
INTRODUCTION
Secondary immunoglobulin diversification by somatic hypermutation and class switch recombination in germinal centers is the major prerequisite for maturation of the humoral adaptive immune response [1, 2]. However, it also includes the danger of genetic instability, as other cellular genes may also be affected by mutagenesis or chromosomal translocations [3, 4]. Accordingly, both the enzymes introducing DNA damage as well as the machinery responsible for DNA repair need to be tightly regulated in germinal center B cells to on the one hand promote their efficient function, but on the other hand prevent their deregulation leading to cellular transformation and hence lymphomagenesis.
AID is the key enzyme initiating hypermutation and class switch recombination by transcription-coupled deamination of cytidines [5–7]. Expression and activity of the AID protein is regulated at multiple levels, including its cellular localization, posttranslational modifications and stability [8–10]. While the main compartment of AID activity is clearly the nucleus, most of the protein is retained in the cytoplasm in the steady state situation based on the coordinate action of heat shock proteins and eEF1A [11–14]. A constant nuclear/cytoplasmic shuttling of AID is mediated by a N-terminal structural nuclear localization sequence (NLS) and a nuclear export sequence (NES) encompassing the C-terminal 14 amino acids [15–18]. Trapping of AID in the nucleus, e.g. by inhibition of CRM1/exportin1 via leptomycin B, leads to its rapid degradation, which involves the proteasome and may occur in both a ubiquitination-dependent as well as -independent manner [9, 19].
AID can also be modified by phosphorylation at serine (S38) and threonine (T27, T140) residues [20–22]. S38 phosphorylation by protein kinase A (PKA) in the nucleus has been shown to be involved in binding of AID to replication protein A (RPA), a process that increases AID processivity on DNA and hence the efficiency of AID-mediated mutagenesis [23]. While AID may be detected at many gene loci [24], PKA activity appears to be specifically targeted to immunoglobulin loci in germinal center B cells [25]. Locus-specificity of somatic hypermutation is also increased by differential DNA repair of AID-induced DNA lesions, which is error-prone in immunoglobulin genes but error-free in many other cellular genes that are not affected by hypermutation in the germinal center [26].
While proficient AID activity is required at hypermutating gene loci in B cells to ensure rapid adaptation of the humoral immune response, a major risk for genetic instability caused by AID activity can occur upon introduction of excessive damage into the genome. This accumulation of DNA lesions may overwhelm the DNA repair capacity of the cell or give rise to multiple strand breaks that can be processed to chromosomal translocations [27, 28]. Such a scenario could be prevented by negative feedback loops by which DNA damage-associated signals avoid introduction of more damage by counteracting nuclear AID activity. However, at least one positive feedback loop has also been identified: introduction of strand breaks promotes AID phosphorylation and hence its activity at the immunoglobulin locus [29, 30].
In the present study, we asked whether DNA damage affects other aspects of posttranslational regulation of AID, in particular its nuclear localization and/or degradation. Unexpectedly, we found that cytotoxic drugs that activate base excision repair (which is also activated by AID) lead to the nuclear accumulation and stabilization of the AID protein. Studies using inhibitors as well as knockout cell lines indicate that activation of poly(ADP-ribose) polymerase (PARP) is required for efficient nuclear AID accumulation and stabilization. These findings define the first molecular pathway that may lead to nuclear accumulation of AID, with interesting potential implications for the regulation of AID function as well as for lymphoma therapy.
RESULTS
Proteasomal nuclear degradation of AID-GFP fusions in human and chicken B cells
To perform our studies on AID localization and degradation in easily tractable hypermutating B cell lines, we first tested whether the degradation of AID in human Raji and chicken DT40 B cell lymphoma lines follows the same mechanism observed before for human BL2 cells [9]. We used human AID fusions carrying a GFP gene either at the C-terminus or N-terminus and containing or lacking the last 14 amino acids comprising the AID NES. In the steady state situation, the wild-type AID fusion proteins were localized in the cytoplasm, while the truncated proteins accumulated in the nucleus (Supplementary Figure S1A). Also, clones with AID fusions containing a NES were on average substantially more highly fluorescent than those lacking it, irrespective of whether GFP was fused to the N- or C-terminus (Supplementary Figure S1B and S1C). Induction of degradation of AID fusions, measured by loss of fluorescence relative to the control sample, required the addition of the nuclear export inhibitor leptomycin B (LMB) or translational inhibition by cycloheximide (CHX) for the wild-type protein; the strongest effect was induced by the combination of these drugs (Supplementary Figure S1D). While the endogenous AID protein disappeared after 8 hours of CHX/LMB treatment, a GFP control protein remained stable (Supplementary Figure S1E). Obviously, the proteasome was involved in nuclear AID degradation, as its inhibition with MG 132 decreased AID degradation (Supplementary Figure S1D). Accordingly, in human Raji cells and chicken DT40 lymphoma cells, the AID protein is degraded by the ubiquitin-proteasome pathway in the nucleus, as shown before for BL2 cells [9]. For further experiments, AID fusions with GFP at the C-terminus were used.
DNA damage increases AID protein stability in the nucleus
To analyze whether (AID-induced) DNA damage might impact on AID degradation, we applied cytotoxic drugs. Intriguingly, methyl methanesulfonate (MMS) and H2O2, both of which similarly to AID initially activate base excision repair, also decreased the degradation of AID fusions trapped in the nucleus (Figure 1A and 1B). Etoposide, a Topoisomerase II inhibitor inducing DNA double strand breaks, caused a very moderate inhibition of AID degradation in chicken lymphoma cells but barely so in human Raji lymphoma cells (Figure 1C). The nucleotide excision repair inducing drug cisplatin did not have this effect at any concentration tested (Figure 1D). All drugs tested caused DNA damage as indicated by Chk1 phosphorylation (Supplementary Figure S2) at the concentrations used. We also observed a restricted AID degradation under MMS and H2O2 in mouse CH12F3 lymphoma cells (Supplementary Figure S3 [31]).
