
Introduction
Aging is hallmarked by the progressive loss of cellular function and integrity that eventually leads to vulnerability and death of organisms [1]. It lowers the quality of life, and is a potent risk factor for cancer, diabetes, and other prevalent diseases [2]. It has been long thought that age-dependent accumulation of stochastic damage of molecules drives aging [3]. Current evidence demonstrate that a programmed event of energy metabolism is a determinant of aging that can be modified to modulate aging [4].
Aging involves reprogramming of energy metabolism
Intermediary metabolism generates ATP from nutrients, providing energy for cellular function and maintenance. Alterations in energy metabolism are linked to the aging process and aging-associated diseases [5]. Substantial evidence has demonstrated that energy production progressively decreases with age in all organisms, mainly due to the decline in the function of mitochondria [6]. Aged organisms also exhibit disrupted homeostasis of carbohydrates, amino acids, and fatty acids [5], major biological fuels [7]. The exact alterations in energy metabolism that are associated with aging, their physiological impact, and their contribution to aging are unclear, impeding the understanding of aging mechanisms and the development of mechanism-based strategies to modulate aging.
The decline in mitochondrial function with age has been attributed to the accumulation of stochastic damage to mitochondrial DNA [3], primarily by reactive oxygen species produced through the electron transport chain (ETC) during ATP production. Although oxidative damage of mitochondrial DNA accumulates with age [8, 9] and leads to reduced gene expression [10, 11], it is inconclusive whether oxidative damage is the cause of aging-associated decline in mitochondrial function or aging [12, 13]. Recently, we have reported that the aging of C. elegans, a genetic model of aging that lives about three weeks, is highlighted by a progressive decline in cytosolic phosphoenolpyruvate carboxykinase (PEPCK-C) after the reproductive peak, and a reciprocal increase in pyruvate kinase (PK) [4]. While PK is an enzyme of glycolysis, PEPCK-C is a metabolic enzyme associated with longevity [4, 14, 15]. A key consequence of this metabolic event is the shunt of energy metabolism from oxidative metabolism to anaerobic glycolysis. In all aerobic species, ATP can be generated both in the presence and absence of oxygen. But 30-36 ATP can be generated from one glucose molecule through oxidative metabolism, while only 2 ATP are produced by anaerobic glycolysis [7]. As a result, reciprocal changes in PEPCK-C and PK with age reduce the efficiency of and total energy production [4].
How do changes in PEPCK-C and PK, two cytosolic enzymes, lead to decline in mitochondrial function? The core of energy metabolism is the tricarboxylic acid (TCA) cycle [7], a series of chemical reactions that oxidize carbohydrates, fats and proteins into carbon dioxide, generating NADH that is used by ETC to produce ATP. Moreover, the TCA cycle intermediates can be withdrawn from mitochondria to the cytosol, a process called cataplerosis. This supplies carbons for the synthesis of glyceride-glycerol, serine, and glucose, as well as metabolites derived from these chemicals [16]. Traditionally viewed as a rate-limiting enzyme of gluconeogenesis and glyceroneogenesis, PEPCK-C is a major cataplerotic enzyme that links the TCA cycle with the metabolism of carbohydrates, fatty acids, amino acids, and other metabolites [16]. At the biochemical level, the activity of PEPCK-C is correlated tightly with the flux of the TCA cycle but not gluconeogenesis [17]. At the organismal level, PEPCK-C is required for the integration of energy metabolism [18], and is critical for the homeostasis of glucose, fatty acids, amino acids and other related metabolites (see ref [16] for review). In C. elegans, PEPCK-C accelerates TCA cycle flux and promotes oxidative metabolism [4], likely via increased cataplerosis (Figure 1A). PEPCK-C also increases mitochondrial respiration and counteracts its decline with age. On the other hand, PK is a key glycolytic enzyme that greatly favors the conversion of phosphoenolpyruvate to pyruvate, promoting glycolysis [7]. During energy production, pyruvate either enters mitochondria for oxidation, or is converted to lactate by lactate dehydrogenase. PEPCK-C shunts glucose metabolism toward oxidation, reducing lactate production [4].
The reciprocal changes in PEPCK-C and PK with age, and their impact on oxidative metabolism and anaerobic glycolysis are likely conserved from C. elegans to humans. First, skeletal muscle of aging humans from 10s to 70s exhibits an aging-associated decrease in PEPCK-C activity, and increases in PK and lactate dehydrogenase that cannot be explained by alterations in numerical ratio of type I and type II muscle fibers with age [19]. In aged skeletal muscle and liver of mammalian animals, PEPCK-C mRNA is decreased and PK mRNA is increased [20, 21]. Second, aging mammals including humans display decreased mitochondrial function and increased glycolysis in many tissues such as liver, skeletal muscle and brain [22-25], as well as elevated lactate in both tissues and serum [26]. Moreover, platelets of aged humans exhibit reduced ATP production by mitochondria, and increased ATP production by anaerobic glycolysis [27], which likely reflects aging-associated changes in energy metabolism of the whole body [27, 28].
Reciprocal changes in PEPCK-C and PK profoundly impact aging organisms
What are the physiological effects of reciprocal changes of PEPCK-C and PK with age? The decline in mitochondrial bioenergetics may subject aging organisms to a relative energy deficiency, although the PK-driven increase in glycolysis likely compensates for some of the reduced energy production. A deficit in energy supply reduces the function and integrity of many cells and tissues, hence the survival of organisms, due to unmatched energy demand and supply (Figure 1B). In support of this view, PEPCK-C promotes physical activity, fertility, autophagy, defense against osmotic and oxidative stresses, and many other energy consuming processes in various animal species [4, 14, 15, 29-34]. During the aging of C. elegans, decline in PEPCK-C is coupled with loss of physical activity, a major energy consumer [35], and genetically enhanced PEPCK-C preserves physical activity and extends lifespan in a dose-dependent manner [4]. Of note, PEPCK-C promotes physical activity to increase ATP turnover, AMP/ATP ratio (a key indicator of cellular energy demand), the activation of 5’ AMP kinase (AMPK, a major mediator of energy homeostasis activated by higher AMP:ATP ratio [36]), fuel oxidation, ATP content, and food intake, both acutely and chronically. Many of these effects of PEPCK-C on energy demand and supply, and cellular function and maintenance require the activation of AMPK [4].
In addition to decline in energy production, the following alterations in energy metabolism are predicted, based on the metabolic roles of PEPCK-C, PK, and the TCA cycle: 1) increase in synthesis and deposition of fats; 2) disrupted homeostasis of glucose and amino acids; 3) reduction in NAD+; and 4) reduction in biosynthesis associated with cataplerosis (Figure 1C).
