
INTRODUCTION
Neuroblastoma (NB) is the most common extracranial solid tumor of childhood. NBs show clinical heterogeneity, from spontaneous regression to relentless progression, and NBs account for a disproportionate number of childhood cancer deaths. We and others have identified different patterns of genomic change that underlie these contrasting clinical behaviors [1–6]. Deletion of the short arm of chromosome 1 (1p) occurs in 35% of primary tumors and 80% of tumor-derived cell lines, representing one of the most characteristic genomic changes in NBs [7–10]. Presumably, 1p deletion reflects loss of a tumor suppressor gene (TSG) from this region. We mapped the smallest region of consistent deletion (SRD) to an approximately 2 Mb region on 1p36.31 [11, 12]. Indeed, the SRD identified by most other groups mapping 1p deletions in NBs overlaps our region [13–16]. We analyzed 23 genes mapping to the maximal SRD on 1p36.31, and we identified CHD5 as the most likely TSG within this region [11, 17, 18].
The CHD5 gene encodes a novel member of the chromodomain helicase DNA binding (CHD) family, and all proteins have nuclear localization signals, paired chromodomains as well as ATP-dependent helicases [19]. Increasing evidence suggests that CHD protein complexes play an important role in regulating development, cell cycle control and oncogenesis through their influence on chromatin structure and gene expression [20]. CHD5 is expressed almost exclusively in the nervous system and in testis, and expression is virtually undetectable in a panel of NB cell lines compared with fetal brain [21–23].
DNA methylation of the CHD5 promoter region has been well documented in NBs and many other tumor types [17, 24–29]. However, we are exploring other important epigenetic mechanisms of CHD5 transcriptional/translational regulation, including microRNAs (miRNAs). MiRNAs are small, non-coding RNA molecules that have a seed region 2-7 nucleotides from the 5’ end of the miRNA. The seed region imperfectly pairs with the 3’-untranslated region (3’-UTR) of the target mRNA [30]. Binding may then result in mRNA cleavage, degradation or reduced translation efficiency at the ribosome [31]. The imperfect pairing between a miRNA and its target allows a given miRNA to bind one or more sites within a 3’-UTR, or to multiple mRNA targets throughout the genome. This allows miRNAs to have a robust influence on gene expression, and it allows simultaneous regulation of multiple components of the signaling network in development and in cancer [32].
In the present study, we investigated the role of miRNAs on downregulation of CHD5 in NB cell lines in vitro. We used computational analysis to predict which miRNAs may base pair with the 3’-UTR of CHD5 and performed a functional assay to confirm which of the miRNAs target CHD5. We also examined the expression of these miRNAs in primary tumors. Finally, we performed functional assays by transfecting miRNA mimics into a cell line with endogenous CHD5 expression to determine if the miRNAs identified by our reporter assay were able to decrease CHD5 protein expression.
RESULTS AND DISCUSSION
We identified 18 miRNAs that were predicted to target CHD5
We used miRanda (www.microrna.org), TargetScan (www.targetscan.org), miRDB (mirdb.org), and DIANA 3.0 (diana.cslab.ece.ntua.gr/microT) to identify miRNAs predicted to target the CHD5 3’-UTR. Each program yielded a unique list, so we focused on the miRNAs identified by at least two of these prediction algorithms. We identified 18 miRNAs that bound to one of three different target regions of the CHD5 3’-UTR: Region 1 bound miR-204 and -211; Region 2 bound miR-216b and -3666; and Region 3 bound miR-17, -19ab, -20ab, -93, -106ab, -130ab, -301ab, -454, and -519d (Figure 1). The miRNAs, as well as their chromosomal location and nucleotide sequence, are shown in Supplementary Table S1. We focused on these miRNAs for further functional analysis in the NLF and SY5Y NB cell lines. Interestingly, others showed that eight of these miRNAs are MYCN driven in NBs, including miR-17, -19a, -19b, -20a, -20b, -93, -106a, and -106b [33–35].

Figure 1: Nucleotide sequence of the 3’ untranslated region (UTR) of CHD5. Three locations that microRNAs are predicted to bind in the CHD5 3’-UTR are shown in blue, and the miRNAs that target each of these regions are shown below. The regions cloned into the targeting vector are highlighted in yellow.