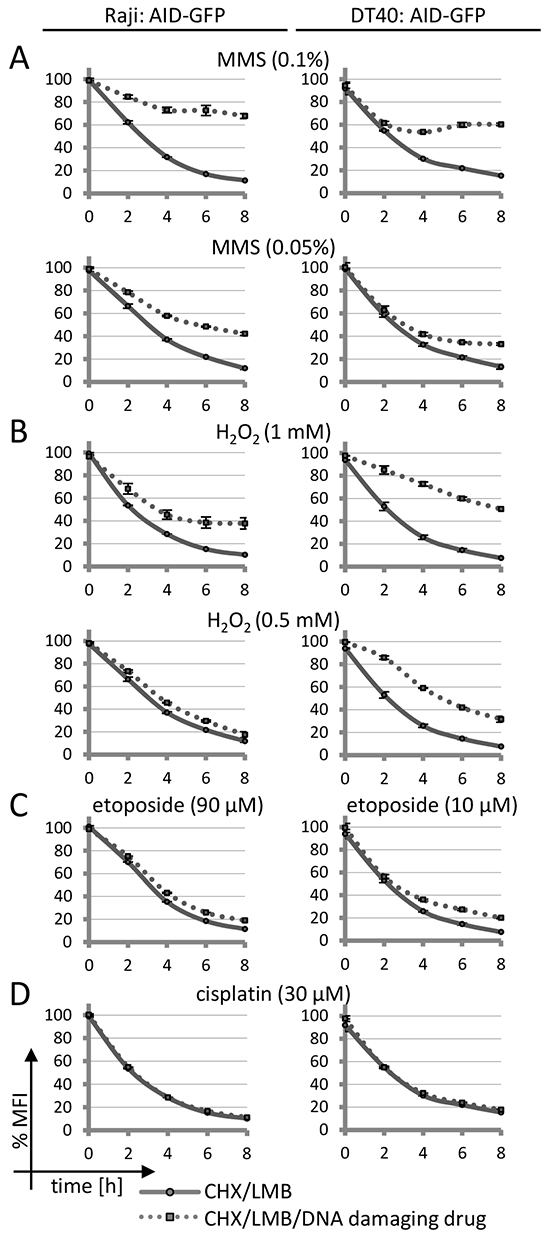
Figure 1: Effects of DNA damage on AID-GFP protein degradation. A–D. FACS analysis of human Raji or chicken DT40 B cell lines stably transfected with an AID-GFP construct: degradation of the AID-GFP proteins trapped in the nucleus upon treatment with the indicated drugs. Untreated cells are set to 100% MFI (geometric mean fluorescence intensity). Relative MFI values are given as a function of time with standard deviation for duplicates. The experiment was performed with two independent transfectants and is representative of more than three independent experiments.
Inhibition of proteasomal AID degradation by DNA damage might indicate that the protein can no longer enter the nucleus in order to be degraded. However, confocal microscopy revealed that in contrast to this assumption, MMS and H2O2 induced a substantial nuclear accumulation of AID-GFP fusions even if no LMB was added to the assay (Figure 2A and 2C). No effect was seen for the GFP protein itself (Figure 2B), implying that the effects are implemented via the AID portion of the fusions. Cisplatin and etoposide did not show significant effects on AID localization, even at etoposide concentrations that induced nuclear fragmentation in many cells. We conclude that drugs activating base excision repair lead to a nuclear accumulation of AID that is coupled to inhibition of nuclear proteasomal AID degradation. These findings imply that a processing intermediate of the damage induced by MMS and H2O2 leads to a reaction that interferes with nuclear AID degradation, and thus causes nuclear AID accumulation.
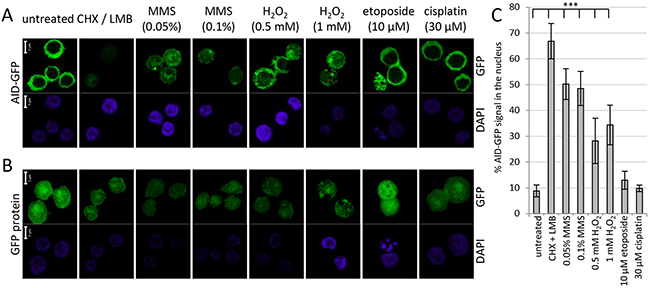
Figure 2: Effects of DNA damage on protein localization of AID-GFP. Localization of AID-GFP A. and the GFP protein itself B. in DT40 lymphoma cells after 6 hours of treatment with the indicated drugs; scale bar: 5 μm. C. Quantification of the experiment shown in A, analyzing 7 to 12 cells for each condition. ***: p < 0.0001 (student’s t-test).
To confirm that a stabilization of AID by DNA damage is also observed for the endogenous protein, we studied AID degradation by Western blots. MMS- and H2O2-derived DNA lesions also caused a stabilization of the endogenous AID protein (Figure 3A), while cisplatin and etoposide had no such effect. Also, fractionation experiments showed that the endogenous AID protein accumulates in the nucleus upon MMS- and H2O2, but not cisplatin treatment (Figure 3B). These results substantiate an altered subcellular localization and an increased protein stability of nuclear AID as a consequence of exogenous DNA damage inducing the base excision repair pathway.
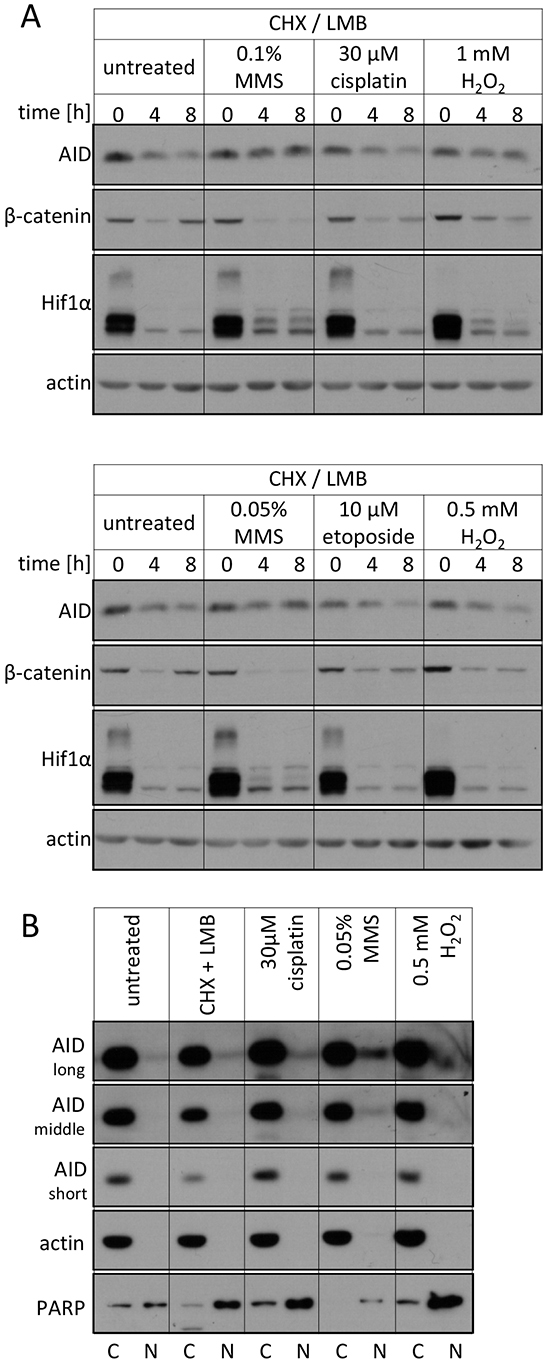
Figure 3: Effects of DNA damage on the endogenous AID protein. A. Western blot analysis of total endogenous AID protein levels in Raji B cells upon treatment with the indicated drugs. Representative blots of 3 independent experiments are shown. β-catenin and Hif1α serve as controls for irrelevant unstable proteins. B. Western blot analysis of endogenous AID protein levels in cytoplasmic and nuclear fractions of Raji B cells upon treatment with the indicated substances for 5 hours. Three different exposures for AID are shown. The quality of separation was tested with the compartment markers actin (cytosolic) and PARP (nuclear). C: cytoplasmic, N: nuclear. Data are representative of two independent experiments.
