
INTRODUCTION
Cytogenetically normal acute myeloid leukemia (CN-AML) comprises the largest percentage of primary AML cases [1]. Although the leukemic blasts do not include detectable chromosome abnormalities in CN-AML patients, they nonetheless hide mutations and aberrantly expressed proteins [2] and microRNAs [3], which are potentially prognostic. Among them, NPM1 [4] and double CEBPA [5] mutations are associated with better outcomes, while FLT3-ITD [6] and RUNX1 mutation [7] are associated with poorer ones. High expression of WT1 [8], BAALC [9], ERG [9], MN1 [10], DNMT3B [11], TCF4 [12], ITPR2 [13] and MAPKBP1 [14] and low expression of LEF1 [15] are also associated with a poor prognosis, as is high expression of miR-155 [16] and miR-188-5p [17] and low expression of let-7a-2-3p [17].
RUNX1 belongs to the Runt-related transcription factor (RUNX) family, which plays a crucial role in normal hematopoiesis, and its abnormal expression is frequently seen in various tumors [18, 19]. In several AML subtypes, for example, chromosomal translocations involving RUNX1 lead to fusion gene formation, RUNX1-RUNX1T1 being the most common type [20]. In addition, RUNX1 mutation leads to a poor outcome in CN-AML [7], and high expression of RUNX1 correlates with a poor prognosis in breast cancer [21]. Notably, although early studies suggested RUNX1 acts as a tumor suppressor gene in AML [22], it is now understood that RUNX1 functions as an oncogene necessary to sustain AML [23–26]. These findings suggest that the prognostic impact of RUNX1 in CN-AML depends on its expression level.
We found that RUNX1 is more strongly expressed in CN-AML patients than in normal bone marrow (NBM), but also was an unfavorable prognostic factor in two large, independent groups of patients with CN-AML. In addition, we provide the first report that RUNX1 expression is linked to particular molecular and clinical characteristics. In order to cast light on the function of RUNX1, we also explored RUNX1-associated genes, microRNAs and important cell signaling pathways.
RESULTS
Expression of RUNX1 in CN-AML BM and NBM
A microarray dataset that included 116 CN-AML samples and 5 NBM samples (GEO accession number GSE1159) was used for the expression analysis [27]. RUNX1 expression was markedly higher in the CN-AML than NBM samples (P < 0.001) (Figure 1A). The overexpression of RUNX1 in CN-AML was further validated using other microarray data, which included 9 CN-AML vs. 10 NBM (P < 0.001) and 9 CN-AML vs. 10 normal peripheral blood (NPB) (P < 0.001). The 9 CN-AML samples consisted of 2 BM and 7 PB samples, GEO accession number GSE9476) [28] (Figure 1B). These findings show that RUNX1 overexpression is widespread among CN-AML patients, and is easy to monitor.
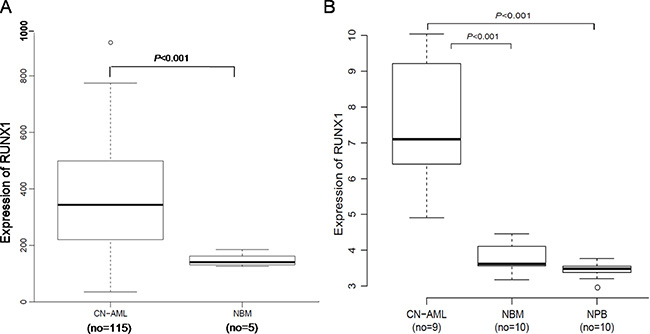
Figure 1: Expression of RUNX1 in CN-AML patients and NBM. (A) Box plot of RUNX1 expression in CN-AML patients (n = 116) and NBM samples (n = 5). (B) Box plot of RUNX1 expression in CN-AML patients (n = 9, including 2 BM and 7 PB samples), NBM samples (n = 10) and NPB samples (n = 10).
Characteristics of patients in the RUNX1high and RUNX1low expression groups
Among the 157 CN-AML patients tested, the RUNX1high group contained significantly more patients with FAB M2 than the RUNX1low (P = 0.001). The RUNX1high patients were also more likely than RUNX1low patients to carry FLT3-ITD and no double CEBPA mutations (P < 0.001, P = 0.003). We found no link between RUNX1 expression and other gene mutations, but RUNX1high patients with CN-AML were more likely to highly express ERG, WT1, DNMT3B, TCF4, MIR155HG, ITPR2 and MAPKBP1 (P < 0.001, P < 0.001, P < 0.001, P < 0.001, P = 0.01, P < 0.001, and P< 0.001, respectively) (Table 1, Supplementary Figure 1).
Table 1: Patients’ characteristics in the testing group of 157 CN-AML patients according to RUNX1 expression levels
Variable |
RUNX1high, n = 78 |
RUNX1low, n = 79 |
P |
|---|---|---|---|
Median age. y (range) |
50 (18~77) |
48 (16~75) |
0.325 |
Female sex, no.(%) |
40 |
33 |
0.27 |
FAB subtype, no. |
|
|
|
M0 |
1 |
2 |
1 |
M1 |
25 |
20 |
0.38 |
M2 |
24 |
8 |
0.001 |
M3 |
1 |
0 |
1 |
M4 |
12 |
12 |
0.5 |
M5 |
14 |
25 |
0.06 |
M6 |
0 |
1 |
1 |
Other |
1 |
11 |
0.005 |
FLT3-ITD, no. |
45 |
21 |
< 0.001 |
FLT3-TKD, no. |
8 |
12 |
0.47 |
NPM1, mutated, no. |
46 |
36 |
0.11 |
Double CEBPA, mutated, no. |
2 |
14 |
0.003 |
N-RAS, mutated, no. |
4 |
9 |
0.25 |
K-RAS, mutated, no. |
0 |
1 |
1 |
IDH1, mutated, no. |
58 |
59 |
0.64 |
IDH2, mutated, no. |
59 |
64 |
0.49 |
ELN genetic group, no. |
|
|
|
Favorable |
13 |
22 |
0.125 |
Intermediate-I |
65 |
57 |
0.12 |
High ERG, no. |
51 |
27 |
< 0.001 |
High BAALC, no. |
43 |
35 |
0.2 |
High LEF1, no. |
33 |
45 |
0.08 |
High MN1, no. |
39 |
39 |
1 |
High WT1, no. |
60 |
18 |
< 0.001 |
High DNMT3B, no. |
58 |
20 |
< 0.001 |
High TCF4, no. |
53 |
25 |
< 0.001 |
High MIR155HG, no. |
47 |
31 |
0.01 |
High ITPR2, no. |
54 |
24 |
< 0.001 |
High MAPKBP1, no. |
54 |
24 |
< 0.001 |
FAB, French-American-British classification; ITD, internal tandem duplication; TKD, tyrosine kinase domain; ELN, European Leukemia Net.