Unlike glucose, fats can only be oxidized to produce ATP [7]. The reduction in oxidative metabolism with age would lead to decreased utilization of fats. On the other hand, increased PK activity and glycolytic flux in aged organisms should produce more pyruvate. In addition to being oxidized in mitochondria or converted to lactate, pyruvate can serve as a precursor of lipogenesis or gluconeogenesis. Because mitochondrial function is reduced, more pyruvate would be shunt to lipogenesis or gluconeogenesis. Indeed, aging involves a shift of fatty metabolism toward lipogenesis in mice [37] and accumulation of fats in all organisms including humans [38]. In mice, decline in PEPCK-C with age underlies aging-associated reduction in lipolysis and the coordinative down regulation of mitochondrial enzymes [39]. Indeed, aged mice over-expressing PEPCK-C exhibited less subcutaneous, visceral, and pericardial fat deposit than even younger control mice fed the same regular chow diet [31]
The shift of energy metabolism from oxidative metabolism to anaerobic glycolysis suggests that aged organisms demand more glucose as energy source. Consistent with this view, gluconeogenesis is elevated in aged yeast [40]. In aged mammals, basal gluconeogenic capacity and blood glucose produced through gluconeogenesis are increased [41], while hepatic incorporation of glucose to glycogen is decreased [42]. In contrast to gluconeogenesis, glucose uptake into skeletal muscle, brain and other energy consuming tissues decreases with age due to reduced insulin signaling [43], decreased insulin sensitivity [44], and reduced glucose transporters [45]. An increased demand and supply of glucose, while a reduction in usable glucose may contribute to the disrupted homeostasis of glucose in aged organisms [46]. Sarcopenia is the progressive loss of muscle in aging humans and animals [47]. The lost muscle mass, primarily proteins, results in increased alanine and glutamine in circulation [48]. A fate of these amino acids is to be oxidized in mitochondria. The shift of energy metabolism away from oxidative metabolism would reduce the disposal of these amino acids, and contribute to the disrupted homeostasis of amino acids in aged organisms [49].
Redox homeostasis is critical for cellular function and integrity [50]. Significantly, aging is accompanied by a progressive decline in intracellular NAD+ in species including humans [51, 52]. NAD+ is an essential cofactor for sirtuin, a deacetylase that promotes longevity and healthy aging [53]. It is also a necessary substrate for poly (ADP-ribose) polymerase [54], a critical enzyme in the DNA repair process [54-56]. Decreased NAD+ increases the vulnerability of cells to accumulation of DNA damage with age [57-59]. The breakdown of glucose to pyruvate consumes NAD+ [7]. NAD+ is regenerated through fermentation that converts pyruvate to lactate. Increased glycolytic flux in aged organisms would lead to increased consumption of NAD+. Insufficient regeneration of NAD+ could result in reduction in NAD+.
The cataplerotic role of PEPCK-C has been recognized in cell proliferation [60], and adaptive response to stresses, such as acidosis [61], inflammation [62], and osmotic stress [30]. Under these circumstances, PEPCK-C supports the synthesis of ribose, glucose, steroid, and glycerol from amino acids and other non-carbohydrates. The released ammonia from the oxidation of amino acids relieves acidosis. Likely, decline in PEPCK-C with age reduces cataplerosis and its related biosynthesis, contributing to reduced proliferation and increased vulnerability associated with aging.
Most cancer cells exhibit the Warburg effect [63], higher glycolysis followed by lactate production, instead of lower glycolysis followed by oxidation. Type II diabetes is associated with metabolic changes paralleling the Warburg effect, including expression of genes involved in anaerobic glycolysis [64] and a decline in mitochondrial function [65]. Thus, the shift of energy metabolism from oxidative metabolism to glycolysis driven by reciprocal changes in PEPCK-C and PK with age may promote tumorigenesis and the prevalence of diabetes [66-68]. For example, metformin, the most commonly prescribed drug treating type II diabetes, may shunt energy metabolism toward oxidative metabolism and away from glycolysis to counteract cancers [68].
Collectively, the cataplerotic role of PEPCK-C acts as a major adaptor of energy metabolism, and a carbon valve for various biosynthetic pathways. Reciprocal changes in PEPCK-C and PK after the reproductive peak are a lead metabolic event associated with aging. This event re-patterns metabolism of aging organisms, decreases their cellular function and integrity, and promotes the onset of aging-associated diseases.
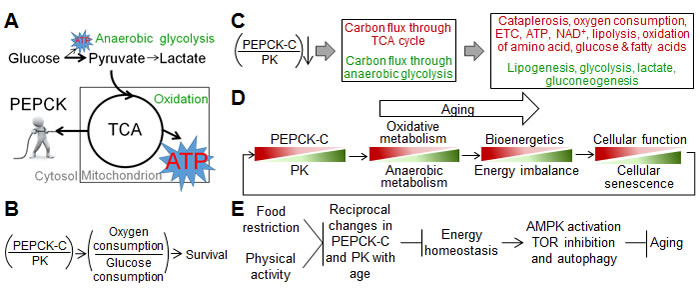
Figure 1: Models for aging-associated changes in energy metabolism and aging. A., “PEPCK-C pulls the strings”, schema illustrating a role of PEPCK-C in energy metabolism. B.-C., Schema illustrating the impact of the ratio of PEPCK-C over PK on energy metabolism and survival. Red, decrease; green increase. D.-E., Models for the role of reciprocal changes in PEPCK-C and PK in aging. These metabolic changes may also promote aging via reduced carbon supply from the TCA cycle, which is needed for various biosynthetic pathways. Direct evidence supporting this view, however, is currently lacking. Panels D-E were originally published in Journal of Biological Chemistry, Yuan, et al., Reciprocal changes in phosphoenolpyruvate carboxykinase and pyruvate kinase with age are a determinant of aging in Caenorhabditis elegans. J Biol Chem, 291: 1307-19. © the American Society for Biochemistry and Molecular Biology.
Reciprocal changes in PEPCK-C and PK with age determine aging
Evidence from mice and C. elegans demonstrate that reciprocal changes in PEPCK-C and PK with age determine aging. First, PEPCK-C counteracts loss of cellular function and integrity with age. Specifically, it extends fertility, retards aging-associated decrease in physical activity, a negative indicator of health span and lifespan [69], and enhances autophagic activity [4, 14]. Autophagy is a cell repair mechanism that removes molecular wastes, and counteracts aging and aging-related diseases [70-73]. Second, PEPCK-C retards cellular senescence [4], assessed by the accumulation of molecular wastes such as lipofuscin and β-galactosidase [74, 75], and the expression of the proliferation restrictive marker cyclin kinase inhibitor [76]. Cellular senescence may contribute to aging [77]. Moreover, reciprocal changes in PEPCK-C and PK with age are necessary and sufficient to limit lifespan and fertility [4]. Last, PEPCK-C activity is correlated with lifespan, and its enzyme level predicts life expectancy [4].
Many effects of PEPCK-C on aging including lifespan extension require the activation of AMPK signaling and/or the inhibition of Target of Rapamycin (TOR) signaling [4]. AMPK and TOR signaling are two major molecular signals that control aging in species including mammals [78-80]. The beneficial impact of activation of AMPK and inhibition of TOR on lifespan necessitates autophagy [81-83]. Consistently, PEPCK-C enhances the activity of autophagy in aged C. elegans, and requires the expression of autophagic genes to promote longevity [4]. Interestingly, AMPK primarily drives catabolism that produces energy and promotes mitochondrial oxidation, and is a negative regulator of glycolysis [84]. On the other hand, TOR signaling is a cellular energy sensor of excessive nutrient and energy that upregulates PK and glycolysis [85]. The anti-aging effects mediated by TOR inhibition involve increased energy metabolism and oxidative phosphorylation complex [86-90].
