
INTRODUCTION
Rheumatoid arthritis (RA) is a common, chronic autoimmune disease, which characterized by nonspecific inflammation of the peripheral joints. It affects 1% of the world’s population, and it occurs more frequently in women than in men [1, 2]. In later disease stages, affected joint and surrounding tissues exhibit progressive destruction, and cause dysfunction of joint and disable. It is generally accepted that RA is associated with both genetic and environmental factors, but the etiology of RA is still not very clear to date.
Genetic factors are known to affect disease susceptibility. In recent years, there are many researches to identify the genetic factors (loci, genes and pathways) related to RA. Genome-wide association studies (GWAS) is one of the main methods in finding genetic variants. Many GWAS have been successfully applied to RA and have identified large number of single nucleotide polymorphisms (SNPs) related to RA [3-8]. But these loci account for a small proportion of genetic variants and can not dissect and explain the complex molecular mechanisms of RA. Besides, some SNPs which do not reach the significant threshold but have modest effects on RA were not identified by GWAS.
Besides genetic association studies, there are still a lot of bioinformatics analysis methods to help us to understand the molecular mechanism of RA. Genome-wide gene expression analysis as a tool to investigate pathogenesis of RA has found many candidate genes for RA. In 2004, Devauchelie et al. identified differential expression genes between RA and osteoarthritis (OA) by DNA microarray analysis [9]. In 2006, Haas et al. found 1,163 significantly differentially expressed genes in RA twin compared with healthy twin [10]. Kim et al. performed a gene expression analysis and found G0S2 plays as a biomarker to predict response to anti-TNF therapy in patients with RA [11]. Xu et al. found P53 may be involved in the progression of RA and osteoarthritis via targeting downstream EGFR and DUSP1 respectively based on gene expression profiles [12]. Several other publications also identified some RA-specific gene signatures by gene expression analysis [13, 14] [15]. These studies show that microarray analysis is a useful tool to detect RA-related genes and could help in identifying novel therapeutic targets. However, some genes that do not change at the level of gene expression but are really associated with RA are still not found.
It is well known that genes do not work in isolation, instead, complex biology pathways and molecular networks are usually involved in disease susceptibility and disease progression [16]. So gene set or pathway association analyses which identify variation in the pathway or biology function related to the disease have been developed. Pathway association analysis can reduce the false positives and uncertainty around causal genes by analyzing associations between pathways and the disease. In addition, the pathways detected by pathway analysis can also help us explain underlying molecular mechanism of the disease.
There are many pathway analyses studies of RA based on a single data type, for example, SNP data or gene expression data, which have successfully revealed some biology pathways associated with RA [17] [2, 18-20]. However, pathway analysis of RA which integrates GWAS and gene expression information is still lacking. Some valuable associations may be ignored by a single data type analyses. For example, some genes with only genetic variations can’t be identified in gene expression studies. Similarly, genes with only expression changes also are discarded in GWAS. It is easily found that the pathway analysis based on a single data type only provide limited understanding about the development of RA. Therefore, integrating GWAS and gene expression information into pathway association analysis can provided more accurate and stable evidence to explain pathogenesis of RA.
In this study, we integrated SNP dataset and gene expression information to identify the pathways related to RA. This multi-leave procedure consists of six computation steps: (1) compute the P-value of a SNP by trend test and assess gene-wise association value based on SNP dataset; (2) compute the gene expression P-value of every gene based on gene expression data; (3) compute the gene association score by integrating GWAS and gene expression; (4) compute the pathway association score based on SNP dataset, gene expression data, and integrated datasets respectively; (5) compute the significance of association between the pathways and RA by permutation test; (6) identify the pathways related to RA after multiple testing corrections. By comparing the results, we found integrative analysis can provide more stable results and also revealed novel pathway associations.
RESULTS
The pathway analysis results based on GWAS
In the WTCCC SNP dataset, there were 370,227 SNPs passed quality control. After mapping SNPs to genes, we found that there were 32,591 genes that contained at least one SNP. We used 1000 genome dataset from Europe to correct the LD and obtained tagSNPs of all 32,591 genes. The smallest P-value of all tagSNPs in a gene was chosen to be the gene-wise association value.
The 234 pathway risk scores were computed by the Fisher’s method. The RA-related pathways were identified by the permutation test and FDR correction. The 19 pathways whose q-values were less than 0.05 after FDR correction were identified as RA-related pathways based on GWAS. The detailed information of these pathways is presented in Table 1.
According to the KEGG classifications, we found these pathways were mainly involved in immune system (n=8), infectious diseases (n=7), signaling molecules and interaction (n=1), transport and catabolism (n=1), endocrine and metabolic diseases (n=1) and cardiovascular diseases (n=1). All of these pathways are reported as RA-related pathway in the previous studies. It is indicated our results are consistent with previously published studies [17, 20-22].
