
Introduction
In addition to morphological change and function loss, another obvious change is cellular energy metabolism during the transition from a normal cell to cancer cell. Deregulated energy metabolism has therefore been recognized as one of the hallmarks of cancer [1]. Abnormal glucose metabolism is well known in tumor cells and characterized with aerobic glycolysis. Glycolysis is a critical catabolic process of the glucose, which breaks down one molecule of glucose to produce two pyruvates together with two ATPs and two reduced nicotinamide adenine dinucleotide (NADH) molecules. The fate of pyruvate largely depends on the supply of oxygen for the cells. Pyruvate is transformed into lactate in the absence of oxygen via an anaerobic glycolysis pathway. In contrast, pyruvate is oxidized to CO2 and H2O in the presence of oxygen through the oxidative phosphorylation (OXPHOS) pathway, resulting in the production of large amounts of ATP. It was found in nearly one century ago that tumor cells have a much higher rate of glucose consumption [2]. Most cancer cells produce large amounts of lactic acid regardless of the availability of oxygen [3]. This phenomenon converted glucose to lactate in the presence of oxygen is known as the Warburg effect or aerobic glycolysis [4]. This review will update the latest progress on the regulatory mechanism of the Warburg effect and discuss the potential targets in this pathway for the tumor therapy.
Pathway of glycolysis
A molecule of glucose is degraded to two molecules of three-carbon pyruvate in ten steps in glycolysis. Glycolysis pathway initiates from the phosphorylation of glucose to glucose-6-phosphate (Figure 1). Glucose-6-phosphate is then converted to fructose-6-phosphate that is further phosphorylated to form fructose-1,6-biphosphate. Two molecules of ATP are consumed in two steps of phosphorylation. Fructose-1,6-biphosphate is then broken to yield two three-carbon tautomeric molecules, dihydroxyacetone phosphate and glyceraldehyde 3-phosphate. Glyceraldehyde 3-phosphate is oxidized and phosphorylated to form 1,3-biphosphoglycerate by inorganic phosphate in the payoff phase of glycolysis. The latter is converted to 3-phosphoglycerate yielding a molecule of ATP via substrate-level phosphorylation. 3-phosphoglycerate is isomerized to 2-phosphoglycerate that is dehydrated to produce phosphoenolpyruvate. Phosphoenolpyruvate is subsequently converted to pyruvate yielding a molecule of ATP again. Two molecules of glyceraldehyde 3-phosphate are oxidized to two pyruvates, in which four molecules of ATP are produced via two steps of substrate-level phosphorylation. The net yield is two molecules of ATP per molecule of glucose oxidized because two ATPs are invested in the preparatory phase. Pyruvate is reduced to lactate under hypoxia, or oxidized to yield acetyl-coenzyme A under aerobic condition and then oxidized completely to CO2 via citric acid cycle.
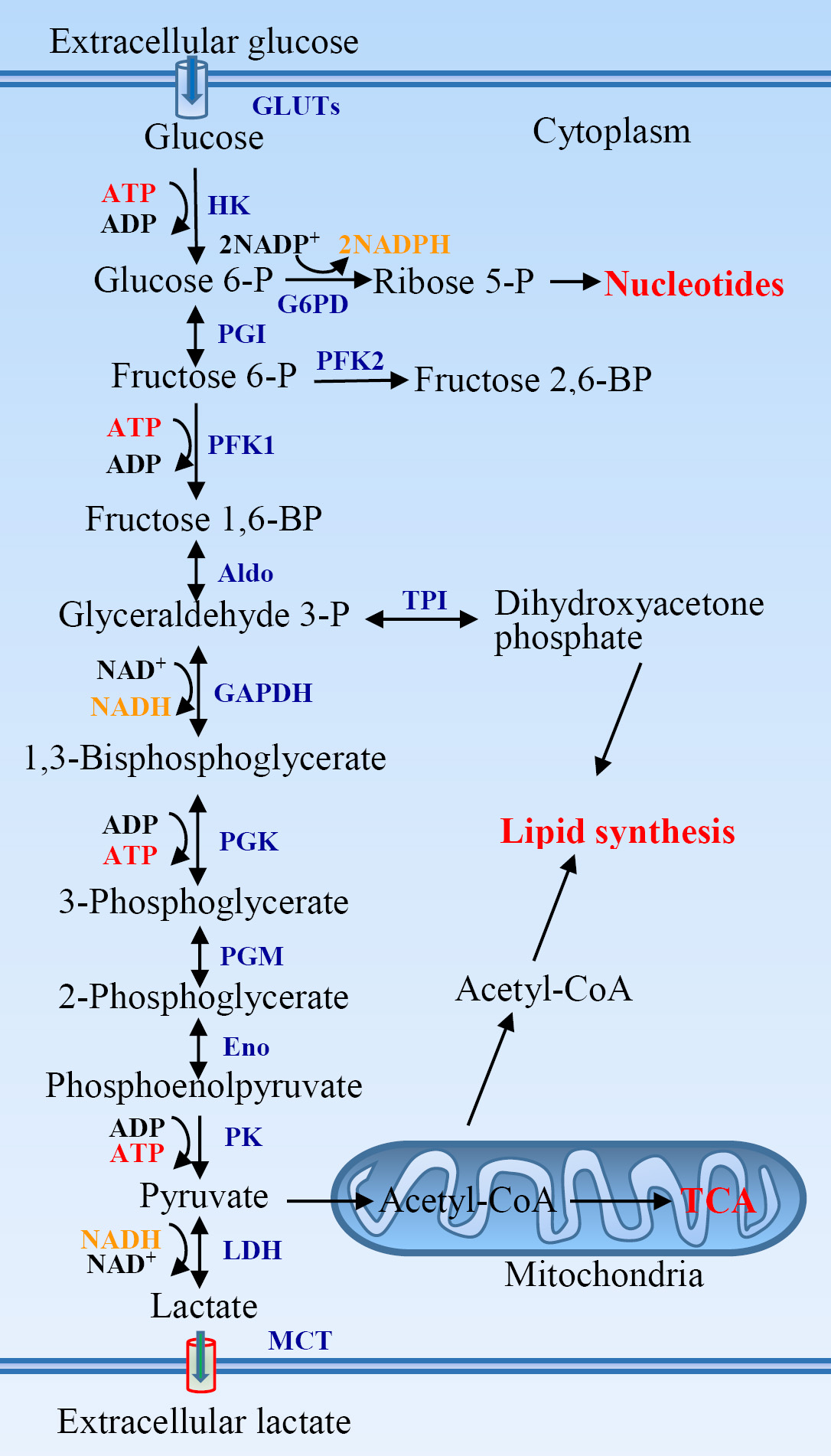
Figure 1: The glycolytic pathway and its association with other metabolic pathways. Aldo, aldolase; Eno, enolase; G6PD, glucose-6-phosphate dehydrogenase; GAPDH, glyceraldehyde 3-phosphate dehydrogenase; GLUTs, glucose transporters; HK, hexokinase; LDH, lactate dehydrogenase; MCT, monocarboxylate transporter; PFK, phosphofructokinase; PGI, phosphoglucose isomerase; PGK, phosphoglycerate kinase; PGM, phosphoglycerate mutase; PK, pyruvate kinase; TCA, tricarboxylic acid cycle; TPI, triose phosphate isomerase.
Aerobic glycolysis in tumors
One of main metabolic characteristics of cancer cells is aerobic glycolysis, the high level of glycolysis even in the presence of oxygen. Hypoxia is supposed to be a main reason driving tumor cells to anaerobic glycolysis pathway. Hypoxia and glucose shortage occur in the inner mass of a growing tumor due to supply of blood. Glycolytic switch is actually acquired very early in carcinogenesis, even before tumors experience hypoxia [3]. It is likely that two early steps in transformation of a normal cell into a tumor cell are the change to dependence on glycolysis for ATP production and the development of tolerance to a low pH in the extracellular environment caused by release of lactate. Lung cancers and leukemic cells are growing in the presence of oxygen. Even so, they still drive glucose into the aerobic glycolysis pathway [5, 6]. Many tumors utilize aerobic glycolysis to meet their metabolic requirements even in normoxic conditions, suggesting that the Warburg effect is not solely adaptive to hypoxia. Actually, glycolysis facilitates tumor growth, and reversing the glycolytic phenotype to OXPHOS in cancer cells can promote cell death [7]. The increased take-up and consumption of glucose in tumors is observed compared with normal tissues, which has been used to identify tumors and metastatic lesions by positron emission tomography [8] and currently new developed methods [9, 10].
It seems reasonable that the metabolic switch from OXPHOS to glycolysis during hypoxia or mitochondrial dysfunction is critical for cancer cell growth [11, 12]. Mitochondrial impairment and subsequent defective OXPHOS is frequently found in cancers [13] . It is clear that mitochondrial defect in cancer cells can cause a shift in energy metabolism, in which hypoxia-inducible factor-1α (HIF-1α) plays an important role as an activator of aerobic glycolysis and lactate production. However, most tumor cells display a normal mitochondrial function including normal capacity for mitochondrial OXPHOS [14-16]. The high glycolytic activity in cancer cells does not mean a reduction in OXPHOS [17]. It was reported that ATP production is 80% oxidative and 20% glycolytic in breast cancer and glioma cells [14-16]. The contribution of aerobic glycolysis in these cells is similar to that in non-transformed cells. In contrast, mtDNA gene mutations reduce colony formation and growth rate of cancer cells and diminish tumorigenicity [18], which is largely discriminated from typical characteristics of cancer cells. Thus glycolytic switch in tumors is subjected to a complex regulation (see discussion below).
It is obvious that glycolysis yields a lower amount of ATP compared to mitochondrial OXPHOS. Why do tumor cells prefer to fuel glucose to the aerobic glycolysis pathway? Several key benefits inherent in aerobic glycolysis drive cancer cells to favor glycolysis over mitochondrial oxidation [13, 19]. First, the glycolysis leads to faster ATP production. The rate of ATP production may be 100 times faster with glycolysis than with OXPHOS [20]. The increased rate of ATP production resulting from glycolysis confers a selective growth advantage to cancer cells [21, 22]. Second, high glycolytic rates likely benefit rapid proliferating cells through the production of glycolytic intermediates to meet the biosynthesis needs of the cells. These intermediates are integrated into various metabolic pathways to generate de novo nucleotides, lipids, amino acids, and NADPH [22-24]. For example, the accumulation of glycolytic intermediates promotes the pentose phosphate pathway (PPP), resulting in the generation of NADPH and ribose-5-phosphate. NADPH acts as a reducing agent for lipid, nucleotide, and amino acid biosynthesis. Ribose-5-phosphate is essential for the biosynthesis of nucleic acids. Dihydroxyacetone phosphate resulting from glycolysis is converted to glycerol-3-phosphate, which is crucial for the biosynthesis of the phospholipids and triacylglycerols required for generation of cell membranes. Finally, the NADPH is also used for the reduction of cellular glutathione (GSH) pools and enables the cancer cells to maintain adequate levels of reduced forms of GSH, which is critical for cancer cells resistance against chemotherapeutic agents.
Proliferating cancer cells adapt several molecular mechanisms to maintain high glycolytic flux [23]. First, they upregulate the expression of phosphofructokinase-2 (PFK2), which produces fructose-2,6-bisphospate as a potent allosteric activator of PFK1 to overcome negative allosteric feedback inhibition of high ATP levels on PFK1, a critical driver of glycolytic flux. Second, generation of NAD+ from NADH via upregulation of LDH is necessary for maintaining glycolytic flux. Finally, cancer cells express higher levels of pyruvate kinase M2 (PKM2) which can be allosterically and covalently inhibited. PKM2 inhibition facilitates glycolytic intermediates upstream of pyruvate into biosynthetic pathways, and phosphoenolpyruvate is subsequently converted to pyruvate through alternative pathways to generate lactate and NAD+ [25].
It must point out that aerobic glycolysis is not a specific marker of tumors. It has been well know that fast growing tissues or cells also depend more on glycolysis than on OXPHOS for energy production. The metabolism of stem cells, including embryonic stem cells, hematopoietic stem cells, and induced pluripotent stem cells, shows the Warburg effect [26-31]. Lymphocytes exhibit a striking metabolic shift upon activation. Glycolysis is significantly upregulated during lymphocyte activation, even in the presence of oxygen [3, 32, 33]. Similar to tumors, the increase in glycolysis generates ATP at a faster rate than OXPHOS and supplies metabolic precursors to meet the metabolic requirements of cell proliferation, while producing less reactive oxygen species (ROS) [3, 34]. However, this metabolic shift may not only be necessary for proliferation or survival, but also specifically required for effector function in T cells [35, 36].
Regulation of glycolysis in tumors
The Warburg effect is a hallmark of cancer. However, the highly glycolytic mechanism of cancer cells remains less clear. Although hypoxia is a main reason resulting in abnormal glycolytic flux in cancer cells, the glycolysis enhances in many tumors under normoxic conditions. The other factors such as oncogenes and related signaling pathways are involved in glycolytic switch in tumors (Table 1). Oncogenes can directly activate hypoxia-inducible factor-1 (HIF-1) and other components of glucose metabolism independently of hypoxia in many cancers (Figure 2).