PARP inhibition prevents AID stabilization and nuclear accumulation
MMS and H2O2 both mainly activate the base excision repair pathway, as does AID itself. This repair pathway is based on the excision of modified bases by different DNA glycosylases chosen depending on the damaged base, followed by incision at the abasic site by apurinic endonucleases (APE1 and 2), and ligation involving re-insertion of the correct base. This last step involves recruitment of PARP to the strand breaks generated, thus triggering poly(ADP-ribosylation) of PARP itself and other proteins, and leading to recruitment of multiple factors for enhanced efficacy of DNA repair. Indeed, of the drugs used in this study, only MMS and H2O2 were able to efficiently induce PARylation of proteins (Supplementary Figure S2).
In order to assess whether intermediates of base excision repair may lead to nuclear AID stabilization, we first analyzed whether inhibitors of PARP affect the process. Indeed, inhibition of PARP activity with several different chemical inhibitors substantially interfered with AID stabilization by MMS treatment in human, chicken and mouse cells (Figure 4A and 4B and Supplementary Figure S4), implying a role for PARP activation in nuclear AID accumulation. Thus, activation of PARP by MMS treatment appears to be responsible for efficient nuclear stabilization and accumulation of AID.
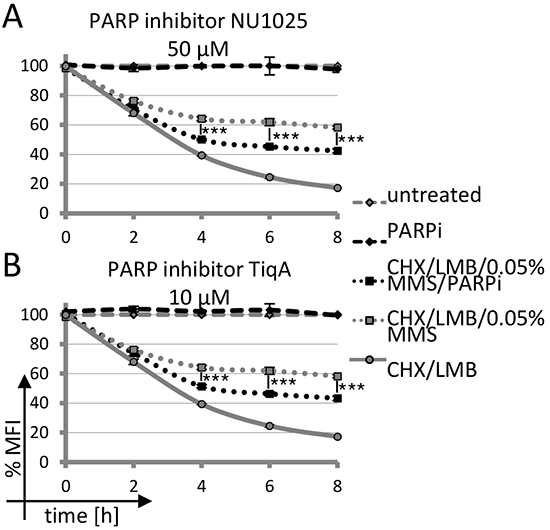
Figure 4: Impact of PARP inhibition on AID-GFP degradation upon DNA damage. A, B. Kinetics of degradation of nuclear AID-GFP fusions in Raji B cells treated with 0.05% MMS and PARP inhibitor. Untreated cells are set to 100% MFI. Relative MFI values are given; significance analysis was performed for six technical replicates (student’s t-test), ***: p < 0.001. Data are representative of three independent experiments.
Nuclear AID stabilization is impaired in PARP-1 knockout cells
To rule out that an off-target activity of the PARP inhibitors caused the observed effect on AID stabilization, we wanted to confirm our results in a clearcut genetic system. In mammalian cells, however, PARP-1 and PARP-2 both contribute to DNA repair, making genetic analyses complicated. We thus resorted to using PARP-1 knockout DT40 B lymphoma cells, as these apparently do not harbor a PARP-2 gene [32]. The kinetics of degradation of AID-GFP fusions trapped in the nucleus by LMB was similar in wild-type and PARP-1-/- DT40 cells (Figure 5A). However, additional MMS- or H2O2-treatment led to a significantly reduced AID stabilization in the PARP-1-/- cells (Figure 5A and 5B) as compared to wild-type cells. In agreement with this, MMS- or H2O2-treatment led to a significantly lower nuclear AID accumulation in PARP-1-/- cells (Figure 5C and 5D). We thus conclude that nuclear activation of PARP, induced here by DNA damage, is capable of promoting nuclear stabilization of the inherently unstable AID protein, leading to its accumulation at its site of action.
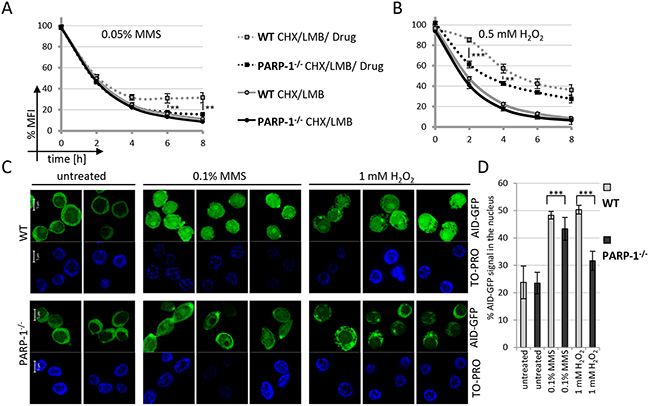
Figure 5: Nuclear AID stabilization is impaired in PARP-1 knockout cells. FACS analysis of nuclear degradation of AID-GFP in wild-type and PARP-1-/- cells and stabilization upon treatment with MMS A. and H2O2 B. Untreated cells are set to 100% MFI. Relative MFI values of five independent clones per condition are given as a function of time with the indicated standard deviation. P-values for wild-type compared to PARP-1-/- cells after CHX / LMB / MMS treatment for different time points are given (student’s t-test), **: p < 0.01, ***: p < 0.001. Data are representative of two independent experiments each. C. Subcellular localization of AID-GFP fusions 4 hours after treatment with MMS and H2O2; scale bar: 5 μm. Data are representative of two independent experiments. Note some focal accumulation of AID at a single spot in the cytoplasm observed in this and some other experiments. D. Quantification of the experiment shown in C, analyzing 15 cells each from two independent clones per condition. ***: p < 0.0001(student’s t-test).
