
Introduction
The mTORC1 complex is an important molecular sensor linking the cellular environment to key processes including proliferation and autophagy [1, 2]. Careful regulation of mTORC1 signaling is required to maintain cellular homeostasis especially following exposure to stress. Suppression of mTORC1 activity simultaneously reduces protein translation while stimulating pro-survival autophagy. SESTRIN 2 (SESN2), a member of the highly conserved stress-inducible Sestrin family, has been linked to mTORC1 regulation via AMP-activated protein kinase (AMPK/PRKA) [3]. SESTRIN 2 interaction with AMPK both activates and tethers AMPK to TSC2 leading to AMPK-mediated phosphorylation of TSC2 and subsequent inhibition of mTORC1 [3].
Stress-mediated induction of SESTRIN 2 has been predominantly associated with P53 signaling [4]. However, stress stimuli such as hypoxia, have been demonstrated to induce SESTRIN 2 via P53-independent means [4]. Recently endoplasmic reticulum (ER) stress has been linked to SESTRIN 2 regulation [5, 6]. ER stress can be induced by various physiological or chemical insults and is characterised by an accumulation of unfolded proteins within the lumen of the ER. Three ER transmembrane receptors IRE1 (ERN1), PERK (EIF2AK3) and ATF6 monitor the health of the ER and upon activation of ER stress act in concert to mediate the unfolded protein response (UPR). The UPR is an integrated signal transduction pathway, which aims to re-establish ER homeostasis through various adaptive mechanisms [7]. IRE1, PERK and ATF6 promote adaptation to protein-folding stress through the transcriptional upregulation of key ER chaperones, enhanced ER-associated degradation (ERAD) and inhibition of general protein translation. However, when ER stress is persistent, the UPR machinery can activate cell death pathways [8, 9]. Recent studies have identified ER stress as a mechanism through which SESTRIN 2 expression can be regulated. The proteasome inhibitor, Bortezomib (Btz), and HIV protease inhibitor, Nelfinavir, have both been demonstrated to induce SESTRIN 2 expression as a consequence of ER stress [6]. Likewise, Park and colleagues reported ER stress-mediated regulation of SESTRIN 2 in hepatocytes treated with palmitic acid [5]. How SESTRIN 2 expression is induced by ER stress is not fully understood although the PERK arm of the UPR appears to be required. Downstream targets of PERK, ATF4 and c/EBPβ (CEBPB), have been reported to regulate ER stress-induced SESTRIN 2 expression [5, 6].
In this study we demonstrate that induction of ER stress following treatment with pharmacological agents or chemotherapeutics such as Methotrexate (Mtx) lead to a P53-independent increase in SESTRIN 2. Our data indicates both IRE1/XBP1 and PERK branches of the UPR contribute to ER stress-induced SESTRIN 2 expression. Knockdown of SESTRIN 2 reduced stress driven autophagy and enhanced cell death. Intriguingly, our results suggest that enhanced stress-induced cell death observed in SESTRIN 2 knockdown cells is due to an exacerbation of ER stress rather than a decrease in autophagy.
Results
ER stress induces SESTRIN 2 independent of P53
Treatment of MCF7 cells with Thapsigargin (Tg) and Brefeldin A (BFA), two classical inducers of ER stress, was optimized to select doses which would permit examination of pro-survival and pro-death signaling over time (0.5 µg/ml BFA and 1 µM Tg) (Figure 1A). Tg and BFA, induced robust expression of SESTRIN 2 and activated the UPR as demonstrated by splicing of XBP1 and PERK phosphorylation (as determined by PERK upshift) (Figure 1B-1C). Similarly, SESTRIN 2 expression was induced by exposure to the ER stress inducer Dithiothreitol (DTT) (Supplemental Figure 1E). ER stress driven increases in SESTRIN 2 were transcriptionally mediated. Treatment with Tg increased SESTRIN 2 mRNA levels (Supplemental Figure 1A) while, addition of Actinomycin D (Act D) prevented Tg-mediated SESTRIN 2 induction (Figure 1D). SESTRIN 2 regulation has been demonstrated to occur via P53 dependent and independent mechanisms [3, 5, 6]. Phosphorylation of P53 (Ser15), while readily detectable in cells treated with Etoposide (Etop), was not observed following either Tg or BFA treatment although a clear increase in the levels of SESTRIN 2 was evident (Figure 1E). Likewise, HCT116 P53+/+ and P53-/- cells displayed Tg-induced SESTRIN 2 up-regulation, irrespective of P53 status. While, Etop-induced SESTRIN 2 expression was only observed in P53+/+ cells (Supplemental Figure 1B-1C). Increased SESTRIN 2 expression was also detected in cells lacking wild-type P53 (HCC1806, K562) post Tg treatment (Figure 1F, Supplemental Figure 1D). Collectively, these data demonstrate the ability of ER stress to induce SESTRIN 2 expression in a P53 independent manner.