Table 1: Major regulators of aerobic glycolysis in tumor
Key modulators |
Via |
References |
Abhd5 |
AMPK, GLUT1, HK I, HK II, LDHA, PKM1, p53 |
[37] |
Caveolin 1 |
Akt-mTOR, GLUT3 |
|
CD147 |
MCT1, MCT4, p53 |
|
Ecdysoneless |
GLUT4 |
[44] |
FAK |
LDH, MCT, PKM2 |
[45] |
GRIM-19 |
HIF-1α, p53, STAT3, HK II, PDK1, PFK1, PKM2 |
[46] |
GRP78 |
HIF-1α, PKM2 |
[47] |
HIF-1α |
GLUT1, HK II, PDK1, PKM2 |
|
HSP40 |
PKM2 |
[53] |
KLF4 |
LDHA |
[54] |
K-Ras |
GLUT1 |
[55] |
KSHV |
PKM2 |
[56] |
LMP1 |
LDHA |
[57] |
miRNAs |
GLUT1, GLUT3, HK II, LDHA, PFK, PFKFB3 |
|
p53 |
AMPK, GLUT1, GLUT3, GLUT4, G6PD, PGM, Pten, TIGAR, TSC2 |
|
P2X7 |
GLUT1, HIF-1α, PKM2 |
[78] |
PI3K-Akt-mTOR |
c-Myc, HIF-1α, NFκB GLUT1, GLUT3, HK II, LDHA, PFK, PKM2 |
|
Skp2 |
Akt, GLUT1 |
[84] |
Survivin |
DNM1L |
[85] |
TRAP1 |
c-Src |
[86] |
Wnt |
PDK1 |
[87] |
ZBTB7A |
GLUT3, PFKP, PKM |
[88] |
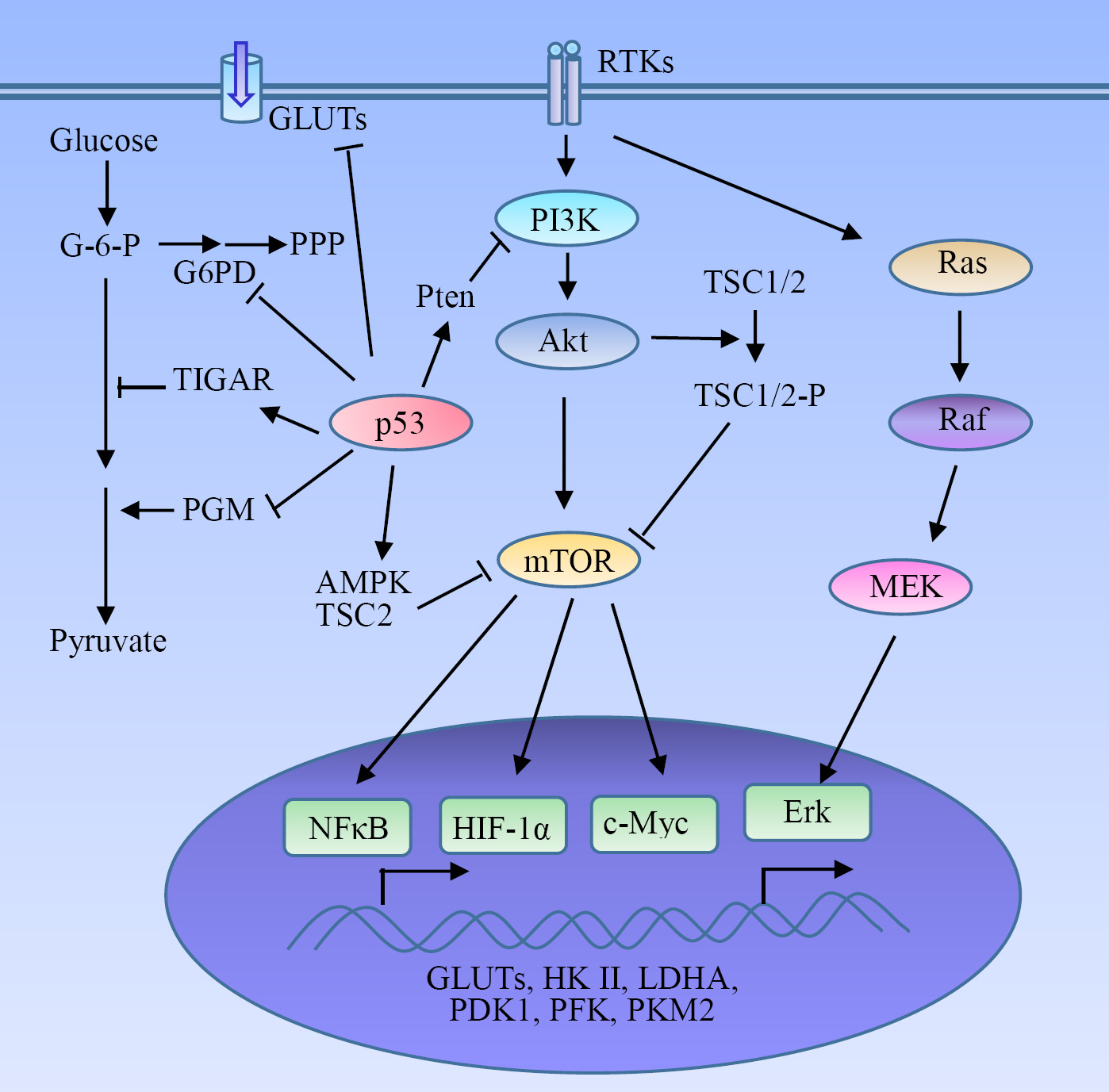
Figure 2: Regulatory mechanism of glycolysis in tumors. HIF-1α serves as a key activator of glycolysis through the induction of GLUTs and many glycolytic enzymes. The receptor tyrosine kinases (RTKs)-mediated PI3K-Akt-mTOR signaling pathway plays a pivotal role in the metabolic switch to aerobic glycolysis in tumor cells via the activation of HIF-1α, NFκB and c-Myc, and the subsequent expression of glycolytic enzymes. Some oncogenes and tumor suppressors such as Ras and p53 are involved in the regulation of the Warburg effect.
HIF-1α
Hypoxia-inducible factor-1α (HIF-1α) contributes greatly to the enhanced glycolysis in tumors. HIF-1α stimulates glycolysis through direct transactivation of glucose transporters (GLUTs) such as GLUT1 and many glycolytic enzymes such as hexokinase II (HK II), pyruvate dehydrogenase kinase 1 (PDK1) and PKM2 [48, 49]. The high glycolytic rate in hypoxic solid tumor is due in part to the greatly increased HIF-1-mediated expression of HK II [50]. Pyruvate dehydrogenase converts pyruvate into acetyl-CoA for tricarboxylic acid (TCA) cycle. PDK1 phosphorylates and inhibits pyruvate dehydrogenase, and subsequently prevents the entry of pyruvate into the TCA cycle [51, 52]. Cancer cells express higher levels of PKM2 over the more catalytically active PKM1, leading to accumulation of cellular carbohydrate metabolites that can be used for the biosynthesis of macromolecules to support the proliferation and rapid growth of tumor cells.
PI3K-Akt-mTOR pathway
Akt, a serine/threonine kinase promoting cancer growth, activates aerobic glycolysis and renders cancer cells dependent on glycolysis for survival [89]. Regulation of Akt on glycolysis involves HK II [90, 91], PFK2 [92], and GLUT1 [91, 93]. Akt signaling induces HK II expression [90, 91], and phosphorylates and activates PFK2 [92]. PFK2 catalyzes the production of fructose-2,6-bisphosphate, which acts as an allosteric activator of PFK1. Activation of the Akt upregulates GLUT1 gene transcription [91, 93]. HK II and PFK1 are rate-controlling enzymes of glycolysis. GLUT1 is the most widely expressed glucose transporter which is translocated to cell surface induced by Akt to promote glucose uptake. Akt promotes a glycolytic switch under normoxic conditions in tumor cells and was coined the ‘Warburg kinase’ [94]. Enhanced aerobic glycolysis mediated by Akt leads to acquired radioresistance of tumor cells [95]. Deficiency of Skp2, an E3 ligase, impairs Akt activation as well as GLUT1 expression, glycolysis, and cancer progression [84]. Glycolysis promotion induced by Akt does not affect the rate of OXPHOS, implying that this effect is not an adaptation to hypoxia rather than to meet the increased needs of metabolic intermediates required for rapid proliferation of tumor cells. Thus the Akt-mediated aerobic glycolysis is critical for growth and survival of tumor cells. Tumor cells bearing an activated form of Akt undergo rapid cell death when shifted to low-glucose conditions [89]. Akt also phosphorylates and inactivates tumor suppressor TSC2, a negative regulator of mammalian target of rapamycin (mTOR), to promote glycolysis (see discussion below) [96].
PI3K-Akt-mTOR is a major signaling pathway involved in cancer development and progression, which is pivotal in the regulation of aerobic glycolysis and tumor growth. A number of receptor tyrosine kinases (RTKs), such as the epidermal growth factor receptor (EGFR), insulin-like growth factor receptor, and platelet-derived growth factor receptor (PDGFR), can activate PI3K at the cell membrane, initiating the signaling cascade. Upon the PI3K activation, Akt is recruited to the cell membrane and activated. mTOR is a serine/threonine kinase downstream of PI3K-Akt and acts through two separate complexes, mTOR complex 1 (mTORC1) and mTORC2, which play a critical role in tumorigenesis and metabolism [97]. RTK-PI3K-Akt-mTOR signaling cascade is a frequently altered pathway in cancer. PDGFR signaling regulates glycolysis in glioma-derived tumor stem-like cells through the activation of Akt [98]. PI3K-Akt-mTOR signaling is indispensable for the EGFR-mediated regulation of aerobic glycolysis in lung cancer cells [99]. Insulin receptor substrate 2-mediated PI3K signaling selectively inhibits glycogen synthase kinase 3β to promotes glucose uptake and aerobic glycolysis [100]. Pten phosphatase is a negative regulator of PI3K, and its deficiency increases HK II mRNA translation in prostate cancer cells through the activation of Akt-mTORC1-4E binding protein 1 axis [101]. Pten inhibits glycolysis through both PI3K/Akt-dependent and -independent pathways [101, 102].
Metabolic switch to aerobic glycolysis in cancer cells involves the mTOR-mediated expression of glycolytic enzymes through the activation of HIF-1α, NFκB, and c-Myc [79-82]. mTOR was identified as a central activator of the Warburg effect by inducing PKM2 under normoxic condition [82]. Upregulation of PKM2 induced by mTOR is critical for aerobic glycolysis and tumor growth [82]. PKM2 is a rate-limiting glycolytic enzyme, and exclusively expressed in embryonic, proliferating, and tumor cells. mTOR stimulates PKM2 expression through HIF-1α-mediated transcription activation and c-Myc-heterogeneous nuclear ribonucleoproteins (hnRNPs)-dependent regulation of PKM2 gene splicing [82]. hnRNPs were identified as splicing repressors for PKM1. Disruption of PKM2 suppresses mTOR-mediated tumorigenesis. Dual suppression of mTOR and glycolysis synergistically blunts the proliferation and tumor development of mTOR hyperactive cells. TSC1/TSC2 complex negatively regulates the expression of GLUT3 [83]. Loss of TSC1/TSC2 upregulates GLUT3 through the activation of the rapamycin-sensitive mTORC1 signaling. mTORC1 upregulates GLUT3 through the activation of NFκB pathway [83]. Depletion of GLUT3 suppresses aerobic glycolysis, inhibits cell proliferation and colony formation, and attenuates the tumorigenic potential of the cells with aberrantly hyper-activated mTORC1 signaling in nude mice. In addition, a SIRT1-mTOR/HIF-1α glycolytic pathway is required for differentiation of myeloid-derived suppressor cells to the M1 phenotype [103]. MUC16, a transmembrane mucin increases glycolysis in pancreatic cancer through the activation of mTOR and c-Myc [104].
K-Ras
Activation of K-Ras (G12V) causes mitochondrial dysfunction, leading to a metabolic switch from OXPHOS to glycolysis. Pre-induction of K-Ras expression in vitro resulting in glycolytic switch enhances the ability of the transformed cells to form tumor in vivo [11]. Mutated K-Ras leads to increased expression of the GLUT1, glucose uptake, glycolysis, and lactate production [55]. K-Ras mutated cells showed increased survival in low-glucose culture condition. They are much sensitive to the toxicity of 3-bromopyruvate (3-BP), a hexokinase inhibitor compared with cells lacking K-Ras mutation [55]. Oncogenic K-Ras has been reported to maintain pancreatic tumors through regulation of glycolysis [105]
p53
p53, a tumor suppressor, induces cell-cycle arrest and cell death after DNA damage, and thus contributes to the maintenance of genomic stability. p53 plays a critical role in promoting mitochondrial OXPHOS and downregulation of glycolysis [106, 107]. First, p53 represses aerobic glycolysis through regulation of glucose transporters and glycolytic enzymes. p53 directly suppresses the expression of GLUT1 and GLUT4 [108], and indirectly downregulates GLUT3 [70]. p53 promotes the ubiquitination-mediated degradation of phosphoglycerate mutase (PGM). Loss of p53 increases PGM level and glycolysis [71]. p53 also negatively regulates glycolysis via TIGAR (TP53-induced glycolysis and apoptosis regulator). TIGAR dephosphorylates fructose-2,6-bisphosphate to fructose-6-phosphate and diverts glucose catabolism to the PPP [72, 73]. Second, p53 regulates glucose metabolism through inhibiting glucose-6-phosphate dehydrogenase (G6PD), the first and rate-limiting enzyme in the PPP [74]. Third, p53 regulates glycolysis through inducing a group of target genes to negatively regulate PI3K-AKT-mTOR pathway. For example, p53 induces Pten to inhibit the PI3K-AKT signaling [75]. p53 activates AMPK, a major upstream negative regulator of mTOR, leading to the downregulation of the mTOR activity [75, 76]. p53 also induces TSC2 to negatively regulate the mTOR activity. It has recently been found that p53 induces its target RRAD, which in turn inhibits the GLUT1 translocation to the plasma membrane and represses glycolysis in lung cancer cells [77].
p53 frequently mutates in tumors. Recent studies have revealed that tumor-associated mutant p53 (mutp53) drives the Warburg effect under normoxia [109]. mutp53 stimulates the Warburg effect in cultured cells through promoting GLUT1 translocation to the plasma membrane, which is mediated by activated RhoA and its downstream effector ROCK. Inhibition of RhoA-ROCK-GLUT1 signaling abolishes role of mutp53 in inducing the aerobic glycolysis, and inhibition of glycolysis in tumor cells greatly compromises mutp53-promoting tumorigenesis [109].