In summary, reciprocal changes in PEPCK-C and PK with age are a determinant of aging. The mechanisms include disrupted energy homeostasis, as well as altered AMPK, TOR signaling, and autophagy. Consistently, mitochondrial bioenergetics and autophagic activity are preserved in fibroblasts of centenarians [91]. Intriguingly, declines in PEPCK-C and physical activity with age promote each other to limit reproductive life and lifespan [4] via a feedback mechanism [4, 14, 92, 93]. These observations indicate that a vicious cycle of reciprocal changes in PEPCK-C and PK, and decline in cellular function and integrity drives aging (Figure 1D). The causality between changes in metabolism and in cellular function and integrity is unknown, and is likely a “chicken and egg” dilemma.
Reciprocal changes in PEPCK-C and PK are a common denominator of aging
Molecular signals, pharmacological reagents, appropriate environmental stresses, and calorie restriction (CR) extend lifespan and may also improve health in various species. Strikingly, most of these maneuvers either increase oxidative metabolism and energy production, inhibit glycolysis, or both. In addition to AMPK and TOR, insulin/IGF signaling (IIS) [94] and sirtuin [53] are molecular signals that affect lifespan. Reduced IIS [95-99] and sirtuin [51, 100-103] increase or stabilize PEPCK-C, promote oxidative metabolism, and inhibit PK and/or glycolysis.
Metformin is an indirect activator of AMPK, and rapamycin is an inhibitor of TOR signaling. Metformin [104, 105], rapamycin [106, 107] and inhibitors of glycolysis, such as D-glucosamine and 2-Deoxy-D-glucose [108, 109], extend lifespan in many species including mammals. D-glucosamine and 2-Deoxy-D-glucose increase mitochondrial respiration, and require AMPK to extend lifespan [108, 109], suggesting that a shunt of energy metabolism toward oxidative metabolism is critical for the observed lifespan extension. Besides pharmacological reagents, lower ambient temperature, and osmotic and oxidative stresses increase PEPCK-C and/or oxidative metabolism, and extend lifespan in C. elegans and other lower organisms [16, 30, 110-112].
CR is the most robust intervention that extends lifespan and improves health in species ranging from yeast to non-human primates, via AMPK-TOR-autophagy axis [80-83]. CR increases PEPCK-C activity and oxidative metabolism while inhibiting PK activity and glycolysis in animals [15, 113-116]. In humans, CR increases mitochondrial biogenesis [117]. A plausible biological reason underling this metabolic shift is to promote efficient energy production and cataplerosis, in order to meet the energy [118] and biosynthetic [119-121] need under limited resources. Significantly, CR counteracts reciprocal changes in PEPCK-C and PK with age to elicit anti-aging effects including longevity in C. elegans [4]. On the other hand, physical activity, which extends life expectancy in humans [122], increases energy expenditure, PEPCK-C and mitochondrial function. Notably, both mice and C. elegans over-expressing PEPCK-C exhibited increased physical activity, ate more, weighed less, had extended fertility, and lived longer [4, 14]. Thus, energy balance, achieved by reduced “energy in” from CR, enhanced “energy out” from enhanced physical activity, or their combination, counteracts reciprocal changes in PEPCK-C and PK with age to retard aging.
In summary, reciprocal changes in PEPCK-C and PK activity with age, and the consequent shift of energy metabolism are a common denominator of aging. These alterations can be retarded by CR, CR mimetics, and other genetic and environmental factors to counteract aging via AMPK and TOR pathways (Figure 1E).
Conclusions and future perspective
Reciprocal changes in PEPCK-C and PK with age are likely part of a bigger reprogramming of energy metabolism that profoundly affects the physiology of aging organisms, thereby impacting the aging process. It is important to obtain a complete picture of changes in metabolism with age, and their influence on decline in cellular function, cellular senescence, lifespan, and other aging traits. Such investigation should focus on metabolic pathways moving carbons into and out of the TCA cycle, and those affecting the homeostasis of glucose, fats and amino acids. This is because both CR and an optimized ratio of macronutrients (carbohydrates, proteins and fats) without reduction in total calorie intake extend lifespan in mice [123-125], suggesting that alterations in these metabolic pathways have significant but complex impact on aging.
Evidence outlined here support the bioenergetics theory of aging [126, 127], which proposes that the decline in bioenergetics with age is the driver of aging. Specifically, the decline in bioenergetics with age is a pacemaker in the aging process, whereas other aging-associated phenomena, such as the accumulation of reactive species and the decline in repair mechanisms, are secondary to the decline in bioenergetics. The sum of all these changes leads to loss of physiological function and eventually vulnerability and death of organisms. The bioenergetics theory of aging also hypothesizes that an unidentified primary genetic program (aging clock) controls the decline in bioenergetics. However, the presence of a vicious cycle between reciprocal changes in PEPCK-C and PK, and decline in cellular function and integrity indicates that a genetically programmed aging clock may [128] or may not [129-131] be needed for the decline in bioenergetics with age. For example, a quasi-programmed hyperfunction, which has been proposed to be associated with development and have harmful impact on organisms, may start this vicious cycle without a genetically programmed aging clock; while the pace of this vicious cycle may be mediated by a genetic program. In either case, it is critical to understand the mechanisms underlying aging-associated changes in PEPCK-C and PK, which are currently unknown. Remarkably, PEPCK-C is acetylated in yeast [132] and human cells [133], and PEPCK-C acetylation leads to its degradation [100]. Sirtuin, which promotes longevity in many species, deacetylates and stabilizes PEPCK-C [100]. Deacetylation mediated by sirtuin shunts energy metabolism away from glycolysis and toward oxidative metabolism [134-137], promoting energy production [138]. Sirtuin expression and activity decrease with age [139, 140], and CR [141] and exercise [142] increase sirtuin to decrease protein acetylation. It will be interesting to examine if PEPCK-C acetylation is involved in its decline with age, if sirtuin slows this decline, and if sirtuin retards aging via altered PEPCK-C stability.
Last, enhancing PEPCK-C is sufficient to delay many key aging-associated metabolic and physiological changes and increase lifespan [4, 14, 15]. Thus interventions that sustain PEPCK-C represent a novel strategy to counteract aging.
Acknowledgments
We thank Transdisciplinary Research on Energetics & Cancer directed by Dr. Nathan A. Berger, Nutrition Obesity Research Center, Case Western Reserve University (U54-CA-116867), Whitehall Foundation (2013-08-82), department fund (ZF), and Ellison Foundation (AG-SS-2420-10)(RWH and NAB) for funding, and Drs. Thomas Sundermeier, Leslie Webster, Hung-Ying Kao, and Johannes Von Lintig for discussion.