High ERG, BAALC, LEF1, MN1, WT1, DNMT3B, TCF4, MIR155HG, ITPR2 and MAPKBP1 expression were defined as an expression level above the median of all samples, respectively.
RUNX1high is associated with poor outcomes
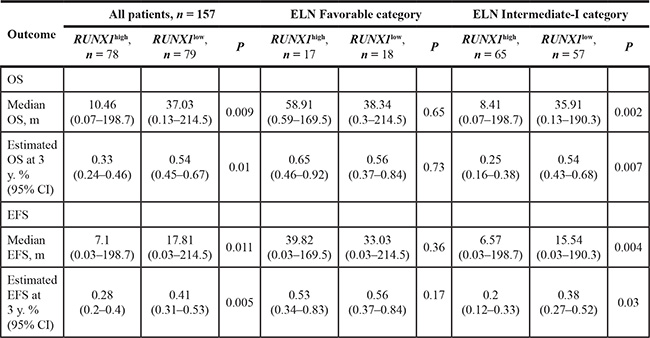
The median overall survival (OS) and event-free survival (EFS) in the RUNX1high group were obviously poorer than that of RUNX1lowgroup (P = 0.009, P = 0.011, respectively, Table 2). This was confirmed comparison using the Log-rank test, which also showed that OS (Figure 2B, P = 0.0025) and EFS (Figure 2A, P = 0.0025) were clearly poorer in the RUNX1high than RUNX1low group.
Table 2: Survival according to RUNX1 expression in the testing group of 157 CN-AML patients
CI, confidence interval.
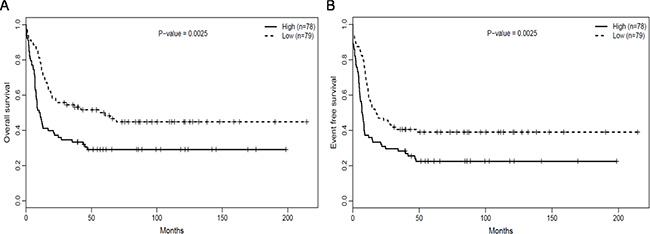
Figure 2: RUNX1high is associated with poorer outcomes. (A) OS and (B) EFS in the testing group of 157 CN-AML patients.
Association of RUNX1 expression with prognostic significance in ELN genetic groups
We assessed the association between RUNX1 expression and prognostic significance separately within the European Leukemia NET (ELN) favorable and Intermediate-I genetic groups. Within the ELN favorable group (n = 35), there was no obvious difference in OS (Figure 3A, P = 0.6976) and EFS (Figure 3B, P = 0.5098) between the RUNX1high and RUNX1lowgroup. In the ELN Intermediate-I group (n = 122), however, the RUNX1high group had poorer OS (Figure 3C, P = 0.0009) and EFS (Figure 3D, P = 0.0014) than the RUNX1low group. The median OS, EFS and estimated survival in the ELN Intermediate-I group (n = 122) also obviously differed between the RUNX1high and RUNX1lowgroups (Table 2).
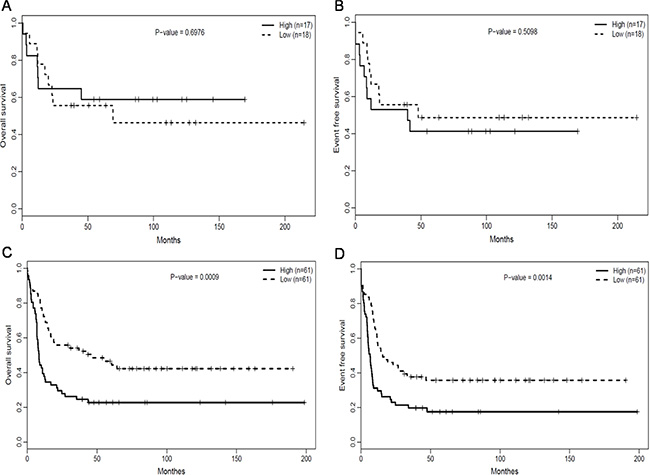
Figure 3: Association of RUNX1 expression with the prognostic significance in ELN genetic groups. (A) OS and (B) EFS of CN-AML patients in the ELN favorable genetic group. (C) OS and (D) EFS of CN-AML patients in the ELN intermediate-I genetic group.
RUNX1 expression is associated with poorer OS and EFS in multivariable analyses
ELN segregated CN-AML patients based on presence of FLT3-ITD, mutations of NPM1 and CEBPA. After adjusting for the impact of these known risk factors, we performed multivariable analyses to confirm the prognostic significance of RUNX1 expression. In a multivariable model, the RUNX1high group had a poorer OS (P = 0.04, Table 3). The other factors associated with poor OS were NPM1 wild-type and FLT3-ITD. The RUNX1high group also had a poorer EFS in a multivariable model (P = 0.02, Table 3). The other factors associated with poor EFS were NPM1 wild-type and FLT3-ITD.