AMPK
Adenosine monophosphate activated protein kinase (AMPK) is a critical cellular energy sensor detecting the balance between ATP production and consumption in eukaryotic cells [110]. Once activated by energetic stress via increases in AMP:ATP and ADP:ATP ratios, AMPK changes the metabolic process from anabolic condition to a catabolic state [111, 112]. AMPK activation switches off most anabolic processes, such as synthesis of lipids, carbohydrates, ribosomal RNA, and proteins [111]. AMPK downregulates protein synthesis through the repression of mTOR-dependent mRNA translation [113-115]. This includes downregulation of ribosomal proteins, HIF-1α, and thus expression of the glycolytic enzymes and transporters required for the Warburg effect [116, 117]. AMPK activation therefore promotes the oxidative metabolism typical of quiescent cells, rather than the aerobic glycolysis observed in most proliferating cells including tumor cells [112, 118]. AMPK is a potential target for cancer prevention and treatment as well as anti-inflammatory drug [119, 120].
c-Myc
c-Myc acts as a transcription factor involved in the control of cell proliferation, differentiation, and apoptosis. By using c-Myc transgenic model, Valera et al. revealed that the expression of glycolysis-related enzymes, such as glucokinase, PFK2, and PK, are increased in the liver of the transgenic mice, suggesting that c-Myc is involved in the control in vivo of carbohydrate metabolism [121]. c-Myc promotes glucose uptake and glycolysis through the upregulation of GLUT1 and many glycolytic enzymes, including HK II and PFK and lactate dehydrogenase A (LDHA) [48]. c-Myc directly transactivates expression of GLUT1 [122], MCT1, and MCT2 [123]. c-Myc can also enhance expression of MCT1 via transcriptionally repressing miR-29a and miR-29c [123]. hnRNPs proteins, polypyrimidine tract binding protein (PTB), hnRNPA1, and hnRNPA2, bind to PKM pre-mRNA and switch PKM splicing to favor PKM2 variant [124, 125]. c-Myc upregulates transcription of PTB, hnRNPA1, and hnRNPA2, ensuring a high PKM2/PKM1 ratio. LDHA was identified as another c-Myc responsive gene [126, 127]. Knockdown of c-Myc reduces the expression of LDHA, lactate production, and glucose consumption [127]. LDHA overexpression is required for c-Myc-mediated transformation and tumor growth [126, 127].
Human pituitary tumor-transforming gene (PTTG), a proto-oncogene, influences glycolysis through regulation of c-Myc [128]. PTTG knockdown in ovarian cancer cells results in the downregulation of c-Myc and several crucial proteins involved in aerobic glycolysis, including PKM2, LDHA, and GLUT1. Overexpression of c-Myc could prevent the PTTG knockdown-induced metabolic shift [128], suggesting that PTTG regulates the metabolic switch via the c-Myc pathway. N-Myc downstream regulated gene 2 (NDRG2), a tumor suppressor gene, significantly suppresses the expression of GLUT1, HK2, PKM2, and LDHA, leading to inhibition of glucose consumption and lactate production in colorectal cancer cells [129]. c-Myc mediates the inhibition of glycolysis by NDRG2. NDRG2 inhibits the expression of c-Myc by suppressing the production of β-catenin, which can transcriptionally activate c-Myc. Therefore NDRG2 functions as an essential regulator in glycolysis via repression of c-Myc [129].
miRNAs
Accumulating evidence suggests that microRNAs (miRNAs) interact with oncogenes/tumor suppressors and induce the aerobic glycolysis in cancer cells. miRNAs can regulate glucose transporters and glycolytic enzymes [130]. miR-144 inhibits GLUT1 expression through targeting its 3’-untranslated region in ovarian cancer cells [64]. miR-22 and miR-1291 also directly target GLUT1 in breast cancer [65] and renal cell carcinoma [66], respectively. miR-195-5p directly targets GLUT3 and suppresses glucose uptake in bladder cancer cells [67]. Several miRNAs regulate glycolysis via targeting HK II. miR-143 inhibits the expression of HK II in head and neck squamous cell carcinoma (HNSCC)-derived cells [68] and colon cancer cells [69]. miR-143, downregulated by mTOR activation, regulates cancer glycolysis and inhibits cancer cell proliferation and tumor formation via targeting HK II in human lung cancer [58]. miR-143 is downregulated in glioma tissues and glioblastoma stem-like cells (GSLCs). It inhibits glycolysis by directly targeting HK II and depletes GSLCs stemness [59]. miR-155 upregulates HK II via repressing miR-143 and activating the signal transducer and activator of transcription 3 (STAT3), a transcriptional activator for HK II [131]. miR-29b negatively regulates Akt expression, causing HK II/PKM2 downregulation and leading to a decreased the Warburg effect and slowed ovarian cancer progression [132]. miR-199a-5p regulates glycolysis and lactate production by targeting HK II [60]. It is downregulated in human liver cancer and is negatively associated with malignancies. The upregulation of HIF-1α under hypoxic conditions suppresses the expression of miR-199a-5p and promotes glycolysis.
LDHA, PFK, and 6-phosphofructo-2-kinase/fructose-2,6-bisphosphatase-3 (PFKFB3) are other targets for miRNAs. LDHA is frequently overexpressed in tumor cells. Knockdown of LDHA results in decreased lactate and ATP production, glucose uptake and cell growth. Several miRNAs target LDHA and regulate glycolysis in cancer cells [61]. miR-320a directly regulates PFK expression and sequential lactate production [63]. miR-26b and miR-206 inhibit tumor growth via the downregulation of PFKFB3-driven glycolysis [62, 133]. Overexpression of miR-26b represses PFKFB3 expression and regulates the expression of LDHA, GLUT1, and markers of invasion and cell cycle. miR-21 diminishes aerobic glycolysis in bladder cancer cells via the Pten/PI3K/AKT/mTOR axis [134].
Other regulators
Wnt signaling regulates glycolysis and angiogenesis through PDK1 [87]. Interference with Wnt signaling in colon cancer cells reduces glycolytic metabolism and suppresses tumor growth. PDK1 overexpression in Wnt-inhibited cancer cells rescues glycolysis and vessel growth. Heat shock proteins (HSPs) including HSP40 [53] and TNF receptor-associated protein 1 (TRAP1) [86], a member of the HSP90 chaperone family, are involved in the regulation of metabolic switch between OXPHOS and aerobic glycolysis in tumors. Focal adhesion kinase (FAK), a key transmitter of growth factor and anchorage stimulation, enhances glycolysis and decreases mitochondrial respiration through the upregulation of glycolytic proteins enolase, PKM2, LDH and MCT [45]. Attenuation of FAK-enhanced glycolysis decreases cell viability and reduces growth of tumor xenografts. HSulf-1, a putative tumor suppressor, is a negative regulator of glycolysis. Silencing of HSulf-1 expression in ovarian cancer cells increases glucose uptake and lactate production by upregulating GLUT1 and glycolytic enzymes HK II and LDHA [135]. The P2X7 receptor (P2X7R), an ATP-gated cation channel, is a key modulator of aerobic glycolysis [78]. Forced expression of P2X7R in HEK293 cells increases phosphorylated Akt and HIF-1α expression; upregulates GLUT1, GAPDH, PFK, PKM2, and PDK1; and inhibits pyruvate dehydrogenase activity.
CD147 is a transmembrane glycoprotein and plays an important role in tumorigenicity, invasion, and metastasis. CD147 promotes glycolysis and tumor progression in epithelial solid tumors through the regulation of the p53-dependent signaling pathway and MCT [41-43]. Plasma membrane-associated protein Caveolin 1 enhances aerobic glycolysis via regulation of Akt-mTORC1-GLUT3 signaling [38-40]. Glucose regulated protein 78 (GRP78) is involved in the modulation of tumor aerobic glycolysis [47]. Overexpression of GRP78 induces inactivation of NF-κB pathway, and subsequently alters the expression of PKM2 and HIF-1α. α/β-hydrolase domain-containing 5 (Abhd5) is an intracellular lipolytic activator. Suppressing Abhd5-mediated intracellular lipolysis stimulates aerobic glycolysis in cancer cells [37]. Decreased expression of the gene associated with retinoid-interferon induced mortality-19 (GRIM-19) promotes aerobic glycolysis and cell proliferation in HNSCC [46]
ZBTB7A, a member of the POK (POZ/BTB and Krüppel) transcription repressor family, acts as a novel tumor suppressor by directly suppressing glycolysis [88]. ZBTB7A-deficient tumors progress very fast and are extremely sensitive to glycolysis inhibition. A novel Krüppel-like factor 4 (KLF4) regulates aerobic glycolysis in pancreatic cancer through negatively regulating the transcription of LDHA [54]. KLF4 overexpression significantly attenuates the aerobic glycolysis and cancer cell growth. Antiapoptotic protein Survivin induces a switch of OXPHOS to aerobic glycolysis in tumor cells and may acts as a target for glycolysis inhibition [85, 136]. Ecdysoneless regulates aerobic glycolysis via GLUT4 in pancreatic cancer cells [44].
Some viruses and their encoded proteins even regulate glucose metabolism in cancer cells. Kaposi’s sarcoma (KS) is a vascular neoplasm caused by infection of Kaposi’s sarcoma-associated herpesvirus (KSHV). KSHV induces aerobic glycolysis through HIF-1-dependent upregulation of PKM2 in Kaposi’s sarcoma [56]. The EBV-encoded latent membrane protein 1 (LMP1) increases cellular uptake of glucose, enhances LDHA activity and lactate production, contributing to aerobic glycolysis [57].
Potential targets for tumor therapy
Although the mechanism triggering the Warburg effect in tumors is not well clear, the oncogenic stress including activation of oncogenes and inactivation of tumor suppressors plays a critical role. Aerobic glycolysis contributes to the diverse aspects of tumor development, progression and prognosis [89, 137, 138]. The reversing the Warburg effect greatly compromises the tumorigenicity of tumor cells [139, 140], suggesting that targeting the metabolic changes could be an effective strategy for cancer treatment. Actually, many attempts have been made and some molecular targets have shown their potential in the cancer therapy. Several small molecules, as a single agent or in combination with other therapeutic modalities, exhibit promising anticancer activity both in vitro and in vivo. Of them, lonidamine, 2-deoxyglucose (2-DG), dichloroacetate, and 3-BP have been clinically tested (Table 2). Malignant cells exhibited the Warburg effect become dependent on de novo lipogenesis, which sustains rapid proliferation and resistance to cellular stress. Anti-tumor activity of some small molecules selectively targets the Warburg effect and lipogenesis [141, 142].
Table 2: The potential targets and the corresponding chemicals
Targets |
Potential chemicals |
Development stage |
Cancer types |
References |
GLUTs |
Fasentin, phloretin, WZB117 |
Animal tested |
Liver, lung |
|
HK II |
2-DG |
Clinical trial terminated |
Lung, osteosarcoma, prostrate |
|
3-BP |
Clinical trial phase I |
Liver, stomach |
||
Lonidamine |
Phase III completed |
Breast, glioblastoma, lung, prostrate |
[150] |
|
FV-429 |
Experimental drugs |
Breast |
[151] |
|
Clotrimazole |
Experimental drugs |
Breast |
[152] |
|
PFK |
3PO |
Experimental drugs |
Bone marrow, epithelium, lung |
|
GAPDH |
3-BP |
Experimental drugs |
Liver |
|
PK |
Shikonin, siRNA |
Experimental drugs |
Multiple cancers |
|
LDHA |
FX11 |
Experimental drugs |
Lymphoma, pancreas |
[162] |
Oxamate |
Experimental drugs |
Breast |
[163] |
|
N-hydroxyindole derivates |
Experimental drugs |
Multiple cancers |
[164] |
|
MCT1 |
α-cyano-4-hydroxy-cinnamic acid |
Experimental drugs |
Glioma cells |
[165] |
PDK |
Dichloroacetate |
Clinical trial phase I |
Multiple cancers |
Glucose transporters (GLUTs)
GLUTs transport glucose into the cancer cells and are considered as putative targets. GLUT1 is upexpressed in many cancer types. The several small molecules are identified to inhibit GLUT1 and kill tumor cells in preclinical models [145, 168]. As GLUTs are ubiquitously expressed proteins, blockade of GLUTs will inevitably disrupt glucose consumption in normal tissues. It would be challenging to inhibit a specific isoform associated with tumor cells within an acceptable therapeutic window. In addition, GLUTs have been explored as receptors to import drug-loaded nanoparticles across the blood-brain barrier [169].