Table 1: The RA-related pathways based on SNP dataset (FDR<0.05)
Pathway ID |
Pathway name |
P-value |
FDR |
Function class |
hsa04145 |
*Phagosome |
0 |
0 |
Cellular Processes; Transport and catabolism |
hsa04514 |
*Cell adhesion molecules (CAMs) |
0 |
0 |
Environmental Information Processing; Signaling molecules and interaction |
hsa04612 |
*Antigen processing and presentation |
0 |
0 |
Organismal Systems; Immune system |
hsa04672 |
*Intestinal immune network for IgA production |
0 |
0 |
Organismal Systems; Immune system |
hsa04940 |
*Type I diabetes mellitus |
0 |
0 |
Human Diseases; Endocrine and metabolic diseases |
hsa05140 |
*Leishmaniasis |
0 |
0 |
Human Diseases; Infectious diseases |
hsa05145 |
*Toxoplasmosis |
0 |
0 |
Human Diseases; Infectious diseases |
hsa05150 |
*Staphylococcus aureus infection |
0 |
0 |
Human Diseases; Infectious diseases |
hsa05168 |
*Herpes simplex infection |
0 |
0 |
Human Diseases; Infectious diseases |
hsa05310 |
*Asthma |
0 |
0 |
Human Diseases; Immune diseases |
hsa05320 |
*Autoimmune thyroid disease |
0 |
0 |
Human Diseases; Immune diseases |
hsa05330 |
*Allograft rejection |
0 |
0 |
Human Diseases; Immune diseases |
hsa05332 |
*Graft-versus-host disease |
0 |
0 |
Human Diseases; Immune diseases |
hsa05416 |
*Viral myocarditis |
0 |
0 |
Human Diseases; Cardiovascular diseases |
hsa05164 |
*Influenza A |
1.00E-05 |
0.00013 |
Human Diseases; Infectious diseases |
hsa05162 |
*Measles |
0.00015 |
0.001831 |
Human Diseases; Infectious diseases |
hsa05323 |
*Rheumatoid arthritis |
0.00038 |
0.004365 |
Human Diseases; Immune diseases |
hsa05152 |
*Tuberculosis |
0.00102 |
0.011064 |
Human Diseases; Infectious diseases |
hsa05322 |
*Systemic lupus erythematosus |
0.00468 |
0.048095 |
Human Diseases; Immune diseases |
* Pathways were reported as RA-related pathways in previous literatures.
The pathway analysis results based on gene expression analysis
The gene expression data of 53 individuals (33 RA and 20 controls) were normalized by “affy” package in R software. Then the probes were mapped to the genes according to platform information. After data preprocessing, 12,752 genes were for following analysis. The differential expression P-values of the 12,752 genes were computed by t-test. Then, we computed the 234 pathway risk scores using the Fisher’s method and identified the RA-related pathways by the permutation test and FDR correction. Finally, the 33 pathways whose q-values were less than 0.05 after FDR correction were identified as RA-related pathways based on gene expression data. The detailed information of these pathways is presented in Table 2.
We found the majority of these 33 pathways are related to immune system (n=17), infectious diseases (n=8), cellular processes (n=3). Among these pathways, there are 24 pathways to be found respectively as RA-related pathways in the pervious published studies [2, 21, 23, 24]. There are 18 pathways to be identified by SNP dataset and 10 novel pathways which were not reported in pervious literatures.