CONFLICTS OF INTEREST
There is no conflict of interest.
References
1. Lopez-Otin C, Blasco MA, Partridge L, Serrano M and Kroemer G. The hallmarks of aging. Cell. 2013; 153:1194-1217.
2. Niccoli T and Partridge L. Ageing as a risk factor for disease. Curr Biol. 2012; 22:R741-752.
3. Miquel J, Economos AC, Fleming J and Johnson JE, Jr. Mitochondrial role in cell aging. Exp Gerontol. 1980; 15:575-591.
4. Yuan Y, Hakimi P, Kao C, Kao A, Liu R, Janocha A, Boyd-Tressler A, Hang X, Alhoraibi H, Slater E, Xia K, Cao P, Shue Q, Ching TT, Hsu AL, Erzurum SC, et al. Reciprocal changes in phosphoenolpyruvate carboxykinase and pyruvate kinase with age are a determinant of aging in C. elegans. J Biol Chem. 2015.
5. Barzilai N, Huffman DM, Muzumdar RH and Bartke A. The critical role of metabolic pathways in aging. Diabetes. 2012; 61:1315-1322.
6. Bratic I and Trifunovic A. Mitochondrial energy metabolism and ageing. Biochim Biophys Acta. 2010; 1797:961-967.
7. Voet D and Voet J. Biochemistry. (New York: John Wiley & Sons, Inc).1995.
8. Sohal RS and Weindruch R. Oxidative stress, caloric restriction, and aging. Science. 1996; 273:59-63.
9. Hamilton ML, Van Remmen H, Drake JA, Yang H, Guo ZM, Kewitt K, Walter CA and Richardson A. Does oxidative damage to DNA increase with age? Proc Natl Acad Sci U S A. 2001; 98:10469-10474.
10. Xanthoudakis S, Miao G, Wang F, Pan YC and Curran T. Redox activation of Fos-Jun DNA binding activity is mediated by a DNA repair enzyme. EMBO J. 1992; 11:3323-3335.
11. Morel Y and Barouki R. Repression of gene expression by oxidative stress. Biochem J. 1999; 342 Pt 3:481-496.
12. Perez VI, Bokov A, Van Remmen H, Mele J, Ran Q, Ikeno Y and Richardson A. Is the oxidative stress theory of aging dead? Biochim Biophys Acta. 2009; 1790:1005-1014.
13. Shokolenko IN, Wilson GL and Alexeyev MF. Aging: A mitochondrial DNA perspective, critical analysis and an update. World J Exp Med. 2014; 4:46-57.
14. Hakimi P, Yang J, Casadesus G, Massillon D, Tolentino-Silva F, Nye CK, Cabrera ME, Hagen DR, Utter CB, Baghdy Y, Johnson DH, Wilson DL, Kirwan JP, Kalhan SC and Hanson RW. Overexpression of the cytosolic form of phosphoenolpyruvate carboxykinase (GTP) in skeletal muscle repatterns energy metabolism in the mouse. J Biol Chem. 2007; 282:32844-32855.
15. Yuan Y, Kadiyala CS, Ching TT, Hakimi P, Saha S, Xu H, Yuan C, Mullangi V, Wang L, Fivenson E, Hanson RW, Ewing R, Hsu AL, Miyagi M and Feng Z. Enhanced energy metabolism contributes to the extended life span of calorie-restricted Caenorhabditis elegans. J Biol Chem. 2012; 287:31414-31426.
16. Yang J, Kalhan SC and Hanson RW. What is the metabolic role of phosphoenolpyruvate carboxykinase? J Biol Chem. 2009; 284:27025-27029.
17. Burgess SC, He T, Yan Z, Lindner J, Sherry AD, Malloy CR, Browning JD and Magnuson MA. Cytosolic phosphoenolpyruvate carboxykinase does not solely control the rate of hepatic gluconeogenesis in the intact mouse liver. Cell Metab. 2007; 5:313-320.
18. She P, Shiota M, Shelton KD, Chalkley R, Postic C and Magnuson MA. Phosphoenolpyruvate carboxykinase is necessary for the integration of hepatic energy metabolism. Mol Cell Biol. 2000; 20:6508-6517.
19. Schlenska GK and Kleine TO. Disorganization of glycolytic and gluconeogenic pathways in skeletal muscle of aged persons studied by histometric and enzymatic methods. Mech Ageing Dev. 1980; 13:143-154.
20. Dhahbi JM, Mote PL, Wingo J, Tillman JB, Walford RL and Spindler SR. Calories and aging alter gene expression for gluconeogenic, glycolytic, and nitrogen-metabolizing enzymes. Am J Physiol. 1999; 277:E352-360.
21. Kil DY, Vester Boler BM, Apanavicius CJ, Schook LB and Swanson KS. Age and diet affect gene expression profiles in canine liver tissue. PLoS One. 2010; 5:e13319.
22. Bowling AC, Mutisya EM, Walker LC, Price DL, Cork LC and Beal MF. Age-dependent impairment of mitochondrial function in primate brain. J Neurochem. 1993; 60:1964-1967.
23. Hagen TM, Yowe DL, Bartholomew JC, Wehr CM, Do KL, Park JY and Ames BN. Mitochondrial decay in hepatocytes from old rats: membrane potential declines, heterogeneity and oxidants increase. Proc Natl Acad Sci U S A. 1997; 94:3064-3069.
24. Gouspillou G, Bourdel-Marchasson I, Rouland R, Calmettes G, Biran M, Deschodt-Arsac V, Miraux S, Thiaudiere E, Pasdois P, Detaille D, Franconi JM, Babot M, Trezeguet V, Arsac L and Diolez P. Mitochondrial energetics is impaired in vivo in aged skeletal muscle. Aging Cell. 2014; 13:39-48.
25. Trounce I, Byrne E and Marzuki S. Decline in skeletal muscle mitochondrial respiratory chain function: possible factor in ageing. Lancet. 1989; 1:637-639.
26. Ross JM, Oberg J, Brene S, Coppotelli G, Terzioglu M, Pernold K, Goiny M, Sitnikov R, Kehr J, Trifunovic A, Larsson NG, Hoffer BJ and Olson L. High brain lactate is a hallmark of aging and caused by a shift in the lactate dehydrogenase A/B ratio. Proc Natl Acad Sci U S A. 2010; 107:20087-20092.
27. Lenaz G, D’Aurelio M, Merlo Pich M, Genova ML, Ventura B, Bovina C, Formiggini G and Parenti Castelli G. Mitochondrial bioenergetics in aging. Biochim Biophys Acta. 2000; 1459:397-404.
28. Holmsen H and Robkin L. Effects of antimycin A and 2-deoxyglucose on energy metabolism in washed human platelets. Thromb Haemost. 1980; 42:1460-1472.
29. Hanson RW and Hakimi P. Born to run; the story of the PEPCK-Cmus mouse. Biochimie. 2008; 90:838-842.
30. Frazier HN, 3rd and Roth MB. Adaptive sugar provisioning controls survival of C. elegans embryos in adverse environments. Curr Biol. 2009; 19:859-863.
