
Introduction
The Skp2 F-box protein is a substrate recognizing component of the SCF (Skp1-Cullin 1-F-box) type of E3 ubiquitin-ligase complex [1]. It is known that the SCF complex consists of four crucial components including the invariable component Skp1, Rbx1 and Cullin1, and the interchangeable substrate-recruiting module, the F-box protein [1]. Among 70 putative F-box proteins, Skp2 is one of the best characterized F-box protein and has been shown to be involved in governing many cellular processes such as cell cycle regulation, cell proliferation, apoptosis, differentiation, and survival, in part through promoting the degradation of its substrate proteins [2, 3]. For example, Skp2 plays an important role in driving the cell cycle through the G1/S transition by promoting the destruction of the p27 tumor suppressor protein, an inhibitor of the CDK (Cyclin-dependent kinase) family of kinases [4, 5]. In addition to p27 [6], recent studies have identified numerous downstream Skp2 substrates including p21, p57, p130, c-Myc, FOXO1 (Forkhead box protein O1) [7-10], and Tob1[11] (Figure 1).
The fact that many Skp2 substrates are negative cell cycle regulators is consistent with the notion that Skp2 mainly functions as a proto-oncogene. Indeed, Skp2 has been found to be frequently overexpressed in a variety of human cancers including lymphomas [12], prostate cancer [13], melanoma [14], nasopharyngeal carcinoma [15], pancreatic cancer [16], and breast carcinomas [17, 18]. More importantly, in support of an oncogenic role for Skp2 in tumor progression, Skp2-/- mice has been found to be resistant to tumor development induced by loss of either the p53 or the PTEN tumor suppressor [19]. Although multiple signaling pathways such as phosphatidylinositol 3-kinase (PI3K)/Akt [20], AR (Androgen receptor) [21], PTEN (Phosphatase and tensin homolog) [13] and STAT1 (Signal transducers and activators of transcription) [22] have been reported to cross-talk with the Skp2 sinaling pathway and subsequently lead to tumorigenesis, the underlying mechanism(s) by which Skp2 is regulated in vivo remains largely elusive. Here, we will discuss the recent advances in our understanding of how Skp2 oncogenic role is governed in vivo by the novel acetylation-dependent mechanism, which is antagonized by the SIRT3 deacetylase.
Skp2 is acetylated by p300
Multiple studies have shown that phosphorylation of Skp2 by Akt at Ser72 protects Skp2 from APC (Anaphase-promoting complex)/Cdh1-mediated proteolysis [23, 24]. However, Ser72 is only present in human and large mammals, but not conserved in mice, suggesting that Akt-mediated Skp2 phosphorylation might be a regulatory mechanism acquired late during the evolution. This implies that additional mechanisms might exist to regulate Skp2 activity. It is noteworthy that besides protein phosphorylation, protein acetylation has been recently demonstrated to emerge as another important type of post-translational modification that modulates many pathways involved in oncogenesis [25, 26]. More interestingly, while PI3K/Akt phosphorylates and activates acetyl-transferase p300 [27], Skp2 binds, but inhibits p300 to block p53-induced apoptosis [28]. Consistently, we found that interaction between p300 and Skp2 under both ectopic overexpression and endogenous co-immunoprecipitation conditions can readily be detected [29]. Furthermore, acetylation of Skp2 is detected using a specific acetyl-lysine antibody after ectopic expression of p300 [29]. Notably, we found that p300 acetylates the Skp2 oncoprotein at both K68 and K71 within its nuclear localization signal (NLS) region, just adjacent to the identified Ser72 Akt site [29]. Moreover, we found that p300-mediated Skp2 acetylation promotes Skp2 dimerization, suggesting that dimerization might affect the Skp2 substrate spectrum. To our knowledge, this is the first example demonstrating acetylation of an F-box protein, thereby suggesting the possibility of acetylation-dependent regulation of F-box protein(s) other than Skp2. In keeping with this note, recent large-scale mass spectrometry analyses have shown that a significant number of cellular proteins are acetylated [30, 31], although it is largely unclear how acetylation functions as a signaling mechanism to modulate downstream signaling and cellular physiology. Therefore, further studies are warranted to explore how analogous to phosphorylation-dependent regulation mechanism, acetylation could be utilized to govern the physiological functions of various F-box proteins.