Table 3: Multivariable analysis with OS and EFS in the testing group of 157 CN-AML patients
Variable |
OS, n = 157 |
EFS, n = 157 |
||
|---|---|---|---|---|
HR (95% CI) |
P |
HR (95% CI) |
P |
|
RUNX1 expression, high VS low |
1.56 (1.01–2.41) |
0.04 |
1.65 (1.10–2.48) |
0.02 |
Age, per 10-y increase |
1.13 (0.98–1.32) |
0.09 |
1.05 (0.92–1.21) |
0.47 |
Sex male VS female |
0.82 (0.54–1.23) |
0.33 |
0.99 (0.67–1.46) |
0.96 |
NPM1, mutated VS wild type |
0.51 (0.32–0.81) |
0.005 |
0.53 (0.34–0.83) |
0.005 |
FLT3-ITD, mutated VS wild type |
1.98 (1.25–3.14) |
0.003 |
1.85 (1.20–2.85) |
0.005 |
CEBPA, mutated VS wild type |
0.71 (0.38–1.35) |
0.3 |
0.78 (0.43–1.41) |
0.41 |
HR, hazards ratio; CI, confidence interval.
Validation in a patient group of 162 CN-AML samples
We also studied a group of 162 previously untreated CN-AML patients. The RUNX1high group contained significantly more patients with FAB M1 than the RUNX1low group (P = 0.0014). In addition, RUNX1high patients with CN-AML were more likely to have higher expression of ERG, BAALC, WT1, DNMT3B, TCF4, ITPR2 and MAPKBP1 (P < 0.001, P = 0.028, P < 0.001, P < 0.001, P < 0.001, P < 0.001, and P < 0.001, respectively) and low LEF1 (P < 0.001) compared with RUNX1low patients (Supplementary Table 1). In addition, RUNX1high patients showed a significant poor OS (n = 81 vs n = 81, P = 0.04; Supplementary Figure 2) than RUNX1low patients.
Genome-wide gene expression profiles associated with RUNX1 expression
To further evaluate the role of RUNX1 in CN-AML, we using microarray analysis to determine RUNX1-associated gene expression profiles. We identified 578 up-regulated genes and 727 down-regulated genes that were significantly associated with RUNX1high (Supplementary Table 2). The up-regulated genes included some of those previously found to be involved in leukemogenesis, including CDK6, which encodes a cyclin kinase; MYCN, MYB and MYC; members of the HOXB gene family (HOXB2, HOXB3, and HOXB4), which encode transcription factors [29]; and c-KIT and FLT3, which encode tyrosine kinases. Several independent unfavorable prognostic factors in CN-AML were also up-regulated, including ERG, WT1, TCF4 and DNMT3B. Also up-regulated were B4GALT6, which is expressed in less differentiated precursors [30]; SOCS2, which is predictive of a poor outcome in pediatric AML [31]; BCL11A and GUCY1A3, which are down-regulated in low ERG expressers [9]; GTF2H2/ABCC5, which correlates with chemotherapy resistance in non-small cell lung cancer [32]; DNTT, which is expressed in early lymphoid precursors [33]; CD109, which is overexpressed in early hematopoietic stem cells [34]; FAM92A1, which enhances cell growth during renal carcinogenesis [35]; and MMP2, which promotes lung cancer metastasis [36]. The down-regulated genes included thanatos-associated protein 2 (THAP2) and CD48, CD86 and ICAM1, all of which are involved in immune function. LEF1, an independent favorable prognostic factor in CN-AML, was also down-regulated (Figure 4A and 4B). These results provided further evidence for the prognostic correlation described above.
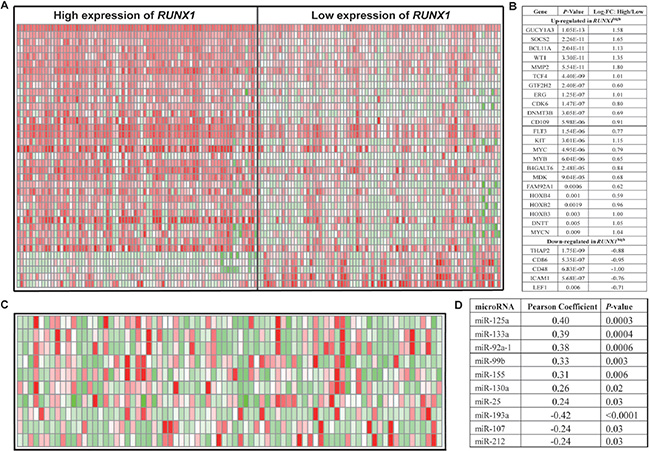
Figure 4: Genome-wide gene/microRNA-expression profiles associated with RUNX1 expression. (A) Expression heat map of associated genes (B) The list of associated genes. (C) Expression heat map of associated microRNAs. (D) The list of associated microRNAs.
Genome-wide microRNA profiles associated with RUNX1 expression
A genome-wide analysis of microRNA profiles revealed that 108 microRNAs were significantly associated with RUNX1 expression (P < 0.05) (Supplementary Table 3). RUNX1high was positively associated with miR-155, miR-125a, miR-99b, miR-133a, miR-130a, miR-25 and miR-92a-1. MiR-155 was previously found to function as an oncogene in CN-AML [16]. MiR-125a and miR-99b were highly expressed in hematopoietic stem cells [37]. MiR-133a was up-regulated in CN-AML along with IDH2 codon R172K [38]. MiR-130a associated with strong expression of WT1, which was consistent with the gene-expression profiles [39]. MiR-25 increases induction of somatic cells into induced pluripotent stem cells [40]. MiR-92a-1 arouses erythroleukemia through down-regulation of p53 [41]. Notably, miR-193a, miR-107 and miR-212 were all down-regulated. We previously found that miR-193a enhanced expression of c-kit [42], which is also consistent with the observed gene-expression profiles. MiR-107 targets NFIX, which competes with CEBPA for binding to the promoter of miR-223, impaired granulocytic differentiation [43, 44]. MiR-212 expression is favorable for survival among molecularly and cytogenetically heterogeneous AMLs [45] (Figure 4C and 4D).