Select MYCN-driven and other miRNAs directly downregulate CHD5 3’-UTR reporter
For our transfection studies, Renilla luciferase signals were first normalized to an internal firefly luciferase transfection control. Then all samples were normalized using the Qiagen Allstars siRNA as a negative control. There was a prior report that miR-211 targeted the CHD5 3’-UTR in colorectal cancer [36], so we used miR211 as a positive control for these studies. Indeed, all transient transfections were screened for a significant reduction in the wild-type (WT) CHD5 3’-UTR compared to no CHD5 3’-UTR and mutant CHD5 3’-UTR constructs when using the miR-211 mimic. Similar values were obtained for all three vectors when transfected with no miRNA mimic (transfection control). For miRNAs that targeted CHD5, we expected to see a lower ratio for the WT CHD5 3’-UTR compared to both the no insert and mutant CHD5 3’-UTR controls. Data from at least three independent experiments, each done in triplicate, were analyzed using the Prism two-way ANOVA method, followed by a Sidak post-test, as described in the Materials and Methods.
We identified seven miRNAs (of the 18 tested) that regulated the CHD5 3’-UTR reporter in at least one cell line, and six were common to both the NLF and SY5Y cell lines (miR-93, -20b, -17, -204, -211, and -3666); this included the miR-211 positive control, which was predicted to bind to the CHD5 3’-UTR. In the NLF line, miR-106b also regulated expression of the CHD5 3’-UTR reporter (Figure 2A, 2B). Of the MYCN driven subset, miR-17, -20b, -93, and -106b all showed a significant downregulation of WT CHD5 3’-UTR, as measured by the luciferase assay (Figure 2B). In the SY5Y cell line, the six common miRNAs mentioned above also significantly downregulated the CHD5 3’-UTR reporter, but miR106b results did not reach statistical significance (Figure 2C, 2D). Nevertheless, the MYCN-driven miRNA -17, -20b and 93 all regulated the WT CHD5 3’-UTR reporter in SY5Y (Figure 2D). Thus, six of the 18 miRNAs predicted to bind the WT CHD5 3’-UTR downregulated expression of the reporter in both cell lines, three of the eight MYCN-driven miRNAs, and a fourth MYCN-driven miRNA (miR-106b) also regulated the reporter in NLF (Figure 3). Relative luciferase expression of all individual microRNAs in both NLF and SY5Y cell lines are shown in Supplementary Figures S1 and S2.
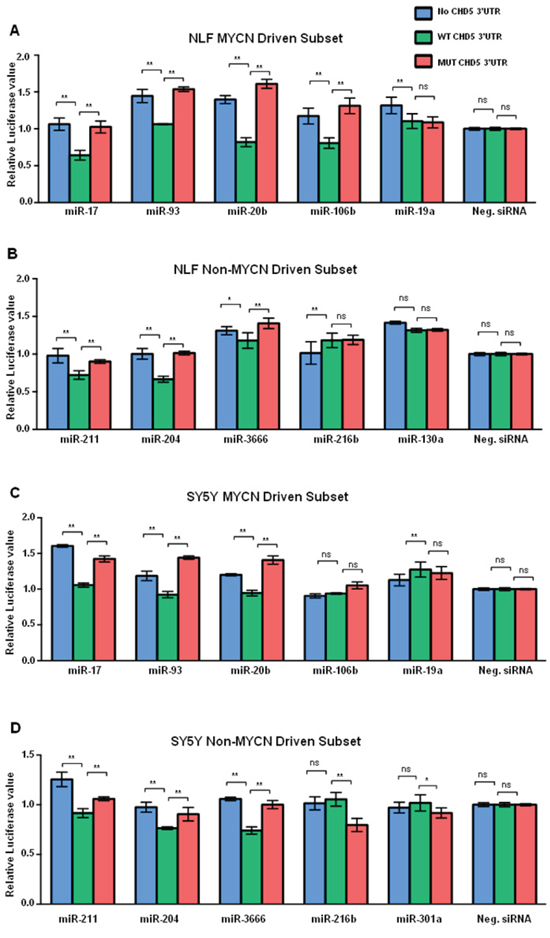
Figure 2: Graphic representation of miRNA regulation of CHD5 3’-RNA reporter construct. Graphical representation of “CHD5 levels” in the NLF cell line after transfection with the MYCN-driven subset of miRNAs. Each sample was transfected with miRNAs from the MYCN driven subset, and either no 3’-UTR inserted (no Insert), wild-type (WT) 3’-UTR insert, or mutated 3’-UTR insert (MUT). Therefore, each value represented in the bar graph reflects the ratio of Renilla to firefly normalized to Allstars siRNA. A. NLF and MYCN-driven miRNAs. B. NLF and non-MYCN-driven miRNAs. C. SY5Y and MYCN-driven miRNAs. D. SY5Y and non-MYCN-driven miRNAs. Each experiment included miR-211 mimic as a positive control, Allstars siRNA (Qiagen) as a negative control, and no miRNA mimic as a transfection control. Each transfection was carried out in triplicate, and each experiment was repeated at least 3 times. Statistical analyses were performed using the Prism two-way ANOVA method followed by a Sidak post-test. Data are expressed as the standard error mean (SEM). Values are the mean of triplicates readings from four independent experiments and p-values were reported (* P<0.05, ** p<0.01 and ns= non-significant).
