
Introduction
The 43 kD cyclic AMP (cAMP)-responsive element binding protein (CREB), first identified 1987, belong to the large family of basic leucine zipper (bZIP)-containing transcription factors (TF) including c-jun, c-fos and c-myc. It is a crucial transcription factor, which regulates a wide range of biological processes to orchestrate proper cell differentiation and cell growth. Genome wide analyses of the CREB-binding sites identified more than 4.000 genes with cAMP-responsive elements (CRE) in their promoters suggesting that CREB controls not only the regulation of immediate early genes as primarily expected [1, 2].
CREB is currently viewed as a multifaceted protein that associates with diverse proteins to direct biological distinct activities in a context-dependent manner. Its importance for basic cellular function and homeostasis is further strengthened by the lethality of CREB knock out mice [3]. In addition, its overexpression is associated with an increased cell proliferation, reduced apoptosis and enhanced migration. Thus, there exists evidence for a causal link between CREB activation, tumor initiation and progression. This review focusses on (i) how CREB expression is controlled and (ii) how protein-protein interactions dynamically regulated in a spatiotemporal manner have endowed the CREB protein with a plethora of functions with particular emphasis on the tumor promoting properties of CREB. In addition, its use as a prognostic biomarker and therapeutic target of tumors is discussed.
Essential features and functions of CREB
CREB is a modular protein consisting of a kinase-inducible domain (KID), two glutamine-rich domains and a bZIP domain. The KID- and glutamine-rich domains are essential for the transactivation and phosphorylation of CREB. The transcriptional activity of CREB is induced upon a reversible phosphorylation at various serine residues, in particular at serine 133 and serine 142, by various kinases, such as protein kinase A (PKA), protein kinase B (PKB/AKT), the mitogen-activated kinase (MAPK) and the 90 kD ribosomal S6 kinase [4-10]. Phosphorylated CREB (pCREB) interacts with diverse transcriptional co-activators including the histone acetyltransferase CREB-binding protein CBP/p300 via the kinase-inducible domain (KID) in CREB and the KID-interacting domain (KIX) in CBP [11-13] thereby subsequently increasing its transcriptional activity. The CREB/CBP complex recruits the transcriptional machinery at the CRE site of gene promoters for the initiation of the CREB-dependent gene transcription [2].
Since CREB is a general transcriptional activator involved in the modulation of the histone H3 and H4 methylation leading to the initiation and maintenance of the chromatin recruitment to the transcriptional apparatus [13], it could regulate a large number of physiological processes dependent on its cellular localization and time-dependent phosphorylation pattern. These include cell proliferation, cell cycle, metabolism, DNA repair, differentiation, inflammation, angiogenesis, immune responses and survival.
Role of CREB in the tumor development
Next to its physiological role CREB is also involved in the malignant transformation of cells, since its frequent and persistent activation is sufficient to convert normal cells into tumor cells. This is mediated by an aberrant activation of components of the cAMP signal transduction relevant pathways, such as G-coupled, receptor tyrosine kinase (RTK) and cytokine/JAK/STAT signaling pathways, but also downstream signaling pathways (Figure 1).
CREB overexpression was found in many solid tumor types like non-small lung carcinoma (NSCLC), glioblastoma, mammary carcinoma, melanoma and diffuse malignant mesothelioma when compared to adjacent normal tissues [8, 10, 14-22] as well as in hematopoietic malignancies [23-26]. This was accompanied by enhanced cell proliferation, reduced sensitivity to undergo apoptosis, increased angiogenesis and radiation-induced differentiation [27]. Furthermore, CREB overexpression is associated with clinicopathological parameters including tumor stage, grade, metastasis, enhanced development of recurrences, a worse prognosis and a reduced survival of tumor patients [17, 28-31]. This was due to a CREB overexpression-mediated upregulation of downstream target genes of CREB carrying CRE elements in their promoters. Chromatin immunoprecipitation (ChIP) and a combination of ChIP with SAGE identified a large number of CREB targets involved in the neoplastic phenotype, clonogenic potential, apoptosis resistance, and abnormal growth properties [32-35]. Furthermore, CREB overexpressing transgenic mice developed myeloproliferative disorders [23].
In addition, CREB has been shown to play a key role in the development of resistances against inhibitors of the Raf-MEK-ERK and PI3K/AKT signal pathways [36, 37]. The resistance against MAPK inhibitors could be enhanced by CREB in mammary carcinoma, which was moreover associated with an altered histone acetylation [36, 38]. Furthermore, downregulation of CREB caused an altered expression of BRAC1 and an increased expression of aromatase, a key enzyme of the estrogen biosynthesis, which is transcriptionally regulated by CREB and associated with the development of resistances to tamoxifen treatment [37].
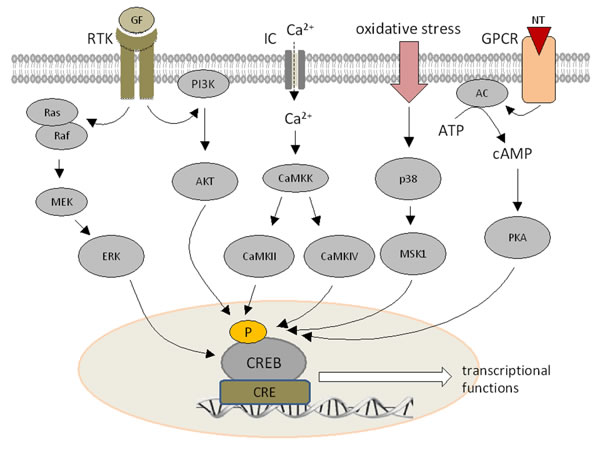
Figure 1: Signal transduction pathways modulating CREB expression. Growth factors (GF) can bind to a membrane-bound receptor, which activates the PI3K-AKT or Ras-MEK-ERK pathways. Ca2+ influx increases the activity of calcium dependent kinases. Hormone receptors and G protein coupled receptors stimulate cAMP synthesis by adenylate cyclase leading to the activation of PKA. All signal transduction pathways can phosphorylate CREB at different serine residues.
Molecular basis of CREB regulation
The expression and activity of CREB is dynamically regulated by diverse mechanisms (Figure 2). CREB responds to cAMP, intracellular Ca2+, various growth factors, such as nerve growth factor (NGF), fibroblast growth factor (FGF) and the insulin growth factor 1 as well as cytokines, like IL-4, IL-10, IL-13 and transforming growth factor (TGF)-β thereby activating gene transcription [39-41]. The FGF- and stress-mediated regulation of CREB occurs via the MAPK kinase pathway [9, 42].