Hexokinase II (HK II)
HKs catalyze the first step of glycolysis, a rate-limiting step. There are four mammalian isoforms (I to IV) that are usually expressed at low levels in cells [170]. Of them, HKs, HK II is specially expressed in insulin-sensitive tissues such as muscle and adipose. It has a high affinity (low Km) for glucose facilitating the glycolysis in low serum glucose level. HK II is a key mediator of aerobic glycolysis and promotes tumor growth [171]. HKII is overexpressed in many tumor cells. Systemic targeting of HK II blocks tumor growth without adverse physiologic consequences [172]. These findings suggest that HK II is another potentially attractive therapeutic target for cancer.
There are different strategies to dysfunction HK II. Glucose analog 2-DG shows promising therapy effects in combination treatments with other anticancer agent [146, 147]. Intracellular accumulation of glucose analogs could inhibit HK via feedback inhibition mechanism. Inhibiting glycolysis with 2-DG reduces tumor cell radioresistance [95]. Some small molecules such as 3-BP and lonidamine are direct inhibitors of HK enzyme. Preclinical studies demonstrate that 3-BP inhibits HK II in human cancer and is a promising anticancer drug targeting glycolysis [148, 149]. Lonidamine has completed phase III clinical trials in breast and lung cancers, and a trend for higher tumor responses and better survival parameters was observed when combined with chemotherapy [150, 173]. FV-429, a newly synthesized flavonoid, inhibits glycolysis in human breast cancer cells through inhibiting Akt-mediated phosphorylation of HK II and downregulation of its activity [151]. Clotrimazole, an azole derivative with promising anti-cancer effects, decreases glucose uptake and inhibits the major glycolytic enzymes, HK, PFK1, and PK in human breast cancer cells [152].
Phosphofructokinase (PFK)
PFK1, another rate-limiting enzyme of glycolysis, is activated by fructose-2,6-bisphosphate. The latter is regulated by the activity of a family of bi-functional enzymes, 6-phosphofructo-2-kinase/fructose-2,6-bisphosphatase isozymes (PFKFB1-4). PFKFB3 isozyme is constitutively expressed by tumor cells and required for the high glycolytic rate. A small molecule inhibitor of PFKFB3, 3-(3-pyridinyl)-1-(4-pyridinyl)-2-propen-1-one (3PO), suppresses glycolytic flux and is cytostatic to tumor cells [153]. A combination of 3-PO with ascorbic acid has been shown synergistic activity in non-small cell lung cancer cells [154].
Glyceraldehyde 3-phosphate dehydrogenase (GAPDH)
In addition to being a glycolytic enzyme, GAPDH has multiple functions such as posttranscriptional control of T cell effector function [35]. GAPDH catalyzes the first step of glycolysis pay-off phase and is an alternative promising therapeutic target. NADH produced during this step plays a critical role in the cellular redox balance. Several GAPDH inhibitors have been tested for their efficacy, and the pyruvate analog 3-BP has been demonstrated to be the most promising one [155]. 3-BP inhibits tumor glycolysis and dramatically reduces intracellular ATP level, with excellent specificity and selectivity for GAPDH [156, 157].
Pyruvate kinase (PK)
Muscle type of pyruvate kinase (PKM) is one of the key mediators of the Warburg effect and tumor metabolism. PKM1 and PKM2 are two major isoforms, which are alternative splice products of the PKM gene. PKM2 is upregulated in many tumors [139, 174]. High expression of PKM2 correlates with shorter recurrence-free survival in pancreatic ductal adenocarcinoma patients. Switching PKM splicing to favor PKM1 variant in drug resistant pancreatic cancer cells rescues sensitivity to gemcitabine and cisplatin, suggesting that PKM2 expression is required to withstand drug-induced genotoxic stress [175]. Some small molecule inhibitors including siRNA specific for PKM2 have been tested, and the preclinical studies demonstrate that PKM2 could be a potential therapeutic target [158-161]. Specific knockdown of the PKM2 results in decreased viability and increased apoptosis in multiple cancer cells and causes substantial tumor regression of established xenografts [159]. However, Inhibiting PKM2 could allow glycolytic intermediates to accumulate and feed biosynthetic pathways, resulting in tumor promotion [23]. PKM2 is regulated by cellular oxidative stress [176]. Increase of intracellular ROS concentration causes inhibition of the PKM2 via oxidation of Cys358, promoting diversion of glycolytic intermediates into the PPP to generate sufficient reducing potential for detoxification of ROS. Oxidation-resistant PKM2 mutant exhibits increased sensitivity to oxidative stress and impaired tumor formation in a xenograft model [176]. The small molecule PKM2 activators may also be used to interfere with cancer cell metabolism for therapeutic purposes.
It was previously thought that PKM1 is specific for non-proliferating tissues and PKM2 for proliferating tissues, and that the isoform switch from PKM1 to PKM2 results in high PKM2 expression in tumors, providing a great advantage to tumor cells [139]. However, this traditional view is challenged by recent studies. It has demonstrated that PKM2 is not specific for tumors [177]. The isoform switch does not occur during tumorigenesis [177] or is tissue specific, only occurred in glioblastomas and not in other tumor types [178]. PKM1 even switches to other isoforms rather than PKM2. Instead of the isoform switch, the upregulation of PKM2 is probably due to elevated transcriptional levels of the entire PKM gene [179]. PKM2 could be non-essential for in vivo tumor growth and maintenance [180].
Lactate dehydrogenase (LDH) and monocarboxylate transporters (MCTs)
LDH is a tetramer composed of two different subunits, LDHA and LDHB. LDH catalyzes the final glycolytic step that converts pyruvate into lactate. The accumulation of lactate and subsequent low intracellular pH is extremely harmful. Inhibition of LDH has demonstrated promising effects in preclinical studies. LDHA inhibitor FX11 lowers intracellular ATP levels, which significantly induces oxidative stress and inhibits tumor progression [162]. Another LDHA inhibitor, oxamate, re-sensitizes taxol-resistant cancer [163]. Some N-hydroxyindole-based LDHA inhibitors show an effective anti-proliferative activity in a series of cancer cells [164]. The lactate is exported into the extracellular space via specific transporters monocarboxylate transporters (MCTs). Blocking lactate export by disrupting MCT1 function leads to an accumulation of intracellular lactate that rapidly disables tumor cell growth and glycolysis [181]. A small molecule inhibitor of lactate transport, α-cyano-4-hydroxy-cinnamic acid, has been shown to inhibit tumor invasiveness and induce tumor necrosis [165].
The significance of LDHB in tumor development is elusive. Expression of LDHB is suppressed in many kinds of cancer due to promoter hypermethylation [182]. LDHB acts as a suppressor of glycolysis and suppresses pancreatic cancer progression [183]. Decreased expression of LDHB in liver cancer enhances cell invasiveness via mitochondrial defects [184]. In contrast, LDHB is required for the growth of K-Ras-dependent lung adenocarcinomas [185] and hyperactive mTOR-mediated tumorigenesis [186]. The tumor suppressor drs regulates glucose metabolism via LDHB [187]. Downregulation of drs may contribute to the enhanced glycolysis via increased LDHB expression, which is closely associated with malignant progression of cancer cells.
miRNAs
Several miRNAs are engaged in glycolysis, providing a potential therapeutic strategy. For example, miR-143 targets HK II to regulate glucose metabolism in multiple cancer cells. miR-143 inhibits proliferation of GSLCs under hypoxic conditions and decreases tumor formation capacity of GSLCs in vivo. miR-143 overexpression promotes differentiation of GSLCs. A combination of miR-143 and glycolysis inhibitor 2-DG has synergistic effects against GSLCs, suggesting that miR-143 is a potential therapeutic target for glioblastoma treatment [59]. miR-199a-5p directly targets the 3’-untranslated region of HK II, indicating that it acts as a suppressor for glucose metabolism [60].
Conclusions
During last 1-2 decades a great progress has made toward understanding the mechanism why tumors have a greater reliance on glycolysis for energy than normal tissues. Some oncogenes and corresponding pathways have been proven to be involved in metabolic switch in tumors. This makes tumor cell glycolysis pathway an attractive target for tumor therapy. Several promising preliminary studies illustrate that small molecule inhibitors targeting glycolytic enzymes exert an effective antitumor activity. However, there is a long way to go to uncover the mystery of the Warburg effect. First, we have to evaluate the exact contribution of individual regulator on the glycolytic shift in tumor. It is therefore of great interest to figure out potential master regulator or key driver from a large amount of participators. Second, in terms of cancer therapy or drug development, the specificity of agents mentioned above remains a critical challenge because many glycolytic enzymes are expressed in normal tissues. It remains to verify whether targeting these enzymes will exhibit an effective antitumor effect without significant toxicity to normal tissues. Although several glycolytic inhibitors are in preclinical and clinical development, but none has reached an approved status. Third, it is believed that a small portion of highly tumorigenic cancer cells with stem-like properties is resistant to chemotherapy and may be responsible for the recurrence of cancer after treatment. These cancer cells highly depend on glycolysis for ATP generation and prefer for hypoxia to maintain their stemness and tumor forming capacity. Combination of standard chemotherapeutic agents with glycolytic inhibition is effective in killing the tumor-initiating cells and inhibits tumor formation in vitro and in vivo. Inhibition of glycolysis is therefore an effective strategy to remove residual cancer stem cells and overcome drug resistance [188, 189].
Abbreviations
Abhd5, α/β-hydrolase domain-containing 5; Aldo, aldolase; AMPK, adenosine monophosphate activated protein kinase; 3-BP, 3-bromopyruvate; 2-DG, 2-deoxyglucose; EGFR, epidermal growth factor receptor; Eno, enolase; FAK, focal adhesion kinase; GAPDH, glyceraldehyde 3-phosphate dehydrogenase; GLUTs, glucose transporters; G6PD, glucose-6-phosphate dehydrogenase; GRIM-19, gene associated with retinoid-interferon induced mortality-19; GRP78, glucose regulated protein 78; GSH, glutathione; GSLCs, glioblastoma stem-like cells; HIF-1α, hypoxia-inducible factor-1α; HK, hexokinase; HK II, hexokinase II; hnRNPs, heterogeneous nuclear ribonucleoproteins; HNSCC, head and neck squamous cell carcinoma; HSPs, heat shock proteins; KLF4, Krüppel-like factor 4; KSHV, Kaposi’s sarcoma-associated herpesvirus; LDH, lactate dehydrogenase; LDHA, lactate dehydrogenase A; LMP1, latent membrane protein 1; MCT, monocarboxylate transporter; miRNAs, microRNAs; mTOR, mammalian target of rapamycin; mTORC1, mTOR complex 1; NADH, nicotinamide adenine dinucleotide; NDRG2, N-Myc downstream regulated gene 2; OXPHOS, oxidative phosphorylation; PDGFR, platelet-derived growth factor receptor; PDK1, pyruvate dehydrogenase kinase 1; PFK, phosphofructokinase; PFKFB3, 6-phosphofructo-2-kinase/fructose-2,6-bisphosphatase-3; PGI, phosphoglucose isomerase; PGK, phosphoglycerate kinase; PGM, phosphoglycerate mutase; PK, pyruvate kinase; PKM, muscle type of pyruvate kinase; PKM2, pyruvate kinase M2; 3PO, 3-(3-pyridinyl)-1-(4-pyridinyl)-2-propen-1-one; PPP, pentose phosphate pathway; PTB, polypyrimidine tract binding protein; PTTG, human pituitary tumor-transforming gene; P2X7R, P2X7 receptor; ROS, reactive oxygen species; RTKs, receptor tyrosine kinases; STAT3, signal transducer and activator of transcription 3; TCA, tricarboxylic acid cycle; TIGAR, TP53-induced glycolysis and apoptosis regulator; TPI, triose phosphate isomerase; TRAP1, TNF receptor-associated protein 1.
Acknowledgments
This study was funded by the National Natural Science Foundation of China (No. 81172485) and the Ph.D. Program Foundation of Ministry of Education of China (No. 20130171110007).
Conflicts of interest
The authors state no conflict of interest.
References
1. Hanahan D and Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011; 144(5):646-674.
2. Warburg O, Wind F and Negelein E. The Metabolism of Tumors in the Body. J Gen Physiol. 1927; 8(6):519-530.
3. Vander Heiden MG, Cantley LC and Thompson CB. Understanding the Warburg effect: the metabolic requirements of cell proliferation. Science. 2009; 324(5930):1029-1033.
4. Warburg O. On the origin of cancer cells. Science. 1956; 123(3191):309-314.
5. Nolop KB, Rhodes CG, Brudin LH, Beaney RP, Krausz T, Jones T and Hughes JM. Glucose utilization in vivo by human pulmonary neoplasms. Cancer. 1987; 60(11):2682-2689.
6. Gottschalk S, Anderson N, Hainz C, Eckhardt SG and Serkova NJ. Imatinib (STI571)-mediated changes in glucose metabolism in human leukemia BCR-ABL-positive cells. Clin Cancer Res. 2004; 10(19):6661-6668.
7. Bonnet S, Archer SL, Allalunis-Turner J, Haromy A, Beaulieu C, Thompson R, Lee CT, Lopaschuk GD, Puttagunta L, Harry G, Hashimoto K, Porter CJ, Andrade MA, Thebaud B and Michelakis ED. A mitochondria-K+ channel axis is suppressed in cancer and its normalization promotes apoptosis and inhibits cancer growth. Cancer Cell. 2007; 11(1):37-51.