Stabilization and nuclear accumulation of AID mutants
A previous study has shown a positive feedback loop of AID activation by phosphorylation, dependent on AID activity-induced DNA damage [30]. To assess whether AID stabilization in the nucleus depends on its activity or phosphorylation, we generated AID mutants defective in either process (Supplementary Figure S5A). Mutagenesis of the zinc coordinating residues H56 and E58 perturbs the cytidine deaminase active center of AID [33]. Enzymatic inactivation is also achieved by mutagenesis of R19 and R24 in a loop that extends close to the active center in wild-type AID but apparently restricts substrate access in the respective mutant [34]. Mutagenesis of S38 impairs PKA-mediated phosphorylation and thus RPA binding of AID, an effect that is exacerbated by additional mutagenesis of T27 [35].
The catalytic (but not the phosphorylation) mutants of the AID-GFP fusion proteins showed a clearly reduced protein expression (Supplementary Figure S5B). However, these differences in protein levels are apparently not due to major differences in protein degradation (Supplementary Figure S5C), as this occurred to a comparable rate in all mutants. Most importantly, though, all the mutant AID proteins were stabilized in the nucleus by MMS treatment, as was the wild-type protein (Supplementary Figure S5D in comparison to S5C). We thus conclude that in the experimental system used here, the AID activity exerted by the AID-GFP fusions alone is not sufficient to promote their stabilization to a degree measurable with our current assays. Also, mutants with defective catalytic function or impaired activation by PKA still accumulate in the nucleus upon exogenously applied DNA damage.
Nuclear AID stabilization as a consequence of cancer therapy
As AID stabilization in the nucleus was mostly observed using cytotoxic drugs, we wished to assess the relevance of this finding to cancer therapy. MMS is a DNA alkylating drug [36], as are other chemotherapy agents commonly used in lymphoma therapy, such as cyclophosphamide [37]. Its active metabolite alkylates N7 of guanine and generates DNA crosslinks, which are resolved by base excision repair [38, 39]. Indeed, we found that 4-hydroperoxy-cyclophosphamide positively affects AID stabilization (Supplementary Figure S6A) as well as nuclear AID accumulation (Supplementary Figure S6B and S6C) in human Burkitt’s lymphoma cells at a concentration at or above that found in cancer patients upon treatment [40, 41]. We conclude that AID may be stabilized in the nucleus during lymphoma therapy using alkylating drugs such as cyclophosphamide.
DISCUSSION
In the present study, we show that PARP inhibition counteracts nuclear stabilization and accumulation of the AID protein upon treatment with alkylating drugs causing exogenous DNA damage. We thereby identify PARP(-1) as the first molecule that, upon induction of DNA damage, may increase the nuclear concentration of this enzyme critical for immunoglobulin diversification and lymphomagenesis. As nuclear AID is highly more active in immunoglobulin mutagenesis than its cytoplasmic variant (Supplementary Figure S7), our data suggest that this mechanism might contribute to increased mutagenesis during lymphoma treatment.
Two previous studies have observed nuclear AID accumulation in adherent (non-B) cells upon induction of DNA strand breaks [42] or treatment with etoposide [43]. In both cases, nuclear AID accumulation affected only a minor proportion of the cells, in contrast to the substantial and homogenous AID accumulation we have observed in all the B cell lines used in this study. Notably, the most potent inducers of nuclear AID accumulation in our study were drugs inducing DNA alkylation and subsequent base excision repair (which is also induced by AID) [36, 38], rather than drugs inducing DNA strand breaks. Also, in case of etoposide treatment, only a very moderate (and PARP-independent, data not shown) nuclear AID stabilization was observed in the chicken lymphoma cell line in association with nuclear fragmentation. These findings may imply different DNA damage responses and different mechanisms of nuclear AID accumulation in B versus non-B cells, and in fact, we did not observe nuclear AID accumulation upon MMS treatment in several adherent cell lines analyzed (data not shown).
For the B cells undergoing immunoglobulin diversification studied here, we show for the first time that nuclear AID accumulation is accompanied by interference with nuclear proteasomal AID degradation, allowing a first glance at the molecular mechanism(s) involved. Also, we identify PARP(-1) as one responsible enzyme for this phenomenon. Presently, the PARP family consists of 17 members and the ubiquitous nuclear PARP-1 [44] is essential for the repair of DNA single-strand breaks via the base excision repair pathway [45] and responsible for around 90% of ADP-ribose polymer synthesis after DNA damage. Together with PARP-2, homo- and heterodimers can be formed and a role for PARP-2 in base excision repair was also detected [46]. Currently, in human cells we cannot distinguish by which PARP enzyme the effects observed are mediated, as the inhibitors used in our studies should target both PARPs. However, PARP-1 is the more likely candidate as deduced from its prominent activity.
It also remains to be seen whether AID stabilization upon PARP activation involves direct or indirect interaction of AID with (activated) PARP, modulation (e.g. poly(ADP-ribosylation)) of a protein involved in proteasomal AID degradation, or e.g. PARP-mediated activation of another DNA processing or DNA damage signaling pathway affecting nuclear AID stability. DNA damage is signaled to the cell by a variety of pathways, including e.g. checkpoint signaling, activation of MAP kinases or induction of NFκB activity [47–49]. In stark contrast to these mostly global DNA damage signals, PARP activation delivers a local signal, and thus triggers highly localized effects [50]. PARP accumulation at sites of DNA damage or at single strand breaks leads to automodification of PARP by long and branched ADP-ribose polymers. These serve as binding sites for DNA repair factors as well as other proteins, thereby enhancing the efficacy of DNA damage processing and repair [50]. Therefore, PARP-1/2, are powerful targets to increase the efficiency of cytotoxic chemotherapeutic agents inducing DNA damage, and PARP inhibitors are thus frequently used in combination therapy with alkylating and other cytotoxic agents.
We presume that in a physiological situation, PARP triggers AID accumulation directly at the site of DNA damage, as its PARylation function is required for the effect. In order to observe a global accumulation of AID in the nucleus of B cells upon PARP activation, we needed to induce substantial global DNA damage using high doses of exogenously applied DNA damaging agents, comparable to a patient’s situation during chemotherapy, but potentially precluding the detection of foci that were apparent in a previous study [42]. Even though AID can be found at many gene loci in B cells undergoing immunoglobulin diversification, only some of these show evidence of DNA damage induction [24, 51]. It is thus not surprising that under steady state conditions in the absence of exogenously induced DNA damage, we cannot detect an influence of PARP on AID degradation. Given the higher AID activity in primary cells or in vivo, it will be worthwhile to study PARP effects on overall nuclear AID stability in other experimental systems, although our knowledge of PARP biology makes strong global effects unlikely.