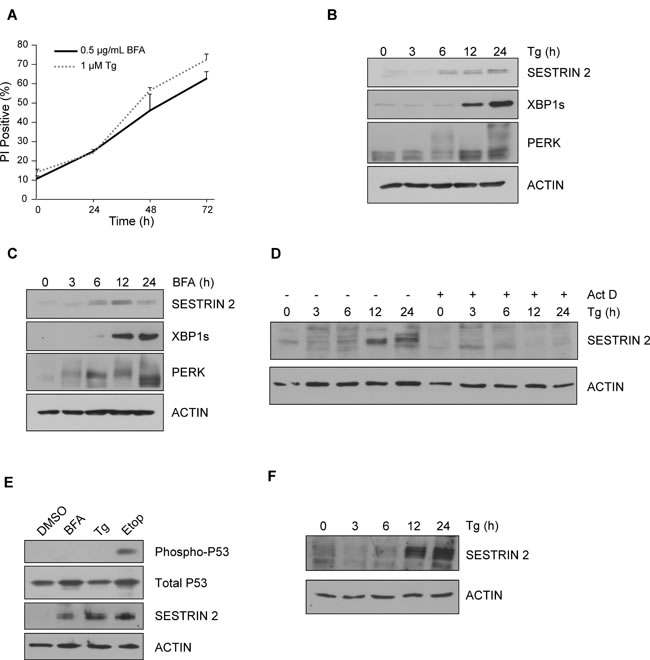
Figure 1: Induction of ER stress leads to upregulation of SESTRIN 2 expression independent of P53. MCF7 cells were treated with A. 0.5 μg/ml of BFA or 1 μM of Tg for the indicated time and viability assessed by propidium iodide (PI) uptake. Error bars represent the mean ± SD. MCF7 cells were treated with 0.5 μg/ml of Tg B. or 1 μM of BFA C. for the indicated time and lysates imunoblotted for SESTRIN 2, PERK, XBP1s. ACTIN was used as a loading control. D. MCF7 cells were treated with 1 μM of Tg alone or in combination with Act D (1 μg/ml) for the indicated time and lysates immunoblotted for SESTRIN 2 and ACTIN. E. MCF7 cells were treated with 0.5 μg/ml BFA, 1 μM Tg or 50 μM Etoposide (Etop) for 24 h after which lysates were immunoblotted for SESTRIN 2, Ser15 p-P53 and total P53. F. HCC1806 cells were treated with 1 μM Tg for the indicated time and cell lysates were then immunoblotted for SESTRIN 2 and ACTIN. Results are representative of at least 3 independent experiments.
UPR mediators contribute to ER stress-mediated induction of SESTRIN 2
To understand how ER stress leads to an increase in SESTRIN 2 expression, we determined the contribution of each signaling arm of the UPR. siRNA knockdown of ATF6, while successful, did not inhibit Tg-induced increase in SESTRIN 2 (Supplemental Figure 2A). Addition of a PERK inhibitor alone, IRE1 inhibitor alone or a combination of both reduced Tg and BFA-mediated induction of SESTRIN 2 (Figure 2A). The functionality of each inhibitor was verified by examining their effect on downstream targets. Treatment with the PERK inhibitor, clearly attenuated ER stress-induced expression of ATF4, while inclusion of the IRE1 inhibitor blocked splicing of XBP1 (Figure 2A). To verify the role of PERK and determine if IRE1-mediated SESTRIN 2 induction is a result of IRE1/XBP1 signaling we examined SESTRIN 2 protein levels following ER stress in Xbp1-/- and Perk-/- mouse embryonic fibroblasts (MEF) cells. PERK-mediated translational inhibition is essential for cell survival following ER stress. Consequently, knockout of Perk renders cells exquisitely sensitive to ER stress-induced death [10]. For this reason Perk+/+ and Perk-/- MEF cells were treated with a lower dose (25nM) of Tg as described previously [11]. Perk-/- MEF cells displayed lower SESTRIN 2 induction upon ER stress than their Perk+/+ counterparts (Figure 2B). In agreement with previous studies, targeting the downstream PERK target ATF4 also reduced Tg-mediated induction of SESTRIN 2 expression (Figure 2B-2C). Likewise, Xbp1-/- MEFs and MCF7 cells transfected with XBP1 siRNA displayed a significant reduction in Tg-induced SESTRIN 2 induction underscoring a previously un-described role for XBP1 in SESTRIN 2 regulation (Figure 2D-2E).
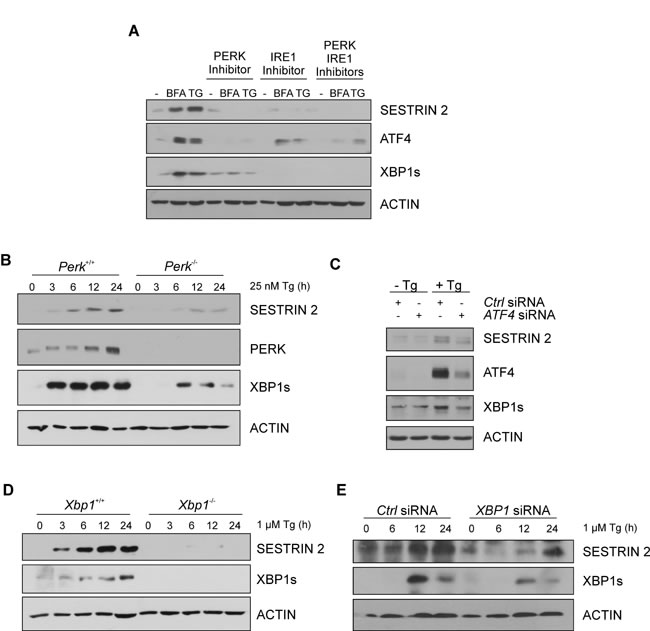
Figure 2: UPR pathways contribute to ER stress-induced enhancement of SESTRIN 2 expression. A. MCF7 cells were treated with 0.5 μg/mL of BFA or 1 μM of Tg for 24 h alone or in combination with 300 nM PERK inhibitor, 10 µM IRE1 inhibitor or both. Levels of SESTRIN 2, ATF4, XBP1s and ACTIN were analysed by immunoblotting. B. Perk+/+ and Perk-/- MEFs were treated with 25 nM Tg for the indicated time and lysates immunoblotted for SESTRIN2, PERK, XBP1s and ACTIN. C. MCF7 cells transfected with ATF4 or Ctrl siRNA were treated with 1 μM Tg for 24 h and lysates immunoblotted for SESTRIN 2, XBP1s and ACTIN. D. Xbp1+/+ and Xbp1-/- MEFs were treated with 1 μM of Tg for the indicated time and lysates immunoblotted for SESTRIN 2, XBP1s and ACTIN. E. MCF7 cells transfected with XBP1 or Ctrl siRNA were treated with 1 μM Tg for the indicated time and lysates immunoblotted for SESTRIN 2, XBP1s and ACTIN. Results are representative of at least 3 independent experiments.