31. Roy D, Gargesha M, Steyer GJ, Hakimi P, Hanson RW and Wilson DL. Multi-scale characterization of the PEPCK-C mouse through 3D cryo-imaging. Int J Biomed Imaging. 2010; 2010:105984.
32. Landis G, Shen J and Tower J. Gene expression changes in response to aging compared to heat stress, oxidative stress and ionizing radiation in Drosophila melanogaster. Aging (Albany NY). 2012; 4:768-789.
33. Choi CY, Min BH, Jo PG and Chang YJ. Molecular cloning of PEPCK and stress response of black porgy (Acanthopagrus schlegeli) to increased temperature in freshwater and seawater. Gen Comp Endocrinol. 2007; 152:47-53.
34. Furukawa F, Tseng YC, Liu ST, Chou YL, Lin CC, Sung PH, Uchida K, Lin LY and Hwang PP. Induction of Phosphoenolpyruvate Carboxykinase (PEPCK) during Acute Acidosis and Its Role in Acid Secretion by V-ATPase-Expressing Ionocytes. Int J Biol Sci. 2015; 11:712-725.
35. Coyle EF. Physical activity as a metabolic stressor. Am J Clin Nutr. 2000; 72:512S-520S.
36. Hardie DG. AMP-activated protein kinase: a key system mediating metabolic responses to exercise. Med Sci Sports Exerc. 2004; 36:28-34.
37. Kuhla A, Blei T, Jaster R and Vollmar B. Aging is associated with a shift of fatty metabolism toward lipogenesis. J Gerontol A Biol Sci Med Sci. 2011; 66:1192-1200.
38. Barzilai N and Gupta G. Interaction between aging and syndrome X: new insights on the pathophysiology of fat distribution. Ann N Y Acad Sci. 1999; 892:58-72.
39. Mennes E, Dungan CM, Frendo-Cumbo S, Williamson DL and Wright DC. Aging-associated reductions in lipolytic and mitochondrial proteins in mouse adipose tissue are not rescued by metformin treatment. J Gerontol A Biol Sci Med Sci. 2014; 69:1060-1068.
40. Lin SS, Manchester JK and Gordon JI. Enhanced gluconeogenesis and increased energy storage as hallmarks of aging in Saccharomyces cerevisiae. J Biol Chem. 2001; 276:36000-36007.
41. Podolin DA, Pagliassotti MJ, Gleeson TT and Mazzeo RS. Influence of endurance training on the age-related decline in hepatic glyconeogenesis. Mech Ageing Dev. 1994; 75:81-93.
42. Satrustegui J, Cuezva JM and Machado A. Increased basal gluconeogenesis in the aged rat. FEBS Lett. 1986; 197:159-163.
43. Muzumdar R, Ma X, Atzmon G, Vuguin P, Yang X and Barzilai N. Decrease in glucose-stimulated insulin secretion with aging is independent of insulin action. Diabetes. 2004; 53:441-446.
44. Karakelides H, Irving BA, Short KR, O’Brien P and Nair KS. Age, obesity, and sex effects on insulin sensitivity and skeletal muscle mitochondrial function. Diabetes. 2010; 59:89-97.
45. dos Santos JM, Benite-Ribeiro SA, Queiroz G and Duarte JA. The effect of age on glucose uptake and GLUT1 and GLUT4 expression in rat skeletal muscle. Cell Biochem Funct. 2012; 30:191-197.
46. Szoke E, Shrayyef MZ, Messing S, Woerle HJ, van Haeften TW, Meyer C, Mitrakou A, Pimenta W and Gerich JE. Effect of aging on glucose homeostasis: accelerated deterioration of beta-cell function in individuals with impaired glucose tolerance. Diabetes Care. 2008; 31:539-543.
47. Doherty TJ. Invited review: Aging and sarcopenia. J Appl Physiol (1985). 2003; 95:1717-1727.
48. Garber AJ, Karl IE and Kipnis DM. Alanine and glutamine synthesis and release from skeletal muscle. I. Glycolysis and amino acid release. J Biol Chem. 1976; 251:826-835.
49. Evans WJ. Exercise, nutrition, and aging. Clin Geriatr Med. 1995; 11:725-734.
50. Ying W. NAD+ and NADH in cellular functions and cell death. Front Biosci. 2006; 11:3129-3148.
51. Mouchiroud L, Houtkooper RH, Moullan N, Katsyuba E, Ryu D, Canto C, Mottis A, Jo YS, Viswanathan M, Schoonjans K, Guarente L and Auwerx J. The NAD(+)/Sirtuin Pathway Modulates Longevity through Activation of Mitochondrial UPR and FOXO Signaling. Cell. 2013; 154:430-441.
52. Massudi H, Grant R, Braidy N, Guest J, Farnsworth B and Guillemin GJ. Age-associated changes in oxidative stress and NAD+ metabolism in human tissue. PLoS One. 2012; 7:e42357.
53. Houtkooper RH, Pirinen E and Auwerx J. Sirtuins as regulators of metabolism and healthspan. Nat Rev Mol Cell Biol. 2012; 13:225-238.
54. Sims JL, Berger SJ and Berger NA. Effects of nicotinamide on NAD and poly(ADP-ribose) metabolism in DNA-damaged human lymphocytes. J Supramol Struct Cell Biochem. 1981; 16:281-288.
55. Berger NA. Poly(ADP-ribose) in the cellular response to DNA damage. Radiat Res. 1985; 101:4-15.
56. Berger NA, Petzold SJ and Berger SJ. Association of poly(ADP-rib) synthesis with cessation of DNA synthesis and DNA fragmentation. Biochim Biophys Acta. 1979; 564:90-104.
57. Rube CE, Fricke A, Widmann TA, Furst T, Madry H, Pfreundschuh M and Rube C. Accumulation of DNA damage in hematopoietic stem and progenitor cells during human aging. PLoS One. 2011; 6:e17487.
58. Garinis GA, van der Horst GT, Vijg J and Hoeijmakers JH. DNA damage and ageing: new-age ideas for an age-old problem. Nat Cell Biol. 2008; 10:1241-1247.
59. Mandavilli BS, Santos JH and Van Houten B. Mitochondrial DNA repair and aging. Mutat Res. 2002; 509:127-151.
60. Montal ED, Dewi R, Bhalla K, Ou L, Hwang BJ, Ropell AE, Gordon C, Liu WJ, DeBerardinis RJ, Sudderth J, Twaddel W, Boros LG, Shroyer KR, Duraisamy S, Drapkin R, Powers RS, et al. PEPCK Coordinates the Regulation of Central Carbon Metabolism to Promote Cancer Cell Growth. Mol Cell. 2015; 60(4):571-583.
61. Boron WF and Boulpaep EL. Medical Physiology: A Cellular And Molecular Approach: (Elsevier/Saunders). 2003.
62. Sadasivam M, Ramatchandirin B, Balakrishnan S, Selvaraj K and Prahalathan C. The role of phosphoenolpyruvate carboxykinase in neuronal steroidogenesis under acute inflammation. Gene. 2014; 552:249-254.