Both in vitro and in vivo studies of tumors and corresponding non-malignant tissues as well as of tumor cell lines demonstrated high levels of CREB expression. The underlying molecular mechanisms of CREB overexpression in tumors have not yet been identified in detail. In contrast to the CREB-binding protein (CBP), which is often mutated in tumors [43, 44] amplifications and/or deletions in CREB have only been rarely detected [21] suggesting that deregulation processes might be the major cause of increased expression and function of CREB in tumors. Although in most tumors a concordant upregulated CREB mRNA and protein expression was found, this linear correlation was not always detected in tumors suggesting additional regulatory mechanisms affecting protein levels [45]. Thus next to the transcriptional regulation, the expression of CREB could be also controlled at the post-transcriptional level.
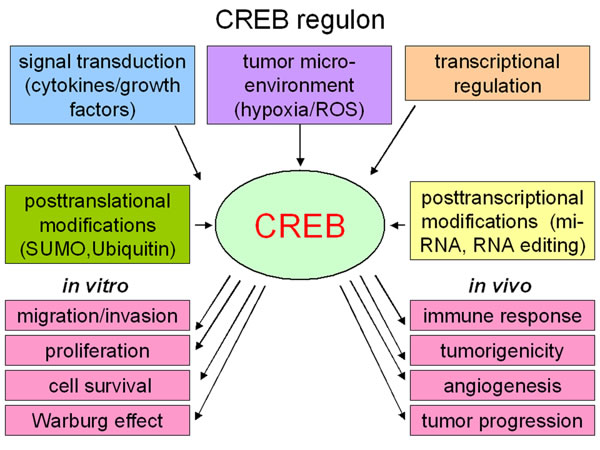
Figure 2: Regulation of CREB (The CREB regulon). Cytokines and growth factors as well as the tumor microenvironment can influence the CREB activity by different signal transduction pathways. Post-transcriptional alterations, like micro-RNAs or RNA-editing, can influence the CREB protein expression, while post-translational modifications regulate the stability or degradation of CREB. In in vitro cell culture CREB increases the migration and invasion potential of the cells and is important for the cell proliferation and cell survival. CREB activity can further induce the Warburg effect. Additionally, in vivo the transcription factor CREB is linked with the immune responses, tumorigenicity as well as angiogenesis and therefore with tumor progression.
Post-transcriptional regulation of CREB by microRNAs
MicroRNAs (miRs) representing small non-coding RNA molecules interact with the 3’ untranslated region (UTR) of their target mRNAs and are involved in the regulation of > 50 % of all genes. Thus, miRs might control many cellular and pathophysiologic processes including the initiation and progression of tumors. During the last years miRs have been identified, which are deregulated by CREB or have CREB as direct target due to binding to its regulatory sequences at the 3’-UTR (Table 1). Using in silico prediction by different algorithms CREB expression could be regulated by different miRs known to be frequently downregulated in tumors, such as miR-181b, miR-128, miR-124, miR-34b, miR-23a, miR-200b, miR-203 and miR-301 [21, 46-49]. In some studies luciferase reporter assays confirmed the interaction of these miRs with the 3’-UTR of CREB. Overexpression of these miRs significantly modulated the expression of CREB, which was associated with altered growth properties of tumor cells thereby suggesting that the miR-mediated deregulation of CREB contributes to tumorigenesis. For example miR-200b, miR-301 and miR-343 have tumor suppressive activity by targeting CREB. Overexpression of these miRs caused an inhibition of tumor cell growth and/or soft agar colony formation in vitro and a reduced tumorigenesis in vivo [49]. This could be associated with suppressed expression levels of CREB target proteins and their related pathways [47]. Furthermore, the inverse expression of CREB and miR-200b had also a prognostic value in astrocytoma [49].
CREB could also regulate the expression of miRs, such as miR-9, which modulates different physiologic and pathophysiologic processes including the differentiation and function of myeloid-derived suppressor cells (MDSC) [50]. Loss of miR-9 suppresses proliferation, promotes migration of progenitor cells in vitro [51] and coordinates the proliferation of glioma cells [21]. In pancreatic cancer, the CREB-dependent induction of miR-373 promotes pancreatic tumor growth in vitro and in vivo [52]. In melanoma cells CREB has been shown to suppress the expression of the RNA-editing enzyme ADAR1 in vitro and in situ, while the restoration of its expression reduced melanoma growth and metastasis formation in vivo. This was accompanied by RNA-editing of miR-455-5p, which occurs in less aggressive, but not in strong aggressive metastatic melanoma [53]. In addition, the RNA-binding protein tristetraprolin (TTP), a tumor suppressor gene, regulates CREB activity suggesting that low TTP levels represent a potential biomarker for human cancers with poor outcome [54].
Table 1: Identification of CREB-regulating miRs in human tumors
Name |
Cell line/Tumor |
Reference |
miRNA-181b |
adenocarcinoma |
[104] |
miRNA-34b |
AML |
[20] |
miRNA-181a |
PC12 (pheochromocytoma) |
[57] |
miRNA-9 |
glioblastoma |
[21] |
miRNA-200b |
glioblastoma |
[45] |
miRNA-372 |
hepatocellular carcinoma |
[105] |
Post-translational modifications
Recently it has been described that CREB expression could be regulated by miRs [20, 21, 55-57]. Next to its post-transcriptional control, CREB expression can be also post-translationally regulated by different extracellular signals, in particular by factors of the tumor microenvironment, like hypoxia, pH and oxidative stress [58]. So far, the best analyzed post-translational modifications (PTMs) of CREB are phosphorylation and ubiquitination, which have been also shown to be altered in tumor cells.
In addition, other PTMs, like methylation, glycosylation and SUMOylation, have also been shown to influence the activity of CREB [55, 59-67]. This might have functional consequences due an altered CREB-regulated gene transcription, including that of nuclear and mitochondrial genes, which is accompanied by changes of protein degradation as well as protein/protein interactions. However up to now, a direct link to tumor initiation and progression has not yet been found.
Phosphorylation
CREB is a substrate of various kinases. Although the phosphorylation of CREB can occur at different serine residues, its phosphorylation at Ser133 and its functional consequences have been investigated in great detail. In particular, CREB Ser133 has been often shown to be overexpressed in human tumors. This modification might result in conformational changes, which might be associated with its functional activity, localization and/or stability. In addition, there exist a number of other serine residues in the KID domain, which could be also phosphorylated. However, neither their expression pattern nor their functions have yet been described in detail. CREBSer108/111 and CREBSer114/117 can be phosphorylated by CK1B [68], CREBSer121 by the ATM kinase/ATR kinase [61] and Ser129 by GSK3beta [69]. In contrast, CREBSer133 could be phosphorylated by various kinases, such as CaMK4, MAPKSPK2, MSK1, p90RSK or PKD1 [63]. It is noteworthy that phosphorylation is not always sufficient to stimulate CREB-dependent transcription suggesting the existence of additional modifications coordinating CREB activity [2, 70]. The identification of an alternative factor, which can bind CREB independent of its phosphorylation status led to the hypothesis that CREB phosphorylation is not essential for all its physiologic activities.