SESTRIN 2 knockdown modulates ER stress-induced autophagy and cell death responses
To determine the relevance of SESTRIN 2 induction, MCF7 cells were transfected with siRNA against SESTRIN 2 and subjected to ER stress. Knockdown was confirmed post Tg or BFA treatment by Western blotting (Figure 3A-3B). The outcome of SESTRIN 2 knockdown on ER stress-induced cell death was determined by PI uptake. Cells transfected with SESTRIN 2 siRNA displayed significantly higher ER stress-induced cell death compared to their control counterparts (Figure 3C-3D). This was especially evident at the earlier time-points, suggesting a role for SESTRIN 2 in the initial protective UPR response. Autophagy, an important pro-survival process, is induced following exposure to a range of stresses including ER stress [12]. Previous work has linked SESTRIN 2 to autophagy induction and cellular survival following genotoxic stress but little is known about its contribution during other types of stress. MTOR dephosphorylation and enhanced LC3-I (MAP1LC3) to LC3-II conversion was evident in cells treated with BFA or Tg suggesting autophagy induction (Supplemental Figure 3A-3B). Examination of GFP-LC3 distribution identified a punctate pattern indicative of autophagy induction upon ER stress (Supplemental Figure 3C). Addition of the autophagy inhibitor Spautin-1 (Spa-1) lowered the number of cells displaying punctate GFP-LC3 distribution following BFA treatment (Supplemental Figure 3C). To determine if SESTRIN 2 contributed to ER stress-induced autophagy, autophagic flux was examined in control and SESTRIN 2 siRNA transfected MCF7 cells. Enhanced LC3-I to II conversion after co-treatment with chloroquine (CQ) was clearly evident in Tg-treated control siRNA cells. However, in SESTRIN 2 siRNA-transfected cells significantly less LC3-I to II conversion was detected in cells treated with both Tg and CQ (Figure 3E). Likewise, SESTRIN 2 knockdown inhibited autophagy (as determined by puncta formation) in BFA treated GFP-LC3 MCF7 (Supplemental Figure 3D).
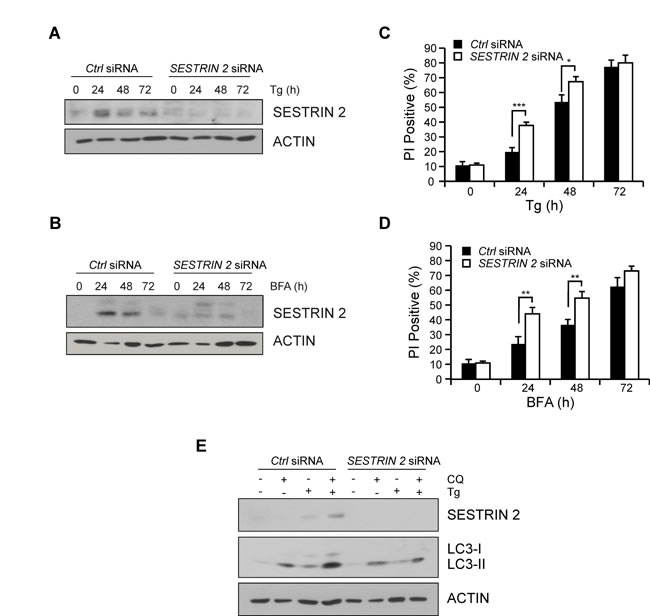
Figure 3: SESTRIN 2 knockdown inhibits ER stress-induced autophagy and enhances ER stress-induced cell death. MCF7 cells transfected with SESTRIN 2 or Ctrl siRNA were treated with 1 μM Tg (A, C) or 0.5 μg/ml BFA (B, D) for the indicated time after which lysates immunoblotted for SESTRIN 2 and ACTIN (A, B) and cell viability determined by PI uptake (C, D). E. Autophagic flux was evaluated in SESTRIN 2 and Ctrl siRNA transfected MCF7 cells treated with 1 μM Tg for 24 h in the presence or absence of 20 μM chloroquine (CQ) and cell lysates immunoblotted for SESTRIN 2, LC3-I/II and ACTIN. Representative image of 3 independent experiments is shown.
The chemotherapeutic Methotrexate induces ER stress and SESTRIN 2 expression via PERK and IRE1 signals
Since our results indicated SESTRIN 2 expression is enhanced by ER stress we sought to determine if commonly used chemotherapeutics also induced SESTRIN 2 expression. Treatment of MCF7 and HCC1806 (triple negative P53-/- breast cancer cell line) cells with a range of chemotherapeutics detected elevated SESTRIN 2 expression following treatment with the anti-folate drug Mtx and the proteosomal inhibitor Btz (Figure 4A-4B). While treatment with Btz clearly induced ER stress (Supplemental Figure 4A) as has previously been reported [13], the induction of ER stress responses by Mtx has not yet been documented. RT-PCR confirmed Mtx, similar to Tg treatment, induced transcriptional upregulation of SESTRIN 2 (Supplemental Figure 1F). Following treatment with Mtx, activation of both the PERK and IRE1 arms of the UPR was observed in MCF7 and HCC1806 cells confirming Mtx treatment, in addition to enhancing SESTRIN 2 expression, also triggered ER stress (Figure 4C-4D).