Local effects of PARP, however, may be highly interesting in the context of immunoglobulin diversification, which requires locus-specific regulation of AID activity as well as of DNA repair processes. PARP is not only activated upon DNA damage to promote DNA repair, but also interacts with several transcription factors and affects transcriptional activity of gene loci [52, 53], among them the hypermutating Bcl-6 locus [54]. In addition, PARP was reported to affect DNA repair fidelity and pathway choice during immunoglobulin diversification [32, 55–60]. It will thus be highly interesting to study whether PARP affects AID stability and activity in a locus-specific manner in B cells undergoing immunoglobulin diversification, and if so, how activation of PARP is regulated in immunoglobulin genes as compared to other genes not undergoing diversification.
While an understanding of a potential physiological AID regulation via PARP will likely require more sophisticated experimental approaches, the form of AID regulation observed here may be of special importance in case of chemotherapy. According to our findings, such treatments may enhance nuclear AID accumulation, with potential impact on AID-induced mutagenesis and tumorigenesis once AID expression becomes deregulated. AID is a potent mutator that can induce aberrant somatic hypermutation of non-immunoglobulin genes and even genome kataegis [61–65]. Simultaneous application of PARP inhibitors appears to reduce nuclear AID accumulation and might thus preserve cells from aberrant AID function. Combined chemotherapy with additional PARP inhibitors is the subject of recent investigations and clinical trials [66, 67], e.g. for cyclophosphamide [68]. Our study shows that PARP(-1) inhibition or inactivation reduces the DNA damage-induced stabilization and accumulation of nuclear AID, providing an additional rationale for this therapeutic approach in lymphoma therapy.
MATERIALs AND METHODS
Cell culture and transfection
The human Burkitt’s lymphoma cell line Raji and the chicken lymphoma cell lines DT40 Cre1 and DT40 ψV- were cultured as described before [69]. The mouse CH12F3 B cells were maintained in RPMI 1640 supplemented with 10% FCS, 0.05 mM 2-mercaptoethanol (Sigma) and 10 mM Hepes (Invitrogen). The DT40 PARP-1 knockout cell set was provided by S. Takeda [32]. Transfections were carried out as described before [69] with a Gene Pulser Xcell (Bio Rad) set at 50 μF and 800 V for DT40 cells, at 850 μF and 250 V for Raji cells and at 400 μF and 400 V for CH12F3 cells. Stable transfectants were selected by addition of 0.5 μg/ml puromycin (Sigma Aldrich) and further tested by flow cytometry and Western blot. For analysis of somatic hypermutation, DT40ψV- AID-/- cells were stably transfected with the respective vectors, the resultant single cell clones were directly cultured for 19 days and stained with anti-chicken-IgM-PE (8310-09, Southern Biotech) before flow cytometry analysis.
Plasmids and site-directed mutagenesis
pCAGGs vectors containing AID-GFP fusions cloned at the EcoRI site, bringing the AID gene under the transcriptional control of the chicken beta-actin promoter [70], were obtained from J. Bachl. For site-directed mutagenesis, the human AID coding sequence was introduced into the EcoRI site of pBluescriptKS (Stratagene). PCR was performed for 30 cycles with primers annealing at 60°C for 7 min and elongation at 72°C for 1 min, using the primers depicted in Supplementary Table S1. Subsequently, the non-mutated strand was cut with DpnI (Fermentas) and the mutated DNA was introduced into E. coli DH5α. The T27A/S38A double mutant was created by the introduction of the T27A mutation into the S38A mutant, while the R19E/R24E and H56R/E58Q double mutants were generated in a single mutagenesis step. Appropriate AID clones were confirmed by sequence analysis and subcloned into the pCAGGs vector.
Induction of DNA damage and analysis of AID localization and degradation
DNA damage was induced by the following agents: etoposide (10 - 90 μM, Sigma Aldrich), cisplatin (30 μM, Ribosepharm), methyl methanesulfonate (MMS, 0.05 - 0.1%, Merck), and H2O2 (0.5 - 1 mM, Sigma-Aldrich). 4-hydroperoxy-cyclophosphamide was purchased from NIOMECH-IIT GmbH in aliquots, and for each experiment a fresh aliquot was dissolved in water and used directly. Protein translation was inhibited by addition of cycloheximide (CHX, 20 μg/ml, Sigma-Aldrich) and AID nuclear export was abrogated with leptomycin B (LMB, 5 ng/ml, Sigma-Aldrich). For additional treatment with inhibitors, the following final concentrations were used: MG132 (Calbiochem®): 10 μM; TiqA (Sigma-Aldrich): 10 μM; NU1025 (Santa Cruz): 50 μM and 3-Aminobenzamide (3-AB, Calbiochem®): 1 mM. For degradation kinetics, cells were analyzed using a CantoII (Becton Dickinson) in two hour intervals for a period of 8 hours followed by data assessment using FlowJo Software. GFP signals of living cells (identified by forward scatter analysis) were calculated as relative MFI (geometric mean fluorescence intensity) percentages, setting the MFI of untreated cells to 100 percent.
For confocal microscopy, cells were treated with the indicated agents for 4 to 6 hours. A total of 5×105 cells in 1 ml were transferred onto cover slips precoated with poly-L-lysine (Sigma-Aldrich) and incubated for 15 min at 37°C, followed by 15 min fixation with 2% paraformaldehyde (Carl Roth) at room temperature. After washing with PBS, cells were permeabilized by 15 min treatment with 0.15% Triton-X-100 in PBS (Sigma-Aldrich) and subsequently stained with 100 ng/ml DAPI (Invitrogen) or 1 μM TO-PRO-3 (ThermoFisher scientific). Samples were scanned with a Zeiss LSM 510 laser scanning confocal device using a 63x Plan-Apochromat oil objective (Carl Zeiss). GFP and DAPI or TO-PRPO-3 were excited by laser light of 488 nm and 405 nm or 633 nm wavelength, respectively. Each signal was scanned independently by the multitracking function of the LSM 510 unit. Within each experiment, the 488 nm laser light was used at constant intensity in order to visualize changes in GFP intensities.
Quantification of confocal data was performed with the ZENblue software of Carl Zeiss Jena. For each cell, 6 ROIs (region of interest) were defined for the nucleus and 6 ROIs for the cytoplasm. The GFP MFI was determined by the software and the arithmetic average of the GFP signal in the nucleus plus the cytoplasm was set to 100%.
Expression analysis of AID and AID mutants
12 to 13 days after electroporation of 1×107 cells, single cell clones were analyzed for GFP signals by FACS analysis. FACS data were analyzed using FlowJo Software (Tree Star Inc., USA), gating for viable cells through scatter analyses. Western blots were done as described previously [71], using the following antibodies: α-AID (clone EK2/5G9, E.K.), α-GFP (B-2, Santa Cruz Biotechnology Inc., USA), α-actin (A-2066, Sigma-Aldrich), α-tubulin (ab59680, Abcam), α-PARP (ab32071, Abcam), α-P-Chk1 (Ser345) (Cell Signalling Technologies #2341), α-PARylation (ALX-202-043, Enzo), α-β-catenin (610154, BD transduction laboratories) and α-Hif1α (Cell Signalling Technologies #3716).