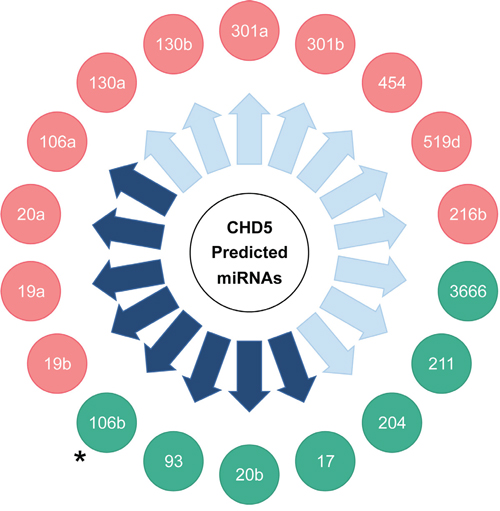
Figure 3: Summary of miRNA regulation of a CHD5 3’-UTR reporter construct in NLF and SY5Y lines with miRNA mimics predicted to target CHD5. miRNAs in green circles caused downregulation of the CHD5 3’-UTR target construct in at least one of the two lines. miRNAs in red circles did not significantly downregulate the CHD5 3’-UTR. Purple arrows indicate miRNAs shown to be upregulated by MYC/MYCN expression. Asterisk (•): miRNA 106b downregulated CHD5 3’-UTR in NLF only, not in SY5Y.
Effect of miRNAs on CHD5 protein expression
In order to further validate the functional regulation of CHD5 expression by miRNAs, we performed transient transfections in the NBLS NB cell line, which has the highest levels of endogenous CHD5 of the lines tested. Cells were transfected with miRNA mimics for each of the seven miRNAs identified as regulating CHD5 expression in our reporter assay, as well as miR-454 as a negative control. Whole cell extracts were subjected to SDS-PAGE followed by western blot analysis. Our western results indicate there was almost complete reduction of CHD5 protein levels in NBLS cells for miR-211, miR-17, miR-93 and miR-20b, whereas no change in CHD4, actin or MYCN protein levels were observed (Figure 4). These results strongly suggest that miR-211, -17, -93 and -20b can dramatically regulate CHD5 protein expression in NBs.
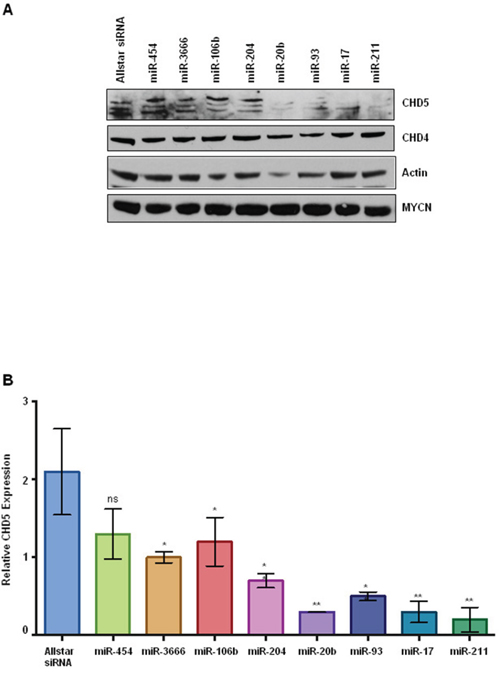
Figure 4: CHD5 protein expression in NBLS cells following transient transfection with miRNA mimics. A. Western blot analysis of transfected cells with indicated microRNAs. Post transfection, cells were washed twice with PBS and isolated cell extracts as described in methods [41]. Whole cell extracts (100 μg) either transfected with indicated miRNAs or mock transfected were subjected to polyacrylamide gel electrophoresis (4-12% SDS-PAGE), using NuPAGE Bis-Tris gels with MOPS-SDS Running Buffer Allstars siRNA and miRNA-454 were used as negative controls. Proteins were transferred on to nitrocellulose membranes (GE Healthcare Life Sciences) and probed with antibodies using rabbit polyclonal CHD5, actin (Santa Cruz Biotechnology, CA 1:1000), rabbit polyclonal CHD4 (Bethyl 1:2000), and MYCN monoclonal (1:5000; BD Biosciences). Almost complete reduction of CHD5 protein levels were observed for miR-20b, miR-93, miR-17, and miR-211 as indicated, but no change in CHD4, actin or MYCN levels were seen. B. Densitometric analysis of CHD5 protein expression in NBLS cell line. The number of pixels from each band was measured, and a bar graph was created using the Prism to indicate the difference in CHD5 expression upon miRNA transfection. Data are expressed as the standard error mean (SEM). Statistical analysis was performed using the Prism one way ANOVA method followed by Tukey’s post-test. Statistical significance relative to the control Allstar siRNA is indicated: *p<0.05; **p<0.01.