63. Warburg O. On the origin of cancer cells. Science. 1956; 123:309-314.
64. Ptitsyn A, Hulver M, Cefalu W, York D and Smith SR. Unsupervised clustering of gene expression data points at hypoxia as possible trigger for metabolic syndrome. BMC Genomics. 2006; 7:318.
65. Petersen KF, Dufour S, Befroy D, Garcia R and Shulman GI. Impaired mitochondrial activity in the insulin-resistant offspring of patients with type 2 diabetes. N Engl J Med. 2004; 350:664-671.
66. Menendez JA, Alarcon T and Joven J. Gerometabolites: the pseudohypoxic aging side of cancer oncometabolites. Cell Cycle. 2014; 13:699-709.
67. Wu LE, Gomes AP and Sinclair DA. Geroncogenesis: metabolic changes during aging as a driver of tumorigenesis. Cancer Cell. 2014; 25:12-19.
68. Menendez JA, Cufi S, Oliveras-Ferraros C, Martin-Castillo B, Joven J, Vellon L and Vazquez-Martin A. Metformin and the ATM DNA damage response (DDR): accelerating the onset of stress-induced senescence to boost protection against cancer. Aging (Albany NY). 2011; 3:1063-1077.
69. Hsu AL, Feng Z, Hsieh MY and Xu XZ. Identification by machine vision of the rate of motor activity decline as a lifespan predictor in C. elegans. Neurobiol Aging. 2009; 30:1498-1503.
70. Bergamini E. Autophagy: a cell repair mechanism that retards ageing and age-associated diseases and can be intensified pharmacologically. Mol Aspects Med. 2006; 27:403-410.
71. Carroll B, Hewitt G and Korolchuk VI. Autophagy and ageing: implications for age-related neurodegenerative diseases. Essays Biochem. 2013; 55:119-131.
72. Madeo F, Zimmermann A, Maiuri MC and Kroemer G. Essential role for autophagy in life span extension. J Clin Invest. 2015; 125:85-93.
73. Kroemer G. Autophagy: a druggable process that is deregulated in aging and human disease. J Clin Invest. 2015; 125:1-4.
74. Jung T, Hohn A and Grune T. Lipofuscin: detection and quantification by microscopic techniques. Methods Mol Biol. 2010; 594:173-193.
75. Lee BY, Han JA, Im JS, Morrone A, Johung K, Goodwin EC, Kleijer WJ, DiMaio D and Hwang ES. Senescence-associated beta-galactosidase is lysosomal beta-galactosidase. Aging Cell. 2006; 5:187-195.
76. Tan W, Gu Z, Shen B, Jiang J, Meng Y, Da Z, Liu H, Tao T and Cheng C. PTEN/Akt-p27(kip1) Signaling Promote the BM-MSCs Senescence and Apoptosis in SLE Patients. J Cell Biochem. 2015; 116:1583-1594.
77. Baker DJ, Wijshake T, Tchkonia T, LeBrasseur NK, Childs BG, van de Sluis B, Kirkland JL and van Deursen JM. Clearance of p16Ink4a-positive senescent cells delays ageing-associated disorders. Nature. 2011; 479:232-236.
78. Jia K, Chen D and Riddle DL. The TOR pathway interacts with the insulin signaling pathway to regulate C. elegans larval development, metabolism and life span. Development. 2004; 131:3897-3906.
79. Apfeld J, O’Connor G, McDonagh T, DiStefano PS and Curtis R. The AMP-activated protein kinase AAK-2 links energy levels and insulin-like signals to lifespan in C. elegans. Genes Dev. 2004; 18:3004-3009.
80. Newgard CB and Pessin JE. Recent progress in metabolic signaling pathways regulating aging and life span. J Gerontol A Biol Sci Med Sci. 2014; 69 Suppl 1:S21-27.
81. Kim J, Kundu M, Viollet B and Guan KL. AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat Cell Biol. 2011; 13:132-141.
82. Hansen M, Chandra A, Mitic LL, Onken B, Driscoll M and Kenyon C. A role for autophagy in the extension of lifespan by dietary restriction in C. elegans. PLoS Genet. 2008; 4:e24.
83. Egan DF, Shackelford DB, Mihaylova MM, Gelino S, Kohnz RA, Mair W, Vasquez DS, Joshi A, Gwinn DM, Taylor R, Asara JM, Fitzpatrick J, Dillin A, Viollet B, Kundu M, Hansen M, et al. Phosphorylation of ULK1 (hATG1) by AMP-activated protein kinase connects energy sensing to mitophagy. Science. 2011; 331:456-461.
84. Faubert B, Boily G, Izreig S, Griss T, Samborska B, Dong Z, Dupuy F, Chambers C, Fuerth BJ, Viollet B, Mamer OA, Avizonis D, DeBerardinis RJ, Siegel PM and Jones RG. AMPK is a negative regulator of the Warburg effect and suppresses tumor growth in vivo. Cell Metab. 2013; 17:113-124.
85. Amiel E, Everts B, Fritz D, Beauchamp S, Ge B, Pearce EL and Pearce EJ. Mechanistic target of rapamycin inhibition extends cellular lifespan in dendritic cells by preserving mitochondrial function. J Immunol. 2014; 193:2821-2830.
86. Kofman AE, McGraw MR and Payne CJ. Rapamycin increases oxidative stress response gene expression in adult stem cells. Aging (Albany NY). 2012; 4:279-289.
87. Khapre RV, Kondratova AA, Patel S, Dubrovsky Y, Wrobel M, Antoch MP and Kondratov RV. BMAL1-dependent regulation of the mTOR signaling pathway delays aging. Aging (Albany NY). 2014; 6:48-57.
88. Leontieva OV and Blagosklonny MV. DNA damaging agents and p53 do not cause senescence in quiescent cells, while consecutive re-activation of mTOR is associated with conversion to senescence. Aging (Albany NY). 2010; 2:924-935.
89. Panieri E, Toietta G, Mele M, Labate V, Ranieri SC, Fusco S, Tesori V, Antonini A, Maulucci G, De Spirito M, Galeotti T and Pani G. Nutrient withdrawal rescues growth factor-deprived cells from mTOR-dependent damage. Aging (Albany NY). 2010; 2:487-503.
90. Pan Y and Shadel GS. Extension of chronological life span by reduced TOR signaling requires down-regulation of Sch9p and involves increased mitochondrial OXPHOS complex density. Aging (Albany NY). 2009; 1:131-145.
91. Sgarbi G, Matarrese P, Pinti M, Lanzarini C, Ascione B, Gibellini L, Dika E, Patrizi A, Tommasino C, Capri M, Cossarizza A, Baracca A, Lenaz G, Solaini G, Franceschi C, Malorni W, et al. Mitochondria hyperfusion and elevated autophagic activity are key mechanisms for cellular bioenergetic preservation in centenarians. Aging (Albany NY). 2014; 6:296-310.