Ubiquitination
Proteasomal degradation represents the primary mechanism of controlled proteolysis and is necessary to maintain cellular function and viability [71, 72]. Targeted proteasomal degradation is also important in the regulation of the expression levels of a number of transcription factors, such as NF-κB and HIF-1 [73]. In addition, CREB can also be targeted for ubiquitination and therefore subsequently degraded by the proteasome. This is important for the quality control of CREB expression and might reflect a mechanism of its fine tuning in response to different stimuli. Recently, a cross talk between different PTMs of CREB has been shown linking hyperphosphorylation of CREB with ubiquitination and its proteasomal degradation [74].
Acetylation
CREB protein has been shown to be acetylated by CBP/p300, which in turn affects its transcriptional activity by promoting its DNA-binding capability thereby enhancing its transactivation activities [75, 76]. Furthermore, acetylation of CREB has been linked to its increased protein stability. Although mutations in p300 have been frequently found in human tumors [77, 78], it has not yet been analyzed whether defects in CBP/p300 have a direct effect on the expression level and function of CREB.
Glycosylation
Different studies suggested that N-acetylglucosamine (O-GlcNAc) glycosylation can regulate the activity of transcription factors and other proteins in the nucleus [79]. CREB has been shown to be modified by O-GlcNAc at Serin 40, which impairs basal and activation-induced CREB-mediated transcriptional activities [80], thereby modulating important cellular functions. Glycosylation can function as a constant repressor of CREB, which controls its basal expression pattern along with the levels of CREB-regulated genes, such as e. g. Wnt2 and c-fos [81], but this mechanism has not yet been linked to tumorigenesis.
SUMOylation
The small ubiquitin-like modifier (SUMO) modification (SUMOylation) is an important mechanism in post-transcriptional control. In most cases SUMOylation suppress the activity of targeted transcriptional activators by altering their sub-compartmentalization and/or protein interaction properties [82, 83]. The short isoform of CREB has been shown to be SUMOylated by the SUMO E3 ligase protein inhibitor of activated STAT1 [55]. Recently, SUMOylation of AKT has been shown to regulate substrate SUMOylation specificity including the targeting of CREB [84].
CREB can also be modified by SUMOylation in response to hypoxia. Overexpression of SUMO1 stabilizes CREB and enhances CREB-dependent gene reporter activity in hypoxia [60]. Lysine residues K304 and K285 of CREB are SUMO1 acceptors demonstrating that SUMOylation represents an important PTM of CREB. The CREB SUMOylation is dependent on its phosphorylation, but lasts longer. The distinct of phosphorylation status and SUMOylation kinetics suggests that CREB phosphorylation is responsible for signal transduction during early responses, while CREB SUMOylation sustains long-term processes [55]. This might also lead to changes within the cellular metabolism by affecting the expression of e.g. mitochondrial genes. Therefore, analysis of the SUMOylation status of CREB in tumor cells is suggested.
Subcellular localization of CREB
Next to the temporal control by post-translational modifications (PTMs) the CREB activity might be also controlled by its subcellular localization. Although primarily localized in the nuclear compartment, CREB can also be found in the cytoplasm as well as in mitochondria [85]. The importance of the compartment-specific translocation of CREB is in particular demonstrated by monitoring CREB expression in tumors of distinct origin as well as in tumor cells cultured under various conditions mimicking the tumor microenvironment, such as hypoxia and altered pH [86, 87]. Furthermore, PTMs have also been linked to the subcellular localization of CREB. This alters CREB function as shown for mitoCREB [88, 89].
CREB downstream targets and their cellular functions in cancer
The transcriptional activator CREB enhances the expression of many target genes, which are involved in various cell functions including metabolism, cell cycle, survival and DNA repair suggesting that CREB is of critical importance for the growth, survival, migration, as well as for viral responses (Figure 3; [90-92]). This raised the question how CREB acts under pathophysiological circumstances e.g. initiation and progression of tumors and viral infection. The effects of CREB, in particular distinct cellular processes, depend on the proper activation of a specific gene expression program. A distinct set of genes is regulated by CREB under different circumstances as demonstrated by chromatin immunoprecipitation and DNA microarray techniques [32]. The CREB-mediated gene transcription is critical in maintaining a homeostatic cellular environment under pathological conditions. Indeed, there exist a large number of putative CREB target genes, which include genes that are involved in signal transduction, cellular structure, differentiation, cell proliferation as well as metabolism. All target genes exhibit the presence of one or more CRE consensus sequences in their promoter regions. Despite these characteristics the CREB target genes only share a few other similarities reflecting the highly variable activities of CREB under different conditions. The functional diversity of CREB target genes has also intriguing mechanistic implications and their activity may not coordinately be turned on when CREB is phosphorylated or turned off when CREB is de-phosphorylated.
Targeting CREB in tumor cells
Based on its central role in the development, maintenance as well as progression of tumors CREB has been suggested as an excellent target structure for the treatment of cancers. This is further underlined by the expression analysis of the early inducible cAMP repressor (ICER; inducible cyclic AMP early repressor), an inhibitor of CREB [93]. ICER is downregulated in bone marrow cells of patients with acute myeloid leukemia (AML) leading to an altered CREB expression level. An advantage of CREB as a target is its regulation by different signal transduction pathways, known to be involved in tumor development. So far, different strategies have been developed to inhibit CREB function in tumor cells (Figure 3). These include the use of dominant-negative CREB mutants (KCREB), which could inhibit the transcription of CREB by heterodimerization of KCREB with wild type CREB. Overexpression of KCREB in metastatic tumor cells leads to a reduced potential of metastasis formation both in vitro and in vivo [94]. Recently, an inhibitor of CREB created by the fusion of the dominant negative inhibitor A-CREB with a photoactive yellow protein was designed controlling CREB function [95]. Thus the link of CREB with optogenetic domains enables the analysis of spatiotemporal control of CREB and its therapeutic use.
Furthermore, a number of CRE „decoy“ oligonucleotides have been established, which not only efficiently inhibit CREB gene transcription, but also tumor growth [96]. Using RNA interference CREB expression was silenced, which was associated with altered growth properties and cell viability. In tumor cells the shRNA-mediated inhibition of CREB caused a reduced tumor cell proliferation and migration anchorage-independent growth, suppression of cell cycle arrest and induction of apoptosis accompanied by a reduced in vivo tumor growth and enhanced tumor immunogenicity [48, 97].