Table 2: The RA-related pathways based on gene expression dataset (FDR<0.05)
Pathway ID |
Pathway name |
P-value |
FDR |
Function class |
hsa04062 |
*Chemokine signaling pathway |
0 |
0 |
Organismal Systems; Immune system |
hsa04145 |
*Phagosome |
0 |
0 |
Cellular Processes; Transport and catabolism |
hsa04514 |
*Cell adhesion molecules (CAMs) |
0 |
0 |
Environmental Information Processing; Signaling molecules and interaction |
hsa04612 |
*Antigen processing and presentation |
0 |
0 |
Organismal Systems; Immune system |
hsa04640 |
*Hematopoietic cell lineage |
0 |
0 |
Organismal Systems; Immune system |
hsa04650 |
*Natural killer cell mediated cytotoxicity |
0 |
0 |
Organismal Systems; Immune system |
hsa04672 |
*Intestinal immune network for IgA production |
0 |
0 |
Organismal Systems; Immune system |
hsa04940 |
*Type I diabetes mellitus |
0 |
0 |
Human Diseases; Endocrine and metabolic diseases |
hsa05140 |
*Leishmaniasis |
0 |
0 |
Human Diseases; Infectious diseases |
hsa05150 |
*Staphylococcus aureus infection |
0 |
0 |
Human Diseases; Infectious diseases |
hsa05152 |
*Tuberculosis |
0 |
0 |
Human Diseases; Infectious diseases |
hsa05162 |
*Measles |
0 |
0 |
Human Diseases; Infectious diseases |
hsa05168 |
*Herpes simplex infection |
0 |
0 |
Human Diseases; Infectious diseases |
hsa05320 |
*Autoimmune thyroid disease |
0 |
0 |
Human Diseases; Immune diseases |
hsa05323 |
*Rheumatoid arthritis |
0 |
0 |
Human Diseases; Immune diseases |
hsa05330 |
*Allograft rejection |
0 |
0 |
Human Diseases; Immune diseases |
hsa05332 |
*Graft-versus-host disease |
0 |
0 |
Human Diseases; Immune diseases |
hsa05340 |
Primary immunodeficiency |
0 |
0 |
Human Diseases; Immune diseases |
hsa05416 |
*Viral myocarditis |
0 |
0 |
Human Diseases; Cardiovascular diseases |
hsa04660 |
*T cell receptor signaling pathway |
1.00E-05 |
9.95E-05 |
Organismal Systems; Immune system |
hsa05310 |
*Asthma |
2.00E-05 |
0.000181 |
Human Diseases; Immune diseases |
hsa05164 |
*Influenza A |
2.00E-05 |
0.000189 |
Human Diseases; Infectious diseases |
hsa04662 |
B cell receptor signaling pathway |
4.00E-05 |
0.000346 |
Organismal Systems; Immune system |
hsa04666 |
Fc gamma R-mediated phagocytosis |
5.00E-05 |
0.000414 |
Organismal Systems; Immune system |
hsa05145 |
*Toxoplasmosis |
9.00E-05 |
0.000716 |
Human Diseases; Infectious diseases |
hsa04620 |
Toll-like receptor signaling pathway |
0.00011 |
0.000842 |
Organismal Systems; Immune system |
hsa04380 |
Osteoclast differentiation |
0.00014 |
0.001031 |
Organismal Systems; Development |
hsa04142 |
Lysosome |
0.00244 |
0.017334 |
Cellular Processes; Transport and catabolism |
hsa03030 |
*DNA replication |
0.00326 |
0.021616 |
Genetic Information Processing; Replication and repair |
hsa05110 |
Vibrio cholerae infection |
0.00317 |
0.021744 |
Human Diseases; Infectious diseases |
hsa04670 |
*Leukocyte transendothelial migration |
0.00431 |
0.027656 |
Organismal Systems; Immune system |
hsa04110 |
*Cell cycle |
0.00521 |
0.032386 |
Cellular Processes; Cell growth and death |
hsa04664 |
Fc epsilon RI signaling pathway |
0.00778 |
0.046896 |
Organismal Systems; Immune system |
* Pathways were reported as RA-related pathways in previous literatures.
Italic pathways were also detected based on GWAS.
The pathway analysis results based on integration GWAS and gene expression data
We chose the 11,922 genes which have both gene-wise association value of GWAS and the differential expression value. We combined gene-wise association value of GWAS and the differential expression value to generate a single gene association score by Fisher’s combined method. Then we translated the gene association score into the of chi-square statistic P-value and defined this P-value as the single gene association P-value after combination GWAS and gene expression data. Then, we computed the 234 KEGG pathway risk scores using the Fisher’s method and identified the RA-related pathways by the permutation test and FDR correction.
Finally, the 28 pathways (FDR< 0.05) after FDR correction were identified as RA-related pathways based on integration GWAS and gene expression data. Among these pathways, they were mainly involved in immune system (n=16), infectious diseases (n=7), signaling molecules and interaction (n=1), transport and catabolism (n=1), endocrine and metabolic diseases (n=1), development (n=1) and cardiovascular diseases (n=1).The detailed information of these pathways is presented in Table 3. There are 18 same pathways to be found by pathway analysis of GWAS and gene expression data respectively. Integrative analysis also predicted 7 novel RA-related pathways which are natural killer cell mediated cytotoxicity, primary immunodeficiency, Hematopoietic cell lineage, Fc gamma R-mediated phagocytosis, B cell receptor signaling pathway, osteoclast differentiation and Toll-like receptor signaling pathway. According to function classification of KEGG, the other six pathways belong to immune system except osteoclast differentiation pathway.
We found that pathways which were identified by pathway analyses based on GWAS and gene expression analysis were not entirely consistent. This is due to that single data type analyses only give limited genetic variation information of RA and maybe ignore some valuable association. Therefore, the pathways identified by single data type analyses are not comprehensive. Our results also indicated pathway analysis of integrating GWAS and gene expression analysis can detect stable and precise pathways which will give us an insight into complex molecular mechanisms of RA.