8. Bomanji JB, Costa DC and Ell PJ. Clinical role of positron emission tomography in oncology. Lancet Oncol. 2001; 2(3):157-164.
9. Rodrigues TB, Serrao EM, Kennedy BW, Hu DE, Kettunen MI and Brindle KM. Magnetic resonance imaging of tumor glycolysis using hyperpolarized 13C-labeled glucose. Nat Med. 2014; 20(1):93-97.
10. Walker-Samuel S, Ramasawmy R, Torrealdea F, Rega M, Rajkumar V, Johnson SP, Richardson S, Goncalves M, Parkes HG, Arstad E, Thomas DL, Pedley RB, Lythgoe MF and Golay X. In vivo imaging of glucose uptake and metabolism in tumors. Nat Med. 2013; 19(8):1067-1072.
11. Hu Y, Lu W, Chen G, Wang P, Chen Z, Zhou Y, Ogasawara M, Trachootham D, Feng L, Pelicano H, Chiao PJ, Keating MJ, Garcia-Manero G and Huang P. K-ras(G12V) transformation leads to mitochondrial dysfunction and a metabolic switch from oxidative phosphorylation to glycolysis. Cell Res. 2012; 22(2):399-412.
12. Lu W, Hu Y, Chen G, Chen Z, Zhang H, Wang F, Feng L, Pelicano H, Wang H, Keating MJ, Liu J, McKeehan W, Luo Y and Huang P. Novel role of NOX in supporting aerobic glycolysis in cancer cells with mitochondrial dysfunction and as a potential target for cancer therapy. PLoS Biol. 2012; 10(5):e1001326.
13. Ganapathy-Kanniappan S and Geschwind JF. Tumor glycolysis as a target for cancer therapy: progress and prospects. Mol Cancer. 2013; 12:152.
14. Guppy M, Leedman P, Zu X and Russell V. Contribution by different fuels and metabolic pathways to the total ATP turnover of proliferating MCF-7 breast cancer cells. Biochem J. 2002; 364(Pt 1):309-315.
15. Martin M, Beauvoit B, Voisin PJ, Canioni P, Guerin B and Rigoulet M. Energetic and morphological plasticity of C6 glioma cells grown on 3-D support; effect of transient glutamine deprivation. J Bioenerg Biomembr. 1998; 30(6):565-578.
16. Pasdois P, Deveaud C, Voisin P, Bouchaud V, Rigoulet M and Beauvoit B. Contribution of the phosphorylable complex I in the growth phase-dependent respiration of C6 glioma cells in vitro. J Bioenerg Biomembr. 2003; 35(5):439-450.
17. Moreno-Sanchez R, Rodriguez-Enriquez S, Marin-Hernandez A and Saavedra E. Energy metabolism in tumor cells. FEBS J. 2007; 274(6):1393-1418.
18. Cavalli LR, Varella-Garcia M and Liang BC. Diminished tumorigenic phenotype after depletion of mitochondrial DNA. Cell Growth Differ. 1997; 8(11):1189-1198.
19. de Souza AC, Justo GZ, de Araujo DR and Cavagis AD. Defining the molecular basis of tumor metabolism: a continuing challenge since Warburg’s discovery. Cell Physiol Biochem. 2011; 28(5):771-792.
20. Locasale JW and Cantley LC. Altered metabolism in cancer. BMC Biol. 2010; 8:88.
21. Gatenby RA and Gillies RJ. Why do cancers have high aerobic glycolysis? Nat Rev Cancer. 2004; 4(11):891-899.
22. Lunt SY and Vander Heiden MG. Aerobic glycolysis: meeting the metabolic requirements of cell proliferation. Annu Rev Cell Dev Biol. 2011; 27:441-464.
23. Hamanaka RB and Chandel NS. Targeting glucose metabolism for cancer therapy. J Exp Med. 2012; 209(2):211-215.
24. Deberardinis RJ, Sayed N, Ditsworth D and Thompson CB. Brick by brick: metabolism and tumor cell growth. Curr Opin Genet Dev. 2008; 18(1):54-61.
25. Vander Heiden MG, Locasale JW, Swanson KD, Sharfi H, Heffron GJ, Amador-Noguez D, Christofk HR, Wagner G, Rabinowitz JD, Asara JM and Cantley LC. Evidence for an alternative glycolytic pathway in rapidly proliferating cells. Science. 2010; 329(5998):1492-1499.
26. Folmes CD, Nelson TJ, Martinez-Fernandez A, Arrell DK, Lindor JZ, Dzeja PP, Ikeda Y, Perez-Terzic C and Terzic A. Somatic oxidative bioenergetics transitions into pluripotency-dependent glycolysis to facilitate nuclear reprogramming. Cell Metab. 2011; 14(2):264-271.
27. Panopoulos AD, Yanes O, Ruiz S, Kida YS, Diep D, Tautenhahn R, Herrerias A, Batchelder EM, Plongthongkum N, Lutz M, Berggren WT, Zhang K, Evans RM, Siuzdak G and Izpisua Belmonte JC. The metabolome of induced pluripotent stem cells reveals metabolic changes occurring in somatic cell reprogramming. Cell Res. 2012; 22(1):168-177.
28. Zhu S, Li W, Zhou H, Wei W, Ambasudhan R, Lin T, Kim J, Zhang K and Ding S. Reprogramming of human primary somatic cells by OCT4 and chemical compounds. Cell Stem Cell. 2010; 7(6):651-655.
29. Fu X, Zhu MJ, Dodson MV and Du M. AMP-activated Protein Kinase Stimulates Warburg-like Glycolysis and Activation of Satellite Cells during Muscle Regeneration. J Biol Chem. 2015; 290(44):26445-26456.
30. Moussaieff A, Rouleau M, Kitsberg D, Cohen M, Levy G, Barasch D, Nemirovski A, Shen-Orr S, Laevsky I, Amit M, Bomze D, Elena-Herrmann B, Scherf T, Nissim-Rafinia M, Kempa S, Itskovitz-Eldor J, et al. Glycolysis-mediated changes in acetyl-CoA and histone acetylation control the early differentiation of embryonic stem cells. Cell Metab. 2015; 21(3):392-402.
31. Du W, Amarachintha S, Wilson AF and Pang Q. SCO2 mediates oxidative stress-induced glycolysis to OXPHOS switch in hematopoietic stem cells. Stem Cells. 2015 Dec 16. doi: 10.1002/stem.2260. [Epub ahead of print]
32. Wang T, Marquardt C and Foker J. Aerobic glycolysis during lymphocyte proliferation. Nature. 1976; 261(5562):702-705.
33. Maciver NJ, Jacobs SR, Wieman HL, Wofford JA, Coloff JL and Rathmell JC. Glucose metabolism in lymphocytes is a regulated process with significant effects on immune cell function and survival. J Leukoc Biol. 2008; 84(4):949-957.
34. Stark H, Fichtner M, Konig R, Lorkowski S and Schuster S. Causes of upregulation of glycolysis in lymphocytes upon stimulation. A comparison with other cell types. Biochimie. 2015; 118:185-194.
35. Chang CH, Curtis JD, Maggi LB, Jr., Faubert B, Villarino AV, O’Sullivan D, Huang SC, van der Windt GJ, Blagih J, Qiu J, Weber JD, Pearce EJ, Jones RG and Pearce EL. Posttranscriptional control of T cell effector function by aerobic glycolysis. Cell. 2013; 153(6):1239-1251.
36. Pearce EL, Poffenberger MC, Chang CH and Jones RG. Fueling immunity: insights into metabolism and lymphocyte function. Science. 2013; 342(6155):1242454.
37. Ou J, Miao H, Ma Y, Guo F, Deng J, Wei X, Zhou J, Xie G, Shi H, Xue B, Liang H and Yu L. Loss of abhd5 promotes colorectal tumor development and progression by inducing aerobic glycolysis and epithelial-mesenchymal transition. Cell Rep. 2014; 9(5):1798-1811.
38. Ha TK and Chi SG. CAV1/caveolin 1 enhances aerobic glycolysis in colon cancer cells via activation of SLC2A3/GLUT3 transcription. Autophagy. 2012; 8(11):1684-1685.
39. Ha TK, Her NG, Lee MG, Ryu BK, Lee JH, Han J, Jeong SI, Kang MJ, Kim NH, Kim HJ and Chi SG. Caveolin-1 increases aerobic glycolysis in colorectal cancers by stimulating HMGA1-mediated GLUT3 transcription. Cancer Res. 2012; 72(16):4097-4109.
40. Tahir SA, Yang G, Goltsov A, Song KD, Ren C, Wang J, Chang W and Thompson TC. Caveolin-1-LRP6 signaling module stimulates aerobic glycolysis in prostate cancer. Cancer Res. 2013; 73(6):1900-1911.
41. Ke X, Fei F, Chen Y, Xu L, Zhang Z, Huang Q, Zhang H, Yang H, Chen Z and Xing J. Hypoxia upregulates CD147 through a combined effect of HIF-1alpha and Sp1 to promote glycolysis and tumor progression in epithelial solid tumors. Carcinogenesis. 2012; 33(8):1598-1607.
42. Huang Q, Li J, Xing J, Li W, Li H, Ke X, Zhang J, Ren T, Shang Y, Yang H, Jiang J and Chen Z. CD147 promotes reprogramming of glucose metabolism and cell proliferation in HCC cells by inhibiting the p53-dependent signaling pathway. J Hepatol. 2014; 61(4):859-866.
43. Huang P, Chang S, Jiang X, Su J, Dong C, Liu X, Yuan Z, Zhang Z and Liao H. RNA interference targeting CD147 inhibits the proliferation, invasiveness, and metastatic activity of thyroid carcinoma cells by down-regulating glycolysis. Int J Clin Exp Pathol. 2015; 8(1):309-318.
44. Dey P, Rachagani S, Chakraborty S, Singh PK, Zhao X, Gurumurthy CB, Anderson JM, Lele S, Hollingsworth MA, Band V and Batra SK. Overexpression of ecdysoneless in pancreatic cancer and its role in oncogenesis by regulating glycolysis. Clin Cancer Res. 2012; 18(22):6188-6198.
45. Zhang J, Gao Q, Zhou Y, Dier U, Hempel N and Hochwald SN. Focal adhesion kinase-promoted tumor glucose metabolism is associated with a shift of mitochondrial respiration to glycolysis. Oncogene. 2015 Jun 29. doi: 10.1038/onc.2015.256. [Epub ahead of print]
46. Zhang XY, Li M, Sun K, Chen XJ, Meng J, Wu L, Zhang P, Tong X and Jiang WW. Decreased expression of GRIM-19 by DNA hypermethylation promotes aerobic glycolysis and cell proliferation in head and neck squamous cell carcinoma. Oncotarget. 2015; 6(1):101-115. doi: 10.18632/oncotarget.2684.
47. Li Z, Wang Y, Newton IP, Zhang L and Ji P. GRP78 is implicated in the modulation of tumor aerobic glycolysis by promoting autophagic degradation of IKKbeta. Cell Signal. 2015; 27(6):1237-45.
48. Dang CV, Kim JW, Gao P and Yustein J. The interplay between MYC and HIF in cancer. Nat Rev Cancer. 2008; 8(1):51-56.
49. Denko NC. Hypoxia, HIF1 and glucose metabolism in the solid tumour. Nat Rev Cancer. 2008; 8(9):705-713.
50. Rempel A, Mathupala SP, Griffin CA, Hawkins AL and Pedersen PL. Glucose catabolism in cancer cells: amplification of the gene encoding type II hexokinase. Cancer Res. 1996; 56(11):2468-2471.
51. Kim JW, Tchernyshyov I, Semenza GL and Dang CV. HIF-1-mediated expression of pyruvate dehydrogenase kinase: a metabolic switch required for cellular adaptation to hypoxia. Cell Metab. 2006; 3(3):177-185.
52. Papandreou I, Cairns RA, Fontana L, Lim AL and Denko NC. HIF-1 mediates adaptation to hypoxia by actively downregulating mitochondrial oxygen consumption. Cell Metab. 2006; 3(3):187-197.
53. Huang L, Yu Z, Zhang T, Zhao X and Huang G. HSP40 interacts with pyruvate kinase M2 and regulates glycolysis and cell proliferation in tumor cells. PLoS One. 2014; 9(3):e92949.
54. Shi M, Cui J, Du J, Wei D, Jia Z, Zhang J, Zhu Z, Gao Y and Xie K. A novel KLF4/LDHA signaling pathway regulates aerobic glycolysis in and progression of pancreatic cancer. Clin Cancer Res. 2014; 20(16):4370-4380.
55. Yun J, Rago C, Cheong I, Pagliarini R, Angenendt P, Rajagopalan H, Schmidt K, Willson JK, Markowitz S, Zhou S, Diaz LA, Jr., Velculescu VE, Lengauer C, Kinzler KW, Vogelstein B and Papadopoulos N. Glucose deprivation contributes to the development of KRAS pathway mutations in tumor cells. Science. 2009; 325(5947):1555-1559.
56. Ma T, Patel H, Babapoor-Farrokhran S, Franklin R, Semenza GL, Sodhi A and Montaner S. KSHV induces aerobic glycolysis and angiogenesis through HIF-1-dependent upregulation of pyruvate kinase 2 in Kaposi’s sarcoma. Angiogenesis. 2015; 18(4):477-88.