In addition, an advantage of CREB as a target is its regulation by different signal transduction pathways, which have been shown to be involved in the tumor development. Since these „proof of concept“ studies demonstrated a therapeutic effect of CREB inhibition, alternative strategies using small molecules have been developed to inhibit the CREB-mediated gene transcription. These include the development of different kinase inhibitors, which inhibit the phosphorylation and thus the activation of CREB as well as of chemical inhibitors, which were able to inhibit the interaction between CREB-CRE or CREB-CBP [98-102]. The inhibitor KG-501 is able to reversibly and dose-dependently inhibit the interaction between the KID domain of CREB and the KIX domain of CBP. This inhibition was already obvious at micromolar concentrations without inhibiting the general transcription machinery. Another option is the use of miRs to inhibit CREB expression and activity. Indeed, miRs directly blocking CREB activity and thus the neoplastic phenotype of tumor cells have been recently identified [30], but so far their implementation in vivo has not yet been established.
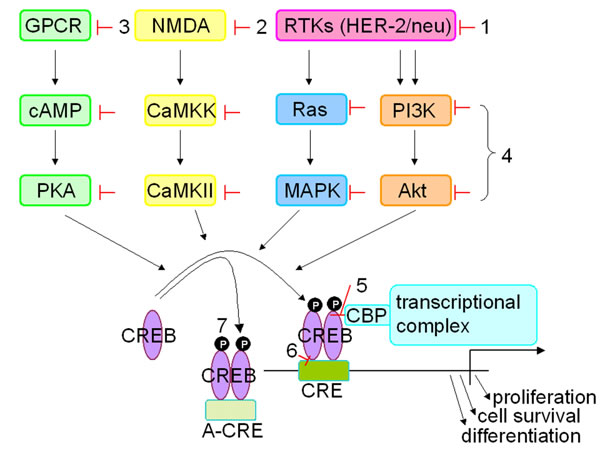
Figure 3: Strategies inhibiting CREB expression. Different approaches were used to inhibit/silence CREB expression and/or activity in vitro and in vivo. These include (i) upstream inhibitors of CREB blocking different receptor tyrosine kinases, like HER-2/neu and EGF-R, with e.g. trastuzumab or lapatinib, (ii) inhibition of the ion transporter (NMDA) by treatment with ketamine, (iii) inactivation of G-protein coupled receptors with beta blockers, (iv) activity of kinases or substrates using various signal transduction inhibitors, (v) inhibition of the interaction between CREB and the co-activators CBP/p300 with KG-501, (vi) influencing binding of CREB at the CRE site by treatment with surfenhydrate and (vii) blocking the interaction with the gene promoter using an artificial CRE element.
Conclusions
It is noteworthy that the disruption of CREB activity has severe consequences and is lethal in mice [103]. The use of sophisticated genetic models might be suitable tools for increasing the knowledge of CREB in survival and maintenance of the cellular fate as well as in its role in many diseases including cancer. The identification of the molecular mechanisms involved in CREB expression and regulation will lead to strategies to inhibit persistent CREB activity in tumors thereby reverting the CREB-induced transformation processes. Thus, CREB might not only serve as a prognostic marker, but also as a therapeutic target for cancers associated with increased activity of signal transduction pathways.
Abbreviations
AML, acute myeloid leukemia; bZIP, basic leucine zipper; cAMP, cyclic AMP; CBP, CREB-binding protein; ChiP, chromatin immunoprecipitation; CRE, cAMP response element; CREB, cAMP response element binding protein; CREM, FGF, fibroblast growth factor; GF, growth factor; ICER, inducible cAMP early repressor; i.e., immediate early; KCREB, dominant-negative CREB; KID, kinase-inducible domain; KIX, KID interacting domain; MAPK, mitogen-activated protein kinase; MDSC, myeloid-derived suppressor cells; miR, microRNA; NSCLC, non-small cell lung carcinoma; O-Glc NAc, N-acetylglucosamine; pCREB, phosphorylated CREB, PI3K, phosphatidylinositol 3-kinases, PKA, protein kinase A; PTM, posttranslational modification; RTK, receptor tyrosine kinase; sh, short hair pin; SUMO, small ubiquitin-like modifier; TF, transcription factor, TGF, transforming growth factor; TTP, tristetraprolin; UTR, untranslated region.
Acknowledgments
We would like to thank Claudia Lennicke for the help with the figure preparation as well as Sylvi Magdeburg and Nicole Ott for excellent secretarial help.
Conflicts of Interest
The authors have not conflicts of interest.
Grant Support
This work was supported by a grant of the Roux program (V1/A9) and the Mildred Scheel Foundation (grant 111091).
References
1. Montminy MR and Bilezikjian LM. Binding of a nuclear protein to the cyclic-AMP response element of the somatostatin gene. Nature. 1987; 328(6126):175-178.
2. Shaywitz AJ and Greenberg ME. CREB: a stimulus-induced transcription factor activated by a diverse array of extracellular signals. Annu Rev Biochem. 1999; 68:821-861.
3. Bleckmann SC, Blendy JA, Rudolph D, Monaghan AP, Schmid W and Schutz G. Activating transcription factor 1 and CREB are important for cell survival during early mouse development. Molecular and cellular biology. 2002; 22(6):1919-1925.
4. Du K and Montminy M. CREB is a regulatory target for the protein kinase Akt/PKB. J Biol Chem. 1998; 273(49):32377-32379.
5. Finkbeiner S. New roles for introns: sites of combinatorial regulation of Ca2+- and cyclic AMP-dependent gene transcription. Sci STKE. 2001; 2001(94):pe1.
6. Johannessen M and Moens U. Multisite phosphorylation of the cAMP response element-binding protein (CREB) by a diversity of protein kinases. Front Biosci. 2007; 12:1814-1832.
7. Mayr B and Montminy M. Transcriptional regulation by the phosphorylation-dependent factor CREB. Nat Rev Mol Cell Biol. 2001; 2(8):599-609.
8. Melnikova VO, Dobroff AS, Zigler M, Villares GJ, Braeuer RR, Wang H, Huang L and Bar-Eli M. CREB inhibits AP-2alpha expression to regulate the malignant phenotype of melanoma. PloS one. 2010; 5(8):e12452.
9. Tan Y, Rouse J, Zhang A, Cariati S, Cohen P and Comb MJ. FGF and stress regulate CREB and ATF-1 via a pathway involving p38 MAP kinase and MAPKAP kinase-2. EMBO J. 1996; 15(17):4629-4642.
10. Wen AY, Sakamoto KM and Miller LS. The role of the transcription factor CREB in immune function. J Immunol. 2010; 185(11):6413-6419.