For cell fractionation, 1×106 cells were resuspended in 1 volume of buffer NE-A (10 mM HEPES, pH 7.6; 30 mM KCl; 2 mM MgCl2; 0.1 mM EDTA;1 mM DTT; supplemented with proteinase inhibitor (Roche)) for 10 min on ice. After addition of another volume of buffer NE-A including 0.2% NP40 and 5 min incubation on ice, nuclei were separated by centrifugation (5 min, 700 rfc, 4°C). The supernatant is the cytoplasmic fraction. Nuclei were washed in PBS, resuspended in buffer NE-B (20 mM HEPES, pH 7.9; 420 mM NaCl; 25% glycerol; 0.2 mM EDTA; 1 mM DTT supplemented with proteinase inhibitor) and incubated on ice for 10 min. Following 3 freeze thaw cycles, the sample was further centrifuged (10 min, 15,000 rpm, 4°C) and the soluble fraction was collected.
ACKNOWLEDGMENTS
We thank S. Takeda for providing the PARP-1-/- DT40 cells, S. Fischer-Burkart for expert technical help, and Stefanie Voigt and Franziska Feiertag for sharing materials and expertise. We are indebted to D. Krappmann, H. Leonhardt, F. Grosse and Z.Q. Wang for stimulating discussions, and to all members of the Jungnickel laboratory for critical reading of the manuscript.
Conflicts of Interest
The authors have no conflict of interest or financial interest.
GRANTS SUPPORT
This work was supported by the Wilhelm Sander foundation [2003/046/2 to B.J.], a Heisenberg fellowship of the Deutsche Forschungsgemeinschaft [JU2690/2-1 to B.J.], a postdoctoral fellowship from the HGF-Wiedereinstiegsfonds [WP-FBG-09-2005 to I.P] and a PhD fellowship of the Ernst-Abbe-Foundation to S.T.
Author contributions
S.T., J.J., I.P., P.H. and B.J. designed experiments and analyzed data; S.T., J.J., K.B., A.S. K.D. and P.M. performed experiments; E.K. provided essential reagents; B.J. and S.T. wrote the manuscript.
REFERENCES
1. Jungnickel B. False moves for survival: error-prone DNA repair in adaptive immunity. Cell Cycle. 2006; 5:2856-2861.
2. Rajewsky K. Clonal selection and learning in the antibody system. Nature. 1996; 381:751-758.
3. Pasqualucci L, Neumeister P, Goossens T, Nanjangud G, Chaganti RS, Kuppers R and Dalla-Favera R. Hypermutation of multiple proto-oncogenes in B-cell diffuse large-cell lymphomas. Nature. 2001; 412:341-346.
4. Kuppers R. Mechanisms of B-cell lymphoma pathogenesis. Nat Rev Cancer. 2005; 5:251-262.
5. Muramatsu M, Sankaranand VS, Anant S, Sugai M, Kinoshita K, Davidson NO and Honjo T. Specific expression of activation-induced cytidine deaminase (AID), a novel member of the RNA-editing deaminase family in germinal center B cells. J Biol Chem. 1999; 274:18470-18476.
6. Okazaki I, Yoshikawa K, Kinoshita K, Muramatsu M, Nagaoka H and Honjo T. Activation-induced cytidine deaminase links class switch recombination and somatic hypermutation. Ann N Y Acad Sci. 2003; 987:1-8.
7. Honjo T, Kinoshita K and Muramatsu M. Molecular mechanism of class switch recombination: linkage with somatic hypermutation. Annu Rev Immunol. 2002; 20:165-196.
8. Di Noia JM and Neuberger MS. Molecular mechanisms of antibody somatic hypermutation. Annu Rev Biochem. 2007; 76:1-22.
9. Aoufouchi S, Faili A, Zober C, D’Orlando O, Weller S, Weill JC and Reynaud CA. Proteasomal degradation restricts the nuclear lifespan of AID. J Exp Med. 2008; 205:1357-1368.
10. Vuong BQ and Chaudhuri J. Combinatorial mechanisms regulating AID-dependent DNA deamination: Interacting proteins and post-translational modifications. Seminars in Immunology. 2012; 24:264-272.
11. Orthwein A, Zahn A, Methot SP, Godin D, Conticello SG, Terada K and Di Noia JM. Optimal functional levels of activation-induced deaminase specifically require the Hsp40 DnaJa1. EMBO J. 2012; 31:679-691.
12. Orthwein A and Di Noia JM. Activation induced deaminase: How much and where? Seminars in Immunology. 2012; 24:246-254.
13. Methot SP, Litzler LC, Trajtenberg F, Zahn A, Robert F, Pelletier J, Buschiazzo A, Magor BG and Di Noia JM. Consecutive interactions with HSP90 and eEF1A underlie a functional maturation and storage pathway of AID in the cytoplasm. The Journal of Experimental Medicine. 2015; 212:581-596.
14. Häsler J, Rada C and Neuberger MS. Cytoplasmic activation-induced cytidine deaminase (AID) exists in stoichiometric complex with translation elongation factor 1α (eEF1A). Proceedings of the National Academy of Sciences. 2011; 108:18366-18371.
15. Patenaude AM and Di Noia JM. The mechanisms regulating the subcellular localization of AID. Nucleus. 2010; 1:325-331.
16. Ito S, Nagaoka H, Shinkura R, Begum N, Muramatsu M, Nakata M and Honjo T. Activation-induced cytidine deaminase shuttles between nucleus and cytoplasm like apolipoprotein B mRNA editing catalytic polypeptide 1. Proc Natl Acad Sci U S A. 2004; 101:1975-1980.
17. Häsler J, Rada C and Neuberger MS. The cytoplasmic AID complex. Seminars in Immunology. 2012; 24:273-280.
18. Patenaude A-M, Orthwein A, Hu Y, Campo VA, Kavli B, Buschiazzo A and Di Noia JM. Active nuclear import and cytoplasmic retention of activation-induced deaminase. Nat Struct Mol Biol. 2009; 16:517-527.
19. Uchimura Y, Barton LF, Rada C and Neuberger MS. REG-γ associates with and modulates the abundance of nuclear activation-induced deaminase. The Journal of Experimental Medicine. 2011; 208:2385-2391.