To determine the relevance of the UPR to Mtx-mediated SESTRIN 2 induction HCC1806 cells were pre-treated with PERK or IRE1 inhibitors prior to Mtx treatment and SESTRIN 2 expression was examined. Addition of either inhibitor reduced SESTRIN 2 expression, with the greatest effect observed in cells pre-treated with IRE1 inhibitor (Figure 4E). Knockdown of XBP1 in HCC1806 cells showed a similar pattern, with reduced Mtx-mediated SESTRIN 2 induction in XBP1 siRNA versus control siRNA-transfected cells (Figure 4F). Likewise in Btz-treated HCC1806 cells knockdown of XBP1 reduced Btz driven expression of SESTRIN 2 (Supplemental Figure 4B). Collectively, our data suggest a model where treatment with chemotherapeutics such as Mtx and Btz induce ER stress, which in turn, via PERK and IRE1/XBP1 signals, drives SESTRIN 2 expression
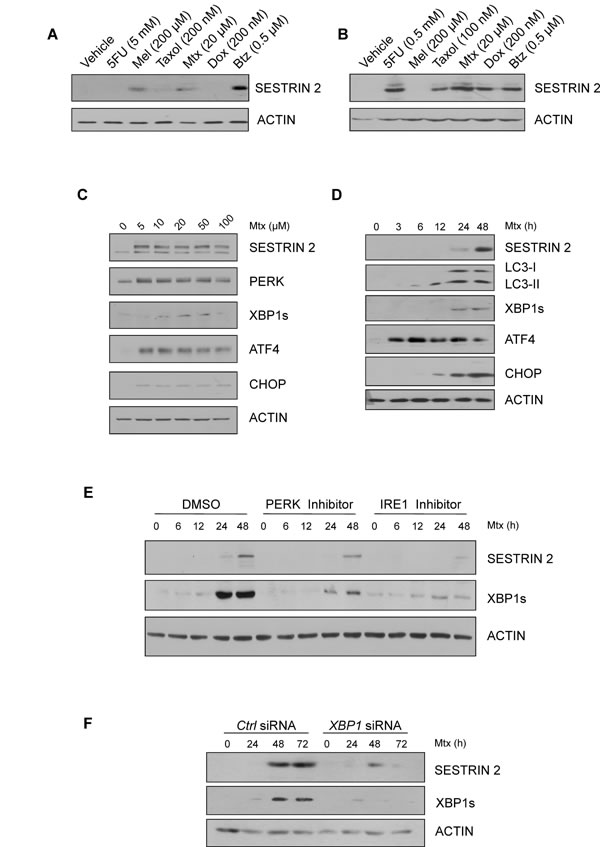
Figure 4: Chemotherapeutics, Methotrexate and Bortezomib, induce ER stress and SESTRIN 2 expression via UPR-mediated signals. A.-B. HCC1806 (A) and MCF7 (B) cells were treated with 5 mM (A) 0.5 mM (B) 5-fluorouracil (5FU), 200 µM melphalan (Mel), 200 nM (A) 100 nM (B) Taxol, 20 µM Methotrexate (Mtx), 200 nM Doxorubicin, 0.5 µM Bortezomib (Btz) for 24 h and lysates immunoblotted for SESTRIN 2 and ACTIN. C. HCC1806 cells were treated with the indicated concentrations of Mtx for 48 h and lysates immunoblotted for SESTRIN 2, PERK, XBP1s, ATF4, CHOP and ACTIN. D. HCC1806 cells were treated with 20 µM Mtx for the indicated time and lysates immunoblotted for SESTRIN 2, LC3-I/II, XBP1s, ATF4, CHOP and ACTIN. E. HCC1806 cells were treated with 20 µM Mtx for the indicated time alone or in combination with 300 nM PERK or 10 µM IRE1 inhibitor and lysates were immunoblotted for SESTRIN 2, XBP1s and ACTIN. A representative image of 3 independent experiments is shown. F. Ctrl and XBP1 siRNA-transfected HCC1806 cells were treated with 20 µM Mtx for the indicated time and lysates immunoblotted for SESTRIN 2, XBP1s and ACTIN.
SESTRIN 2 knockdown enhances Mtx-induced death in P53-/- HCC1806 cells
Since our earlier results, using specific ER stress-inducing agents, indicated knockdown of SESTRIN 2 expression enhanced ER stress-induced cell death, we examined the outcome of SESTRIN 2 knockdown on Mtx-induced death in HCC1806 cells. Examination of SESTRIN 2 expression following Mtx treatment confirmed knockdown over the 72 h time-course (Figure 5A). Similar to Tg and BFA, reduction of SESTRIN 2 expression via siRNA increased cell death in response to Mtx (Figure 5B). Owing to the lack of P53, HCC1806 cells are relatively resistant to Mtx-induced death. To examine the long term outcome of SESTRIN 2 knockdown, HCC1806 cells were transfected with SESTRIN 2 siRNA, followed by Mtx treatment for 72 h. After drug removal cells were left for an additional 10 days and colony formation was evaluated by crystal violet staining. While colonies were abundant in cells transfected with the control siRNA, the same was not true for cells transfected with the SESTRIN 2 siRNA, where few if any colonies were visible (Figure 5C). This data indicates that SESTRIN 2 knockdown enhances Mtx-induced death in HCC1806 cells in both the short and long term.
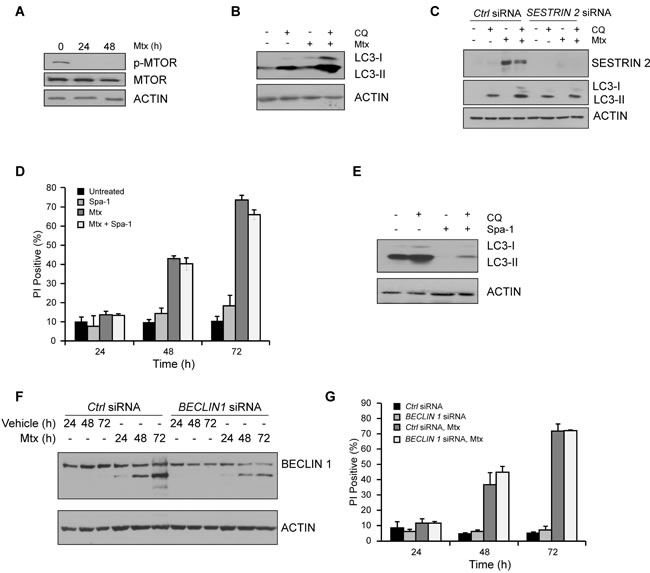
Figure 6: Impairment in autophagy does not account for enhanced Mtx-induced death observed in SESTRIN 2 knockdown cells. A. HCC1806 cells were treated with 20 µM Mtx for the indicated time after which lysates were immunoblotted for phospho-MTOR, total MTOR and ACTIN. B. Autophagic flux was evaluated in HCC1806 cells treated with 20 μM CQ alone, 20 μM Mtx alone, or a combination of 20 μM CQ and 20 μM Mtx for 48 h and cell lysates immunoblotted for SESTRIN 2, LC3-I/II and ACTIN. C. Ctrl and SESTRIN 2 siRNAs transfected HCC1806 cells were treated with 20 µM Mtx ± 20 µM CQ for 48 h. Lysates were immunoblotted for SESTRIN 2, LC3-I/II and ACTIN. D. HCC1806 cells were treated with 20 µM Mtx alone, 10 µM Spautin-1 (Spa-1) alone or a combination of Mtx and Spa-1 for up to 72 h after which cell viability was assessed by PI uptake. E. HCC1806 cells were treated with 20 µM CQ alone, 10 µM Spa-1 alone or a combination of CQ and Spa-1 for 24 h and lysates immunoblotted for LC3-I/II and ACTIN. F.-G. Ctrl and BECLIN1 siRNA transfected HCC1806 cells were treated with 20 µM Mtx for the indicated time. (F) Lysates were immunoblotted for SESTRIN 2 and ACTIN and (G) Cell viability assessed at each time-point by PI uptake.