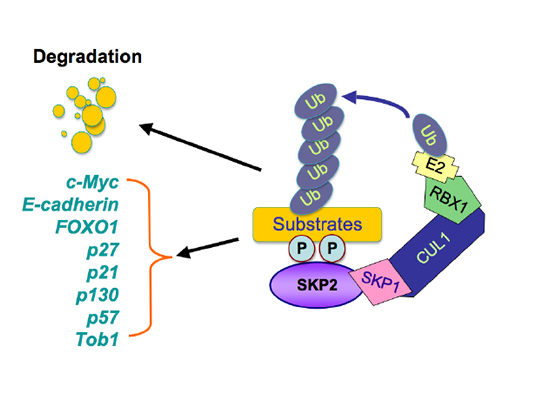
Figure 1: Illustrated pathway of Skp2-mediated degradation of its substrates. The SCF (Skp1-Cullin 1-F-box) complex consists of four components: Skp1, Rbx1, Cullin1, and the F-box protein. While Skp2 recognizes its downstream substrates, cullin1-Rbx1 complex catalyzes the ubiquitin transfers from E2 to the substrates for targeted degradation by 26S-proteasome. The multiple substrates including p21, p27, p57, p130, Tob1, FOXO1, E-cadherin, and c-Myc have been identified.
Interestingly, Akt activates p300 acetyl-transferase activity to influence the Skp2 acetylation. However, p300 exerts its function independent of the Akt-Ser72-Skp2 pathway [29]. Therefore it is critical to further understand the possible redundancy or cross-talks between these two upstream regulatory pathways, Akt-mediated phosphorylation of Ser72 versus p300-mediated acetylation of Skp2, in terms of promoting Skp2 oncogenic signaling. It is possible that p300 and Akt are activated in response to distinct upstream signals to modulate Skp2 activity in specific settings. Alternatively, they share redundant functions with the p300 pathway being the ancestral mechanism of regulation, and Akt-mediated regulation acquired later in evolution. Obviously additional studies will be required to fully dissect the potential intercommunication between the p300 and Akt signal transduction pathways that modulate Skp2 activity.
Skp2 is deacetylated by the SIRT3 tumor suppressor
The Sirtuin (SIRT) family of deacetylases have recently gained tremendous amount of attention due to their critical roles in many cellular functions [9]. The Sirtuins play important roles in a variety of cellular processes including aging, cellular metabolism and tumorigenesis [32]. Interestingly, Sirtuins are located in different cellular compartments, which may dictate the specific cellular function of each Sirtuin protein through promoting the deacetylation of various target proteins such as FOXO3a [33, 34], PPARγ (Peroxisome proliferators-activated receptor gamma) [35] and p53 [36]. For example, SIRT1, the best-characterized member of the mammalian Sirtuins, is located predominately in the nucleus [37, 38], whereas SIRT2 is found in the cytosol [39, 40]. SIRT3, SIRT4 and SIRT5, on the other hand, are localized mainly in the mitochondria [41, 42], [43].
Recent studies have also shown that SIRT3 has seemingly dichotomous role as either tumor promoter or tumor suppressor in cancer biology. For example, SIRT3 expression was found significantly higher in oral cancer cell lines and human oral cancer samples than in normal control [10]. In contrast, SIRT3 was reported to suppress tumor growth via induction of growth arrest and apoptosis in colorectal carcinoma, osteosarcoma cells, ovarian cancer, prostate cancer, suggesting that SIRT3 is tumor suppressor in these cancers [9]. Several published papers demonstrated a tumor suppressor role for SIRT3 via the ability of SIRT3 to negatively regulate ROS (reactive oxygen species) and HIF1-α (hypoxia inducible factor-1 α) [44, 45]. However, the exact mechanisms how SIRT3 is involved in cancer are largely unclear.