57. Lo AK, Dawson CW, Young LS, Ko CW, Hau PM and Lo KW. Activation of the FGFR1 signalling pathway by the Epstein-Barr virus-encoded LMP1 promotes aerobic glycolysis and transformation of human nasopharyngeal epithelial cells. J Pathol. 2015 Jun 15. doi: 10.1002/path.4575. [Epub ahead of print]
58. Fang R, Xiao T, Fang Z, Sun Y, Li F, Gao Y, Feng Y, Li L, Wang Y, Liu X, Chen H, Liu XY and Ji H. MicroRNA-143 (miR-143) regulates cancer glycolysis via targeting hexokinase 2 gene. J Biol Chem. 2012; 287(27):23227-23235.
59. Zhao S, Liu H, Liu Y, Wu J, Wang C, Hou X, Chen X, Yang G, Zhao L, Che H, Bi Y, Wang H, Peng F and Ai J. miR-143 inhibits glycolysis and depletes stemness of glioblastoma stem-like cells. Cancer Lett. 2013; 333(2):253-260.
60. Guo W, Qiu Z, Wang Z, Wang Q, Tan N, Chen T, Chen Z, Huang S, Gu J, Li J, Yao M, Zhao Y and He X. MiR-199a-5p is negatively associated with malignancies and regulates glycolysis and lactate production by targeting hexokinase 2 in liver cancer. Hepatology. 2015; 62(4):1132-44.
61. Wang J, Wang H, Liu A, Fang C, Hao J and Wang Z. Lactate dehydrogenase A negatively regulated by miRNAs promotes aerobic glycolysis and is increased in colorectal cancer. Oncotarget. 2015; 6(23):19456-19468. doi: 10.18632/oncotarget.3318.
62. Du JY, Wang LF, Wang Q and Yu LD. miR-26b inhibits proliferation, migration, invasion and apoptosis induction via the downregulation of 6-phosphofructo-2-kinase/fructose-2,6-bisphosphatase-3 driven glycolysis in osteosarcoma cells. Oncol Rep. 2015; 33(4):1890-1898.
63. Tang H, Lee M, Sharpe O, Salamone L, Noonan EJ, Hoang CD, Levine S, Robinson WH and Shrager JB. Oxidative stress-responsive microRNA-320 regulates glycolysis in diverse biological systems. FASEB J. 2012; 26(11):4710-4721.
64. Fan JY, Yang Y, Xie JY, Lu YL, Shi K and Huang YQ. MicroRNA-144 mediates metabolic shift in ovarian cancer cells by directly targeting Glut1. Tumour Biol. 2015 Dec 11. [Epub ahead of print]
65. Chen B, Tang H, Liu X, Liu P, Yang L, Xie X, Ye F, Song C and Wei W. miR-22 as a prognostic factor targets glucose transporter protein type 1 in breast cancer. Cancer Lett. 2015; 356(2 Pt B):410-417.
66. Yamasaki T, Seki N, Yoshino H, Itesako T, Yamada Y, Tatarano S, Hidaka H, Yonezawa T, Nakagawa M and Enokida H. Tumor-suppressive microRNA-1291 directly regulates glucose transporter 1 in renal cell carcinoma. Cancer Sci. 2013; 104(11):1411-1419.
67. Fei X, Qi M, Wu B, Song Y, Wang Y and Li T. MicroRNA-195-5p suppresses glucose uptake and proliferation of human bladder cancer T24 cells by regulating GLUT3 expression. FEBS Lett. 2012; 586(4):392-397.
68. Peschiaroli A, Giacobbe A, Formosa A, Markert EK, Bongiorno-Borbone L, Levine AJ, Candi E, D’Alessandro A, Zolla L, Finazzi Agro A and Melino G. miR-143 regulates hexokinase 2 expression in cancer cells. Oncogene. 2012; 32(6):797-802.
69. Gregersen LH, Jacobsen A, Frankel LB, Wen J, Krogh A and Lund AH. MicroRNA-143 down-regulates Hexokinase 2 in colon cancer cells. BMC Cancer. 2012; 12:232.
70. Kawauchi K, Araki K, Tobiume K and Tanaka N. p53 regulates glucose metabolism through an IKK-NF-kappaB pathway and inhibits cell transformation. Nat Cell Biol. 2008; 10(5):611-618.
71. Kondoh H, Lleonart ME, Gil J, Wang J, Degan P, Peters G, Martinez D, Carnero A and Beach D. Glycolytic enzymes can modulate cellular life span. Cancer Res. 2005; 65(1):177-185.
72. Madan E, Gogna R, Bhatt M, Pati U, Kuppusamy P and Mahdi AA. Regulation of glucose metabolism by p53: emerging new roles for the tumor suppressor. Oncotarget. 2011; 2(12):948-957. doi: 10.18632/oncotarget.389.
73. Bensaad K, Tsuruta A, Selak MA, Vidal MN, Nakano K, Bartrons R, Gottlieb E and Vousden KH. TIGAR, a p53-inducible regulator of glycolysis and apoptosis. Cell. 2006; 126(1):107-120.
74. Jiang P, Du W, Wang X, Mancuso A, Gao X, Wu M and Yang X. p53 regulates biosynthesis through direct inactivation of glucose-6-phosphate dehydrogenase. Nat Cell Biol. 2011; 13(3):310-316.
75. Feng Z, Hu W, de Stanchina E, Teresky AK, Jin S, Lowe S and Levine AJ. The regulation of AMPK beta1, TSC2, and PTEN expression by p53: stress, cell and tissue specificity, and the role of these gene products in modulating the IGF-1-AKT-mTOR pathways. Cancer Res. 2007; 67(7):3043-3053.
76. Budanov AV and Karin M. p53 target genes sestrin1 and sestrin2 connect genotoxic stress and mTOR signaling. Cell. 2008; 134(3):451-460.
77. Zhang C, Liu J, Wu R, Liang Y, Lin M, Chan CS, Hu W and Feng Z. Tumor suppressor p53 negatively regulates glycolysis stimulated by hypoxia through its target RRAD. Oncotarget. 2014; 5(14):5535-5546. doi: 10.18632/oncotarget.2137.
78. Amoroso F, Falzoni S, Adinolfi E, Ferrari D and Di Virgilio F. The P2X7 receptor is a key modulator of aerobic glycolysis. Cell Death Dis. 2012; 3:e370.
79. Csibi A and Blenis J. Appetite for destruction: the inhibition of glycolysis as a therapy for tuberous sclerosis complex-related tumors. BMC Biol. 2011; 9:69.
80. Jiang X, Kenerson H, Aicher L, Miyaoka R, Eary J, Bissler J and Yeung RS. The tuberous sclerosis complex regulates trafficking of glucose transporters and glucose uptake. Am J Pathol. 2008; 172(6):1748-1756.
81. Duvel K, Yecies JL, Menon S, Raman P, Lipovsky AI, Souza AL, Triantafellow E, Ma Q, Gorski R, Cleaver S, Vander Heiden MG, MacKeigan JP, Finan PM, Clish CB, Murphy LO and Manning BD. Activation of a metabolic gene regulatory network downstream of mTOR complex 1. Mol Cell. 2010; 39(2):171-183.
82. Sun Q, Chen X, Ma J, Peng H, Wang F, Zha X, Wang Y, Jing Y, Yang H, Chen R, Chang L, Zhang Y, Goto J, Onda H, Chen T, Wang MR, et al. Mammalian target of rapamycin up-regulation of pyruvate kinase isoenzyme type M2 is critical for aerobic glycolysis and tumor growth. Proc Natl Acad Sci U S A. 2011; 108(10):4129-4134.
83. Zha X, Hu Z, Ji S, Jin F, Jiang K, Li C, Zhao P, Tu Z, Chen X, Di L, Zhou H and Zhang H. NFkappaB up-regulation of glucose transporter 3 is essential for hyperactive mammalian target of rapamycin-induced aerobic glycolysis and tumor growth. Cancer Lett. 2015; 359(1):97-106.
84. Chan CH, Li CF, Yang WL, Gao Y, Lee SW, Feng Z, Huang HY, Tsai KK, Flores LG, Shao Y, Hazle JD, Yu D, Wei W, Sarbassov D, Hung MC, Nakayama KI, et al. The Skp2-SCF E3 ligase regulates Akt ubiquitination, glycolysis, herceptin sensitivity, and tumorigenesis. Cell. 2012; 149(5):1098-1111.
85. Hagenbuchner J, Kuznetsov AV, Obexer P and Ausserlechner MJ. BIRC5/Survivin enhances aerobic glycolysis and drug resistance by altered regulation of the mitochondrial fusion/fission machinery. Oncogene. 2013; 32(40):4748-4757.
86. Yoshida S, Tsutsumi S, Muhlebach G, Sourbier C, Lee MJ, Lee S, Vartholomaiou E, Tatokoro M, Beebe K, Miyajima N, Mohney RP, Chen Y, Hasumi H, Xu W, Fukushima H, Nakamura K, et al. Molecular chaperone TRAP1 regulates a metabolic switch between mitochondrial respiration and aerobic glycolysis. Proc Natl Acad Sci U S A. 2013; 110(17):E1604-1612.
87. Pate KT, Stringari C, Sprowl-Tanio S, Wang K, TeSlaa T, Hoverter NP, McQuade MM, Garner C, Digman MA, Teitell MA, Edwards RA, Gratton E and Waterman ML. Wnt signaling directs a metabolic program of glycolysis and angiogenesis in colon cancer. EMBO J. 2014; 33(13):1454-1473.
88. Liu XS, Haines JE, Mehanna EK, Genet MD, Ben-Sahra I, Asara JM, Manning BD and Yuan ZM. ZBTB7A acts as a tumor suppressor through the transcriptional repression of glycolysis. Genes Dev. 2014; 28(17):1917-1928.
89. Elstrom RL, Bauer DE, Buzzai M, Karnauskas R, Harris MH, Plas DR, Zhuang H, Cinalli RM, Alavi A, Rudin CM and Thompson CB. Akt stimulates aerobic glycolysis in cancer cells. Cancer Res. 2004; 64(11):3892-3899.
90. Zhuo B, Li Y, Li Z, Qin H, Sun Q, Zhang F, Shen Y, Shi Y and Wang R. PI3K/Akt signaling mediated Hexokinase-2 expression inhibits cell apoptosis and promotes tumor growth in pediatric osteosarcoma. Biochem Biophys Res Commun. 2015; 464(2):401-406.
91. Li W, Peng C, Lee MH, Lim D, Zhu F, Fu Y, Yang G, Sheng Y, Xiao L, Dong X, Ma W, Bode AM, Cao Y and Dong Z. TRAF4 is a critical molecule for Akt activation in lung cancer. Cancer Res. 2013; 73(23):6938-6950.
92. Deprez J, Vertommen D, Alessi DR, Hue L and Rider MH. Phosphorylation and activation of heart 6-phosphofructo-2-kinase by protein kinase B and other protein kinases of the insulin signaling cascades. J Biol Chem. 1997; 272(28):17269-17275.
93. Barthel A, Okino ST, Liao J, Nakatani K, Li J, Whitlock JP, Jr. and Roth RA. Regulation of GLUT1 gene transcription by the serine/threonine kinase Akt1. J Biol Chem. 1999; 274(29):20281-20286.
94. Robey RB and Hay N. Is Akt the “Warburg kinase”?-Akt-energy metabolism interactions and oncogenesis. Semin Cancer Biol. 2009; 19(1):25-31.
95. Shimura T, Noma N, Sano Y, Ochiai Y, Oikawa T, Fukumoto M and Kunugita N. AKT-mediated enhanced aerobic glycolysis causes acquired radioresistance by human tumor cells. Radiother Oncol. 2014; 112(2):302-307.
96. Inoki K, Li Y, Zhu T, Wu J and Guan KL. TSC2 is phosphorylated and inhibited by Akt and suppresses mTOR signalling. Nat Cell Biol. 2002; 4(9):648-657.
97. Inoki K, Corradetti MN and Guan KL. Dysregulation of the TSC-mTOR pathway in human disease. Nat Genet. 2005; 37(1):19-24.
98. Ran C, Liu H, Hitoshi Y and Israel MA. Proliferation-independent control of tumor glycolysis by PDGFR-mediated AKT activation. Cancer Res. 2013; 73(6):1831-1843.
99. Makinoshima H, Takita M, Saruwatari K, Umemura S, Obata Y, Ishii G, Matsumoto S, Sugiyama E, Ochiai A, Abe R, Goto K, Esumi H and Tsuchihara K. Signaling through the Phosphatidylinositol 3-Kinase (PI3K)/Mammalian Target of Rapamycin (mTOR) Axis Is Responsible for Aerobic Glycolysis mediated by Glucose Transporter in Epidermal Growth Factor Receptor (EGFR)-mutated Lung Adenocarcinoma. J Biol Chem. 2015; 290(28):17495-17504.
100. Landis J and Shaw LM. Insulin receptor substrate 2-mediated phosphatidylinositol 3-kinase signaling selectively inhibits glycogen synthase kinase 3beta to regulate aerobic glycolysis. J Biol Chem. 2014; 289(26):18603-18613.
101. Wang L, Xiong H, Wu F, Zhang Y, Wang J, Zhao L, Guo X, Chang LJ, You MJ, Koochekpour S, Saleem M, Huang H, Lu J and Deng Y. Hexokinase 2-mediated Warburg effect is required for PTEN- and p53-deficiency-driven prostate cancer growth. Cell Rep. 2014; 8(5):1461-1474.