SESTRIN 2 knockdown reduces Mtx-induced autophagy independently of its pro-survival function
To understand how SESTRIN 2 knockdown enhanced Mtx-induced cell death we examined the contribution of autophagy. Following Mtx treatment, both MTOR dephosphorylation and enhanced LC3-I to II conversion was evident in HCC1806 cells (Figure 6A-6B). Knockdown of SESTRIN 2, while enhancing Mtx-induced cell death (Figure 5A-5B), also decreased Mtx-induced autophagy (Figure 6C). Likewise, SESTRIN 2 induction in Btz-treated HCC1806 cells was associated with enhanced autophagy (Supplemental Figure 4C). Similarly to Mtx-treated cells knockdown of SESTRIN 2 both inhibited Btz-induced autophagy and enhanced cell death (Supplemental Figure 4D-4E). To determine if the reduced autophagy observed in Mtx-treated SESTRIN 2 knockdown cells directly impacted upon cell death, an inhibitor of autophagy, Spa-1, was employed. Pre-treatment of cells with Spa-1, while clearly reducing autophagy, did not increase Mtx-induced cell death (Figure 6D-6E). In addition, knockdown of BECLIN1 (BECN1) failed to sensitize cells to Mtx-induced death (Figure 6F-6G). Collectively, these observations suggest inhibition of autophagy is unlikely to contribute to the enhanced death observed in Mtx-treated HCC1806 cells following SESTRIN 2 knockdown.
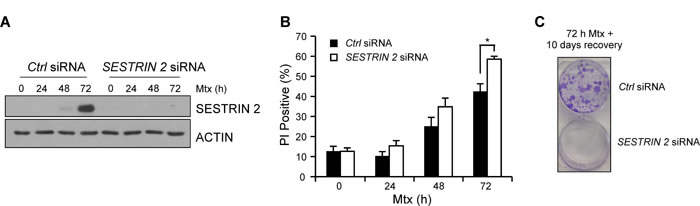
Figure 5: Knockdown of SESTRIN 2 enhances Methotrexate-induced death. Ctrl and SESTRIN 2 siRNAs transfected HCC1806 cells were treated 20 µM Mtx for the indicated time. A. Lysates were immunoblotted with SESTRIN 2 and ACTIN B. cell viability was determined by PI uptake. Mean of three independent experiments is shown ± SD. Statistical analysis was determined using t-Test. C. After transfection with Ctrl or SESTRIN 2 siRNA HCC1806 cells were treated with 20 µM Mtx for 72 h. Mtx was removed and the colonies were left for 10 days before crystal violet staining. Representative image of two independent experiments is shown.
SESTRIN 2 controls ER homeostasis by regulating MTOR activation in response to Mtx
Recently it has been reported that SESTRIN 2 expression is critical for inhibition of protein translation during ER stress [5]. It was demonstrated that SESTRIN 2 deficiency resulted in persistent protein synthesis under ER stress conditions resulting in enhanced ER stress-induced death [5]. To examine if this was also the case in our model, levels of MTOR phosphorylation were examined in HCC1806 cells following Mtx treatment. Clear inactivation, as determined by MTOR dephosphorylation, was observed in Mtx and Btz-treated HCC1806 cells 24 h post treatment (Figure 6A, Supplemental Figure 4F). Knockdown of SESTRIN 2 abrogated MTOR dephosphorylation and prolonged P70 S6K (RPS6KB1) phosphorylation following Mtx treatment (Figure 7A). Lower SESTRIN 2 expression has been previously linked to an increase in UPR signals and in particular enhanced activation of the PERK arm of the UPR [5]. To determine whether SESTRIN 2 knockdown potentiates PERK-mediated signaling following Mtx-induced ER stress we examined ATF4, CHOP (DDIT3) and EIF2α (EIF2S1) phosphorylation in Mtx-treated control versus SESTRIN 2 siRNA transfected cells. Prolonged activation of PERK signaling was observed in Mtx-treated cells transfected with SESTRIN 2 siRNA, suggesting SESTRIN 2 knockdown potentiates Mtx-induced ER stress in HCC1806 cells (Figure 7A-7B). Similar results were also observed with Tg demonstrating this is due to increased ER stress per se and not specifically Mtx treatment (Supplemental Figure 5A). To ascertain if the enhancement in ER stress-induced markers observed in SESTRIN 2 siRNA cells was a direct result of sustained MTOR activity we utilised an inhibitor of MTOR, rapamycin. Treatment of SESTRIN 2 siRNA HCC1806 cells with rapamycin reduced both Mtx-induced mTORC1 activity, as determined by phospho-P70 S6K, and also reduced SESTRIN 2 knockdown-mediated enhancement of PERK signaling (Figure 7B).