20. Basu U, Chaudhuri J, Alpert C, Dutt S, Ranganath S, Li G, Schrum JP, Manis JP and Alt FW. The AID antibody diversification enzyme is regulated by protein kinase A phosphorylation. Nature. 2005; 438:508-511.
21. Stavnezer J. Complex regulation and function of activation-induced cytidine deaminase. Trends Immunol. 2011; 32:194-201.
22. McBride KM, Gazumyan A, Woo EM, Schwickert TA, Chait BT and Nussenzweig MC. Regulation of class switch recombination and somatic mutation by AID phosphorylation. J Exp Med. 2008; 205:2585-2594.
23. Chaudhuri J, Khuong C and Alt FW. Replication protein A interacts with AID to promote deamination of somatic hypermutation targets. Nature. 2004; 430:992-998.
24. Yamane A, Resch W, Kuo N, Kuchen S, Li Z, Sun H-w, Robbiani DF, McBride K, Nussenzweig MC and Casellas R. Deep-sequencing identification of the genomic targets of the cytidine deaminase AID and its cofactor RPA in B lymphocytes. Nat Immunol. 2011; 12:62-69.
25. Vuong BQ, Lee M, Kabir S, Irimia C, Macchiarulo S, McKnight GS and Chaudhuri J. Specific recruitment of protein kinase A to the immunoglobulin locus regulates class-switch recombination. Nat Immunol. 2009; 10:420-426.
26. Liu M, Duke JL, Richter DJ, Vinuesa CG, Goodnow CC, Kleinstein SH and Schatz DG. Two levels of protection for the B cell genome during somatic hypermutation. Nature. 2008; 451:841-845.
27. de Yébenes VG and Ramiro AR. Activation-induced deaminase: light and dark sides. Trends in Molecular Medicine. 2006; 12:432-439.
28. Ramiro AR, Jankovic M, Eisenreich T, Difilippantonio S, Chen-Kiang S, Muramatsu M, Honjo T, Nussenzweig A and Nussenzweig MC. AID Is Required for c-myc/IgH Chromosome Translocations In Vivo. Cell. 2004; 118:431-438.
29. McBride KM, Gazumyan A, Woo EM, Schwickert TA, Chait BT and Nussenzweig MC. Regulation of class switch recombination and somatic mutation by AID phosphorylation. The Journal of Experimental Medicine. 2008; 205:2585-2594.
30. Vuong BQ, Herrick-Reynolds K, Vaidyanathan B, Pucella JN, Ucher AJ, Donghia NM, Gu X, Nicolas L, Nowak U, Rahman N, Strout MP, Mills KD, Stavnezer J and Chaudhuri J. A DNA break- and phosphorylation-dependent positive feedback loop promotes immunoglobulin class-switch recombination. Nat Immunol. 2013; 14:1183-1189.
31. Nakamura M, Kondo S, Sugai M, Nazarea M, Imamura S and Honjo T. High frequency class switching of an lgM+ B lymphoma clone CH12F3 to lgA+ cells. International Immunology. 1996; 8:193-201.
32. Hochegger H, Dejsuphong D, Fukushima T, Morrison C, Sonoda E, Schreiber V, Zhao GY, Saberi A, Masutani M, Adachi N, Koyama H, de Murcia G and Takeda S. Parp-1 protects homologous recombination from interference by Ku and Ligase IV in vertebrate cells. EMBO J. 2006; 25:1305-1314.
33. Papavasiliou FN and Schatz DG. The activation-induced deaminase functions in a postcleavage step of the somatic hypermutation process. J Exp Med. 2002; 195:1193-1198.
34. Prochnow C, Bransteitter R, Klein MG, Goodman MF and Chen XS. The APOBEC-2 crystal structure and functional implications for the deaminase AID. Nature. 2007; 445:447-451.
35. Pasqualucci L, Kitaura Y, Gu H and Dalla-Favera R. PKA-mediated phosphorylation regulates the function of activation-induced deaminase (AID) in B cells. Proc Natl Acad Sci U S A. 2006; 103:395-400.
36. Lundin C, North M, Erixon K, Walters K, Jenssen D, Goldman ASH and Helleday T. Methyl methanesulfonate (MMS) produces heat-labile DNA damage but no detectable in vivo DNA double-strand breaks. Nucleic Acids Research. 2005; 33:3799-3811.
37. Emadi A, Jones RJ and Brodsky RA. Cyclophosphamide and cancer: golden anniversary. Nat Rev Clin Oncol. 2009; 6:638-647.
38. Xu Y, Hansen WK, Rosenquist TA, Williams DA, Limp-Foster M and Kelley MR. Protection of mammalian cells against chemotherapeutic agents thiotepa, 1,3-N,N’-bis(2-chloroethyl)-N-nitrosourea, and mafosfamide using the DNA base excision repair genes Fpg and alpha-hOgg1: implications for protective gene therapy applications. The Journal of pharmacology and experimental therapeutics. 2001; 296:825-831.
39. Yadav L, Khan S, Shekh K and Jena GB. Influence of 3-aminobenzamide, an inhibitor of poly(ADP-ribose)polymerase, in the evaluation of the genotoxicity of doxorubicin, cyclophosphamide and zidovudine in female mice. Mutation Research/Genetic Toxicology and Environmental Mutagenesis. 2014; 770:6-15.
40. Chen T-L, Kennedy MJ, Anderson LW, Kiraly SB, Black KC, Colvin OM and Grochow LB. Nonlinear Pharmacokinetics of Cyclophosphamide and 4-Hydroxycyclophosphamide/Aldophosphamide in Patients with Metastatic Breast Cancer Receiving High-dose Chemotherapy Followed by Autologous Bone Marrow Transplantation. Drug Metabolism and Disposition. 1997; 25:544-551.
41. Hassan M, Ljungman P, Ringden O, Hassan Z, Oberg G, Nilsson C, Bekassy A, Bielenstein M, Abdel-Rehim M, Georen S and Astner L. The effect of busulphan on the pharmacokinetics of cyclophosphamide and its 4-hydroxy metabolite: time interval influence on therapeutic efficacy and therapy-related toxicity. Bone Marrow Transplant. 2000; 25:915-924.
42. Brar SS, Watson M and Diaz M. Activation-induced Cytosine Deaminase (AID) Is Actively Exported out of the Nucleus but Retained by the Induction of DNA Breaks. Journal of Biological Chemistry. 2004; 279:26395-26401.
43. Lambert LJ, Walker S, Feltham J, Lee HJ, Reik W and Houseley J. Etoposide Induces Nuclear Re-Localisation of AID. PLoS ONE. 2013; 8:e82110.