Genome-wide methylation profiling associated with RUNX1 expression
It has been suggested that control of gene expression through methylation of the gene promoter or body plays a pivotal role in determining the behavior of cancer cells [46, 47]. Moreover, gene promoter methylation can be predictive of clinical outcome in AML patients [48, 49]. Because RUNX1 expression correlated positively with DNMT3B expression, we compared the patterns of gene methylation in RUNX1high (n = 37) and RUNX1low (n = 37) CN-AML from TCGA [50]. However, we found no significant differences in patterns of RUNX1 methylation in any of these analyses (Supplementary Figure 3A and 3B).
Cell signaling pathways associated with RUNX1 expression
We used MSigDB [51] to evaluate the cell signaling pathways underlying the biological features associated with RUNX1. Signaling pathways involved in DNA_REPLICATION, RNA_POLYMERASE and CELL_CYCLE were significantly up-regulated, while NATURAL_KILLER_CELL_MEDIATED_CYTOTOXICI TY, ANTIGEN_PROCESSING_AND_PRESENTATION and APOPTOSIS were down-regulated (Table 4). These findings were consistent with the above-noted dysregulated genes involved in the development of CN-AML.
Table 4: Cell signalling pathways associated with RUNX1 expression levels
Pathway name |
According to high expression of RUNX1 |
|
|---|---|---|
Regulation |
P |
|
KEGG_DNA_REPLICATION |
Up |
0.00424 |
KEGG_RNA_POLYMERASE |
Up |
0.01575 |
KEGG_CELL_CYCLE |
Up |
0.02204 |
KEGG_NATURAL_KILLER_CELL_MEDIATED_CYTOTOXICITY |
Down |
0.00000 |
KEGG_ANTIGEN_PROCESSING_AND_PRESENTATION |
Down |
0.00000 |
KEGG_APOPTOSIS |
Down |
0.00217 |
DISCUSSION
CN-AML is the largest cytogenetic subset in AML patients and lacks sensitive prognostic biomarkers, so identification of universal prognostic biomarkers is a very important field in CN-AML research. RUNX1 plays a crucial role in the development of normal hematopoiesis. Traditionally, loss of RUNX1 leads to impaired differentiation and is followed by leukemia development [52]. However, several recent studies found that RUNX1 plays a prosurvival role by supporting leukemia cell proliferation [23–26]. Based on earlier studies of RUNX1, the following conclusions can be made: 1) RUNX1 plays an important dual role in myeloid leukemogenesis, depending on the level of its expression; 2) normal expression of RUNX1 works as a tumor suppressor, inhibiting cell proliferation and promoting differentiation of hematopoietic progenitor cells; 3) Partial deactivation of RUNX1 leads to amplification of myeloid progenitors and subsequent development of AML; and 4) further reduction of RUNX1 expression causes cell cycle arrest and cell death [23–26].
Extending the studies outlined above, ours is the first study to show the prognostic relevance of RUNX1 expression in CN-AML patients and that RUNX1high is associated with poorer OS and EFS in CN-AML patients. RUNX1 is up-regulated in CN-AML patients compared with NBM. In our study, the RUNX1high group contained significantly more patients from the M1 (validating group) and M2 (testing group) FAB subgroups than did the RUNX1low group, which suggests leukemic cells from RUNX1high patients derive from relatively more immature cells. In addition, we found that RUNX1high was associated with FLT3-ITD, non double CEBPA mutation and higher ERG, WT1, DNMT3B, TCF4, MIR155HG, ITPR2, MAPKBP1 expression, all of which are unfavorable molecular characteristics in CN-AML patients. Furthermore, the association of RUNX1high with poorer OS and EFS was confirmed in multivariable analyses adjusting for the most important clinical and molecular prognosticators in CN-AML patients. RUNX1high was associated with wild-type NPM1 and FLT3-ITD, both of which are unfavorable molecular characteristics in CN-AML patients. These results suggest RUNX1high may be a surrogate marker for other unfavorable mutations. Our results also suggest that the prognostic impact of RUNX1 expression is most pronounced in the ELN intermediate-I genetic group, and thus RUNX1 expression may be used to further refine risk stratification for these patients.
The mechanisms underlying the association between RUNX1high and poorer treatment outcomes are unclear. In our present study, we analyzed gene and microRNA expression, DNA methylation profiles, and cell signaling pathways to identify biological mechanisms associated with RUNX1 expression in CN-AML patients. Gene sets related to cell proliferation and cell cycle regulation, particularly c-KIT, FLT3, MYCN, MYB, MYC and CDK6, were up-regulated in the CN-AML patients with RUNX1high, while gene sets related to independent unfavorable prognostic factors, particularly ERG, WT1 and DNMT3B, were also up-regulated, and gene sets related to apoptosis, immune activation of NK cell and independent superior prognostic factor were down-regulated. Acting collectively, these features may lead to CN-AML.
The RUNX1-associated microRNA profile was also noteworthy, as it included the miR-155 and miR-130a families, which were expressed with RUNX1. The up-regulation of miR-155 was associated with an unfavorable clinical outcome independently in CN-AML. MiR-130a was associated with high expression of WT1. The down-regulation of miR-193a was associated with high expression of c-KIT. This new finding of RUNX1-associated alterations in microRNA expression may contribute to leukemogenesis.
Current studies suggest that hypermethylation of the gene promoter and hypomethylation of gene body contribute to the development of tumors [46, 47]. However, we found no significant association between RUNX1 expression and the methylation levels in its promoter region or gene body. Therefore, although RUNX1high is a predictive marker poorer outcome in CN-AML, epigenetic regulation may not play an important role in RUNX1high CN-AML development.
Several important signaling pathways that promote cell proliferation in tumors or contribute to leukemogenesis, including DNA_REPLICATION, RNA_POLYMERASE and CELL_CYCLE were up-regulated, and NATURAL_KILLER_CELL_MEDIATED_CYTOTO XICITY, ANTIGEN_PROCESSING_AND_PRESENTA TION, all lead to immune escape, while APOPTOSIS was down-regulated in the RUNX1high CN-AML. These changes may contribute to a poor outcome.