Table 3: The RA-related pathways identified by integrating GWAS and gene expression (FDR<0.05)
Pathway ID |
Pathway name |
Combined_P-value |
FDR |
Function class |
hsa04062 |
*Chemokine signaling pathway |
0 |
0 |
Organismal Systems; Immune system |
hsa04145 |
*Phagosome |
0 |
0 |
Cellular Processes; Transport and catabolism |
hsa04514 |
*Cell adhesion molecules (CAMs) |
0 |
0 |
Environmental Information Processing; Signaling molecules and interaction |
hsa04612 |
*Antigen processing and presentation |
0 |
0 |
Organismal Systems; Immune system |
hsa04650 |
Natural killer cell mediated cytotoxicity |
0 |
0 |
Organismal Systems; Immune system |
hsa04672 |
*Intestinal immune network for IgA production |
0 |
0 |
Organismal Systems; Immune system |
hsa04940 |
*Type I diabetes mellitus |
0 |
0 |
Human Diseases; Endocrine and metabolic diseases |
hsa05140 |
*Leishmaniasis |
0 |
0 |
Human Diseases; Infectious diseases |
hsa05145 |
*Toxoplasmosis |
0 |
0 |
Human Diseases; Infectious diseases |
hsa05150 |
*Staphylococcus aureus infection |
0 |
0 |
Human Diseases; Infectious diseases |
hsa05152 |
*Tuberculosis |
0 |
0 |
Human Diseases; Infectious diseases |
hsa05162 |
*Measles |
0 |
0 |
Human Diseases; Infectious diseases |
hsa05164 |
*Influenza A |
0 |
0 |
Human Diseases; Infectious diseases |
hsa05168 |
*Herpes simplex infection |
0 |
0 |
Human Diseases; Infectious diseases |
hsa05310 |
*Asthma |
0 |
0 |
Human Diseases; Immune diseases |
hsa05320 |
*Autoimmune thyroid disease |
0 |
0 |
Human Diseases; Immune diseases |
hsa05323 |
*Rheumatoid arthritis |
0 |
0 |
Human Diseases; Immune diseases |
hsa05330 |
*Allograft rejection |
0 |
0 |
Human Diseases; Immune diseases |
hsa05332 |
*Graft-versus-host disease |
0 |
0 |
Human Diseases; Immune diseases |
hsa05340 |
Primary immunodeficiency |
0 |
0 |
Human Diseases; Immune diseases |
hsa05416 |
*Viral myocarditis |
0 |
0 |
Human Diseases; Cardiovascular diseases |
hsa04660 |
*T cell receptor signaling pathway |
2.00E-05 |
0.000175 |
Organismal Systems; Immune system |
hsa04640 |
Hematopoietic cell lineage |
3.00E-05 |
0.000251 |
Organismal Systems; Immune system |
hsa04666 |
Fc gamma R-mediated phagocytosis |
0.00029 |
0.002325 |
Organismal Systems; Immune system |
hsa04662 |
B cell receptor signaling pathway |
0.00042 |
0.003233 |
Organismal Systems; Immune system |
hsa04380 |
Osteoclast differentiation |
0.00074 |
0.005477 |
Organismal Systems; Development |
hsa04620 |
Toll-like receptor signaling pathway |
0.00111 |
0.007912 |
Organismal Systems; Immune system |
hsa04670 |
*Leukocyte transendothelial migration |
0.00651 |
0.044744 |
Organismal Systems; Immune system |
* Pathways were reported as RA-related pathways in previous literatures.
DISCUSSION
RA is a complex and chronic autoimmune disease which is affected by multiple genes and environmental factors. Recently, many pathway analysis based on a single data type appeared. However, the results of these studies are varied. To identify the comprehensive and stable pathways, we carried out a pathway analysis by integrating GWAS and gene expression analysis.
In the pervious genetics studies, the authors usually selected the significant SNPs or genes by the given threshold and performed pathway analysis by using SNPs or genes which reached the threhold. The pathways identified by this method may be really associated with RA. However, some SNPs or genes which did not reach the threshold but were related to RA were usually ignored. Therefore, the moderate-effect pathways which contained many modest-effect SNPs or genes can not be identified by threshold methods. In order to overcome this limitation, we didn’t scan the SNPs or genes by the threshold, but used all of genes for the following analysis.
We have not only identified some pathways which were reported to be associated with RA in the previous studies, but also some hitherto unrecognized cellular pathways, including natural killer cell mediated cytotoxicity, primary immunodeficiency, hematopoietic cell lineage, Fc gamma R-mediated phagocytosis, B cell receptor signaling pathway, osteoclast differentiation and toll-like receptor signaling pathway. These novel pathways belonged to immune system according to KEGG function classification. At present, there was no evidence of association between these pathways and RA in previous studies. But it is known that RA is a typical autoimmune disease, so we infer RA may be associated with these immune pathways very likely. Therefore, it is easy to see that the integration analysis of GWAS and gene expression can provide more comprehensive results.