Analysis of miRNA, MYCN and CHD5 levels of primary NB tumors
In order to understand possible in vivo relationships between miRNA, MYCN and CHD5 expression, we analyzed 160 primary NB tumor samples. Subcellular protein extraction and western analysis were performed as described previously [37, 38]. Expression level data were available for the following miRNAs, miR-130ab, -93, -17, -106ab, -301ab, -216b, -3666, -211, -204, -20b, and -519d. We found a significant inverse relationship between MYCN expression and CHD5 expression, as reported previously (data not shown) [17, 25]. We found a positive correlation between MYCN and three miRNAs, miR-93, -106b and -130b, two of which are considered MYCN-driven miRNAs. Next, we compared the CHD5 expression levels in all 160 samples to the expression level of each miRNA. The most significant correlation we were able to find was of that between CHD5 and miRNA 130a (data not shown). The lack of a clear correlation between the miRNAs identified here and the primary tumor data may reflect the complexities of epigenetic regulation of CHD5 expression in primary NBs.
miRNA regulation of CHD5 expression in neuroblastoma and other cancers
Cai and colleagues published a study showing that miR-211 regulated CHD5 expression in vitro and in vivo in colorectal cancer [36]. The prediction algorithms we used also identified miR-211 as targeting the CHD5 3’-UTR, and our reporter assay confirmed function regulation in both NB cell lines tested. Yu and coworkers recently identified miR-454 as a regulator of CHD5 expression in hepatocellular carcinoma [39]. However, our studies did not identify activity of miR-454 in NBs, even though this miRNA was also predicted to bind to the CHD5 3’-UTR. Nevertheless, our reporter assay was performed using transient transfection assays, compared to permanent transfection used by Yu for their study. Also, there may be differences in the miRNAs that regulate CHD5 in different cell types (hepatocellular carcinoma vs. NB). Nevertheless, we validated the effect of miR-211 and identified six additional miRNAs that regulate CHD5 expression, including four MYCN-driven miRNAs. Interestingly, there is an inverse relationship between MYCN amplification/overexpression and CHD5 expression in neuroblastomas [11, 17, 25, 40]]. Therefore, increased expression of MYCN could drive expression of miRNAs, which in turn could reduce the expression of CHD5 mRNA and protein.
We used four different prediction algorithms to identify miRNAs that were likely to bind to the CHD5 3’-UTR, and we focused on the 18 miRNAs that were identified by at least two programs. Indeed, we provide functional evidence that at least seven of these miRNAs (including miRNA-211) regulate CHD5 expression in at least one NB line in our reporter assay. Furthermore, four of the seven miRNAs also dramatically reduced CHD5 protein expression in an NB line with exogenous CHD5 expression. However, prediction algorithms are imperfect, and we may have missed other important miRNA regulators of CHD5 expression.
Clearly there are a number of potential mechanisms of epigenetic regulation of genes, including DNA methylation, histone modification, and microRNA regulation. These may be particularly important for silencing of the remaining allele of TSGs in cancers in which one copy is deleted, but the remaining allele is not structurally inactivated. Our data strongly support miRNA regulation as an additional mechanism of CHD5 regulation. Therapy targeting these miRNAs thus represents a potentially viable approach to reactivate expression of the remaining unmutated CHD5 gene and restore growth control in NBs.