In summary, our study is the first to provide evidence that RUNX1high is associated with poorer outcomes in CN-AML patients, even after adjusting for known molecular risk factors. In the validating group, earlier findings demonstrated that the microarray expression data for LEF1 was in good agreement with quantitative real time PCR (qPCR) [15]. This shows to some degree the consistency and validity of the microarray expression data. Because RUNX1 is widely expressed at a higher level in CN-AML patients than NBM, RUNX1 expression can be easily measured. This may therefore be a valuable new marker for risk stratification of CN-AML patients. Moreover, our gene/microRNA expression data and cell signaling pathways from tested CN-AML patients offers insight into the biological changes associated with different RUNX1 expression levels.
MATERIALS AND METHODS
Patients and treatment
In the testing group, 157 patients with previously untreated CN-AML (median age, 50 years; range, 16–77 years) were studied. All patients received uniform therapeutic treatment based on study protocols of the Dutch-Belgian Hemato-Oncology Cooperative Group (HOVON) between 1990 and 2008 (The details of therapeutic protocol are available at http://www.hovon.nl) [53] (Supplementary Figure 4). One hundred thirty patients (83%) were aged < 60 years (younger patients) and 27 patients (17%) were ≥ 60 years (older patients). The diagnosis of normal karyotype AML was based on conventional cytogenetic examination of at least 20 metaphases from BM. Patients were assessed for NPM1, CEBPA, N-RAS, K-RAS, IDH1, and IDH2 mutations, FLT3-ITD, and tyrosine kinase domain mutations (FLT3-TKD [D835]). Clinical, cytogenetic and molecular information, as well as the gene expression profiles for all primary AML cases, can be publicly downloaded from the Gene Expression Omnibus (www.ncbi.nlm.nih.gov/geo, accession number GSE6891) [53]. This research was approved by the institutional review boards at Weill Cornell Medical College and Erasmus University Medical Center, and written informed consent was obtained from all patients in accordance with the Declaration of Helsinki. Another independent validation group of 162 CN-AML patients received uniform therapeutic treatment provided as part the multicenter AMLCG-1999 trial, which was used to validate our findings. These patients received intensive double induction and consolidation chemotherapy. Gene expression data are publicly available (http://www.ncbi.nlm.nih.gov/geo/, accession number GSE12417) [54]. The AMLCG-1999 clinical trials were approved by the local institutional review boards, and informed consent from all patients was obtained in accordance with the Declaration of Helsinki [54].
Microarray analyses
Gene expression and methylation data have been previously published (accession number GSE1159 [27], GSE9476 [28], GSE6891 [53] and GSE12417 [54] for expression, The Cancer Genome Atlas (TCGA) [50] for methylation). Briefly, gene expression data were obtained using Affymetrix Human Genome 133 plus 2.0 and U133A Gene Chips. All the designs and quality control for microarray experiment were according to the standard Affymetrix protocols. Expression data for microRNA were from TCGA obtained using whole-genome high-throughput sequencing, which provided 79 CN-AML patients [50]. In addition, genome-wide methylation levels in these patients were determined using Illumina 450K chips [50]. Patients with RUNX1 expression values above the median of all patients were classified as having RUNX1high, and the others were considered to have RUNX1low. Levels of ERG, BAALC, LEF1, MN1, WT1, DNMT3B, TCF4, MIR155HG, ITPR2 and MAPKBP1 expression were also determined from the microarray data.
Statistical analyses
The time from the date of diagnosis to death due to any cause defined OS, and the time from the date of diagnosis to removal from the study due to the absence of complete remission, relapse or death defined EFS. Because we found that RUNX1 expression is normally distributed, a distribution of the cohort based on the highest 50% (RUNX1high) and the lowest 50% RUNX1 expression (RUNX1low) was used for further analysis (Supplementary Figure 5). The Kaplan-Meier method was then used to estimate the association between RUNX1 expression and the OS and EFS, which were further validated using the log-rank test. To investigate the associations between RUNX1 expression levels and clinical, molecular characteristics, the Fisher exact and Wilcoxon rank-sum tests were used for hypothesis testing with categorical and continuous variables, respectively. In addition, multivariable Cox proportional hazard models were used to study how RUNX1 expression levels were associated with OS and EFS in the presence of other known risk factors. With the two groups divided based on RUNX1 expression levels, Student’s t-test and multiple hypothesis correction (False Discovery Rate, FDR) was used to identify differences in gene/microRNA expression and DNA methylation profiles. The statistical cutoff values were an absolute fold-change (FC) ≥ 1.5 and an adjusted P-value ≤ 0.05. All analyses were performed using the R 3.1.1 software packages.
ACKNOWLEDGMENTS
This work was supported by grants from the National Natural Science Foundation of China (81500118, 61501519, 81172245)
Authors’ contributions
L. Fu and L. Tian designed the study and wrote the manuscript, H.P Fu, K.M Xu, K. Hu and J. Wang performed statistical analyses, J.J. Wang and H.M. Jing analyzed the data, J.L. Shi and X.Y. Ke coordinated the study over the entire time. All authors approved the final manuscript.
CONFLICTS OF INTEREST
The authors report no potential conflicts of interest.
REFERENCES
1. Mrozek K, Heerema NA, Bloomfield CD. Cytogenetics in acute leukemia. Blood reviews. 2004; 18:115–136. doi: 10.1016/S0268-960X(03)00040-7.
2. Mrozek K, Marcucci G, Paschka P, Whitman SP, Bloomfield CD. Clinical relevance of mutations and gene-expression changes in adult acute myeloid leukemia with normal cytogenetics: are we ready for a prognostically prioritized molecular classification? Blood. 2007; 109:431–448. doi: 10.1182/blood-2006-06-001149.
3. Marcucci G, Mrozek K, Radmacher MD, Garzon R, Bloomfield CD. The prognostic and functional role of microRNAs in acute myeloid leukemia. Blood. 2011; 117:1121–1129. doi: 10.1182/blood-2010-09-191312.
4. Dohner K, Schlenk RF, Habdank M, Scholl C, Rucker FG, Corbacioglu A, Bullinger L, Frohling S, Dohner H. Mutant nucleophosmin (NPM1) predicts favorable prognosis in younger adults with acute myeloid leukemia and normal cytogenetics: interaction with other gene mutations. Blood. 2005; 106:3740–3746. doi: 10.1182/blood-2005-05-2164.