92. Novak CM, Escande C, Gerber SM, Chini EN, Zhang M, Britton SL, Koch LG and Levine JA. Endurance capacity, not body size, determines physical activity levels: role of skeletal muscle PEPCK. PLoS One. 2009; 4:e5869.
93. Novak CM, Escande C, Burghardt PR, Zhang M, Barbosa MT, Chini EN, Britton SL, Koch LG, Akil H and Levine JA. Spontaneous activity, economy of activity, and resistance to diet-induced obesity in rats bred for high intrinsic aerobic capacity. Horm Behav. 2010; 58:355-367.
94. Yu S and Driscoll M. EGF signaling comes of age: promotion of healthy aging in C. elegans. Exp Gerontol. 2011; 46:129-134.
95. Dong MQ, Venable JD, Au N, Xu T, Park SK, Cociorva D, Johnson JR, Dillin A and Yates JR, 3rd. Quantitative mass spectrometry identifies insulin signaling targets in C. elegans. Science. 2007; 317:660-663.
96. Depuydt G, Xie F, Petyuk VA, Smolders A, Brewer HM, Camp DG, 2nd, Smith RD and Braeckman BP. LC-MS proteomics analysis of the insulin/IGF-1-deficient Caenorhabditis elegans daf-2(e1370) mutant reveals extensive restructuring of intermediary metabolism. J Proteome Res. 2014; 13:1938-1956.
97. Zarse K, Schmeisser S, Groth M, Priebe S, Beuster G, Kuhlow D, Guthke R, Platzer M, Kahn CR and Ristow M. Impaired insulin/IGF1 signaling extends life span by promoting mitochondrial L-proline catabolism to induce a transient ROS signal. Cell Metab. 2012; 15:451-465.
98. Bharill P, Ayyadevara S, Alla R and Shmookler Reis RJ. Extreme Depletion of PIP3 Accompanies the Increased Life Span and Stress Tolerance of PI3K-null C. elegans Mutants. Front Genet. 2013; 4:34.
99. McElwee JJ, Schuster E, Blanc E, Thornton J and Gems D. Diapause-associated metabolic traits reiterated in long-lived daf-2 mutants in the nematode Caenorhabditis elegans. Mech Ageing Dev. 2006; 127:458-472.
100. Jiang W, Wang S, Xiao M, Lin Y, Zhou L, Lei Q, Xiong Y, Guan KL and Zhao S. Acetylation regulates gluconeogenesis by promoting PEPCK1 degradation via recruiting the UBR5 ubiquitin ligase. Mol Cell. 2011; 43:33-44.
101. Sack MN and Finkel T. Mitochondrial metabolism, sirtuins, and aging. Cold Spring Harb Perspect Biol. 2012; 4.
102. Kok SH, Hou KL, Hong CY, Chao LH, Hsiang-Hua Lai E, Wang HW, Yang H, Shun CT, Wang JS and Lin SK. Sirtuin 6 Modulates Hypoxia-induced Apoptosis in Osteoblasts via Inhibition of Glycolysis: Implication for Pathogenesis of Periapical Lesions. J Endod. 2015; 41:1631-1637.
103. Liu TF, Vachharajani VT, Yoza BK and McCall CE. NAD+-dependent sirtuin 1 and 6 proteins coordinate a switch from glucose to fatty acid oxidation during the acute inflammatory response. J Biol Chem. 2012; 287:25758-25769.
104. De Haes W, Frooninckx L, Van Assche R, Smolders A, Depuydt G, Billen J, Braeckman BP, Schoofs L and Temmerman L. Metformin promotes lifespan through mitohormesis via the peroxiredoxin PRDX-2. Proc Natl Acad Sci U S A. 2014; 111:E2501-2509.
105. Martin-Montalvo A, Mercken EM, Mitchell SJ, Palacios HH, Mote PL, Scheibye-Knudsen M, Gomes AP, Ward TM, Minor RK, Blouin MJ, Schwab M, Pollak M, Zhang Y, Yu Y, Becker KG, Bohr VA, et al. Metformin improves healthspan and lifespan in mice. Nat Commun. 2013; 4:2192.
106. Robida-Stubbs S, Glover-Cutter K, Lamming DW, Mizunuma M, Narasimhan SD, Neumann-Haefelin E, Sabatini DM and Blackwell TK. TOR signaling and rapamycin influence longevity by regulating SKN-1/Nrf and DAF-16/FoxO. Cell Metab. 2012; 15:713-724.
107. Harrison DE, Strong R, Sharp ZD, Nelson JF, Astle CM, Flurkey K, Nadon NL, Wilkinson JE, Frenkel K, Carter CS, Pahor M, Javors MA, Fernandez E and Miller RA. Rapamycin fed late in life extends lifespan in genetically heterogeneous mice. Nature. 2009; 460:392-395.
108. Schulz TJ, Zarse K, Voigt A, Urban N, Birringer M and Ristow M. Glucose restriction extends Caenorhabditis elegans life span by inducing mitochondrial respiration and increasing oxidative stress. Cell Metab. 2007; 6:280-293.
109. Weimer S, Priebs J, Kuhlow D, Groth M, Priebe S, Mansfeld J, Merry TL, Dubuis S, Laube B, Pfeiffer AF, Schulz TJ, Guthke R, Platzer M, Zamboni N, Zarse K and Ristow M. D-Glucosamine supplementation extends life span of nematodes and of ageing mice. Nat Commun. 2014; 5:3563.
110. Xiao R, Zhang B, Dong Y, Gong J, Xu T, Liu J and Xu XZ. A genetic program promotes C. elegans longevity at cold temperatures via a thermosensitive TRP channel. Cell. 2013; 152:806-817.
111. Kaeberlein M, Andalis AA, Fink GR and Guarente L. High osmolarity extends life span in Saccharomyces cerevisiae by a mechanism related to calorie restriction. Mol Cell Biol. 2002; 22:8056-8066.
112. Ito Y, Oumi S, Nagasawa T and Nishizawa N. Oxidative stress induces phosphoenolpyruvate carboxykinase expression in H4IIE cells. Biosci Biotechnol Biochem. 2006; 70:2191-2198.
113. Feuers RJ, Duffy PH, Leakey JA, Turturro A, Mittelstaedt RA and Hart RW. Effect of chronic caloric restriction on hepatic enzymes of intermediary metabolism in the male Fischer 344 rat. Mech Ageing Dev. 1989; 48:179-189.
114. Lin SJ, Kaeberlein M, Andalis AA, Sturtz LA, Defossez PA, Culotta VC, Fink GR and Guarente L. Calorie restriction extends Saccharomyces cerevisiae lifespan by increasing respiration. Nature. 2002; 418:344-348.
115. Moroz N, Carmona JJ, Anderson E, Hart AC, Sinclair DA and Blackwell TK. Dietary restriction involves NAD(+) -dependent mechanisms and a shift toward oxidative metabolism. Aging Cell. 2014; 13:1075-1085.