MATERIALS AND METHODS
Reagents
Cell culture media RPMI-1640 (Roswell Park Memorial Institute medium), antibiotics and fetal bovine serum (FBS) were obtained from Invitrogen Inc. (Grand Island, NY). Parental NB cell lines were maintained in our lab but are also available from the American Type Culture Collection (ATCC, Manassas, VA). 1X Dulbecco’s Phosphate Buffered Saline (PBS) and Opti-MEM were purchased from Life Technologies (Grand Island, NY). Restriction enzymes and other molecular biology reagents were purchased from Roche Applied Sciences (Indianapolis, IN), Promega Inc. (Madison, WI), and New England Biolabs Inc. (Beverly, MA). NuPAGE gels (4-12%), buffers, and prestained Rainbow molecular weight markers were obtained from Invitrogen (Grand Island, NY). The Dual-Luciferase Reporter Assay reagent (E1910) was from Promega Inc. (Madison, WI).
Antibodies
Rabbit polyclonal CHD5 (sc-68390), rabbit polyclonal CHD4 (Bethyl 1:2000), and mouse monoclonal MYCN (1:5000; BD Biosciences) antibodies were used in this study. HRP conjugated secondary antibodies (1:3000) were from (GE Healthcare Life Sciences, Piscataway, NJ).
Cell culture
We cultured NLF and SY5Y NB lines as described previously [17, 41]. NLF, SH-SY5Y neuroblastoma parental cell lines were obtained from ATCC (Manassas, VA) and cultured in RPMI-1640 with 10% FBS, 1% L-glutamine and Pen Strep (Life Technologies, Grand Island, NY) according to the provider instructions. Both cell lines were maintained at 37°C in 5% CO2 and 95% humidity. Cell authenticity was checked on annual basis by utilizing PCR techniques for mycoplasmas, bacterial and other viral contaminations as well as for genetic variations. These tests were performed at the cell center facility of the University of Pennsylvania.
Transfections
We transfected NLF (MYCN amplified) and SH-SY5Y (non-amplified) neuroblastoma cell lines using commercially available lipid based reagent Lipofectamine 3000 as per the manufacturers recommendations (Invitrogen, Grand Island, NY). Briefly, cells (1x105 per/well) were plated in triplicate in 24-well tissue culture plates. The next day, cells were transfected with the following: Psicheck™-2 plasmid, either wild type CHD5 3’-UTR, or mutated CHD5 3’-UTR miRNA along with various miRNA mimics (Sigma-Aldrich, St. Louis, MO) separately. PcDNA3.1 (Invitrogen, Grand Island, NY) was added as filler to match the uniform DNA concentration in each transfection mix. Transfections in triplicate were performed using Opti-MEM medium and were optimized to each cell line. Each transfection included, miR-211 mimic as a positive control, Allstars siRNA (Qiagen, Valencia, CA) as a negative control, and a no miRNA mimic condition as a transfection control. After at least 36 hours of transfection cells were washed twice with PBS, harvested cells with commercially available reagents (ThermoFisher Scientific, Waltham, MA) [41] and extracts were analyzed using the Dual-Luciferase Reporter Assay (Promega, Madison, WI) as per the manufacturer’s recommendation. Each transfection was carried out in triplicate and each experiment was performed at least 3 times.
Whole cell extract preparation
Whole cell extracts for luciferase assays and western blot analysis were prepared using commercially available kits (Promega Inc. Madison, WI and ThermoFisher Scientific, Waltham, MA) as per the manufacturer’s instructions and also as described previously in Kolla et al. [41]. Protein concentrations were determined using Bradford Protein Assay Reagent with SmartSpec Plus spectrophotometer (BIO-RAD Laboratories, Hercules, CA) as described earlier by Laemmli [37].
Luciferase reporter assay
A luciferase reporter assays were performed to verify the binding of various microRNAs that bind 3’-UTR of CHD5 mRNA. A portion of the 3’ untranslated region of CHD5 was cloned in a PsiCHECK, a dual reporter (Firefly and Renilla) luciferase vector (Promega Inc. Madison, WI). Transfections were performed as described above. Triplicate values of dual luciferase expressions were measured post 48 hours of transfection using a chemiluminescence plate reader (BioTek, Synergy2 multi-mode plate reader). Firefly luciferase expression values were normalized with reporter Renilla luciferase numbers to normalize the transfection efficiency in each well.