Our recent study showed that SIRT3 interacts with Skp2 and subsequently deacetylates Skp2 to suppress tumorigenesis [29]. Specifically, only SIRT3 and SIRT4 specifically interact with Skp2 among the various Sirtuin family members [29]. As SIRT3 is a tumor suppressor protein [45, 46], we further examined a potential role for SIRT3 in regulation of Skp2 acetylation. We detected a specific interaction between ectopically expressed as well as endogenous Skp2 and SIRT3. Moreover, depletion of SIRT3 caused an increase in endogenous Skp2 acetylation [29]. Additionally, depletion of SIRT3 also leads to a moderate increase in Skp2 abundance, which correlates with decreased expression of the Skp2 substrates p27 and p21. More importantly, the inverse correlation between Skp2 and SIRT3 immunohistochemical staining was observed in breast cancer tissues [29]. As SIRT3 has been implicated to possess tumor suppressor function [45, 46], this result suggests that loss of SIRT3 may lead to elevated Skp2 expression in breast cancers. In support of a tumor suppressor function for SIRT3, it has also been reported previously that SIRT3-/- cells displayed elevated ability to form tumors in a xenograft model and loss of SIRT3 has been identified as a frequent event in breast cancer cases [45, 46]. Using the xenograft model, we further showed that depletion of Skp2 retarded the in vivo tumorigenesis of SIRT3-/- cells [29]. Altogether, our results suggest that in our experimental settings, SIRT3 inhibited tumor growth mainly through deacetylation of Skp2 oncoprotein. Future study is directed to mechanistically understand how and whether SIRT3 exerts its tumor suppressor function by inactivating the Skp2 oncogenic pathway solely in a deacetylase-dependent manner.
Skp2 acetylation governs its oncogenic function
The regulation of proteins by acetylation/deacetylation is considered as a significant post-translational regulatory mechanism to modify the specific enzyme’s activity [47]. Hence, we intended to examine whether acetylation of Skp2 controls its oncogenic functions. Notably, we found that acetylation of Skp2 positively regulates its oncogenic activity partly through modulating its stability [29]. Moreover, acetylation of Skp2 exerts its function through promoting the destruction of its downstream targets such as p21 and FOXO1. To further support the role of Skp2 acetylation in tumorigenesis, depletion of endogenous SIRT3, which leads to increased acetylation of Skp2, promotes cell growth [29].
A canonical NLS was identified at the Skp2 amino terminus, and its function is highlighted by the fact that over-expression of an active Akt allele relocalizes a pool of Skp2 to the cytoplasm [23, 24]. Acetylation of lysines within an NLS has been reported to influence cellular localization [48-51]. Therefore, we detected whether acetylation of Skp2 NLS influences its localization. Consistent with the fact that Skp2 acetylation sites are found within its NLS, p300 promotes Skp2 cytoplasmic localization [29]. Interestingly, p300-induced Skp2 cellular localization is independent of Akt-mediated Skp2 phosphorylation [29]. In support of this notion, Skp2 cytoplasmic localization has been observed in many clinical tumor samples and is correlated with aggressive malignancy and poor diagnosis [17, 52-54]. Taken together, our results demonstrate that p300-mediated acetylation of Skp2 affects its stability and cytoplasmic localization, which in turn can influence its oncogenic activity. These results suggest that acetylation-mediated post-translational modifications might also control the function of other F-box proteins.
Skp2 regulates cell migration through promoting E-cadherin destruction
It has been well accepted that enhanced cell migration is a critical phenotype required for cancer progression that leads to invasion and ultimately metastatic dissemination of tumor cells. In addition to controlling cell cycle progression, Skp2 has also been implicated in the regulation of cell migration. For example, a study showed that ectopic expression of Skp2 fused with an extra copy of an NES rescues the deficiency of cell motility in Skp2-/- MEFs, in the absence of p27 degradation [23], indicating that cytoplasmic Skp2 may have a distinct function related to cell migration, independent of its major role in cell cycle regulation. Indeed, a correlation between elevated Skp2 protein expression and tumor metastasis has been noted in multiple tumors, including melanoma, lymphoma and breast carcinoma [55-57]. Furthermore, several studies have demonstrated that cytosolic Skp2 can positively regulate cell migration, although the molecular mechanisms are largely unknown [23]. As we have shown that acetylation promotes cytosolic localization, it became increasingly necessary to explore whether acetylation could influence Skp2-governed cellular migration.