We identified 16 pathways which were related to immune system. In the present studies, there was no evidence to elucidate the relationship between Chemokine signaling pathway and RA until 2015. In 2015, zhang et al. used real-time polymerase chain reaction to determine gene expressions and found the chemokine signaling pathway was involved in CCL2 expression in RA patient peripheral blood and synovial tissues [25]. The same year, Lv et al. used a random walk strategy based on the subpathway- subpathway interaction network to prioritize risk subpathways and also found chemokine signaling pathway was a risk pathway to RA [26]. We also found the association between this pathway and RA by integration analysis. B cell receptor signaling pathway is an immune pathway. To date, there were no studies to indicate the relationship between B cell receptor signaling pathway and RA. But Xu et al. investigated the microRNA (miRNA) expression pattern in synovial fluid from patients with knee osteoarthritis (OA) and found B cell receptor signaling pathway was primarily unregulated [27]. Wu et al. performed a differential expression analysis of microRNA expression profiles of extracellular vesicles in nasal mucus from patients with allergic rhinitis and identified 35 differential expressed microRNAs and 32 significantly enriched KEGG pathways. Among 32 pathways, B cell receptor signaling pathway (P = 3.709E-09) was one of the most prominent pathways [28]. Guo et al. carried out a microRNA analysis in peripheral blood lymphocytes in experimental autoimmune uveitis (EAU) rats versus control and found B cell receptor signaling pathway was closely associated with EAU [29]. The evidence suggests that B cell receptor signaling pathway may be associated with autoimmune disease and inflammatory disorder. Further genetics researches or animal experiments are required to confirm the relation between B cell receptor signaling pathway and RA. We believe that our results will contribute to perform future genetic studies in RA pathogenesis and may promote the development of new therapeutic strategies by targeting these pathways.
We identified 7 pathways which were related to infectious disease. These seven pathways are identified respectively by GWAS, gene expression analysis and integration analysis. Among these pathways, Influenza-A pathway was not found as RA-related pathway in previously published studies until 2013. Sharma et al. performed a multistage integrative pathway analysis on RA and firstly detected a relation between the influenza-A pathway and RA (P = 2.0E-04) [21]. Besides, he also found Measles pathway was significantly associated with RA (P = 1.0E-03) [21]. Peter et al. developed a model-based approach to pathway analysis and found the relationship between RA and “Measles” pathway genes taken part in immune responses caused by measles infection [30]. In the same year, Liu et al. performed a pathway and network analysis of large-scale RA GWAS and also identified Measles pathway as the most significant pathway associated with RA (P = 1.57E-08) [24]. In addition, there was no evidence to indicate the links between Measles pathway and RA in the previous studies. We also identified Measles pathway as RA-related pathway by integrative analysis of GWAS and gene expression analysis. Further researches are required to uncover the mechanism of the interaction between Measles and RA.
In our research, we performed a pathway analysis by integrating GWAS and gene expression analysis and detected some pathways related to RA. We found integrative analysis did not only detect RA-associated pathways which had confirmed in previous studies, but also can identify some novel RA-related pathways. However, in order to reveal the pathogenesis of RA, we are required to integrate more types of data into pathway analysis and perform genetic studies or experiments to prove the relationship between the novel pathways and RA in further research.
MATERIALS AND METHODS
Data
GWAS SNP data came from WTCCC RA datasets. The dataset included 459,446 autosomal SNPs from 12,468 subjects (1,860 cases and 10,608 controls). The individuals from the dataset are Caucasian. SNPs with a minor allele frequency (MAF) <0.01 or a P-value <0.001 for Hardy-Weinberg equilibrium (HWE) or a genotype percent <0.25 were excluded from the analysis [20]. After quality control, WTCCC dataset included 448,033 SNPs.
Gene expression data are from three microarray datasets in Gene Expression Omnibus (GEO) database. The three microarray datasets are GSE55235, GSE55457 and GSE55584. Their platform is GPL96 (Affymetrix HG-U133A). The individuals of three datasets are Caucasian. In this study, 53 individuals are contained, including 33 RA patients and 20 healthy individuals. The detailed information of individuals is shown in Table 4.
Gene data were downloaded at the National Center for Biotechnology Information (ftp://ftp.ncbi.nlm.nih.gov/genomes/MapView/Homo_sapiens/sequence/BUILD.37.3/). There are altogether 34,402 autosomal genes for the following analysis. The detailed information of these genes was extracted, such as ‘gene ID’, ‘chromosome ID’, ‘gene start position’, ‘gene end position’ and so on. Then we map SNPs to genes according to the information.
Pathway data were obtained from the Kyoto Encyclopedia of Genes and Genomes (KEGG) [31]. The KEGG pathway database is a common bioinformatics resource that provides path graph of molecular interactions, chemical reactions, and gene relations. It has been widely used for the systematic analysis of gene functions that involve molecular interaction in complex diseases. There are in total 253 pathways in KEGG. To avoid testing overly narrow or broad function categories, we selected 234 human KEGG pathways that contained at least 5 and at most 200 genes [32].