11. Radhakrishnan I, Perez-Alvarado GC, Parker D, Dyson HJ, Montminy MR and Wright PE. Solution structure of the KIX domain of CBP bound to the transactivation domain of CREB: a model for activator:coactivator interactions. Cell. 1997; 91(6):741-752.
12. Thakur JK, Yadav A and Yadav G. Molecular recognition by the KIX domain and its role in gene regulation. Nucleic Acids Res. 2014; 42(4):2112-2125.
13. Wang F, Marshall CB and Ikura M. Transcriptional/epigenetic regulator CBP/p300 in tumorigenesis: structural and functional versatility in target recognition. Cell Mol Life Sci. 2013; 70(21):3989-4008.
14. Abramovitch R, Tavor E, Jacob-Hirsch J, Zeira E, Amariglio N, Pappo O, Rechavi G, Galun E and Honigman A. A pivotal role of cyclic AMP-responsive element binding protein in tumor progression. Cancer research. 2004; 64(4):1338-1346.
15. Antonescu CR, Nafa K, Segal NH, Dal Cin P and Ladanyi M. EWS-CREB1: a recurrent variant fusion in clear cell sarcoma—association with gastrointestinal location and absence of melanocytic differentiation. Clin Cancer Res. 2006; 12(18):5356-5362.
16. Braeuer RR, Zigler M, Villares GJ, Dobroff AS and Bar-Eli M. Transcriptional control of melanoma metastasis: the importance of the tumor microenvironment. Semin Cancer Biol. 2011; 21(2):83-88.
17. Chhabra A, Fernando H, Watkins G, Mansel RE and Jiang WG. Expression of transcription factor CREB1 in human breast cancer and its correlation with prognosis. Oncol Rep. 2007; 18(4):953-958.
18. Fan CF, Mao XY and Wang EH. Elevated p-CREB-2 (ser 245) expression is potentially associated with carcinogenesis and development of breast carcinoma. Mol Med Rep. 2012; 5(2):357-362.
19. Jean D and Bar-Eli M. Regulation of tumor growth and metastasis of human melanoma by the CREB transcription factor family. Mol Cell Biochem. 2000; 212(1-2):19-28.
20. Pigazzi M, Manara E, Bresolin S, Tregnago C, Beghin A, Baron E, Giarin E, Cho EC, Masetti R, Rao DS, Sakamoto KM and Basso G. MicroRNA-34b promoter hypermethylation induces CREB overexpression and contributes to myeloid transformation. Haematologica. 2013; 98(4):602-610.
21. Tan X, Wang S, Yang B, Zhu L, Yin B, Chao T, Zhao J, Yuan J, Qiang B and Peng X. The CREB-miR-9 negative feedback minicircuitry coordinates the migration and proliferation of glioma cells. PloS one. 2012 (b); 7(11):e49570.
22. Yamada T, Amann JM, Fukuda K, Takeuchi S, Fujita N, Uehara H, Iwakiri S, Itoi K, Shilo K, Yano S and Carbone DP. Akt Kinase-Interacting Protein 1 Signals through CREB to Drive Diffuse Malignant Mesothelioma. Cancer research. 2015; 75(19):4188-4197.
23. Cheng JC, Kinjo K, Judelson DR, Chang J, Wu WS, Schmid I, Shankar DB, Kasahara N, Stripecke R, Bhatia R, Landaw EM and Sakamoto KM. CREB is a critical regulator of normal hematopoiesis and leukemogenesis. Blood. 2008; 111(3):1182-1192.
24. Cho EC, Mitton B and Sakamoto KM. CREB and leukemogenesis. Crit Rev Oncog. 2011; 16(1-2):37-46.
25. Slamon DJ, Leyland-Jones B, Shak S, Fuchs H, Paton V, Bajamonde A, Fleming T, Eiermann W, Wolter J, Pegram M, Baselga J and Norton L. Use of chemotherapy plus a monoclonal antibody against HER2 for metastatic breast cancer that overexpresses HER2. The New England journal of medicine. 2001; 344(11):783-792.
26. Zimmer SN, Lemieux ME, Karia BP, Day C, Zhou T, Zhou Q, Kung AL, Suresh U, Chen Y, Kinney MC, Bishop AJ and Rebel VI. Mice heterozygous for CREB binding protein are hypersensitive to gamma-radiation and invariably develop myelodysplastic/myeloproliferative neoplasm. Exp Hematol. 2012; 40(4):295-306 e295.
27. Suarez CD, Deng X and Hu CD. Targeting CREB inhibits radiation-induced neuroendocrine differentiation and increases radiation-induced cell death in prostate cancer cells. Am J Cancer Res. 2014; 4(6):850-861.
28. Cheng JC, Esparza S, Sandoval S, Shankar D, Fu C and Sakamoto KM. Potential role of CREB as a prognostic marker in acute myeloid leukemia. Future Oncol. 2007; 3(4):475-480.
29. Deng X, Liu H, Huang J, Cheng L, Keller ET, Parsons SJ and Hu CD. Ionizing radiation induces prostate cancer neuroendocrine differentiation through interplay of CREB and ATF2: implications for disease progression. Cancer research. 2008; 68(23):9663-9670.
30. Sakamoto KM and Frank DA. CREB in the pathophysiology of cancer: implications for targeting transcription factors for cancer therapy. Clin Cancer Res. 2009; 15(8):2583-2587.
31. Seo HS, Liu DD, Bekele BN, Kim MK, Pisters K, Lippman SM, Wistuba, II and Koo JS. Cyclic AMP response element-binding protein overexpression: a feature associated with negative prognosis in never smokers with non-small cell lung cancer. Cancer research. 2008; 68(15):6065-6073.
32. Impey S, McCorkle SR, Cha-Molstad H, Dwyer JM, Yochum GS, Boss JM, McWeeney S, Dunn JJ, Mandel G and Goodman RH. Defining the CREB regulon: a genome-wide analysis of transcription factor regulatory regions. Cell. 2004; 119(7):1041-1054.
33. Pigazzi M, Ricotti E, Germano G, Faggian D, Arico M and Basso G. cAMP response element binding protein (CREB) overexpression CREB has been described as critical for leukemia progression. Haematologica. 2007; 92(10):1435-1437.
34. Shankar DB, Cheng JC and Sakamoto KM. Role of cyclic AMP response element binding protein in human leukemias. Cancer. 2005; 104(9):1819-1824.
35. Zhang X, Odom DT, Koo SH, Conkright MD, Canettieri G, Best J, Chen H, Jenner R, Herbolsheimer E, Jacobsen E, Kadam S, Ecker JR, Emerson B, Hogenesch JB, Unterman T, Young RA, et al. Genome-wide analysis of cAMP-response element binding protein occupancy, phosphorylation, and target gene activation in human tissues. Proceedings of the National Academy of Sciences of the United States of America. 2005; 102(12):4459-4464.