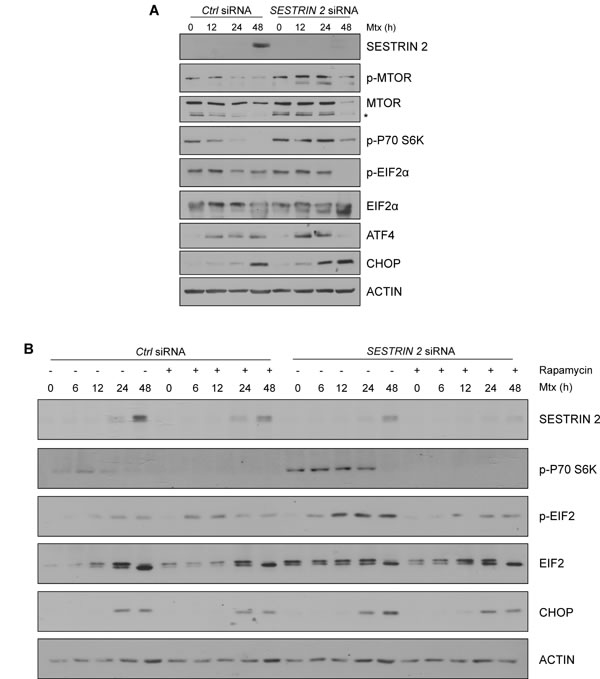
Figure 7: Knockdown of SESTRIN 2 delays Mtx-induced dephosphorylation of MTOR and potentiates ER stress. Ctrl and SESTRIN 2 siRNAs transfected HCC1806 cells were treated with 20 µM Mtx for the indicated time. A. Lysates were immunoblotted with SESTRIN 2, phospho-MTOR, total MTOR, phospho-P70 S6K, phospho-EIF2α, total EIF2α, ATF4, CHOP and ACTIN. B. Ctrl and SESTRIN 2 siRNAs transfected HCC1806 cells were treated 20 µM Mtx ± 100 nM Rapamycin for the indicated time. Lysates were immunoblotted for SESTRIN 2, phospho-P70 S6K, phospho-EIF2α, total EIF2α, CHOP and ACTIN. A representative image of 3 independent experiments is shown.* Denotes non-specific band.
Discussion
Microarray studies within our laboratory had previously identified SESTRIN 2 as an ER stress regulated gene [14]. In this study, we demonstrate ER stress regulation of SESTRIN 2 expression independent of P53. How SESTRIN 2 is induced following ER stress is not entirely understood. Park and colleagues reported PERK-mediated SESTRIN 2 induction via c/EBPβ, while Bruning et al found SESTRIN 2 induction to be ATF4 dependent [5, 6]. In our system ER stress-mediated upregulation of SESTRIN 2 was dependent on both IRE1 and PERK signals. Reduced SESTRIN 2 expression was evident in both Perk and Xbp1 deficient MEF cells upon exposure to ER stress. Knockdown of XBP1 in MCF7 and HCC1806 cells attenuated Tg and Mtx-mediated induction of SESTRIN 2. This data suggests enhancement of SESTRIN 2 expression upon ER stress requires both IRE1/XBP1 and PERK-mediated signals. Such a co-operative induction is unsurprising as ER stress sensors rarely function in a singular distinct way but rather in a highly co-ordinated manner. For example, we observed decreased XBP1 splicing in both Perk-/- MEFs and cells treated with PERK inhibitor following ER stress, indicating PERK signaling is required for robust activation of the XBP1. Indeed reduced Xbp1 mRNA expression has been previously reported in Perk-/- MEFs following tunicamycin treatment [15]. Based on these observations it seems likely that reducing/blocking one branch of the UPR can have far reaching effects on other branches of this signaling pathway.
To examine the therapeutic relevance of targeting SESTRIN 2 we examined the ability of a panel of commonly used chemotherapeutics to induce SESTRIN 2 expression in MCF7 cells and the P53 null triple negative breast cancer cell line HCC1806. Treatment with Mtx and or Btz potently induced SESTRIN 2 expression in both cell lines. As the triple negative HCC1806 cell line is devoid of P53 we chose to use this cell line for all further experiments. Examination of both Mtx and Btz-treated cells revealed, that in addition to inducing SESTRIN 2, both drugs triggered ER stress. It has been well established that Btz is a potent inducer of ER stress [13]. However, a role for Mtx as a novel inducer of ER stress has not previously been characterised. Again, as observed with Tg and BFA treatment of MCF7 cells, knockdown of SESTRIN 2 expression decreased Mtx-induced autophagy and enhanced cell death in HCC1806 cells. Since autophagy is predominantly considered a pro-survival process we reasoned enhanced Mtx-induced death in SESTRIN 2 knockdown cells is a direct consequence of lowered autophagy. However, studies using the autophagy inhibitor Spautin-1 or knockdown of BECLIN1 argued against this. Following siRNA knockdown of SESTRIN 2 Mtx-induced dephosphorylation of MTOR was reduced. Associated with this we observed an enhancement of PERK-mediated signaling indicating a potentiation of ER stress. Inhibition of MTOR signaling in Mtx-treated SESTRIN 2 siRNA cells, by addition of rapamycin, lowered PERK driven signals as determined by attenuated EIF2α phosphorylation and CHOP induction. Unexpectedly, addition of rapamycin to control and SESTRIN 2 siRNA cells treated with Mtx also lowered SESTRIN 2 expression (Figure 7B). This reduction may be a consequence of reduced protein translation by rapamycin-mediated inhibition of mTORC1.
In agreement with our findings, exacerbation of ER stress as a consequence of SESTRIN 2 deficiency has recently been reported. Park and colleagues demonstrated sustained mTORC1 activity leading to elevated ATF4 and CHOP expression in Sestrin 2 deficient cells upon induction of ER stress [5]. Increased ATF4 and CHOP expression has been demonstrated to antagonize translational inhibition by inducing genes involved in protein synthesis triggering oxidative stress and ultimately cell death [16]. Enhancement of SESTRIN 2 expression, by ER stress, blocks protein translation via mTORC1 inactivation promoting cellular adaption and restoration of ER homeostasis. This supports the hypothesis that SESTRIN 2 expression protects breast cancer cells from Mtx-induced cell death by means of inhibiting protein synthesis through inhibition of mTORC1 rather than autophagy induction (Figure 8).
This study highlights the pro-survival role of SESTRIN 2 and demonstrates targeting of SESTRIN 2 in breast cancer cells represents a mechanism to potentiate ER stress-induced cell death. SESTRIN 2 induction has been reported in other cancer models including skin cancer where it was also linked cell survival [17]. These results, together with the results of our study, highlight the importance SESTRIN 2 induction during cell stress.