Table 4: The number of the samples in three microarray datasets
Dataset |
RA |
Control |
Total |
Platform |
GSE55235 |
10 |
10 |
20 |
Affymetrix HG-U133A |
GSE55457 |
13 |
10 |
23 |
Affymetrix HG-U133A |
GSE55584 |
10 |
0 |
10 |
Affymetrix HG-U133A |
Total |
33 |
20 |
53 |
Assessing gene-wise association value based on SNP dataset
We used the Cochran-Armitage trend test to calculate P-values of all SNPs in the WTCCC datasets. Then we mapped a SNP to a gene if it was located within the gene or 50kb immediately upstream or downstream. All the SNPs from WTCCC RA datasets were mapped to 34,402 autosomal genes and their extended gene intervals.
Linkage disequilibrium is an important problem that should be considered in pathway analysis. In order to make the results more accurate, we carried out a LD correction. Because our WTCCC SNP data are not raw SNP genotype data, we chose 1000 genomes data from Europe as a reference dataset to correct LD. We obtained the tagSNPs of each gene by Haploview software when r2>0.8. We chose the smallest P-value of all tagSNPs in this gene to assess the gene-wise association value. See part I of workflow (Figure 1).
Computing the differential expression value based on gene expression data
We downloaded the cel files of the three microarray datasets (GSE55235, GSE55457 and GSE55584) from GEO database. Then we mixed the three datasets together and obtained 53 individuals, including 33 RA patients and 20 controls. The data were normalized by affy package in R software. Then the probes were mapped to the genes according to platform information.
The differential expression score reflects the degree of a differentially expressed gene between cases and controls. There are many statistical methods to compute the differential expression score, such as t-test, analysis of variance and so on. In this study, we chose t-test method to analyze the microarray data after normalization and compute the differential expression P-values of all genes. See part II of workflow (Figure 1).
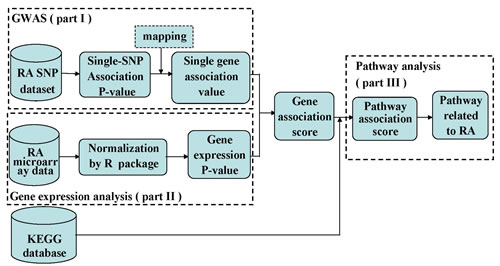
Figure 1: The workflow of integrative GWAS and gene expression analysis.
Calculating the gene association P-value by integrating GWAS and gene expression analysis
We chose the genes which have both gene-wise association value based on SNP dataset and the differential expression value. We combined gene-wise association value of GWAS and the differential expression value to generate a single gene association score by Fisher’s combined method. For example:
![]() (1)
(1)
Where gi is a single gene association score of i-th gene by integrating GWAS and gene expression. Pi1, Pi2 are gene-wise association value of GWAS and the differential expression value based on gene expression data.
Because gi follows X2(2K) distribution [33], we transformed the gi into the probability of chi-square distribution. The probability represented gene association P-value by integrating GWAS and gene expression and marked as Pi.
Calculating the pathway association score
It was known that the pathways were composed of genes. The cumulative effect of DNA variation in the pathway leads to the association of the pathway with susceptibility to disease. Thus, we calculated pathway association scores based on integrative gene association P-value. We still adopted Fisher’s combination method to calculate pathway association scores for 234 human pathways. See part III of workflow (Figure 1).
Permutation test
In order to further determine whether a pathway was significantly associated with RA, we performed a permutation test by shuffling the pathway labels. The null hypothesis is that there is no association between the pathway and RA. We counted the number of non-redundant genes that had association P-value in all 234 pathways. Then, the 234 pathway association scores were calculated by Fisher’s method. Specifically, for a pathway, the pathway association score was denoted as Strue. Next, we randomly chose the same number of genes as the number of genes involved in calculating this pathway association score from all non-redundant genes, calculated the random pathway association score by Fisher’s method and denoted it as Srandom. This process was repeated 100,000 times in order to achieve sufficient randomization. The random pathway association score (Srandom) formed an estimated background distribution of this pathway. The smaller gene association P-value, the pathway association score is greater. The greater the pathway association score, the more associated the pathway is with RA. A Ppermutation was computed for each pathway by counting the number of permutations that have Srandom more than Strue divided by the total number of permutations.
Multiple testing corrections
Multiple hypothesis tests had been used to identify pathways related to RA, but they could increase type I errors and the false-positive error rate. Therefore, we adopted multiple testing corrections to solve this problem. A threshold 0.05 of q-value (FDR) was used to identify RA-realted pathways.
The workflow of this study was shown in Fig.1.