5. Li HY, Deng DH, Huang Y, Ye FH, Huang LL, Xiao Q, Zhang B, Ye BB, Lai YR, Mo ZN, Liu ZF. Favorable prognosis of biallelic CEBPA gene mutations in acute myeloid leukemia patients: a meta-analysis. European journal of haematology. 2015; 94:439–48. doi: 10.1111/ejh.12450.
6. Santos FP, Jones D, Qiao W, Cortes JE, Ravandi F, Estey EE, Verma D, Kantarjian H, Borthakur G. Prognostic value of FLT3 mutations among different cytogenetic subgroups in acute myeloid leukemia. Cancer. 2011; 117:2145–2155. doi: 10.1002/cncr.25670.
7. Mendler JH, Maharry K, Radmacher MD, Mrozek K, Becker H, Metzeler KH, Schwind S, Whitman SP, Khalife J, Kohlschmidt J, Nicolet D, Powell BL, Carter TH, et al. RUNX1 mutations are associated with poor outcome in younger and older patients with cytogenetically normal acute myeloid leukemia and with distinct gene and MicroRNA expression signatures. Journal of clinical oncology. 2012; 30:3109–3118. doi: 10.1200/JCO.2011.40.6652.
8. Lyu X, Xin Y, Mi R, Ding J, Wang X, Hu J, Fan R, Wei X, Song Y, Zhao RY. Overexpression of Wilms tumor 1 gene as a negative prognostic indicator in acute myeloid leukemia. PloS one. 2014; 9:e92470. doi: 10.1371/journal.pone.0092470.
9. Schwind S, Marcucci G, Maharry K, Radmacher MD, Mrozek K, Holland KB, Margeson D, Becker H, Whitman SP, Wu YZ, Metzeler KH, Powell BL, Kolitz JE, et al. BAALC and ERG expression levels are associated with outcome and distinct gene and microRNA expression profiles in older patients with de novo cytogenetically normal acute myeloid leukemia: a Cancer and Leukemia Group B study. Blood. 2010; 116:5660–5669. doi: 10.1182/blood-2010-06-290536.
10. Schwind S, Marcucci G, Kohlschmidt J, Radmacher MD, Mrozek K, Maharry K, Becker H, Metzeler KH, Whitman SP, Wu YZ, Powell BL, Baer MR, Kolitz JE, et al. Low expression of MN1 associates with better treatment response in older patients with de novo cytogenetically normal acute myeloid leukemia. Blood. 2011; 118:4188–4198. doi: 10.1182/blood-2011-06-357764.
11. Niederwieser C, Kohlschmidt J, Volinia S, Whitman SP, Metzeler KH, Eisfeld AK, Maharry K, Yan P, Frankhouser D, Becker H, Schwind S, Carroll AJ, Nicolet D, et al. Prognostic and biologic significance of DNMT3B expression in older patients with cytogenetically normal primary acute myeloid leukemia. Leukemia. 2015; 29:567–575. doi: 10.1038/leu.2014.267.
12. In ‘t Hout FE, van der Reijden BA, Monteferrario D, Jansen JH, Huls G. High expression of transcription factor 4 (TCF4) is an independent adverse prognostic factor in acute myeloid leukemia that could guide treatment decisions. Haematologica. 2014; 99:e257–259. doi: 10.3324/haematol.2014.110437.
13. Shi JL, Fu L, Wang WD. High expression of inositol 1,4,5-trisphosphate receptor, type 2 (ITPR2) as a novel biomarker for worse prognosis in cytogenetically normal acute myeloid leukemia. Oncotarget. 2015; 6:5299–5309. doi: 10.18632/oncotarget.3024.
14. Fu L, Shi J, Hu K, Wang J, Wang W, Ke X. Mitogen-activated protein kinase binding protein 1 (MAPKBP1) is an unfavorable prognostic biomarker in cytogenetically normal acute myeloid leukemia. Oncotarget. 2015; 6:8144–8154. doi: 10.18632/oncotarget.3519.
15. Metzeler KH, Heilmeier B, Edmaier KE, Rawat VP, Dufour A, Dohner K, Feuring-Buske M, Braess J, Spiekermann K, Buchner T, Sauerland MC, Dohner H, Hiddemann W, et al. High expression of lymphoid enhancer-binding factor-1 (LEF1) is a novel favorable prognostic factor in cytogenetically normal acute myeloid leukemia. Blood. 2012; 120:2118–2126. doi: 10.1182/blood-2012-02-411827.
16. Marcucci G, Maharry KS, Metzeler KH, Volinia S, Wu YZ, Mrozek K, Nicolet D, Kohlschmidt J, Whitman SP, Mendler JH, Schwind S, Becker H, Eisfeld AK, et al. Clinical role of microRNAs in cytogenetically normal acute myeloid leukemia: miR-155 upregulation independently identifies high-risk patients. Journal of clinical oncology. 2013; 31:2086–2093. doi: 10.1200/JCO.2012.45.6228.
17. Shi Jinlong, Fu Lin, Li Yonghui, Yu Li, Weidong W. Identification of let-7a-2-3p or/and miR-188-5p as Prognostic Biomarkers in Cytogenetically Normal Acute Myeloid Leukemia. PLOS One. 2015 3;10:e0118099. doi: 10.1371/journal.pone.0118099.
18. Lam K, Zhang DE. RUNX1 and RUNX1-ETO: roles in hematopoiesis and leukemogenesis. Front Biosci (Landmark Ed). 2012; 17:1120–1139. doi: 10.2741/3977.
19. Ge T, Yin M, Yang M, Liu T, Lou G. MicroRNA-302b Suppresses Human Epithelial Ovarian Cancer Cell Growth by Targeting RUNX1. Cellular physiology and biochemistry: international journal of experimental cellular physiology, biochemistry, and pharmacology. 2014; 34:2209–2220. doi: 10.1159/000369664.