To this end, E-cadherin is considered as a major player in EMT (epithelial to mesenchymal transition) [58]. It is accepted that after EMT, cells lose epithelial features including down-regulation of E-cadherin and gain mesenchymal characteristics such as upregulation of N-cadherin, fibronectin, and vimentin, leading to enhanced cell migration, invasion and metastasis [44, 59, 60]. Consistent with this notion, loss of E-cadherin is frequently observed in high-grade breast tumor samples [61-63]. However, it remains unclear whether enhanced proteolysis of E-cadherin also contributes to the reduced E-cadherin abundance in high-grade breast tumors. On the other hand, Skp2 is found to be frequently overexpressed in various types of cancers including breast cancer [1]. We therefore propose that aberrant Skp2 signaling may lead to elevated E-cadherin destruction that in turn profoundly affects cell migration and potentially EMT. In keeping with this hypothesis, our study indicates that Skp2 promotes cellular migration partly through promotion of E-cadherin destruction [29]. Furthermore, we observed an inverse correlation between Skp2 and E-cadherin expression in an array of breast cancer clinical samples [29]. Interestingly, we found that although ectopic expression of Skp2 in LNCaP cells leads to decreased expression of E-cadherin and α-E-catenin, other molecular markers for EMT were not significantly altered [29]. These results argue that cytoplasmic Skp2 preferentially promotes E-cadherin destruction to regulate cellular migration, but has nominal effects on other cell adhesion proteins, implying that additional oncogenic signaling, which is not currently fully underscored, is required for promoting full EMT. Therefore, additional studies are required to pinpoint these genetic alterations that might synergize with elevated Skp2 oncogenic signaling in facilitating cellular transformation and metastasis.
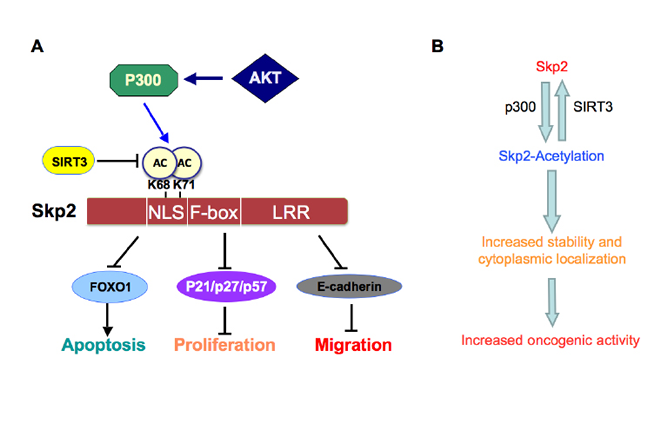
Figure 2: Proposed model for how Skp2 oncogenic role is regulated by p300 and SIRT3. A. Skp2 is acetylated by the p300 acetyl-transferase at both K68 and K71, leading to increased Skp2 stability and its oncogenic functions partly through promoting its cytoplasmic localization. Moreover, Skp2 promotes tumor cell migration via governing E-cadherin degradation. Furthermore, SIRT3 interacts with and deacetylates Skp2 to antagonize the acetylation by p300. B. Schematic illustration of how p300-dependent acetylation of Skp2, a process that can be antagonized by the SIRT3 deacetylase, leads to elevated Skp2 oncogenic functions in part by stabilizing Skp2 as well as promoting Skp2 cytoplasmic localization.
Conclusion
In conclusion, Skp2 plays an oncogenic role in the development and progression of human cancers through degradation of its downstream target proteins that control a variety of cellular processes such as cell proliferation, apoptosis, migration, invasion and metastasis. More importantly, we recently identified that Skp2 is acetylated by p300, resulting in its cytoplasmic localization and enhanced stability. We further demonstrate that the SIRT3 tumor suppressor interacts with and deacetylates Skp2 (Figure 2), suggesting that targeting Skp2 or the p300/SIRT3 axis could be a novel approach for the treatment of human cancers, especially those with up-regulation of Skp2. However, we recognize that although these studies provide the molecular basis for targeting Skp2 as novel anti-cancer therapeutic options, further in-depth studies are required to provide further insights to guide the design of effective therapeutics targeting Skp2 acetylation events to combat human cancers.
Acknowledgements
In this article, we apologize to those authors whose work was not cited due to space restriction. This work was supported by grants from the National Institute of General Medicine, NIH (GM089763, GM094777) to W.W, and from the NCI grants (CA118762 and CA156744) to Y.S. W.W is an American Cancer Society Scholar. Z. W is supported by NIH NRSA fellowship. H.I is supported by NIH K01 (AG041218).