ACKNOWLEDGMENTS
This work was supported in part by grants from the National Natural Science Foundation of China (Grant Nos. 81172842, 31200934, 61300116) and the Natural Science Foundation of Heilongjiang Province (Grant Nos. QC2013C063, F200802).
CONFLICTS OF INTEREST
The authors declare no conflicts of interest.
REFERENCES
1. Plenge RM. Rheumatoid arthritis genetics: 2009 update. Curr Rheumatol Rep. 2009; 11:351-356.
2. Heruth DP, Gibson M, Grigoryev DN, Zhang LQ and Ye SQ. RNA-seq analysis of synovial fibroblasts brings new insights into rheumatoid arthritis. Cell Biosci. 2012; 2:43.
3. Genome-wide association study of 14,000 cases of seven common diseases and 3,000 shared controls. Nature. 2007; 447:661-678.
4. Negi S, Juyal G, Senapati S, Prasad P, Gupta A, Singh S, Kashyap S, Kumar A, Kumar U, Gupta R, Kaur S, Agrawal S, Aggarwal A, Ott J, Jain S, Juyal RC, et al. A genome-wide association study reveals ARL15, a novel non-HLA susceptibility gene for rheumatoid arthritis in North Indians. Arthritis Rheum. 2013; 65:3026-3035.
5. Freudenberg J, Lee HS, Han BG, Shin HD, Kang YM, Sung YK, Shim SC, Choi CB, Lee AT, Gregersen PK and Bae SC. Genome-wide association study of rheumatoid arthritis in Koreans: population-specific loci as well as overlap with European susceptibility loci. Arthritis Rheum. 2011; 63:884-893.
6. Julia A, Ballina J, Canete JD, Balsa A, Tornero-Molina J, Naranjo A, Alperi-Lopez M, Erra A, Pascual-Salcedo D, Barcelo P, Camps J and Marsal S. Genome-wide association study of rheumatoid arthritis in the Spanish population: KLF12 as a risk locus for rheumatoid arthritis susceptibility. Arthritis Rheum. 2008; 58:2275-2286.
7. Stahl EA, Raychaudhuri S, Remmers EF, Xie G, Eyre S, Thomson BP, Li Y, Kurreeman FA, Zhernakova A, Hinks A, Guiducci C, Chen R, Alfredsson L, Amos CI, Ardlie KG, Barton A, et al. Genome-wide association study meta-analysis identifies seven new rheumatoid arthritis risk loci. Nat Genet. 2010; 42:508-514.
8. Liu Y, Helms C, Liao W, Zaba LC, Duan S, Gardner J, Wise C, Miner A, Malloy MJ, Pullinger CR, Kane JP, Saccone S, Worthington J, Bruce I, Kwok PY, Menter A, et al. A genome-wide association study of psoriasis and psoriatic arthritis identifies new disease loci. PLoS Genet. 2008; 4:e1000041.
9. Devauchelle V, Marion S, Cagnard N, Mistou S, Falgarone G, Breban M, Letourneur F, Pitaval A, Alibert O, Lucchesi C, Anract P, Hamadouche M, Ayral X, Dougados M, Gidrol X, Fournier C, et al. DNA microarray allows molecular profiling of rheumatoid arthritis and identification of pathophysiological targets. Genes Immun. 2004; 5:597-608.
10. Haas CS, Creighton CJ, Pi X, Maine I, Koch AE, Haines GK, Ling S, Chinnaiyan AM and Holoshitz J. Identification of genes modulated in rheumatoid arthritis using complementary DNA microarray analysis of lymphoblastoid B cell lines from disease-discordant monozygotic twins. Arthritis Rheum. 2006; 54:2047-2060.
11. Kim TH, Choi SJ, Lee YH, Song GG and Ji JD. Gene expression profile predicting the response to anti-TNF treatment in patients with rheumatoid arthritis; analysis of GEO datasets. Joint Bone Spine. 2014; 81:325-330.
12. Xu Y, Huang Y, Cai D, Liu J and Cao X. Analysis of differences in the molecular mechanism of rheumatoid arthritis and osteoarthritis based on integration of gene expression profiles. Immunol Lett. 2015.
13. Nzeusseu Toukap A, Galant C, Theate I, Maudoux AL, Lories RJ, Houssiau FA and Lauwerys BR. Identification of distinct gene expression profiles in the synovium of patients with systemic lupus erythematosus. Arthritis Rheum. 2007; 56:1579-1588.
14. Julia A, Barcelo M, Erra A, Palacio C and Marsal S. Identification of candidate genes for rituximab response in rheumatoid arthritis patients by microarray expression profiling in blood cells. Pharmacogenomics. 2009; 10:1697-1708.
15. van Baarsen LG, Wijbrandts CA, Timmer TC, van der Pouw Kraan TC, Tak PP and Verweij CL. Synovial tissue heterogeneity in rheumatoid arthritis in relation to disease activity and biomarkers in peripheral blood. Arthritis Rheum. 2010; 62:1602-1607.