20. Peterson LF, Zhang DE. The 8;21 translocation in leukemogenesis. Oncogene. 2004; 23:4255–4262. doi: 10.1038/sj.onc.1207727.
21. Ferrari N, Mohammed ZM, Nixon C, Mason SM, Mallon E, McMillan DC, Morris JS, Cameron ER, Edwards J, Blyth K. Expression of RUNX1 correlates with poor patient prognosis in triple negative breast cancer. PloS one. 2014; 9:e100759. doi: 10.1371/journal.pone.0100759.
22. Silva FP, Morolli B, Storlazzi CT, Anelli L, Wessels H, Bezrookove V, Kluin-Nelemans HC, Giphart-Gassler M. Identification of RUNX1/AML1 as a classical tumor suppressor gene. Oncogene. 2003; 22:538–547. doi: 10.1038/sj.onc.1206141.
23. Ben-Ami O, Friedman D, Leshkowitz D, Goldenberg D, Orlovsky K, Pencovich N, Lotem J, Tanay A, Groner Y. Addiction of t(8;21) and inv(16) acute myeloid leukemia to native RUNX1. Cell reports. 2013; 4:1131–1143. doi: 10.1016/j.celrep.2013.08.020.
24. Goyama S, Schibler J, Cunningham L, Zhang Y, Rao Y, Nishimoto N, Nakagawa M, Olsson A, Wunderlich M, Link KA, Mizukawa B, Grimes HL, Kurokawa M, et al. Transcription factor RUNX1 promotes survival of acute myeloid leukemia cells. The Journal of clinical investigation. 2013; 123:3876–3888. doi: 10.1172/JCI68557.
25. Goyama S, Huang G, Kurokawa M, Mulloy JC. Posttranslational modifications of RUNX1 as potential anticancer targets. Oncogene. 2015; 34:3483–92. doi: 10.1038/onc.2014.305.
26. Wilkinson AC, Ballabio E, Geng H, North P, Tapia M, Kerry J, Biswas D, Roeder RG, Allis CD, Melnick A, de Bruijn MF, Milne TA. RUNX1 is a key target in t(4;11) leukemias that contributes to gene activation through an AF4-MLL complex interaction. Cell reports. 2013; 3:116–127. doi: 10.1016/j.celrep.2012.12.016.
27. Valk PJ, Verhaak RG, Beijen MA, Erpelinck CA, Barjesteh van Waalwijk van Doorn-Khosrovani S, Boer JM, Beverloo HB, Moorhouse MJ, van der Spek PJ, Lowenberg B, Delwel R. Prognostically useful gene-expression profiles in acute myeloid leukemia. The New England journal of medicine. 2004; 350:1617–1628. doi: 10.1056/NEJMoa040465.
28. Stirewalt DL, Meshinchi S, Kopecky KJ, Fan W, Pogosova-Agadjanyan EL, Engel JH, Cronk MR, Dorcy KS, McQuary AR, Hockenbery D, Wood B, Heimfeld S, Radich JP. Identification of genes with abnormal expression changes in acute myeloid leukemia. Genes, chromosomes & cancer. 2008; 47:8–20. doi: 10.1002/gcc.20500.
29. Shah N, Sukumar S. The Hox genes and their roles in oncogenesis. Nature reviews Cancer. 2010; 10:361–371. doi: 10.1038/nrc2826.
30. Toren A, Bielorai B, Jacob-Hirsch J, Fisher T, Kreiser D, Moran O, Zeligson S, Givol D, Yitzhaky A, Itskovitz-Eldor J, Kventsel I, Rosenthal E, Amariglio N, et al. CD133-positive hematopoietic stem cell “stemness” genes contain many genes mutated or abnormally expressed in leukemia. Stem Cells. 2005; 23:1142–1153. doi: 10.1634/stemcells.2004-0317.
31. Laszlo GS, Ries RE, Gudgeon CJ, Harrington KH, Alonzo TA, Gerbing RB, Raimondi SC, Hirsch BA, Gamis AS, Meshinchi S, Walter RB. High expression of suppressor of cytokine signaling-2 predicts poor outcome in pediatric acute myeloid leukemia: a report from the Children’s Oncology Group. Leukemia & lymphoma. 2014; 55:2817–2821. doi: 10.3109/10428194.2014.893305.
32. Weaver DA, Crawford EL, Warner KA, Elkhairi F, Khuder SA, Willey JC. ABCC5, ERCC2, XPA and XRCC1 transcript abundance levels correlate with cisplatin chemoresistance in non-small cell lung cancer cell lines. Molecular cancer. 2005; 4:18. doi: 10.1186/1476-4598-4-18.
33. Santos PM, Borghesi L. Molecular resolution of the B cell landscape. Current opinion in immunology. 2011; 23:163–170. doi: 10.1016/j.coi.2010.11.014.
34. Murray LJ, Bruno E, Uchida N, Hoffman R, Nayar R, Yeo EL, Schuh AC, Sutherland DR. CD109 is expressed on a subpopulation of CD34+ cells enriched in hematopoietic stem and progenitor cells. Experimental hematology. 1999; 27:1282–1294. doi: 10.1016/S0301-472X(99)00071-5.
35. Liang S, Gong F, Zhao X, Wang X, Shen G, Xu Y, Yang H, Ruan X, Wei Y. Prokaryotic expression, purification of a new tumor-relative protein FAM92A1-289 and its characterization in renal cell carcinoma. Cancer letters. 2009; 276:81–87. doi: 10.1016/j.canlet.2008.10.043.
36. Kuo HY, Huang YS, Tseng CH, Chen YC, Chang YW, Shih HM, Wu CW. PML represses lung cancer metastasis by suppressing the nuclear EGFR-mediated transcriptional activation of MMP2. Cell Cycle. 2014; 13:3132–3142. doi: 10.4161/15384101.2014.949212.
37. Gerrits A, Walasek MA, Olthof S, Weersing E, Ritsema M, Zwart E, van Os R, Bystrykh LV, de Haan G. Genetic screen identifies microRNA cluster 99b/let-7e/125a as a regulator of primitive hematopoietic cells. Blood. 2012; 119:377–387. doi: 10.1182/blood-2011-01-331686.