116. Lopez-Lluch G, Hunt N, Jones B, Zhu M, Jamieson H, Hilmer S, Cascajo MV, Allard J, Ingram DK, Navas P and de Cabo R. Calorie restriction induces mitochondrial biogenesis and bioenergetic efficiency. Proc Natl Acad Sci U S A. 2006; 103:1768-1773.
117. Civitarese AE, Carling S, Heilbronn LK, Hulver MH, Ukropcova B, Deutsch WA, Smith SR and Ravussin E. Calorie restriction increases muscle mitochondrial biogenesis in healthy humans. PLoS Med. 2007; 4:e76.
118. Martin-Montalvo A and de Cabo R. Mitochondrial metabolic reprogramming induced by calorie restriction. Antioxid Redox Signal. 2013; 19:310-320.
119. Owen OE, Smalley KJ, D’Alessio DA, Mozzoli MA and Dawson EK. Protein, fat, and carbohydrate requirements during starvation: anaplerosis and cataplerosis. Am J Clin Nutr. 1998; 68:12-34.
120. Goldrick RB and Hirsch J. Serial Studies on the Metabolism of Human Adipose Tissue. Ii. Effects of Caloric Restriction and Refeeding on Lipogenesis, and the Uptake and Release of Free Fatty Acids in Obese and Nonobese Individuals. J Clin Invest. 1964; 43:1793-1804.
121. Savendahl L and Underwood LE. Fasting increases serum total cholesterol, LDL cholesterol and apolipoprotein B in healthy, nonobese humans. J Nutr. 1999; 129:2005-2008.
122. Moore SC, Patel AV, Matthews CE, Berrington de Gonzalez A, Park Y, Katki HA, Linet MS, Weiderpass E, Visvanathan K, Helzlsouer KJ, Thun M, Gapstur SM, Hartge P and Lee IM. Leisure time physical activity of moderate to vigorous intensity and mortality: a large pooled cohort analysis. PLoS Med. 2012; 9:e1001335.
123. Solon-Biet SM, Mitchell SJ, Coogan SC, Cogger VC, Gokarn R, McMahon AC, Raubenheimer D, de Cabo R, Simpson SJ and Le Couteur DG. Dietary Protein to Carbohydrate Ratio and Caloric Restriction: Comparing Metabolic Outcomes in Mice. Cell Rep. 2015; 11:1529-1534.
124. Solon-Biet SM, Walters KA, Simanainen UK, McMahon AC, Ruohonen K, Ballard JW, Raubenheimer D, Handelsman DJ, Le Couteur DG and Simpson SJ. Macronutrient balance, reproductive function, and lifespan in aging mice. Proc Natl Acad Sci U S A. 2015; 112:3481-3486.
125. Solon-Biet SM, McMahon AC, Ballard JW, Ruohonen K, Wu LE, Cogger VC, Warren A, Huang X, Pichaud N, Melvin RG, Gokarn R, Khalil M, Turner N, Cooney GJ, Sinclair DA, Raubenheimer D, et al. The ratio of macronutrients, not caloric intake, dictates cardiometabolic health, aging, and longevity in ad libitum-fed mice. Cell Metab. 2014; 19:418-430.
126. Trubitsyn AG. The Lag of the Proliferative Aging Clock Underlies the Lifespan-Extending Effect of Calorie Restriction. Curr Aging Sci. 2015; 8:220-226.
127. Trubitsyn AG. Bioenergetics Theory of Aging. In: Clark K, ed. Bioenergetics. (Rijeka, Croatia: InTech). 2012; pp. 63-94.
128. Blagosklonny MV. M(o)TOR of aging: MTOR as a universal molecular hypothalamus. Aging (Albany NY). 2013; 5:490-494.
129. Blagosklonny MV. Aging and immortality: quasi-programmed senescence and its pharmacologic inhibition. Cell Cycle. 2006; 5:2087-2102.
130. Blagosklonny MV. Aging is not programmed: genetic pseudo-program is a shadow of developmental growth. Cell Cycle. 2013; 12:3736-3742.
131. Gems D and de la Guardia Y. Alternative Perspectives on Aging in Caenorhabditis elegans: Reactive Oxygen Species or Hyperfunction? Antioxid Redox Signal. 2013; 19:321-329.
132. Lin YY, Lu JY, Zhang J, Walter W, Dang W, Wan J, Tao SC, Qian J, Zhao Y, Boeke JD, Berger SL and Zhu H. Protein acetylation microarray reveals that NuA4 controls key metabolic target regulating gluconeogenesis. Cell. 2009; 136:1073-1084.
133. Zhao S, Xu W, Jiang W, Yu W, Lin Y, Zhang T, Yao J, Zhou L, Zeng Y, Li H, Li Y, Shi J, An W, Hancock SM, He F, Qin L, et al. Regulation of cellular metabolism by protein lysine acetylation. Science. 2010; 327:1000-1004.
134. Hallows WC, Yu W and Denu JM. Regulation of glycolytic enzyme phosphoglycerate mutase-1 by Sirt1 protein-mediated deacetylation. J Biol Chem. 2012; 287:3850-3858.
135. Lim JH, Lee YM, Chun YS, Chen J, Kim JE and Park JW. Sirtuin 1 modulates cellular responses to hypoxia by deacetylating hypoxia-inducible factor 1alpha. Mol Cell. 2010; 38:864-878.
136. Shi T, Wang F, Stieren E and Tong Q. SIRT3, a mitochondrial sirtuin deacetylase, regulates mitochondrial function and thermogenesis in brown adipocytes. J Biol Chem. 2005; 280:13560-13567.
137. Guarente L. Calorie restriction and sirtuins revisited. Genes Dev. 2013; 27:2072-2085.
138. Ho L, Titus AS, Banerjee KK, George S, Lin W, Deota S, Saha AK, Nakamura K, Gut P, Verdin E and Kolthur-Seetharam U. SIRT4 regulates ATP homeostasis and mediates a retrograde signaling via AMPK. Aging (Albany NY). 2013; 5:835-849.
139. Zeng L, Yang Y, Hu Y, Sun Y, Du Z, Xie Z, Zhou T and Kong W. Age-related decrease in the mitochondrial sirtuin deacetylase Sirt3 expression associated with ROS accumulation in the auditory cortex of the mimetic aging rat model. PLoS One. 2014; 9:e88019.
140. Poulose N and Raju R. Sirtuin regulation in aging and injury. Biochim Biophys Acta. 2015; 1852(11):2442-2455.
141. Schwer B, Eckersdorff M, Li Y, Silva JC, Fermin D, Kurtev MV, Giallourakis C, Comb MJ, Alt FW and Lombard DB. Calorie restriction alters mitochondrial protein acetylation. Aging Cell. 2009; 8:604-606.
142. Marton O, Koltai E, Nyakas C, Bakonyi T, Zenteno-Savin T, Kumagai S, Goto S and Radak Z. Aging and exercise affect the level of protein acetylation and SIRT1 activity in cerebellum of male rats. Biogerontology. 2010; 11:679-686.