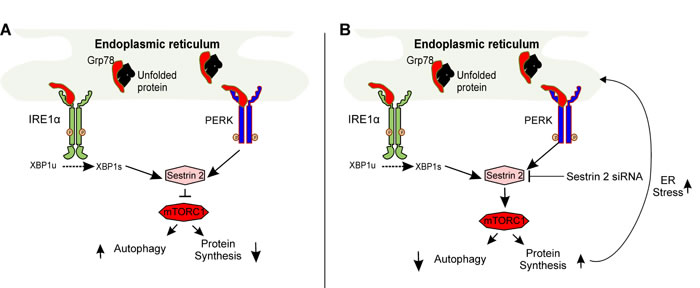
Figure 8: Schematic representation of SESTRIN 2-mediated signaling during ER stress. A. Accumulation of unfolded proteins within the ER lumen (ER stress) triggers activation of IRE1 and PERK upregulating SESTRIN 2 expression. SESTRIN 2 via interaction with AMPK and TSC2 inhibits MTORC1 activity decreasing protein translation and increasing autophagy. B. Knockdown of SESTRIN 2 prevents MTORC1 inhibition resulting in sustained protein translation and autophagy inhibition. This in turn, places further stress on the ER exacerbating ER stress and leading to enhanced cell death.
Materials and methods
Cell culture and treatments
MCF7 (ATCC), HCC1806 (ATCC), K562 (ATCC) cells were cultured in either Dulbecco’s modified Eagle’s medium (DMEM) (Sigma #D6429) (MCF-7) or Roswel Park Memorial Institute medium (RPMI 1640) (Sigma #R0883) (HCC1806, K562) supplemented with 10% heat inactivated fetal bovine serum (HI FBS, Sigma #7524), 100 U/ml penicillin/100 mg/ml streptomycin (Sigma #P0781) and 2 mM L-glutamine (Sigma #G7513). HCT116 colon cancer cells were cultured in McCOYs 5A medium modified (Sigma #M9309) supplemented with 10% HI FBS, 100 U/ml penicillin/100 mg/ml streptomycin. Xbp1−/− and Xbp1+/+ control MEFs (Dr. Laurie Glimcher, Harvard Medical School, USA) were maintained in DMEM medium supplemented with 10% HI FBS, 2 mM L-glutamine, 1 mM sodium pyruvate (Sigma #S8636), non-essential amino acid solution (Sigma #M7145). Perk+/+ and Perk−/− MEFs (Prof. David Ron Institute of Metabolic Science, University of Cambridge, UK) were maintained in DMEM medium supplemented with 10% HI FBS, 2 mM L-glutamine, 1 mM sodium pyruvate, non-essential amino acid solution, 55 μM β-mercaptoethanol (βME), 1% penicillin/streptomycin. All cells were cultured at 37°C, 5% CO2 in a humidified incubator. Cells were seeded at an appropriate number 24 h prior to treatment. 5-Fluorouracil (Sigma #F6627), Paclitaxel (Sigma #T7402), Doxorubicin (LC laboratories #D-4000), Melphalan (Sigma #M2011), Bortezomib (Selleck Chemicals #PS-341), Methotrexate (Sigma #M9929), Brefeldin A (Sigma #B7651), Thapsigargin (Sigma #T9033), Etoposide (Sigma #E1383), Rapamycin (LC-laboratories #R-5000), IRE1 inhibitor, MKC-8866, (Mannkind Corporation), Spautin-1 (Sigma #SML0440) and PERK inhibitor, GSKG797800 (Toronto Research Chemicals).
Flow cytometry
Cells were collected by trypsinization and subsequent centrifugation, resuspended in ice-cold PBS containing 0.7 µg/ml of propidium iodide (Sigma #P4170) and analysed using FACSCalibur flow cytometer (Becton Dickinson).
Western blotting
Cells were washed once in ice-cold PBS and lysed in whole cell lysis buffer (4% SDS, 120 mM Tris HCl, 10% glycerol, 100 mM DTT and dash of bromphenol blue) and boiled at 95°C for 5 min. Equal amounts of protein samples were run on an SDS polyacrylamide gel. The proteins were transferred onto nitrocellulose membrane and blocked with 5% milk in PBS-0.1% Tween. For detection of protein expression the following antibodies were used: ACTIN (Sigma #A5060), LC3 (Sigma #L7543), SESTRIN 2 (Sigma #WH0083667M3), XBP1 (Abcam #619502), ATF4 (CST #11815), CHOP (CST #2895), MTOR (CST #2983) p-MTOR (CST #2974), PERK (CST #3192), p-P70 S6K (CST #9234). All the secondary antibodies were purchased from Jackson and the signal was visualized using Western Blotting Luminol Reagent (SantaCruz sc-2048).
Gene silencing and overexpression
MCF7 and HCC1806 cells were transfected using Lipofectamine 2000 (Life Technologies, #11668019) according to the manufacturer’s protocol. siRNAs (ON-TARGET plus smart pool) were obtained from Dharmacon SESTRIN 2 (L-019134-02-0005); XBP1 (L-009552-00-0005); ATF6 (L-009917-00-0005); ATF4 (L-005125-00-0005); BECLIN1 (L-010552-00-0005); Non-coding siRNA (D-001810-01-20). Media was changed 4 h post-transfection and cells were left to recover for 24 h. After the recovery, cells were reseeded for the experiment that was performed 36 h post-transfection.
GFP-LC3 plasmid was transiently transfected into MCF7 cells according to manufacturers protocol using Lipofectamine 2000 (Life Technologies, #11668019). 24 h post transfection cells were treated as indicated and GFP-LC3 positive punctate versus diffuse cells (3 fields of 100 cells) were counted.
RNA extraction and cDNA synthesis
Total RNA was isolated from cells using TRI Reagent (Invitrogen) according to the manufactures protocol. To synthesize cDNA, 2 μg of RNA was subjected to DNase treatment and incubated for 15 min at room temperature. The RNA was then reverse transcribed into cDNA using Superscript III first strand RT-PCR system and random hexamers (Invitrogen). The cDNA product was subjected to PCR using the forward primer 5′-CAGAGGGCACAGGAAAGAAG-3′ and the reverse primer 5′- GAACTAGGATTCGGGCAACA-3′ for the detection of human SESTRIN 2. GAPDH was used as an endogenous control using the forward primer 5′-ACCACAGTCCATGCCATC-3′ and reverse 5′-TCCACCACCTGTTGCTG-3′.