102. Garcia-Cao I, Song MS, Hobbs RM, Laurent G, Giorgi C, de Boer VC, Anastasiou D, Ito K, Sasaki AT, Rameh L, Carracedo A, Vander Heiden MG, Cantley LC, Pinton P, Haigis MC and Pandolfi PP. Systemic elevation of PTEN induces a tumor-suppressive metabolic state. Cell. 2012; 149(1):49-62.
103. Liu G, Bi Y, Shen B, Yang H, Zhang Y, Wang X, Liu H, Lu Y, Liao J, Chen X and Chu Y. SIRT1 limits the function and fate of myeloid-derived suppressor cells in tumors by orchestrating HIF-1alpha-dependent glycolysis. Cancer Res. 2014; 74(3):727-737.
104. Shukla SK, Gunda V, Abrego J, Haridas D, Mishra A, Souchek J, Chaika NV, Yu F, Sasson AR, Lazenby AJ, Batra SK and Singh PK. MUC16-mediated activation of mTOR and c-Myc reprograms pancreatic cancer metabolism. Oncotarget. 2015; 6(22):19118-19131. doi: 10.18632/oncotarget.4078.
105. Ying H, Kimmelman AC, Lyssiotis CA, Hua S, Chu GC, Fletcher-Sananikone E, Locasale JW, Son J, Zhang H, Coloff JL, Yan H, Wang W, Chen S, Viale A, Zheng H, Paik JH, et al. Oncogenic Kras maintains pancreatic tumors through regulation of anabolic glucose metabolism. Cell. 2012; 149(3):656-670.
106. Liang Y, Liu J and Feng Z. The regulation of cellular metabolism by tumor suppressor p53. Cell Biosci. 2013; 3(1):9.
107. Matoba S, Kang JG, Patino WD, Wragg A, Boehm M, Gavrilova O, Hurley PJ, Bunz F and Hwang PM. p53 regulates mitochondrial respiration. Science. 2006; 312(5780):1650-1653.
108. Schwartzenberg-Bar-Yoseph F, Armoni M and Karnieli E. The tumor suppressor p53 down-regulates glucose transporters GLUT1 and GLUT4 gene expression. Cancer Res. 2004; 64(7):2627-2633.
109. Zhang C, Liu J, Liang Y, Wu R, Zhao Y, Hong X, Lin M, Yu H, Liu L, Levine AJ, Hu W and Feng Z. Tumour-associated mutant p53 drives the Warburg effect. Nat Commun. 2013; 4:2935.
110. Oakhill JS, Steel R, Chen ZP, Scott JW, Ling N, Tam S and Kemp BE. AMPK is a direct adenylate charge-regulated protein kinase. Science. 2011; 332(6036):1433-1435.
111. Hardie DG, Ross FA and Hawley SA. AMPK: a nutrient and energy sensor that maintains energy homeostasis. Nat Rev Mol Cell Biol. 2012; 13(4):251-262.
112. Hardie DG. Molecular Pathways: Is AMPK a Friend or a Foe in Cancer? Clin Cancer Res. 2015; 21(17):3836-3840.
113. Inoki K, Zhu T and Guan KL. TSC2 mediates cellular energy response to control cell growth and survival. Cell. 2003; 115(5):577-590.
114. Gwinn DM, Shackelford DB, Egan DF, Mihaylova MM, Mery A, Vasquez DS, Turk BE and Shaw RJ. AMPK phosphorylation of raptor mediates a metabolic checkpoint. Mol Cell. 2008; 30(2):214-226.
115. Green AS, Chapuis N, Maciel TT, Willems L, Lambert M, Arnoult C, Boyer O, Bardet V, Park S, Foretz M, Viollet B, Ifrah N, Dreyfus F, Hermine O, Moura IC, Lacombe C, et al. The LKB1/AMPK signaling pathway has tumor suppressor activity in acute myeloid leukemia through the repression of mTOR-dependent oncogenic mRNA translation. Blood. 2010; 116(20):4262-4273.
116. Hoppe S, Bierhoff H, Cado I, Weber A, Tiebe M, Grummt I and Voit R. AMP-activated protein kinase adapts rRNA synthesis to cellular energy supply. Proc Natl Acad Sci U S A. 2009; 106(42):17781-17786.
117. Dandapani M and Hardie DG. AMPK: opposing the metabolic changes in both tumour cells and inflammatory cells? Biochem Soc Trans. 2013; 41(2):687-693.
118. Grahame Hardie D. AMP-activated protein kinase: a key regulator of energy balance with many roles in human disease. J Intern Med. 2014; 276(6):543-559.
119. Sui X, Xu Y, Yang J, Fang Y, Lou H, Han W, Zhang M, Chen W, Wang K, Li D, Jin W, Lou F, Zheng Y, Hu H, Gong L, Zhou X, et al. Use of metformin alone is not associated with survival outcomes of colorectal cancer cell but AMPK activator AICAR sensitizes anticancer effect of 5-fluorouracil through AMPK activation. PLoS One. 2014; 9(5):e97781.
120. Shafaee A, Dastyar DZ, Islamian JP and Hatamian M. Inhibition of tumor energy pathways for targeted esophagus cancer therapy. Metabolism. 2015; 64(10):1193-1198.
121. Valera A, Pujol A, Gregori X, Riu E, Visa J and Bosch F. Evidence from transgenic mice that myc regulates hepatic glycolysis. FASEB J. 1995; 9(11):1067-1078.
122. Osthus RC, Shim H, Kim S, Li Q, Reddy R, Mukherjee M, Xu Y, Wonsey D, Lee LA and Dang CV. Deregulation of glucose transporter 1 and glycolytic gene expression by c-Myc. J Biol Chem. 2000; 275(29):21797-21800.
123. Gan L, Xiu R, Ren P, Yue M, Su H, Guo G, Xiao D, Yu J, Jiang H, Liu H, Hu G and Qing G. Metabolic targeting of oncogene MYC by selective activation of the proton-coupled monocarboxylate family of transporters. Oncogene. 2015 Oct 5. doi: 10.1038/onc.2015.360. [Epub ahead of print]
124. David CJ, Chen M, Assanah M, Canoll P and Manley JL. HnRNP proteins controlled by c-Myc deregulate pyruvate kinase mRNA splicing in cancer. Nature. 2010; 463(7279):364-368.
125. Luan W, Wang Y, Chen X, Shi Y, Wang J, Zhang J, Qian J, Li R, Tao T, Wei W, Hu Q, Liu N and You Y. PKM2 promotes glucose metabolism and cell growth in gliomas through a mechanism involving a let-7a/c-Myc/hnRNPA1 feedback loop. Oncotarget. 2015; 6(15):13006-13018. doi: 10.18632/oncotarget.3514.
126. Shim H, Dolde C, Lewis BC, Wu CS, Dang G, Jungmann RA, Dalla-Favera R and Dang CV. c-Myc transactivation of LDH-A: implications for tumor metabolism and growth. Proc Natl Acad Sci U S A. 1997; 94(13):6658-6663.
127. He TL, Zhang YJ, Jiang H, Li XH, Zhu H and Zheng KL. The c-Myc-LDHA axis positively regulates aerobic glycolysis and promotes tumor progression in pancreatic cancer. Med Oncol. 2015; 32(7):187.
128. Wang X, Duan W, Li X, Liu J, Li D, Ye L, Qian L, Yang A, Xu Q, Liu H, Fu Q, Wu E, Ma Q and Shen X. PTTG regulates the metabolic switch of ovarian cancer cells via the c-myc pathway. Oncotarget. 2015; 6(38):40959-40969. doi: 10.18632/oncotarget.5726.
129. Xu X, Li J, Sun X, Guo Y, Chu D, Wei L, Li X, Yang G, Liu X, Yao L, Zhang J and Shen L. Tumor suppressor NDRG2 inhibits glycolysis and glutaminolysis in colorectal cancer cells by repressing c-Myc expression. Oncotarget. 2015; 6(28):26161-26176. doi: 10.18632/oncotarget.4544.
130. Chen B, Li H, Zeng X, Yang P, Liu X, Zhao X and Liang S. Roles of microRNA on cancer cell metabolism. J Transl Med. 2012; 10:228.
131. Jiang S, Zhang LF, Zhang HW, Hu S, Lu MH, Liang S, Li B, Li Y, Li D, Wang ED and Liu MF. A novel miR-155/miR-143 cascade controls glycolysis by regulating hexokinase 2 in breast cancer cells. EMBO J. 2012; 31(8):1985-1998.
132. Teng Y, Zhang Y, Qu K, Yang X, Fu J, Chen W and Li X. MicroRNA-29B (mir-29b) regulates the Warburg effect in ovarian cancer by targeting AKT2 and AKT3. Oncotarget. 2015; 6(38):40799-40814. doi: 10.18632/oncotarget.5695.
133. Ge X, Lyu P, Cao Z, Li J, Guo G, Xia W and Gu Y. Overexpression of miR-206 suppresses glycolysis, proliferation and migration in breast cancer cells via PFKFB3 targeting. Biochem Biophys Res Commun. 2015; 463(4):1115-1121.
134. Yang X, Cheng Y, Li P, Tao J, Deng X, Zhang X, Gu M, Lu Q and Yin C. A lentiviral sponge for miRNA-21 diminishes aerobic glycolysis in bladder cancer T24 cells via the PTEN/PI3K/AKT/mTOR axis. Tumour Biol. 2015; 36(1):383-391.
135. Mondal S, Roy D, Camacho-Pereira J, Khurana A, Chini E, Yang L, Baddour J, Stilles K, Padmabandu S, Leung S, Kalloger S, Gilks B, Lowe V, Dierks T, Hammond E, Dredge K, et al. HSulf-1 deficiency dictates a metabolic reprograming of glycolysis and TCA cycle in ovarian cancer. Oncotarget. 2015; 6(32):33705-19. doi: 10.18632/oncotarget.5605.
136. Hagenbuchner J, Kiechl-Kohlendorfer U, Obexer P and Ausserlechner MJ. BIRC5/Survivin as a target for glycolysis inhibition in high-stage neuroblastoma. Oncogene. 2015 Jul 6. doi: 10.1038/onc.2015.264. [Epub ahead of print]
137. Shiraishi T, Verdone JE, Huang J, Kahlert UD, Hernandez JR, Torga G, Zarif JC, Epstein T, Gatenby R, McCartney A, Elisseeff JH, Mooney SM, An SS and Pienta KJ. Glycolysis is the primary bioenergetic pathway for cell motility and cytoskeletal remodeling in human prostate and breast cancer cells. Oncotarget. 2015; 6(1):130-143. doi: 10.18632/oncotarget.2766.
138. Guillaumond F, Leca J, Olivares O, Lavaut MN, Vidal N, Berthezene P, Dusetti NJ, Loncle C, Calvo E, Turrini O, Iovanna JL, Tomasini R and Vasseur S. Strengthened glycolysis under hypoxia supports tumor symbiosis and hexosamine biosynthesis in pancreatic adenocarcinoma. Proc Natl Acad Sci U S A. 2013; 110(10):3919-3924.
139. Christofk HR, Vander Heiden MG, Harris MH, Ramanathan A, Gerszten RE, Wei R, Fleming MD, Schreiber SL and Cantley LC. The M2 splice isoform of pyruvate kinase is important for cancer metabolism and tumour growth. Nature. 2008; 452(7184):230-233.
140. Fantin VR, St-Pierre J and Leder P. Attenuation of LDH-A expression uncovers a link between glycolysis, mitochondrial physiology, and tumor maintenance. Cancer Cell. 2006; 9(6):425-434.
141. Flaveny CA, Griffett K, El-Gendy Bel D, Kazantzis M, Sengupta M, Amelio AL, Chatterjee A, Walker J, Solt LA, Kamenecka TM and Burris TP. Broad Anti-tumor Activity of a Small Molecule that Selectively Targets the Warburg Effect and Lipogenesis. Cancer Cell. 2015; 28(1):42-56.
142. Rajeshkumar NV, Dutta P, Yabuuchi S, de Wilde RF, Martinez GV, Le A, Kamphorst JJ, Rabinowitz JD, Jain SK, Hidalgo M, Dang CV, Gillies RJ and Maitra A. Therapeutic Targeting of the Warburg Effect in Pancreatic Cancer Relies on an Absence of p53 Function. Cancer Res. 2015; 75(16):3355-3364.
143. Wood TE, Dalili S, Simpson CD, Hurren R, Mao X, Saiz FS, Gronda M, Eberhard Y, Minden MD, Bilan PJ, Klip A, Batey RA and Schimmer AD. A novel inhibitor of glucose uptake sensitizes cells to FAS-induced cell death. Mol Cancer Ther. 2008; 7(11):3546-3555.
144. Wu CH, Ho YS, Tsai CY, Wang YJ, Tseng H, Wei PL, Lee CH, Liu RS and Lin SY. In vitro and in vivo study of phloretin-induced apoptosis in human liver cancer cells involving inhibition of type II glucose transporter. Int J Cancer. 2009; 124(9):2210-2219.
145. Liu Y, Cao Y, Zhang W, Bergmeier S, Qian Y, Akbar H, Colvin R, Ding J, Tong L, Wu S, Hines J and Chen X. A small-molecule inhibitor of glucose transporter 1 downregulates glycolysis, induces cell-cycle arrest, and inhibits cancer cell growth in vitro and in vivo. Mol Cancer Ther. 2012; 11(8):1672-1682.
146. Dwarakanath B and Jain V. Targeting glucose metabolism with 2-deoxy-D-glucose for improving cancer therapy. Future Oncol. 2009; 5(5):581-585.