36. Johannessen CM, Johnson LA, Piccioni F, Townes A, Frederick DT, Donahue MK, Narayan R, Flaherty KT, Wargo JA, Root DE and Garraway LA. A melanocyte lineage program confers resistance to MAP kinase pathway inhibition. Nature. 2013; 504(7478):138-142.
37. Phuong NT, Lim SC, Kim YM and Kang KW. Aromatase induction in tamoxifen-resistant breast cancer: Role of phosphoinositide 3-kinase-dependent CREB activation. Cancer Lett. 2014; 351(1):91-99.
38. Donnelly SM, Paplomata E, Peake BM, Sanabria E, Chen Z and Nahta R. P38 MAPK contributes to resistance and invasiveness of HER2- overexpressing breast cancer. Curr Med Chem. 2014; 21(4):501-510.
39. Ruffell D, Mourkioti F, Gambardella A, Kirstetter P, Lopez RG, Rosenthal N and Nerlov C. A CREB-C/EBPbeta cascade induces M2 macrophage-specific gene expression and promotes muscle injury repair. Proceedings of the National Academy of Sciences of the United States of America. 2009; 106(41):17475-17480.
40. Strempel JM and Vercelli D. Functional dissection identifies a conserved noncoding sequence-1 core that mediates IL13 and IL4 transcriptional enhancement. J Biol Chem. 2007; 282(6):3738-3746.
41. Zuloaga R, Fuentes EN, Molina A and Valdes JA. The cAMP response element binding protein (CREB) is activated by insulin-like growth factor-1 (IGF-1) and regulates myostatin gene expression in skeletal myoblast. Biochem Biophys Res Commun. 2013; 440(2):258-264.
42. Gee K, Angel JB, Mishra S, Blahoianu MA and Kumar A. IL-10 regulation by HIV-Tat in primary human monocytic cells: involvement of calmodulin/calmodulin-dependent protein kinase-activated p38 MAPK and Sp-1 and CREB-1 transcription factors. J Immunol. 2007; 178(2):798-807.
43. Green MR, Kihira S, Liu CL, Nair RV, Salari R, Gentles AJ, Irish J, Stehr H, Vicente-Duenas C, Romero-Camarero I, Sanchez-Garcia I, Plevritis SK, Arber DA, Batzoglou S, Levy R and Alizadeh AA. Mutations in early follicular lymphoma progenitors are associated with suppressed antigen presentation. Proceedings of the National Academy of Sciences of the United States of America. 2015; 112(10):E1116-1125.
44. Malinowska-Ozdowy K, Frech C, Schonegger A, Eckert C, Cazzaniga G, Stanulla M, zur Stadt U, Mecklenbrauker A, Schuster M, Kneidinger D, von Stackelberg A, Locatelli F, Schrappe M, Horstmann MA, Attarbaschi A, Bock C, et al. KRAS and CREBBP mutations: a relapse-linked malicious liaison in childhood high hyperdiploid acute lymphoblastic leukemia. Leukemia. 2015; 29(8):1656-1667.
45. Peng B, Hu S, Jun Q, Luo D, Zhang X, Zhao H and Li D. MicroRNA-200b targets CREB1 and suppresses cell growth in human malignant glioma. Mol Cell Biochem. 2013; 379(1-2):51-58.
46. Noguchi S, Kumazaki M, Mori T, Baba K, Okuda M, Mizuno T and Akao Y. Analysis of microRNA-203 function in CREB/MITF/RAB27a pathway: comparison between canine and human melanoma cells. Vet Comp Oncol. 2014.
47. Pigazzi M, Manara E, Baron E and Basso G. miR-34b targets cyclic AMP-responsive element binding protein in acute myeloid leukemia. Cancer research. 2009; 69(6):2471-2478.
48. Tan X, Wang S, Zhu L, Wu C, Yin B, Zhao J, Yuan J, Qiang B and Peng X. cAMP response element-binding protein promotes gliomagenesis by modulating the expression of oncogenic microRNA-23a. Proceedings of the National Academy of Sciences of the United States of America. 2012 (a); 109(39):15805-15810.
49. Zhang JQ, Yao QH, Kuang YQ, Ma Y, Yang LB, Huang HD, Cheng JM, Yang T, Liu EY, Liang L, Fan KX, Zhao K, Xia X and Gu JW. Prognostic value of coexistence of abnormal expression of micro-RNA-200b and cyclic adenosine monophosphate-responsive element-binding protein 1 in human astrocytoma. Hum Pathol. 2014; 45(10):2154-2161.
50. Tian J, Rui K, Tang X, Ma J, Wang Y, Tian X, Zhang Y, Xu H, Lu L and Wang S. MicroRNA-9 Regulates the Differentiation and Function of Myeloid-Derived Suppressor Cells via Targeting Runx1. J Immunol. 2015; 195(3):1301-1311.
51. Senyuk V, Zhang Y, Liu Y, Ming M, Premanand K, Zhou L, Chen P, Chen J, Rowley JD, Nucifora G and Qian Z. Critical role of miR-9 in myelopoiesis and EVI1-induced leukemogenesis. Proceedings of the National Academy of Sciences of the United States of America. 2013; 110(14):5594-5599.
52. Zhang Y, Yang J, Cui X, Chen Y, Zhu VF, Hagan JP, Wang H, Yu X, Hodges SE, Fang J, Chiao PJ, Logsdon CD, Fisher WE, Brunicardi FC, Chen C, Yao Q, et al. A novel epigenetic CREB-miR-373 axis mediates ZIP4-induced pancreatic cancer growth. EMBO Mol Med. 2013; 5(9):1322-1334.
53. Shoshan E, Mobley AK, Braeuer RR, Kamiya T, Huang L, Vasquez ME, Salameh A, Lee HJ, Kim SJ, Ivan C, Velazquez-Torres G, Nip KM, Zhu K, Brooks D, Jones SJ, Birol I, et al. Reduced adenosine-to-inosine miR-455-5p editing promotes melanoma growth and metastasis. Nat Cell Biol. 2015; 17(3):311-321.
54. Fallahi M, Amelio AL, Cleveland JL and Rounbehler RJ. CREB targets define the gene expression signature of malignancies having reduced levels of the tumor suppressor tristetraprolin. PloS one. 2014; 9(12):e115517.
55. Chen YC, Hsu WL, Ma YL, Tai DJ and Lee EH. CREB SUMOylation by the E3 ligase PIAS1 enhances spatial memory. J Neurosci. 2014; 34(29):9574-9589.
56. Huang W, Liu X, Cao J, Meng F, Li M, Chen B and Zhang J. miR-134 Regulates Ischemia/Reperfusion Injury-Induced Neuronal Cell Death by Regulating CREB Signaling. J Mol Neurosci. 2015; 55:821-9.