Conflict of Interest
The authors declare that they have no conflict of interest.
Reference
1. Frescas D and Pagano M. Deregulated proteolysis by the F-box proteins SKP2 and beta-TrCP: tipping the scales of cancer. Nat Rev Cancer. 2008; 8(6):438-449.
2. Shi L, Wang S, Zangari M, Xu H, Cao TM, Xu C, Wu Y, Xiao F, Liu Y, Yang Y, Salama M, Li G, Tricot G and Zhan F. Over-expression of CKS1B activates both MEK/ERK and JAK/STAT3 signaling pathways and promotes myeloma cell drug-resistance. Oncotarget. 2010; 1(1):22-33.
3. Wang Z, Gao D, Fukushima H, Inuzuka H, Liu P, Wan L, Sarkar FH and Wei W. Skp2: a novel potential therapeutic target for prostate cancer. Biochim Biophys Acta. 2012; 1825(1):11-17.
4. Carrano AC, Eytan E, Hershko A and Pagano M. SKP2 is required for ubiquitin-mediated degradation of the CDK inhibitor p27. Nat Cell Biol. 1999; 1(4):193-199.
5. Sutterluty H, Chatelain E, Marti A, Wirbelauer C, Senften M, Muller U and Krek W. p45SKP2 promotes p27Kip1 degradation and induces S phase in quiescent cells. Nat Cell Biol. 1999; 1(4):207-214.
6. Jonason JH, Gavrilova N, Wu M, Zhang H and Sun H. Regulation of SCF(SKP2) ubiquitin E3 ligase assembly and p27(KIP1) proteolysis by the PTEN pathway and cyclin D1. Cell Cycle. 2007; 6(8):951-961.
7. Zhang H. Skip the nucleus, AKT drives Skp2 and FOXO1 to the same place? Cell Cycle. 2010; 9(5):868-869.
8. Wang H, Cui J, Bauzon F and Zhu L. A comparison between Skp2 and FOXO1 for their cytoplasmic localization by Akt1. Cell Cycle. 2010; 9(5):1021-1022.
9. Huang H, Regan KM, Wang F, Wang D, Smith DI, van Deursen JM and Tindall DJ. Skp2 inhibits FOXO1 in tumor suppression through ubiquitin-mediated degradation. Proc Natl Acad Sci U S A. 2005; 102(5):1649-1654.
10. von der Lehr N, Johansson S and Larsson LG. Implication of the ubiquitin/proteasome system in Myc-regulated transcription. Cell Cycle. 2003; 2(5):403-407.
11. Cardozo T and Pagano M. The SCF ubiquitin ligase: insights into a molecular machine. Nat Rev Mol Cell Biol. 2004; 5(9):739-751.
12. Lim MS, Adamson A, Lin Z, Perez-Ordonez B, Jordan RC, Tripp S, Perkins SL and Elenitoba-Johnson KS. Expression of Skp2, a p27(Kip1) ubiquitin ligase, in malignant lymphoma: correlation with p27(Kip1) and proliferation index. Blood. 2002; 100(8):2950-2956.
13. Yang G, Ayala G, De Marzo A, Tian W, Frolov A, Wheeler TM, Thompson TC and Harper JW. Elevated Skp2 protein expression in human prostate cancer: association with loss of the cyclin-dependent kinase inhibitor p27 and PTEN and with reduced recurrence-free survival. Clin Cancer Res. 2002; 8(11):3419-3426.
14. Rose AE, Wang G, Hanniford D, Monni S, Tu T, Shapiro RL, Berman RS, Pavlick AC, Pagano M, Darvishian F, Mazumdar M, Hernando E and Osman I. Clinical relevance of SKP2 alterations in metastatic melanoma. Pigment Cell Melanoma Res. 2011; 24(1):197-206.
15. Xu HM, Liang Y, Chen Q, Wu QN, Guo YM, Shen GP, Zhang RH, He ZW, Zeng YX, Xie FY and Kang TB. Correlation of Skp2 overexpression to prognosis of patients with nasopharyngeal carcinoma from South China. Chin J Cancer. 2011; 30(3):204-212.