Western analysis
Whole cell extracts (100 μg), either transfected with indicated miRNAs or mock transfected, were subjected to polyacrylamide gel electrophoresis (4-12% SDS-PAGE), using NuPAGE Bis-Tris gels with MOPS-SDS Running Buffer (Invitrogen, Grand Island, NY). Allstars siRNA and miRNA-454 were used as negative controls. Proteins were transferred on to nitrocellulose membranes (GE Healthcare Life Sciences) and probed with antibodies using rabbit polyclonal CHD5, actin (Santa Cruz Biotechnology, CA 1:1000), CHD4 (Bethyl laboratories, Montgomery, TX 1:2000), and MYCN monoclonal (1:5000; BD Biosciences, San Jose, CA). HRP conjugated secondary antibodies (1:3000) were from (GE Healthcare Life Sciences, Piscataway, NJ).
Statistical analysis
Statistical analyses were performed using the Prism two-way ANOVA method followed by a Sidak post-test. Each experiment was performed at least three times and triplicate readings were used and reported all p-values. Data are expressed as the standard error mean (SEM). Values are the mean of triplicates readings from four independent experiments and p-values were reported (* P<0.05, ** p<0.01 and ns= non-significant).
ACKNOWLEDGMENTS
Authors thank Dr. Andre T. Tikhonenko lab members for their expert help in conducting luciferase assays.
FUNDING
*This work was supported by National Institutes of Health Grant (R01-CA39771), Alex’s Lemonade Stand Foundation, and the Audrey E. Evans Chair in Molecular Oncology (GMB).
CONFLICTS OF INTEREST
The authors declare no conflicts of interest.
Authors’ contributions
KN designed and performed all experiments. VK assisted, supervised the experiments, helped write and edit the manuscript. TZ, MH RI, assisted in conducting experiments and or suggestions. HK purified all plasmids used in the study. ERO performed initial experimental design and cloning of 3’-UTR of CHD5. GAB is a collaborator and provided valuable suggestions. GMB conceived the initial experimental design, supervised the experiments, analyzed the data, interpreted results, and helped write and edit the manuscript to its final form.
REFERENCES
1. Bilke S, Chen QR, Westerman F, Schwab M, Catchpoole D, Khan J. Inferring a tumor progression model for neuroblastoma from genomic data. J Clin Oncol. 2005; 23:7322-7331.
2. Brodeur GM. Neuroblastoma: biological insights into a clinical enigma. Nat Rev Cancer. 2003; 3:203-216.
3. Brodeur GM, Hogarty MD, Mosse YP, Maris JM. (2011). Neuroblastoma. In: Pizzo PA and Poplack DG, eds. Principles and Practice of Pediatric Oncology. (Philadelphia: Lippincott, Williams and Wilkins), pp. 886-922.
4. George RE, Attiyeh EF, Li S, Moreau LA, Neuberg D, Li C, Fox EA, Meyerson M, Diller L, Fortina P, Look AT, Maris JM. Genome-wide analysis of neuroblastomas using high-density single nucleotide polymorphism arrays. PLoS One. 2007; 2:e255.
5. Mosse YP, Greshock J, Margolin A, Naylor T, Cole K, Khazi D, Hii G, Winter C, Shahzad S, Asziz MU, Biegel JA, Weber BL, Maris JM. High-resolution detection and mapping of genomic DNA alterations in neuroblastoma. Genes Chromosomes Cancer. 2005; 43:390-403.
6. Tomioka N, Oba S, Ohira M, Misra A, Fridlyand J, Ishii S, Nakamura Y, Isogai E, Hirata T, Yoshida Y, Todo S, Kaneko Y, Albertson DG, et al. Novel risk stratification of patients with neuroblastoma by genomic signature, which is independent of molecular signature. Oncogene. 2008; 27:441-449.
7. Fong CT, Dracopoli NC, White PS, Merrill PT, Griffith RC, Housman DE, Brodeur GM. Loss of heterozygosity for the short arm of chromosome 1 in human neuroblastomas: correlation with N-myc amplification. Proc Natl Acad Sci U S A. 1989; 86:3753-3757.
8. Maris JM, White PS, Beltinger CP, Sulman EP, Castleberry RP, Shuster JJ, Look AT, Brodeur GM. Significance of chromosome 1p loss of heterozygosity in neuroblastoma. Cancer Res. 1995; 55:4664-4669.
