-VEGF-A CrossMab, a novel bispecific human IgG1 antibody blocking VEGF-A and Ang-2 functions simultaneously, mediates potent antitumor, antiangiogenic, and antimetastatic efficacy. Clin Cancer Res. 2013; 19: 6730-40.
88. Koh YJ, Kim HZ, Hwang SI, Lee JE, Oh N, Jung K, Kim M, Kim KE, Kim H, Lim NK, Jeon CJ, Lee GM, Jeon BH, et al. Double antiangiogenic protein, DAAP, targeting VEGF-A and angiopoietins in tumor angiogenesis, metastasis, and vascular leakage. Cancer Cell. 2010; 18: 171-84.
89. Zhao C, Yang H, Shi H, Wang X, Chen X, Yuan Y, Lin S, Wei Y. Distinct contributions of angiogenesis and vascular co-option during the initiation of primary microtumors and micrometastases. Carcinogenesis. 2011; 32: 1143-50.
90. Paez-Ribes M, Allen E, Hudock J, Takeda T, Okuyama H, Vinals F, Inoue M, Bergers G, Hanahan D, Casanovas O. Antiangiogenic therapy elicits malignant progression of tumors to increased local invasion and distant metastasis. Cancer Cell. 2009; 15: 220-31.
91. Roodink I, Leenders WP. Targeted therapies of cancer: angiogenesis inhibition seems not enough. Cancer Lett. 2010; 299: 1-10.