57. Liu Y, Zhao Z, Yang F, Gao Y, Song J and Wan Y. microRNA-181a is involved in insulin-like growth factor-1-mediated regulation of the transcription factor CREB1. J Neurochem. 2013; 126(6):771-780.
58. Ahmed BY, Husnain O, Stafford R, Howard M, Gujar AS, Moradiya V, Patel KK and Sihotra S. Hyperphosphorylation of CREB in human dopaminergic neurons: a kinetic study of cellular distribution of total CREB and phospho-CREB following oxidative stress. Neuroreport. 2013; 24(13):757-762.
59. Angel L, Isingrini M, Bouazzaoui B, Taconnat L, Allan K, Granjon L and Fay S. The amount of retrieval support modulates age effects on episodic memory: evidence from event-related potentials. Brain Res. 2010; 1335:41-52.
60. Comerford KM, Leonard MO, Karhausen J, Carey R, Colgan SP and Taylor CT. Small ubiquitin-related modifier-1 modification mediates resolution of CREB-dependent responses to hypoxia. Proceedings of the National Academy of Sciences of the United States of America. 2003; 100(3):986-991.
61. Dodson GE and Tibbetts RS. DNA replication stress-induced phosphorylation of cyclic AMP response element-binding protein mediated by ATM. J Biol Chem. 2006; 281(3):1692-1697.
62. Galardo MN, Riera MF, Regueira M, Pellizzari EH, Cigorraga SB and Meroni SB. Different signal transduction pathways elicited by basic fibroblast growth factor and interleukin 1beta regulate CREB phosphorylation in Sertoli cells. J Endocrinol Invest. 2013; 36(5):331-338.
63. Johannessen M, Delghandi MP and Moens U. What turns CREB on? Cell Signal. 2004; 16(11):1211-1227.
64. Kaleem A, Hoessli DC, Haq IU, Walker-Nasir E, Butt A, Iqbal Z, Zamani Z, Shakoori AR and Nasir ud D. CREB in long-term potentiation in hippocampus: role of post-translational modifications-studies In silico. J Cell Biochem. 2011; 112(1):138-146.
65. Lin XP, Feng L, Xie CG, Chen DB, Pei Z, Liang XL, Xie QY, Li XH and Pan SY. Valproic acid attenuates the suppression of acetyl histone H3 and CREB activity in an inducible cell model of Machado-Joseph disease. Int J Dev Neurosci. 2014; 38:17-22.
66. Shi Y, Venkataraman SL, Dodson GE, Mabb AM, LeBlanc S and Tibbetts RS. Direct regulation of CREB transcriptional activity by ATM in response to genotoxic stress. Proceedings of the National Academy of Sciences of the United States of America. 2004; 101(16):5898-5903.
67. Sun B, Rong R, Jiang H, Zhang H, Wang Y, Bai X, Zhang M, Ma J, Xia S, Shu W, Zhang L and Leng J. Prostaglandin E2 receptor EP1 phosphorylate CREB and mediates MMP2 expression in human cholangiocarcinoma cells. Mol Cell Biochem. 2013; 378(1-2):195-203.
68. Shanware NP, Williams LM, Bowler MJ and Tibbetts RS. Non-specific in vivo inhibition of CK1 by the pyridinyl imidazole p38 inhibitors SB 203580 and SB 202190. BMB Rep. 2009; 42(3):142-147.
69. Hansen T, Rehfeld JF and Nielsen FC. GSK-3beta reduces cAMP-induced cholecystokinin gene expression by inhibiting CREB binding. Neuroreport. 2004; 15(5):841-845.
70. Conkright MD, Canettieri G, Screaton R, Guzman E, Miraglia L, Hogenesch JB and Montminy M. TORCs: transducers of regulated CREB activity. Mol Cell. 2003; 12(2):413-423.
71. Bochtler M, Ditzel L, Groll M, Hartmann C and Huber R. The proteasome. Annu Rev Biophys Biomol Struct. 1999; 28:295-317.
72. DeMartino GN and Slaughter CA. The proteasome, a novel protease regulated by multiple mechanisms. J Biol Chem. 1999; 274(32):22123-22126.
73. Taylor CT, Furuta GT, Synnestvedt K and Colgan SP. Phosphorylation-dependent targeting of cAMP response element binding protein to the ubiquitin/proteasome pathway in hypoxia. Proceedings of the National Academy of Sciences of the United States of America. 2000; 97(22):12091-12096.
74. Mu Y, Yu Y, Yue X, Musarat I, Gong R, Zhu C, Liu Y, Liu F, Zhu Y and Wu J. The X protein of HBV induces HIV-1 long terminal repeat transcription by enhancing the binding of C/EBPbeta and CREB1/2 regulatory proteins to the long terminal repeat of HIV-1. Virus Res. 2011; 156(1-2):81-90.
75. Barral S, Reitz C, Small SA and Mayeux R. Genetic variants in a ‘cAMP element binding protein’ (CREB)-dependent histone acetylation pathway influence memory performance in cognitively healthy elderly individuals. Neurobiology of aging. 2014; 35(12):2881 e2887-2881 e2810.
76. Li X, Zhang B, Wu Q, Ci X, Zhao R, Zhang Z, Xia S, Su D, Chen J, Ma G, Fu L and Dong JT. Interruption of KLF5 acetylation converts its function from tumor suppressor to tumor promoter in prostate cancer cells. International journal of cancer. 2015; 136(3):536-546.
77. Haery L, Lugo-Pico JG, Henry RA, Andrews AJ and Gilmore TD. Histone acetyltransferase-deficient p300 mutants in diffuse large B cell lymphoma have altered transcriptional regulatory activities and are required for optimal cell growth. Molecular cancer. 2014; 13:29.
78. Peifer M, Fernandez-Cuesta L, Sos ML, George J, Seidel D, Kasper LH, Plenker D, Leenders F, Sun R, Zander T, Menon R, Koker M, Dahmen I, Muller C, Di Cerbo V, Schildhaus HU, et al. Integrative genome analyses identify key somatic driver mutations of small-cell lung cancer. Nature genetics. 2012; 44(10):1104-1110.
79. Zachara NE and Hart GW. The emerging significance of O-GlcNAc in cellular regulation. Chem Rev. 2002; 102(2):431-438.
80. Lamarre-Vincent N and Hsieh-Wilson LC. Dynamic glycosylation of the transcription factor CREB: a potential role in gene regulation. J Am Chem Soc. 2003; 125(22):6612-6613.