16. Schuler S, Diersch S, Hamacher R, Schmid RM, Saur D and Schneider G. SKP2 confers resistance of pancreatic cancer cells towards TRAIL-induced apoptosis. Int J Oncol. 2011; 38(1):219-225.
17. Radke S, Pirkmaier A and Germain D. Differential expression of the F-box proteins Skp2 and Skp2B in breast cancer. Oncogene. 2005; 24(21):3448-3458.
18. Zheng WQ, Zheng JM, Ma R, Meng FF and Ni CR. Relationship between levels of Skp2 and P27 in breast carcinomas and possible role of Skp2 as targeted therapy. Steroids. 2005; 70(11):770-774.
19. Lin HK, Chen Z, Wang G, Nardella C, Lee SW, Chan CH, Yang WL, Wang J, Egia A, Nakayama KI, Cordon-Cardo C, Teruya-Feldstein J and Pandolfi PP. Skp2 targeting suppresses tumorigenesis by Arf-p53-independent cellular senescence. Nature. 2010; 464(7287):374-379.
20. van Duijn PW and Trapman J. PI3K/Akt signaling regulates p27(kip1) expression via Skp2 in PC3 and DU145 prostate cancer cells, but is not a major factor in p27(kip1) regulation in LNCaP and PC346 cells. Prostate. 2006; 66(7):749-760.
21. Chuu CP, Kokontis JM, Hiipakka RA, Fukuchi J, Lin HP, Lin CY, Huo C, Su LC and Liao S. Androgen Suppresses Proliferation of Castration-Resistant LNCaP 104-R2 Prostate Cancer Cells via Androgen Receptor, Skp2, and c-Myc. Cancer Sci. 2011.
22. Wang S, Raven JF and Koromilas AE. STAT1 represses Skp2 gene transcription to promote p27Kip1 stabilization in Ras-transformed cells. Mol Cancer Res. 2010; 8(5):798-805.
23. Lin HK, Wang G, Chen Z, Teruya-Feldstein J, Liu Y, Chan CH, Yang WL, Erdjument-Bromage H, Nakayama KI, Nimer S, Tempst P and Pandolfi PP. Phosphorylation-dependent regulation of cytosolic localization and oncogenic function of Skp2 by Akt/PKB. Nat Cell Biol. 2009; 11(4):420-432.
24. Gao D, Inuzuka H, Tseng A, Chin RY, Toker A and Wei W. Phosphorylation by Akt1 promotes cytoplasmic localization of Skp2 and impairs APCCdh1-mediated Skp2 destruction. Nat Cell Biol. 2009; 11(4):397-408.
25. Iyer NG, Ozdag H and Caldas C. p300/CBP and cancer. Oncogene. 2004; 23(24):4225-4231.
26. Yang XJ. The diverse superfamily of lysine acetyltransferases and their roles in leukemia and other diseases. Nucleic Acids Res. 2004; 32(3):959-976.
27. Huang WC and Chen CC. Akt phosphorylation of p300 at Ser-1834 is essential for its histone acetyltransferase and transcriptional activity. Mol Cell Biol. 2005; 25(15):6592-6602.
28. Kitagawa M, Lee SH and McCormick F. Skp2 suppresses p53-dependent apoptosis by inhibiting p300. Mol Cell. 2008; 29(2):217-231.
29. Inuzuka H, Gao D, Finley LW, Yang W, Wan L, Fukushima H, Chin YR, Zhai B, Shaik S, Lau AW, Wang Z, Gygi SP, Nakayama K, Teruya-Feldstein J, Toker A, Haigis MC, et al. Acetylation-dependent regulation of Skp2 function. Cell. 2012; 150(1):179-193.
30. Choudhary C, Kumar C, Gnad F, Nielsen ML, Rehman M, Walther TC, Olsen JV and Mann M. Lysine acetylation targets protein complexes and co-regulates major cellular functions. Science. 2009; 325(5942):834-840.
31. Zhao S, Xu W, Jiang W, Yu W, Lin Y, Zhang T, Yao J, Zhou L, Zeng Y, Li H, Li Y, Shi J, An W, Hancock SM, He F, Qin L, et al. Regulation of cellular metabolism by protein lysine acetylation. Science. 2010; 327(5968):1000-1004.