9. Maris JM, Weiss MJ, Guo C, Gerbing RB, Stram DO, White PS, Hogarty MD, Sulman EP, Thompson PM, Lukens JN, Matthay KK, Seeger RC, Brodeur GM. Loss of heterozygosity at 1p36 independently predicts for disease progression but not decreased overall survival probability in neuroblastoma patients: a Children’s Cancer Group study. J Clin Oncol. 2000; 18:1888-1899.
10. White PS, Maris JM, Beltinger C, Sulman E, Marshall HN, Fujimori M, Kaufman BA, Biegel JA, Allen C, Hilliard C, et al. A region of consistent deletion in neuroblastoma maps within human chromosome 1p36.2-36.3. Proc Natl Acad Sci U S A. 1995; 92:5520-5524.
11. Okawa ER, Gotoh T, Manne J, Igarashi J, Fujita T, Silverman KA, Xhao H, Mosse YP, White PS, Brodeur GM. Expression and sequence analysis of candidates for the 1p36.31 tumor suppressor gene deleted in neuroblastomas. Oncogene. 2008; 27:803-810.
12. White PS, Thompson PM, Gotoh T, Okawa ER, Igarashi J, Kok M, Winter C, Gregory SG, Hogarty MD, Maris JM, Brodeur GM. Definition and characterization of a region of 1p36.3 consistently deleted in neuroblastoma. Oncogene. 2005; 24:2684-2694.
13. Bauer A, Savelyeva L, Claas A, Praml C, Berthold F, Schwab M. Smallest region of overlapping deletion in 1p36 in human neuroblastoma: a 1 Mbp cosmid and PAC contig. Genes Chromosomes Cancer. 2001; 31:228-239.
14. Caron H, Spieker N, Godfried M, Veenstra M, van Sluis P, de Kraker J, Voute P, Versteeg R. Chromosome bands 1p35-36 contain two distinct neuroblastoma tumor suppressor loci, one of which is imprinted. Genes Chromosomes Cancer. 2001; 30:168-174.
15. Cheng NC, Van Roy N, Chan A, Beitsma M, Westerveld A, Speleman F, Versteeg R. Deletion mapping in neuroblastoma cell lines suggests two distinct tumor suppressor genes in the 1p35-36 region, only one of which is associated with N-myc amplification. Oncogene. 1995; 10:291-297.
16. Martinsson T, Sjoberg RM, Hallstensson K, Nordling M, Hedborg F, Kogner P. Delimitation of a critical tumour suppressor region at distal 1p in neuroblastoma tumours. Eur J Cancer. 1997; 33:1997-2001.
17. Fujita T, Igarashi J, Okawa ER, Gotoh T, Manne J, Kolla V, Kim J, Zhao H, Pawel BR, London WB, Maris JM, White PS, Brodeur GM. CHD5, a tumor suppressor gene deleted from 1p36.31 in neuroblastomas. J Natl Cancer Inst. 2008; 100:940-949.
18. Thompson PM, Gotoh T, Kok M, White PS, Brodeur GM. CHD5, a new member of the chromodomain gene family, is preferentially expressed in the nervous system. Oncogene. 2003; 22:1002-1011.
19. Woodage T, Basrai MA, Baxevanis AD, Hieter P, Collins FS. Characterization of the CHD family of proteins. Proc Natl Acad Sci U S A. 1997; 94:11472-11477.
20. Kolla V, Zhuang T, Higashi M, Naraparaju K, Brodeur GM. Role of CHD5 in Human Cancers: 10 Years Later. Cancer Res. 2014; 74:652-658.
21. Bergs JW, Neuendorff N, van der Heijden G, Wassenaar E, Rexin P, Elsasser HP, Moll R, Baarends WM, Brehm A. Differential Expression and Sex Chromosome Association of CHD3/4 and CHD5 during Spermatogenesis. PLoS One. 2014; 9:e98203.
22. Li W, Wu J, Kim SY, Zhao M, Hearn SA, Zhang MQ, Meistrich ML, Mills AA. Chd5 orchestrates chromatin remodelling during sperm development. Nature communications. 2014; 5:3812.
23. Zhuang T, Hess RA, Kolla V, Higashi M, Raabe TD, Brodeur GM. CHD5 is required for spermiogenesis and chromatin condensation. Mechanisms of development. 2014; 131:35-46.
24. Chen Y, Tsai YH, Tseng SH. Inhibition of cyclin-dependent kinase 1-induced cell death in neuroblastoma cells through the microRNA-34a-MYCN-survivin pathway. Surgery. 2013; 153:4-16.