44. Kraus WL. PARPs and ADP-Ribosylation: 50 Years and Counting. Molecular Cell. 2015; 58:902-910.
45. Peralta-Leal A, Rodríguez-Vargas JM, Aguilar-Quesada R, Rodríguez MI, Linares JL, de Almodóvar MR and Oliver FJ. PARP inhibitors: New partners in the therapy of cancer and inflammatory diseases. Free Radical Biology and Medicine. 2009; 47:13-26.
46. Schreiber V, Amé J-C, Dollé P, Schultz I, Rinaldi B, Fraulob V, Ménissier-de Murcia J and de Murcia G. Poly(ADP-ribose) Polymerase-2 (PARP-2) Is Required for Efficient Base Excision DNA Repair in Association with PARP-1 and XRCC1. Journal of Biological Chemistry. 2002; 277:23028-23036.
47. Zhou B-BS and Elledge SJ. The DNA damage response: putting checkpoints in perspective. Nature. 2000; 408:433-439.
48. Bender K, Göttlicher M, Whiteside S, Rahmsdorf HJ and Herrlich P. Sequential DNA damage-independent and -dependent activation of NF-κB by UV. The EMBO Journal. 1998; 17:5170-5181.
49. Roos WP and Kaina B. DNA damage-induced cell death by apoptosis. Trends in Molecular Medicine. 2006; 12:440-450.
50. Kim MY, Zhang T and Kraus WL. Poly(ADP-ribosyl)ation by PARP-1: ‘PAR-laying’ NAD+ into a nuclear signal. Genes & Development. 2005; 19:1951-1967.
51. Liu M and Schatz DG. Balancing AID and DNA repair during somatic hypermutation. Trends in Immunology. 2009; 30:173-181.
52. Kraus WL and Lis JT. PARP Goes Transcription. Cell. 2003; 113:677-683.
53. Kraus WL. Transcriptional control by PARP-1: chromatin modulation, enhancer-binding, coregulation, and insulation. Current Opinion in Cell Biology. 2008; 20:294-302.
54. Ambrose HE, Papadopoulou V, Beswick RW and Wagner SD. Poly-(ADP-ribose) polymerase-1 (Parp-1) binds in a sequence-specific manner at the Bcl-6 locus and contributes to the regulation of Bcl-6 transcription. Oncogene. 2007; 26:6244-6252.
55. Ambrose HE, Willimott S, Beswick RW, Dantzer F, De Murcia JM, Yelamos J and Wagner SD. Poly(ADP-ribose) polymerase-1 (Parp-1)-deficient mice demonstrate abnormal antibody responses. Immunology. 2009; 127:178-186.
56. Sugimura K, Takebayashi S-i, Taguchi H, Takeda S and Okumura K. PARP-1 ensures regulation of replication fork progression by homologous recombination on damaged DNA. The Journal of Cell Biology. 2008; 183:1203-1212.
57. Shockett P and Stavnezer J. Inhibitors of poly(ADP-ribose) polymerase increase antibody class switching. The Journal of Immunology. 1993; 151:6962-6976.
58. Paddock MN, Buelow BD, Takeda S and Scharenberg AM. The BRCT Domain of PARP-1 Is Required for Immunoglobulin Gene Conversion. PLoS Biol. 2010; 8:e1000428.
59. Rosado MM, Bennici E, Novelli F and Pioli C. Beyond dna repair,the immunological role of parp-1 and its siblings. Immunology. 2013; 139:428-437.
60. Robert I, Dantzer F and Reina-San-Martin B. Parp1 facilitates alternative NHEJ, whereas Parp2 suppresses IgH/c-myc translocations during immunoglobulin class switch recombination. The Journal of Experimental Medicine. 2009; 206:1047-1056.
61. Mechtcheriakova D, Svoboda M, Meshcheryakova A and Jensen-Jarolim E. Activation-induced cytidine deaminase (AID) linking immunity, chronic inflammation, and cancer. Cancer Immunology, Immunotherapy. 2012; 61:1591-1598.
62. Krokan HE, Sætrom P, Aas PA, Pettersen HS, Kavli B and Slupphaug G. Error-free versus mutagenic processing of genomic uracil-Relevance to cancer. DNA Repair. 2014; 19:38-47.
63. Lada A, Dhar A, Boissy R, Hirano M, Rubel A, Rogozin I and Pavlov Y. AID/APOBEC cytosine deaminase induces genome-wide kataegis. Biology Direct. 2012; 7:47.
64. Pettersen HS, Galashevskaya A, Doseth B, Sousa MML, Sarno A, Visnes T, Aas PA, Liabakk N-B, Slupphaug G, Sætrom P, Kavli B and Krokan HE. AID expression in B-cell lymphomas causes accumulation of genomic uracil and a distinct AID mutational signature. DNA Repair. 2015; 25:60-71.
65. Qian J, Wang Q, Dose M, Pruett N, Kieffer-Kwon K-R, Resch W, Liang G, Tang Z, Mathé E, Benner C, Dubois W, Nelson S, Vian L, Oliveira Thiago Y, Jankovic M, Hakim O, et al. B Cell Super-Enhancers and Regulatory Clusters Recruit AID Tumorigenic Activity. Cell. 2014; 159:1524-1537.
66. Li M and Yu X. The role of poly(ADP-ribosyl)ation in DNA damage response and cancer chemotherapy. Oncogene. 2015; 34:3349-3356.
67. Feng Felix Y, de Bono Johann S, Rubin Mark A and Knudsen Karen E. Chromatin to Clinic: The Molecular Rationale for PARP1 Inhibitor Function. Molecular Cell. 2015; 58:925-934.
68. Tan AR, Toppmeyer D, Stein MN, Moss RA, Gounder M, Lindquist DC, Ji JJ, Chen AP, Egorin MJ, Kiesel B, Beumer JH and DiPaola RS. Phase I trial of veliparib, (ABT-888), a poly(ADP-ribose) polymerase (PARP) inhibitor, in combination with doxorubicin and cyclophosphamide in breast cancer and other solid tumors. J Clin Oncol (Meeting Abstracts). 2011; 29:3041.
69. Frankenberger S, Davari K, Fischer-Burkart S, Böttcher K, Tomi N-S, Zimber-Strobl U and Jungnickel B. Checkpoint kinase 1 negatively regulates somatic hypermutation. Nucleic Acids Research. 2014; 42:3666-3674.
70. Hitoshi N, Ken-ichi Y and Jun-ichi M. Efficient selection for high-expression transfectants with a novel eukaryotic vector. Gene. 1991; 108:193-199.
71. Greiner A, Tobollik S, Buettner M, Jungnickel B, Herrmann K, Kremmer E and Niedobitek G. Differential expression of activation-induced cytidine deaminase (AID) in nodular lymphocyte-predominant and classical Hodgkin lymphoma. J Pathol. 2005; 205:541-547.