32. Haigis MC and Sinclair DA. Mammalian sirtuins: biological insights and disease relevance. Annu Rev Pathol. 2010; 5:253-295.
33. Brunet A, Sweeney LB, Sturgill JF, Chua KF, Greer PL, Lin Y, Tran H, Ross SE, Mostoslavsky R, Cohen HY, Hu LS, Cheng HL, Jedrychowski MP, Gygi SP, Sinclair DA, Alt FW, et al. Stress-dependent regulation of FOXO transcription factors by the SIRT1 deacetylase. Science. 2004; 303(5666):2011-2015.
34. Motta MC, Divecha N, Lemieux M, Kamel C, Chen D, Gu W, Bultsma Y, McBurney M and Guarente L. Mammalian SIRT1 represses forkhead transcription factors. Cell. 2004; 116(4):551-563.
35. Picard F, Kurtev M, Chung N, Topark-Ngarm A, Senawong T, Machado De Oliveira R, Leid M, McBurney MW and Guarente L. Sirt1 promotes fat mobilization in white adipocytes by repressing PPAR-gamma. Nature. 2004; 429(6993):771-776.
36. Vaziri H, Dessain SK, Ng Eaton E, Imai SI, Frye RA, Pandita TK, Guarente L and Weinberg RA. hSIR2(SIRT1) functions as an NAD-dependent p53 deacetylase. Cell. 2001; 107(2):149-159.
37. Chen W. Accelerating cancer evolution: a dark side of SIRT1 in genome maintenance. Oncotarget. 2012; 3(4):363-364.
38. Herranz D, Iglesias G, Munoz-Martin M and Serrano M. Limited role of Sirt1 in cancer protection by dietary restriction. Cell Cycle. 2011; 10(13):2215-2217.
39. Inoue T, Hiratsuka M, Osaki M and Oshimura M. The molecular biology of mammalian SIRT proteins: SIRT2 in cell cycle regulation. Cell Cycle. 2007; 6(9):1011-1018.
40. Inoue T, Nakayama Y, Yamada H, Li YC, Yamaguchi S, Osaki M, Kurimasa A, Hiratsuka M, Katoh M and Oshimura M. SIRT2 downregulation confers resistance to microtubule inhibitors by prolonging chronic mitotic arrest. Cell Cycle. 2009; 8(8):1279-1291.
41. Sundaresan NR, Samant SA, Pillai VB, Rajamohan SB and Gupta MP. SIRT3 is a stress-responsive deacetylase in cardiomyocytes that protects cells from stress-mediated cell death by deacetylation of Ku70. Mol Cell Biol. 2008; 28(20):6384-6401.
42. Scher MB, Vaquero A and Reinberg D. SirT3 is a nuclear NAD+-dependent histone deacetylase that translocates to the mitochondria upon cellular stress. Genes Dev. 2007; 21(8):920-928.
43. Haigis MC, Mostoslavsky R, Haigis KM, Fahie K, Christodoulou DC, Murphy AJ, Valenzuela DM, Yancopoulos GD, Karow M, Blander G, Wolberger C, Prolla TA, Weindruch R, Alt FW and Guarente L. SIRT4 inhibits glutamate dehydrogenase and opposes the effects of calorie restriction in pancreatic beta cells. Cell. 2006; 126(5):941-954.
44. Jordan NV, Johnson GL and Abell AN. Tracking the intermediate stages of epithelial-mesenchymal transition in epithelial stem cells and cancer. Cell Cycle. 2011; 10(17):2865-2873.
45. Finley LW, Carracedo A, Lee J, Souza A, Egia A, Zhang J, Teruya-Feldstein J, Moreira PI, Cardoso SM, Clish CB, Pandolfi PP and Haigis MC. SIRT3 opposes reprogramming of cancer cell metabolism through HIF1alpha destabilization. Cancer Cell. 2011; 19(3):416-428.
46. Kim HS, Patel K, Muldoon-Jacobs K, Bisht KS, Aykin-Burns N, Pennington JD, van der Meer R, Nguyen P, Savage J, Owens KM, Vassilopoulos A, Ozden O, Park SH, Singh KK, Abdulkadir SA, Spitz DR, et al. SIRT3 is a mitochondria-localized tumor suppressor required for maintenance of mitochondrial integrity and metabolism during stress. Cancer Cell. 2010; 17(1):41-52.