25. Koyama H, Zhuang T, Light JE, Kolla V, Higashi M, McGrady PW, London WB, Brodeur GM. Mechanisms of CHD5 Inactivation in neuroblastomas. Clin Cancer Res. 2012; 18:1588-1597.
26. Mulero-Navarro S, Esteller M. Chromatin remodeling factor CHD5 is silenced by promoter CpG island hypermethylation in human cancer. Epigenetics. 2008; 3:210-215.
27. Wang J, Chen H, Fu S, Xu ZM, Sun KL, Fu WN. The involvement of CHD5 hypermethylation in laryngeal squamous cell carcinoma. Oral Oncol. 2011; 47:601-608.
28. Wang X, Lau KK, So LK, Lam YW. CHD5 is down-regulated through promoter hypermethylation in gastric cancer. J Biomed Sci. 2009; 16:95.
29. Zhao R, Yan Q, Lv J, Huang H, Zheng W, Zhang B, Ma W. CHD5, a tumor suppressor that is epigenetically silenced in lung cancer. Lung Cancer. 2012; 76:324-331.
30. Lewis BP, Burge CB, Bartel DP. Conserved seed pairing, often flanked by adenosines, indicates that thousands of human genes are microRNA targets. Cell. 2005; 120:15-20.
31. Grimson A, Farh KK, Johnston WK, Garrett-Engele P, Lim LP, Bartel DP. MicroRNA targeting specificity in mammals: determinants beyond seed pairing. Mol Cell. 2007; 27:91-105.
32. Jansson MD, Lund AH. MicroRNA and cancer. Mol Oncol. 2012; 6:590-610.
33. Beckers A, Van Peer G, Carter DR, Mets E, Althoff K, Cheung BB, Schulte JH, Mestdagh P, Vandesompele J, Marshall GM, De Preter K, Speleman F. MYCN-targeting miRNAs are predominantly downregulated during MYCNdriven neuroblastoma tumor formation. Oncotarget. 2015; 6:5204-5216. doi: 10.18632/oncotarget.2477.
34. Bray I, Bryan K, Prenter S, Buckley PG, Foley NH, Murphy DM, Alcock L, Mestdagh P, Vandesompele J, Speleman F, London WB, McGrady PW, Higgins DG, O’Meara A, O’Sullivan M, Stallings RL. Widespread dysregulation of MiRNAs by MYCN amplification and chromosomal imbalances in neuroblastoma: association of miRNA expression with survival. PLoS One. 2009; 4:e7850.
35. Schulte JH, Schowe B, Mestdagh P, Kaderali L, Kalaghatgi P, Schlierf S, Vermeulen J, Brockmeyer B, Pajtler K, Thor T, de Preter K, Speleman F, Morik K, Eggert A, Vandesompele J, Schramm A. Accurate prediction of neuroblastoma outcome based on miRNA expression profiles. Int J Cancer. 2010; 127:2374-2385.
36. Cai C, Ashktorab H, Pang X, Zhao Y, Sha W, Liu Y, Gu X. MicroRNA-211 expression promotes colorectal cancer cell growth in vitro and in vivo by targeting tumor suppressor CHD5. PLoS One. 2012; 7:e29750.
37. Laemmli UK. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature. 1970; 227:680-685.
38. Hong W, Nakazawa M, Chen YY, Kori R, Vakoc CR, Rakowski C, Blobel GA. FOG-1 recruits the NuRD repressor complex to mediate transcriptional repression by GATA-1. Embo J. 2005; 24:2367-2378.
39. Yu L, Gong X, Sun L, Yao H, Lu B, Zhu L. miR-454 functions as an oncogene by inhibiting CHD5 in hepatocellular carcinoma. Oncotarget. 2015; 6:39225-34. doi: 10.18632/oncotarget.4407.
40. Garcia I, Mayol G, Rodriguez E, Sunol M, Gershon TR, Rios J, Cheung NK, Kieran MW, George RE, Perez-Atayde AR, Casala C, Galvan P, de Torres C, Mora J, Lavarino C. Expression of the neuron-specific protein CHD5 is an independent marker of outcome in neuroblastoma. Mol Cancer. 2010; 9:277.
41. Kolla V, Naraparaju K, Zhuang T, Higashi M, Kolla S, Blobel GA, Brodeur GM. The tumour suppressor CHD5 forms a NuRD-type chromatin remodelling complex. Biochem J. 2015; 468:345-352.