147. Maschek G, Savaraj N, Priebe W, Braunschweiger P, Hamilton K, Tidmarsh GF, De Young LR and Lampidis TJ. 2-deoxy-D-glucose increases the efficacy of adriamycin and paclitaxel in human osteosarcoma and non-small cell lung cancers in vivo. Cancer Res. 2004; 64(1):31-34.
148. Xian SL, Cao W, Zhang XD and Lu YF. 3-Bromopyruvate inhibits human gastric cancer tumor growth in nude mice via the inhibition of glycolysis. Oncol Lett. 2015; 9(2):739-744.
149. Jae HJ, Chung JW, Park HS, Lee MJ, Lee KC, Kim HC, Yoon JH, Chung H and Park JH. The antitumor effect and hepatotoxicity of a hexokinase II inhibitor 3-bromopyruvate: in vivo investigation of intraarterial administration in a rabbit VX2 hepatoma model. Korean J Radiol. 2009; 10(6):596-603.
150. Talekar M, Boreddy SR, Singh A and Amiji M. Tumor aerobic glycolysis: new insights into therapeutic strategies with targeted delivery. Expert Opin Biol Ther. 2014; 14(8):1145-1159.
151. Zhou Y, Lu N, Qiao C, Ni T, Li Z, Yu B, Guo Q and Wei L. FV-429 induces apoptosis and inhibits glycolysis by inhibiting Akt-mediated phosphorylation of hexokinase II in MDA-MB-231 cells. Mol Carcinog. 2015 Aug 10. doi: 10.1002/mc.22374. [Epub ahead of print]
152. Furtado CM, Marcondes MC, Sola-Penna M, de Souza ML and Zancan P. Clotrimazole preferentially inhibits human breast cancer cell proliferation, viability and glycolysis. PLoS One. 2012; 7(2):e30462.
153. Clem B, Telang S, Clem A, Yalcin A, Meier J, Simmons A, Rasku MA, Arumugam S, Dean WL, Eaton J, Lane A, Trent JO and Chesney J. Small-molecule inhibition of 6-phosphofructo-2-kinase activity suppresses glycolytic flux and tumor growth. Mol Cancer Ther. 2008; 7(1):110-120.
154. Vuyyuri SB, Rinkinen J, Worden E, Shim H, Lee S and Davis KR. Ascorbic acid and a cytostatic inhibitor of glycolysis synergistically induce apoptosis in non-small cell lung cancer cells. PLoS One. 2013; 8(6):e67081.
155. Ganapathy-Kanniappan S, Kunjithapatham R and Geschwind JF. Glyceraldehyde-3-phosphate dehydrogenase: a promising target for molecular therapy in hepatocellular carcinoma. Oncotarget. 2012; 3(9):940-953. doi: 10.18632/oncotarget.623.
156. Ganapathy-Kanniappan S, Kunjithapatham R and Geschwind JF. Anticancer efficacy of the metabolic blocker 3-bromopyruvate: specific molecular targeting. Anticancer Res. 2013; 33(1):13-20.
157. Ganapathy-Kanniappan S, Vali M, Kunjithapatham R, Buijs M, Syed LH, Rao PP, Ota S, Kwak BK, Loffroy R and Geschwind JF. 3-bromopyruvate: a new targeted antiglycolytic agent and a promise for cancer therapy. Curr Pharm Biotechnol. 2010; 11(5):510-517.
158. Vander Heiden MG, Christofk HR, Schuman E, Subtelny AO, Sharfi H, Harlow EE, Xian J and Cantley LC. Identification of small molecule inhibitors of pyruvate kinase M2. Biochem Pharmacol. 2010; 79(8):1118-1124.
159. Goldberg MS and Sharp PA. Pyruvate kinase M2-specific siRNA induces apoptosis and tumor regression. J Exp Med. 2012; 209(2):217-224.
160. Chen J, Xie J, Jiang Z, Wang B, Wang Y and Hu X. Shikonin and its analogs inhibit cancer cell glycolysis by targeting tumor pyruvate kinase-M2. Oncogene. 2011; 30(42):4297-4306.
161. Shi HS, Li D, Zhang J, Wang YS, Yang L, Zhang HL, Wang XH, Mu B, Wang W, Ma Y, Guo FC and Wei YQ. Silencing of pkm2 increases the efficacy of docetaxel in human lung cancer xenografts in mice. Cancer Sci. 2010; 101(6):1447-1453.
162. Le A, Cooper CR, Gouw AM, Dinavahi R, Maitra A, Deck LM, Royer RE, Vander Jagt DL, Semenza GL and Dang CV. Inhibition of lactate dehydrogenase A induces oxidative stress and inhibits tumor progression. Proc Natl Acad Sci U S A. 2010; 107(5):2037-2042.
163. Zhou M, Zhao Y, Ding Y, Liu H, Liu Z, Fodstad O, Riker AI, Kamarajugadda S, Lu J, Owen LB, Ledoux SP and Tan M. Warburg effect in chemosensitivity: targeting lactate dehydrogenase-A re-sensitizes taxol-resistant cancer cells to taxol. Mol Cancer. 2010; 9:33.
164. Granchi C, Roy S, Giacomelli C, Macchia M, Tuccinardi T, Martinelli A, Lanza M, Betti L, Giannaccini G, Lucacchini A, Funel N, Leon LG, Giovannetti E, Peters GJ, Palchaudhuri R, Calvaresi EC, et al. Discovery of N-hydroxyindole-based inhibitors of human lactate dehydrogenase isoform A (LDH-A) as starvation agents against cancer cells. J Med Chem. 2011; 54(6):1599-1612.
165. Colen CB, Shen Y, Ghoddoussi F, Yu P, Francis TB, Koch BJ, Monterey MD, Galloway MP, Sloan AE and Mathupala SP. Metabolic targeting of lactate efflux by malignant glioma inhibits invasiveness and induces necrosis: an in vivo study. Neoplasia. 2011; 13(7):620-632.
166. Dunbar EM, Coats BS, Shroads AL, Langaee T, Lew A, Forder JR, Shuster JJ, Wagner DA and Stacpoole PW. Phase 1 trial of dichloroacetate (DCA) in adults with recurrent malignant brain tumors. Invest New Drugs. 2014; 32(3):452-464.
167. Lin G, Hill DK, Andrejeva G, Boult JK, Troy H, Fong AC, Orton MR, Panek R, Parkes HG, Jafar M, Koh DM, Robinson SP, Judson IR, Griffiths JR, Leach MO, Eykyn TR, et al. Dichloroacetate induces autophagy in colorectal cancer cells and tumours. Br J Cancer. 2014; 111(2):375-385.
168. Chan DA, Sutphin PD, Nguyen P, Turcotte S, Lai EW, Banh A, Reynolds GE, Chi JT, Wu J, Solow-Cordero DE, Bonnet M, Flanagan JU, Bouley DM, Graves EE, Denny WA, Hay MP, et al. Targeting GLUT1 and the Warburg effect in renal cell carcinoma by chemical synthetic lethality. Sci Transl Med. 2011; 3(94):94ra70.
169. Jiang X, Xin H, Ren Q, Gu J, Zhu L, Du F, Feng C, Xie Y, Sha X and Fang X. Nanoparticles of 2-deoxy-D-glucose functionalized poly(ethylene glycol)-co-poly(trimethylene carbonate) for dual-targeted drug delivery in glioma treatment. Biomaterials. 2014; 35(1):518-529.
170. Wilson JE. Isozymes of mammalian hexokinase: structure, subcellular localization and metabolic function. J Exp Biol. 2003; 206(Pt 12):2049-2057.
171. Wolf A, Agnihotri S, Micallef J, Mukherjee J, Sabha N, Cairns R, Hawkins C and Guha A. Hexokinase 2 is a key mediator of aerobic glycolysis and promotes tumor growth in human glioblastoma multiforme. J Exp Med. 2011; 208(2):313-326.
172. Ros S and Schulze A. Glycolysis back in the limelight: systemic targeting of HK2 blocks tumor growth. Cancer Discov. 2013; 3(10):1105-1107.
173. Cervantes-Madrid D, Romero Y and Duenas-Gonzalez A. Reviving Lonidamine and 6-Diazo-5-oxo-L-norleucine to Be Used in Combination for Metabolic Cancer Therapy. Biomed Res Int. 2015; 2015:690492.
174. Spoden GA, Mazurek S, Morandell D, Bacher N, Ausserlechner MJ, Jansen-Durr P, Eigenbrodt E and Zwerschke W. Isotype-specific inhibitors of the glycolytic key regulator pyruvate kinase subtype M2 moderately decelerate tumor cell proliferation. Int J Cancer. 2008; 123(2):312-321.
175. Calabretta S, Bielli P, Passacantilli I, Pilozzi E, Fendrich V, Capurso G, Fave GD and Sette C. Modulation of PKM alternative splicing by PTBP1 promotes gemcitabine resistance in pancreatic cancer cells. Oncogene. 2015 Aug 3. doi: 10.1038/onc.2015.270. [Epub ahead of print]
176. Anastasiou D, Poulogiannis G, Asara JM, Boxer MB, Jiang JK, Shen M, Bellinger G, Sasaki AT, Locasale JW, Auld DS, Thomas CJ, Vander Heiden MG and Cantley LC. Inhibition of pyruvate kinase M2 by reactive oxygen species contributes to cellular antioxidant responses. Science. 2011; 334(6060):1278-1283.
177. Bluemlein K, Gruning NM, Feichtinger RG, Lehrach H, Kofler B and Ralser M. No evidence for a shift in pyruvate kinase PKM1 to PKM2 expression during tumorigenesis. Oncotarget. 2011; 2(5):393-400. doi: 10.18632/oncotarget.278.
178. Desai S, Ding M, Wang B, Lu Z, Zhao Q, Shaw K, Yung WK, Weinstein JN, Tan M and Yao J. Tissue-specific isoform switch and DNA hypomethylation of the pyruvate kinase PKM gene in human cancers. Oncotarget. 2014; 5(18):8202-8210. doi: 10.18632/oncotarget.1159
179. Zhan C, Yan L, Wang L, Ma J, Jiang W, Zhang Y, Shi Y and Wang Q. Isoform switch of pyruvate kinase M1 indeed occurs but not to pyruvate kinase M2 in human tumorigenesis. PLoS One. 2015; 10(3):e0118663.
180. Cortes-Cros M, Hemmerlin C, Ferretti S, Zhang J, Gounarides JS, Yin H, Muller A, Haberkorn A, Chene P, Sellers WR and Hofmann F. M2 isoform of pyruvate kinase is dispensable for tumor maintenance and growth. Proc Natl Acad Sci U S A. 2013; 110(2):489-494.
181. Doherty JR, Yang C, Scott KE, Cameron MD, Fallahi M, Li W, Hall MA, Amelio AL, Mishra JK, Li F, Tortosa M, Genau HM, Rounbehler RJ, Lu Y, Dang CV, Kumar KG, et al. Blocking lactate export by inhibiting the Myc target MCT1 Disables glycolysis and glutathione synthesis. Cancer Res. 2014; 74(3):908-920.
182. Leiblich A, Cross SS, Catto JW, Phillips JT, Leung HY, Hamdy FC and Rehman I. Lactate dehydrogenase-B is silenced by promoter hypermethylation in human prostate cancer. Oncogene. 2006; 25(20):2953-2960.
183. Cui J, Quan M, Jiang W, Hu H, Jiao F, Li N, Jin Z, Wang L and Wang Y. Suppressed expression of LDHB promotes pancreatic cancer progression via inducing glycolytic phenotype. Med Oncol. 2015; 32(5):143.
184. Kim JH, Kim EL, Lee YK, Park CB, Kim BW, Wang HJ, Yoon CH, Lee SJ and Yoon G. Decreased lactate dehydrogenase B expression enhances claudin 1-mediated hepatoma cell invasiveness via mitochondrial defects. Exp Cell Res. 2011; 317(8):1108-1118.
185. McCleland ML, Adler AS, Deming L, Cosino E, Lee L, Blackwood EM, Solon M, Tao J, Li L, Shames D, Jackson E, Forrest WF and Firestein R. Lactate dehydrogenase B is required for the growth of KRAS-dependent lung adenocarcinomas. Clin Cancer Res. 2013; 19(4):773-784.
186. Zha X, Wang F, Wang Y, He S, Jing Y, Wu X and Zhang H. Lactate dehydrogenase B is critical for hyperactive mTOR-mediated tumorigenesis. Cancer Res. 2011; 71(1):13-18.
187. Tambe Y, Hasebe M, Kim CJ, Yamamoto A and Inoue H. The drs tumor suppressor regulates glucose metabolism via lactate dehydrogenase-B. Mol Carcinog. 2015; 55(1):52-63.
188. Zhou Y, Shingu T, Feng L, Chen Z, Ogasawara M, Keating MJ, Kondo S and Huang P. Metabolic alterations in highly tumorigenic glioblastoma cells: preference for hypoxia and high dependency on glycolysis. J Biol Chem. 2011; 286(37):32843-32853.
189. Nakano A, Tsuji D, Miki H, Cui Q, El Sayed SM, Ikegame A, Oda A, Amou H, Nakamura S, Harada T, Fujii S, Kagawa K, Takeuchi K, Sakai A, Ozaki S, Okano K, et al. Glycolysis inhibition inactivates ABC transporters to restore drug sensitivity in malignant cells. PLoS One. 2011; 6(11):e27222.