Clonogenic assay
HCC1806 cells were transfected with SESTRIN 2 and non-coding control siRNAs as described above. 36 h later they were treated with 20 µM of Mtx for 72 h. After the treatment, media was removed and cells were left to recover for 10 days. Colonies were stained with 2% crystal violet in 20% methanol.
Statistical analysis
Cell death and data is expressed as mean ± SD for three independent experiments. Real Time data is expressed as mean ± SEM for three independent experiments. Differences between the treatment groups were assessed using Graphpad’s Two-tailed unpaired student’s t-tests. The values with *p < 0.05 is considered statistically significant, **p < 0.01, ***p < 0.001.
Acknowledgments
We would like to thank Drs. Sanjeev Gupta and Shane Deegan, with the advice and feedback during the initial stages of the project. The authors acknowledge the facilities and scientific and technical assistance of the NCBES Flow Cytometry core facility at the National University of Ireland Galway, a facility that is funded by NUIG and the Irish Government’s Programme for Research in Third Level Institutions, Cycles 4 and 5, National Development Plan 2007-2013.
Conflicts of interest
JP is an employee and director of MannKind Corporation. AS is a co-founder and director of Aquila Bioscience Ltd. A.S., S.L., K.M. and K.P.Z. are all co-founders of Cell Stress Discoveries Ltd.
Grant Support
This work was funded in part by Health Research Board (grant number HRA/2009/59; HRA-POR-2014-643), Belgium Grant (IAP 7/32) and Breast Cancer Campaign grant (2010NovPR13) to A.S. A Science Foundation Ireland (SFI) grant co-funded under the European Regional Development Fund under Grant Number 13/RC/2073. P.C was funded by an Irish Cancer Society Scholarship (CRS11CLE). K.M. is funded by an Irish Research Council Fellowship (grant number GOIPD/2014/53). P.C. and S.S. were also funded by Thomas Crawford Hayes.
References
1. Kim J, Kundu M, Viollet B and Guan K-L. AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nature cell biology. 2011; 13:132-141.
2. Sengupta S, Peterson TR and Sabatini DM. Regulation of the mTOR complex 1 pathway by nutrients, growth factors, and stress. Molecular cell. 2010; 40:310-322.
3. Budanov AV and Karin M. p53 Target Genes Sestrin1 and Sestrin2 Connect Genotoxic Stress and mTOR Signaling. Cell. 2008; 134:451-460.
4. Budanov AV, Shoshani T, Faerman A, Zelin E, Kamer I, Kalinski H, Gorodin S, Fishman A, Chajut A, Einat P, Skaliter R, Gudkov AV, Chumakov PM and Feinstein E. Identification of a novel stress-responsive gene Hi95 involved in regulation of cell viability. Oncogene. 2002; 21:6017-6031.
5. Park H-W, Park H, Ro S-H, Jang I, Semple IA, Kim DN, Kim M, Nam M, Zhang D, Yin L and Lee JH. Hepatoprotective role of Sestrin2 against chronic ER stress. Nature communications. 2014; 5:4233.
6. Bruning A, Rahmeh M and Friese K. Nelfinavir and bortezomib inhibit mTOR activity via ATF4-mediated sestrin-2 regulation. Molecular oncology. 2013; 7:1012-1018.
7. Gorman AM, Healy SJM, Jäger R and Samali A. Stress management at the ER: Regulators of ER stress-induced apoptosis. Pharmacology & Therapeutics. 2012; 134:306-316.
8. Logue SE, Cleary P, Saveljeva S and Samali A. New directions in ER stress-induced cell death. Apoptosis. 2013; 18:537-546.
9. Jäger R, Bertrand MJM, Gorman AM, Vandenabeele P and Samali A. The unfolded protein response at the crossroads of cellular life and death during endoplasmic reticulum stress. Biology of the Cell. 2012; 104:259-270.
10. Harding HP, Zhang Y, Bertolotti A, Zeng H and Ron D. Perk is essential for translational regulation and cell survival during the unfolded protein response. Molecular cell. 2000; 5:897-904.
11. Gupta S, Giricz Z, Natoni A, Donnelly N, Deegan S, Szegezdi E and Samali A. NOXA contributes to the sensitivity of PERK-deficient cells to ER stress. FEBS letters. 2012; 586:4023-4030.
12. Deegan S, Saveljeva S, Gorman A and Samali A. Stress-induced self-cannibalism: on the regulation of autophagy by endoplasmic reticulum stress. Cellular and Molecular Life Sciences. 2013; 70:2425-2441.
13. Fribley A, Zeng Q and Wang CY. Proteasome inhibitor PS-341 induces apoptosis through induction of endoplasmic reticulum stress-reactive oxygen species in head and neck squamous cell carcinoma cells. Molecular and cellular biology. 2004; 24:9695-9704.
14. Deegan S, Koryga I, Glynn SA, Gupta S, Gorman AM and Samali A. A close connection between the PERK and IRE arms of the UPR and the transcriptional regulation of autophagy. Biochemical and Biophysical Research Communications. 2015; 456:305-311.
15. Teske BF, Wek SA, Bunpo P, Cundiff JK, McClintick JN, Anthony TG and Wek RC. The eIF2 kinase PERK and the integrated stress response facilitate activation of ATF6 during endoplasmic reticulum stress. Molecular biology of the cell. 2011; 22:4390-4405.
16. Han J, Back SH, Hur J, Lin YH, Gildersleeve R, Shan J, Yuan CL, Krokowski D, Wang S, Hatzoglou M, Kilberg MS, Sartor MA and Kaufman RJ. ER-stress-induced transcriptional regulation increases protein synthesis leading to cell death. Nature cell biology. 2013; 15:481-490.
17. Zhao B, Shah P, Budanov AV, Qiang L, Ming M, Aplin A, Sims DM and He Y-Y. Sestrin2 Protein Positively Regulates AKT Enzyme Signaling and Survival in Human Squamous Cell Carcinoma and Melanoma Cells. Journal of Biological Chemistry. 2014; 289:35806-35814.