47. Lindemann RK, Gabrielli B and Johnstone RW. Histone-deacetylase inhibitors for the treatment of cancer. Cell Cycle. 2004; 3(6):779-788.
48. Santos-Rosa H, Valls E, Kouzarides T and Martinez-Balbas M. Mechanisms of P/CAF auto-acetylation. Nucleic Acids Res. 2003; 31(15):4285-4292.
49. Dietschy T, Shevelev I, Pena-Diaz J, Huhn D, Kuenzle S, Mak R, Miah MF, Hess D, Fey M, Hottiger MO, Janscak P and Stagljar I. p300-mediated acetylation of the Rothmund-Thomson-syndrome gene product RECQL4 regulates its subcellular localization. J Cell Sci. 2009; 122(Pt 8):1258-1267.
50. Faiola F, Wu YT, Pan S, Zhang K, Farina A and Martinez E. Max is acetylated by p300 at several nuclear localization residues. Biochem J. 2007; 403(3):397-407.
51. di Bari MG, Ciuffini L, Mingardi M, Testi R, Soddu S and Barila D. c-Abl acetylation by histone acetyltransferases regulates its nuclear-cytoplasmic localization. EMBO Rep. 2006; 7(7):727-733.
52. Drobnjak M, Melamed J, Taneja S, Melzer K, Wieczorek R, Levinson B, Zeleniuch-Jacquotte A, Polsky D, Ferrara J, Perez-Soler R, Cordon-Cardo C, Pagano M and Osman I. Altered expression of p27 and Skp2 proteins in prostate cancer of African-American patients. Clin Cancer Res. 2003; 9(7):2613-2619.
53. Dowen SE, Scott A, Mukherjee G and Stanley MA. Overexpression of Skp2 in carcinoma of the cervix does not correlate inversely with p27 expression. Int J Cancer. 2003; 105(3):326-330.
54. Signoretti S, Di Marcotullio L, Richardson A, Ramaswamy S, Isaac B, Rue M, Monti F, Loda M and Pagano M. Oncogenic role of the ubiquitin ligase subunit Skp2 in human breast cancer. J Clin Invest. 2002; 110(5):633-641.
55. Traub F, Mengel M, Luck HJ, Kreipe HH and von Wasielewski R. Prognostic impact of Skp2 and p27 in human breast cancer. Breast Cancer Res Treat. 2006; 99(2):185-191.
56. Yokoi S, Yasui K, Mori M, Iizasa T, Fujisawa T and Inazawa J. Amplification and overexpression of SKP2 are associated with metastasis of non-small-cell lung cancers to lymph nodes. Am J Pathol. 2004; 165(1):175-180.
57. Li JQ, Wu F, Masaki T, Kubo A, Fujita J, Dixon DA, Beauchamp RD, Ishida T, Kuriyama S and Imaida K. Correlation of Skp2 with carcinogenesis, invasion, metastasis, and prognosis in colorectal tumors. Int J Oncol. 2004; 25(1):87-95.
58. Thiery JP, Acloque H, Huang RY and Nieto MA. Epithelial-mesenchymal transitions in development and disease. Cell. 2009; 139(5):871-890.
59. Grego-Bessa J, Diez J, Timmerman L and de la Pompa JL. Notch and epithelial-mesenchyme transition in development and tumor progression: another turn of the screw. Cell Cycle. 2004; 3(6):718-721.
60. Gregory PA, Bracken CP, Bert AG and Goodall GJ. MicroRNAs as regulators of epithelial-mesenchymal transition. Cell Cycle. 2008; 7(20):3112-3118.
61. Schmalhofer O, Brabletz S and Brabletz T. E-cadherin, beta-catenin, and ZEB1 in malignant progression of cancer. Cancer Metastasis Rev. 2009; 28(1-2):151-166.
62. Giehl K and Menke A. Microenvironmental regulation of E-cadherin-mediated adherens junctions. Front Biosci. 2008; 13:3975-3985.
63. Huber MA, Kraut N and Beug H. Molecular requirements for epithelial-mesenchymal transition during tumor progression. Curr Opin Cell Biol. 2005; 17(5):548-558.