16. Wang K, Li M and Hakonarson H. Analysing biological pathways in genome-wide association studies. Nat Rev Genet. 2010; 11:843-854.
17. Beyene J, Hu P, Hamid JS, Parkhomenko E, Paterson AD and Tritchler D. Pathway-based analysis of a genome-wide case-control association study of rheumatoid arthritis. BMC Proc. 2009; 3 Suppl 7:S128.
18. Torkamani A, Topol EJ and Schork NJ. Pathway analysis of seven common diseases assessed by genome-wide association. Genomics. 2008; 92:265-272.
19. Zhang MM, Jiang YS, Lv HC, Mu HB, Li J, Shang ZW and Zhang RJ. Pathway-based association analysis of two genome-wide screening data identifies rheumatoid arthritis-related pathways. Genes Immun. 2014; 15:487-494.
20. Ballard DH, Aporntewan C, Lee JY, Lee JS, Wu Z and Zhao H. A pathway analysis applied to Genetic Analysis Workshop 16 genome-wide rheumatoid arthritis data. BMC Proc. 2009; 3 Suppl 7:S91.
21. Buchel F, Mittag F, Wrzodek C, Zell A, Gasser T and Sharma M. Integrative pathway-based approach for genome-wide association studies: identification of new pathways for rheumatoid arthritis and type 1 diabetes. PLoS One. 2013; 8:e78577.
22. Eleftherohorinou H, Wright V, Hoggart C, Hartikainen AL, Jarvelin MR, Balding D, Coin L and Levin M. Pathway analysis of GWAS provides new insights into genetic susceptibility to 3 inflammatory diseases. PLoS One. 2009; 4:e8068.
23. Yi CQ, Ma CH, Xie ZP, Cao Y, Zhang GQ, Zhou XK and Liu ZQ. Comparative genome-wide gene expression analysis of rheumatoid arthritis and osteoarthritis. Genet Mol Res. 2013; 12:3136-3145.
24. Liu G, Jiang Y, Chen X, Zhang R, Ma G, Feng R, Zhang L, Liao M, Miao Y, Chen Z, Zeng R and Li K. Measles contributes to rheumatoid arthritis: evidence from pathway and network analyses of genome-wide association studies. PLoS One. 2013; 8:e75951.
25. Zhang L, Yu M, Deng J, Lv X, Liu J, Xiao Y, Yang W, Zhang Y and Li C. Chemokine Signaling Pathway Involved in CCL2 Expression in Patients with Rheumatoid Arthritis. Yonsei Med J. 2015; 56:1134-1142.
26. Lv W, Wang Q, Chen H, Jiang Y, Zheng J, Shi M, Xu Y, Han J, Li C and Zhang R. Prioritization of rheumatoid arthritis risk subpathways based on global immune subpathway interaction network and random walk strategy. Mol Biosyst. 2015; 11:2986-2997.
27. Xu JF, Zhang SJ, Zhao C, Qiu BS, Gu HF, Hong JF, Cao L, Chen Y, Xia B, Bi Q and Wang YP. Altered microRNA Expression Profile in Synovial Fluid from Patients with Knee Osteoarthritis with Treatment of Hyaluronic Acid. Mol Diagn Ther. 2015; 19:299-308.
28. Wu G, Yang G, Zhang R, Xu G, Zhang L, Wen W, Lu J, Liu J and Yu Y. Altered microRNA Expression Profiles of Extracellular Vesicles in Nasal Mucus From Patients With Allergic Rhinitis. Allergy Asthma Immunol Res. 2015; 7:449-457.
29. Guo D, Li J, Liu Z, Tang K, Song H and Bi H. Characterization of microRNA expression profiling in peripheral blood lymphocytes in rats with experimental autoimmune uveitis. Inflamm Res. 2015; 64:683-696.
30. Carbonetto P and Stephens M. Integrated enrichment analysis of variants and pathways in genome-wide association studies indicates central role for IL-2 signaling genes in type 1 diabetes, and cytokine signaling genes in Crohn’s disease. PLoS Genet. 2013; 9:e1003770.
31. Kanehisa M and Goto S. KEGG: kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 2000; 28:27-30.
32. de las Fuentes L, Yang W, Davila-Roman VG and Gu C. Pathway-based genome-wide association analysis of coronary heart disease identifies biologically important gene sets. Eur J Hum Genet. 2012; 20:1168-1173.
33. Peng G, Luo L, Siu H, Zhu Y, Hu P, Hong S, Zhao J, Zhou X, Reveille JD, Jin L, Amos CI and Xiong M. Gene and pathway-based second-wave analysis of genome-wide association studies. Eur J Hum Genet. 2010; 18:111-117.