38. Marcucci G, Maharry K, Wu YZ, Radmacher MD, Mrozek K, Margeson D, Holland KB, Whitman SP, Becker H, Schwind S, Metzeler KH, Powell BL, Carter TH, et al. IDH1 and IDH2 gene mutations identify novel molecular subsets within de novo cytogenetically normal acute myeloid leukemia: a Cancer and Leukemia Group B study. Journal of clinical oncology. 2010; 28:2348–2355. doi: 10.1200/JCO.2009.27.3730.
39. Havelange V, Stauffer N, Heaphy CC, Volinia S, Andreeff M, Marcucci G, Croce CM, Garzon R. Functional implications of microRNAs in acute myeloid leukemia by integrating microRNA and messenger RNA expression profiling. Cancer. 2011; 117:4696–4706. doi: 10.1002/cncr.26096.
40. Lu D, Davis MP, Abreu-Goodger C, Wang W, Campos LS, Siede J, Vigorito E, Skarnes WC, Dunham I, Enright AJ, Liu P. MiR-25 regulates Wwp2 and Fbxw7 and promotes reprogramming of mouse fibroblast cells to iPSCs. PloS one. 2012; 7:e40938. doi: 10.1371/journal.pone.0040938.
41. Li Y, Vecchiarelli-Federico LM, Li YJ, Egan SE, Spaner D, Hough MR, Ben-David Y. The miR-17-92 cluster expands multipotent hematopoietic progenitors whereas imbalanced expression of its individual oncogenic miRNAs promotes leukemia in mice. Blood. 2012; 119:4486–4498. doi: 10.1182/blood-2011-09-378687.
42. Gao XN, Lin J, Li YH, Gao L, Wang XR, Wang W, Kang HY, Yan GT, Wang LL, Yu L. MicroRNA-193a represses c-kit expression and functions as a methylation-silenced tumor suppressor in acute myeloid leukemia. Oncogene. 2011; 30:3416–3428. doi: 10.1038/onc.2011.62.
43. Fazi F, Rosa A, Fatica A, Gelmetti V, De Marchis ML, Nervi C, Bozzoni I. A minicircuitry comprised of microRNA-223 and transcription factors NFI-A and C/EBPalpha regulates human granulopoiesis. Cell. 2005; 123:819–831. doi: 10.1016/j.cell.2005.09.023.
44. Garzon R, Pichiorri F, Palumbo T, Visentini M, Aqeilan R, Cimmino A, Wang H, Sun H, Volinia S, Alder H, Calin GA, Liu CG, Andreeff M, et al. MicroRNA gene expression during retinoic acid-induced differentiation of human acute promyelocytic leukemia. Oncogene. 2007; 26:4148–4157. doi: 10.1038/sj.onc.1210186.
45. Sun SM, Rockova V, Bullinger L, Dijkstra MK, Dohner H, Lowenberg B, Jongen-Lavrencic M. The prognostic relevance of miR-212 expression with survival in cytogenetically and molecularly heterogeneous AML. Leukemia. 2013; 27:100–106. doi: 10.1038/leu.2012.158.
46. Esteller M, Fraga MF, Paz MF, Campo E, Colomer D, Novo FJ, Calasanz MJ, Galm O, Guo M, Benitez J, Herman JG. Cancer epigenetics and methylation. Science. 2002; 297:1807–1808; discussion 1807–1808. doi: 10.1126/science.297.5588.1807d.
47. Yang X, Han H, De Carvalho DD, Lay FD, Jones PA, Liang G. Gene body methylation can alter gene expression and is a therapeutic target in cancer. Cancer cell. 2014; 26:577–590. doi: 10.1016/j.ccr.2014.07.028.
48. Figueroa ME, Lugthart S, Li Y, Erpelinck-Verschueren C, Deng X, Christos PJ, Schifano E, Booth J, van Putten W, Skrabanek L, Campagne F, Mazumdar M, Greally JM, et al. DNA methylation signatures identify biologically distinct subtypes in acute myeloid leukemia. Cancer cell. 2010; 17:13–27. doi: 10.1016/j.ccr.2009.11.020.
49. Alvarez S, Suela J, Valencia A, Fernandez A, Wunderlich M, Agirre X, Prosper F, Martin-Subero JI, Maiques A, Acquadro F, Rodriguez Perales S, Calasanz MJ, Roman-Gomez J, et al. DNA methylation profiles and their relationship with cytogenetic status in adult acute myeloid leukemia. PloS one. 2010; 5:e12197. doi: 10.1371/journal.pone.0012197.
50. Cancer Genome Atlas Research Network. Genomic and epigenomic landscapes of adult de novo acute myeloid leukemia. The New England journal of medicine. 2013; 368:2059–2074. doi: 10.1056/NEJMoa1301689
51. Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub TR, Lander ES, Mesirov JP. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proceedings of the National Academy of Sciences of the United States of America. 2005; 102:15545–15550. doi:10.1073/pnas.0506580102
52. Osato M. Point mutations in the RUNX1/AML1 gene: another actor in RUNX leukemia. Oncogene. 2004; 23:4284–4296. doi: 10.1038/sj.onc.1207779.
53. Verhaak RG, Wouters BJ, Erpelinck CA, Abbas S, Beverloo HB, Lugthart S, Lowenberg B, Delwel R, Valk PJ. Prediction of molecular subtypes in acute myeloid leukemia based on gene expression profiling. Haematologica. 2009; 94:131–134. doi: 10.3324/haematol.13299.
54. Metzeler KH, Hummel M, Bloomfield CD, Spiekermann K, Braess J, Sauerland MC, Heinecke A, Radmacher M, Marcucci G, Whitman SP, Maharry K, Paschka P, Larson RA, et al. An 86-probe-set gene-expression signature predicts survival in cytogenetically normal acute myeloid leukemia. Blood. 2008; 112:4193–4201. doi: 10.1182/blood-2008-02-134411.