81. Rexach JE, Clark PM, Mason DE, Neve RL, Peters EC and Hsieh-Wilson LC. Dynamic O-GlcNAc modification regulates CREB-mediated gene expression and memory formation. Nat Chem Biol. 2012; 8(3):253-261.
82. Girdwood DW, Tatham MH and Hay RT. SUMO and transcriptional regulation. Semin Cell Dev Biol. 2004; 15(2):201-210.
83. Johnson ES. Protein modification by SUMO. Annu Rev Biochem. 2004; 73:355-382.
84. Lin CH, Liu SY and Lee EH. SUMO modification of Akt regulates global SUMOylation and substrate SUMOylation specificity through Akt phosphorylation of Ubc9 and SUMO1. Oncogene. 2016; 35:595-60.
85. Shibanuma M, Ishikawa F, Kobayashi M, Katayama K, Miyoshi H, Wakamatsu M, Mori K and Nose K. Critical roles of the cAMP-responsive element-binding protein-mediated pathway in disorganized epithelial phenotypes caused by mitochondrial dysfunction. Cancer Sci. 2012; 103(10):1803-1810.
86. Meyuhas R, Pikarsky E, Tavor E, Klar A, Abramovitch R, Hochman J, Lago TG and Honigman A. A Key role for cyclic AMP-responsive element binding protein in hypoxia-mediated activation of the angiogenesis factor CCN1 (CYR61) in Tumor cells. Molecular cancer research. 2008; 6(9):1397-1409.
87. Riemann A, Schneider B, Ihling A, Nowak M, Sauvant C, Thews O and Gekle M. Acidic environment leads to ROS-induced MAPK signaling in cancer cells. PloS one. 2011; 6(7):e22445.
88. Choi EM. Luteolin protects osteoblastic MC3T3-E1 cells from antimycin A-induced cytotoxicity through the improved mitochondrial function and activation of PI3K/Akt/CREB. Toxicology in vitro : an international journal published in association with BIBRA. 2011; 25(8):1671-1679.
89. Franko A, Mayer S, Thiel G, Mercy L, Arnould T, Hornig-Do HT, Wiesner RJ and Goffart S. CREB-1alpha is recruited to and mediates upregulation of the cytochrome c promoter during enhanced mitochondrial biogenesis accompanying skeletal muscle differentiation. Molecular and cellular biology. 2008; 28(7):2446-2459.
90. Chae HD, Mitton B, Lacayo NJ and Sakamoto KM. Replication factor C3 is a CREB target gene that regulates cell cycle progression through the modulation of chromatin loading of PCNA. Leukemia. 2015; 29(6):1379-1389.
91. Fang M, Pak ML, Chamberlain L, Xing W, Yu H and Green MR. The CREB Coactivator CRTC2 Is a Lymphoma Tumor Suppressor that Preserves Genome Integrity through Transcription of DNA Mismatch Repair Genes. Cell reports. 2015; 11(9):1350-1357.
92. Kew VG, Yuan J, Meier J and Reeves MB. Mitogen and stress activated kinases act co-operatively with CREB during the induction of human cytomegalovirus immediate-early gene expression from latency. PLoS pathogens. 2014; 10(6):e1004195.
93. Pigazzi M, Manara E, Baron E and Basso G. ICER expression inhibits leukemia phenotype and controls tumor progression. Leukemia. 2008; 22(12):2217-2225.
94. Linnerth NM, Baldwin M, Campbell C, Brown M, McGowan H and Moorehead RA. IGF-II induces CREB phosphorylation and cell survival in human lung cancer cells. Oncogene. 2005; 24(49):7310-7319.
95. Ali AM, Reis JM, Xia Y, Rashid AJ, Mercaldo V, Walters BJ, Brechun KE, Borisenko V, Josselyn SA, Karanicolas J and Woolley GA. Optogenetic Inhibitor of the Transcription Factor CREB. Chemistry & biology. 2015; 22(11):1531-1539.
96. Alper O, Bergmann-Leitner ES, Abrams S and Cho-Chung YS. Apoptosis, growth arrest and suppression of invasiveness by CRE-decoy oligonucleotide in ovarian cancer cells: protein kinase A downregulation and cytoplasmic export of CRE-binding proteins. Mol Cell Biochem. 2001; 218(1-2):55-63.
97. Steven A, Leisz S, Massa C, Iezzi M, Lattanzio R, Lamolinara A, Bukur J, Muller A, Hiebl B, Holzhausen HJ and Seliger B. HER-2/neu mediates oncogenic transformation via altered CREB expression and function. Molecular cancer research. 2013; 11(11):1462-1477.
98. Best JL, Amezcua CA, Mayr B, Flechner L, Murawsky CM, Emerson B, Zor T, Gardner KH and Montminy M. Identification of small-molecule antagonists that inhibit an activator: coactivator interaction. Proceedings of the National Academy of Sciences of the United States of America. 2004; 101(51):17622-17627.
99. Jiang M, Li BX, Xie F, Delaney F and Xiao X. Design, synthesis, and biological evaluation of conformationally constrained analogues of naphthol AS-E as inhibitors of CREB-mediated gene transcription. Journal of medicinal chemistry. 2012; 55(8):4020-4024.
100. Li BX, Yamanaka K and Xiao X. Structure-activity relationship studies of naphthol AS-E and its derivatives as anticancer agents by inhibiting CREB-mediated gene transcription. Bioorg Med Chem. 2012; 20(23):6811-6820.
101. Xie F, Li BX, Broussard C and Xiao X. Identification, synthesis and evaluation of substituted benzofurazans as inhibitors of CREB-mediated gene transcription. Bioorg Med Chem Lett. 2013; 23(19):5371-5375.
102. Xie F, Li BX, Kassenbrock A, Xue C, Wang X, Qian DZ, Sears RC and Xiao X. Identification of a Potent Inhibitor of CREB-Mediated Gene Transcription with Efficacious in Vivo Anticancer Activity. Journal of medicinal chemistry. 2015; 58(12):5075-5087.
103. Rudolph D, Tafuri A, Gass P, Hammerling GJ, Arnold B and Schutz G. Impaired fetal T cell development and perinatal lethality in mice lacking the cAMP response element binding protein. Proceedings of the National Academy of Sciences of the United States of America. 1998; 95(8):4481-4486.
104. Chen L, Yang Q, Kong WQ, Liu T, Liu M, Li X and Tang H. MicroRNA-181b targets cAMP responsive element binding protein 1 in gastric adenocarcinomas. IUBMB life. 2012 (a); 64(7):628-635.
105. Wang J, Liu X, Wu H, Ni P, Gu Z, Qiao Y, Chen N, Sun F and Fan Q. CREB up-regulates long non-coding RNA, HULC expression through interaction with microRNA-372 in liver cancer. Nucleic acids research. 2010; 38(16):5366-5383.