
Introduction
Cancer metastasis remains the main cause of cancer related mortality and contributes to poor prognosis in the majority of cancer types. Metastases decrease the likelihood of survival; this is best represented by the significant decline in five year survival rates in prostate and breast cancer patients with metastasis upon presentation [1]. Surgical intervention and other conventional treatments have limited efficacy once cancer cells have spread to distant sites, owing to the heterogeneity and aggressiveness of the disease, high chance of recurrence and the difficulty in targeting multiple sites after metastatic spread. Research efforts have been directed at interfering with key mediators important in metastatic development as alternative strategies for cancer treatment. Over the past few decades, distinct chemokine-receptor signaling pathways have emerged as attractive targets for therapy due to their key roles in the metastatic process.
Chemokines and their receptors mediate acute inflammation and were initially described in the context of their chemoattractant function for leukocytes; chemokines, induced at sites of inflammation, provide directional cues during migration of leukocytes to damaged or infected tissues [2, 3]. However, elevated expression of chemokines, leading to alterations in chemokine-receptor signaling can contribute to chronic inflammation and malignancy [4, 5]. Cancer cells and host stromal cells in the tumor microenvironment including endothelial cells, fibroblasts, mesenchymal stem cells and infiltrating leukocytes produce a wide range of chemokines that exert numerous biological functions during tumor progression and metastasis [6, 7]. Of these, CCL2 together with its cognate receptor CCR2 have been shown to play key roles in cancer metastasis by sustaining cancer cell proliferation and survival, stimulating cancer cell migration and invasion, and inducing deleterious inflammation and angiogenesis. Here we provide an overview of CCL2-CCR2 signaling during the metastatic process, and draw upon experimental and clinical studies to highlight their significant contributions to metastatic development and progression. Finally, we discuss the relevance and efficacy of targeting this signaling pair as a means of therapeutic intervention.
Biology of CCL2 and CCR2
CCL2 belongs to a group of low molecular weight cytokines with chemoattractant activity, collectively known as chemokines. As a prototypic chemokine, CCL2 orchestrates immune cell recruitment to specific sites, and is expressed constitutively for homeostatic functions such as regulating lymphocyte trafficking from blood to lymph nodes, and is induced during inflammatory responses when leukocytes are required for tissue defense and repair [3]. CCL2 is expressed by a wide range of cells including endothelial, epithelial, myeloid and smooth muscle cells and fibroblasts, either constitutively or after induction, and is a potent chemoattractant for monocytes, basophils, T lymphocytes and NK cells [8].
CCL2 was initially categorized as monocyte chemotactic protein 1 (MCP-1) due to its structural similarity with other MCPs, including MCP-2 (CCL8), MCP-3 (CCL7), MCP-4 (CCL13) and MCP-5 (CCL12). These MCPs share high sequence homology and highly conserved secondary structures of two adjacent N-terminal cysteine residues that are important for receptor binding [9]. Binding of a chemokine to its cognate receptor is required to trigger signal transduction pathways and exert biological effects such as chemotaxis. These cell surface G protein-coupled receptors are characterized by an N-terminal extracellular domain, 7 conserved transmembrane domains linked by three intracellular and extracellular loops, and a serine/threonine-rich C-terminal intracellular domain, the latter of which is coupled to a heterotrimeric G-protein for signal transduction.
CCL2 preferentially binds the CCR2 receptor, which is expressed in various tissues including blood, brain, heart, kidney, liver, lung, ovary, pancreas, spinal cord, spleen and thymus. CCR2 is expressed as two isoforms due to alternative splicing: CCR2A and CCR2B, which differ by 50 base pair in the C-terminal domain [10]. CCR2B is the predominant isoform of CCR2 surface receptors, highly expressed by monocytes and NK cells, and accounting for 90% of all CCR2 expressed [11, 12]. CCR2A is expressed by a small subset of mononuclear and smooth muscle cells [13]. It is important to note that binding of CCL2 to the CCR2A isoform induces different biological responses from binding to the CCR2B isoform, as demonstrated in Jurkat T cells [14]. Cell-specific expression of the CCR2 isoforms may serve as a means of functional regulation.
Apart from CCR2, CCL2 can also bind other chemokine receptors. CCL2 has been reported to bind CCR4 on cytotoxic T lymphocytes, resulting in their recruitment to melanoma cells [15] thus implicating an immune-mediated protective role in cancer. However, CCL2-CCR4 signaling also recruits T regulatory cells to tumor sites, which may result in cytotoxic T cell suppression [16]. CCL2 has also been shown to bind two atypical receptors, ACKR1 and ACKR2. These atypical receptors do not signal through G proteins and are regarded as decoys or scavenger receptors as they lack chemotactic activity. Scavenger receptors are likely to compete for CCL2 binding with other conventional G-protein coupled chemokine receptors [17], enabling them to modulate free CCL2 levels [18].
CCR2 is a promiscuous receptor that binds other chemokines, particularly other MCPs including MCP-2 (CCL8), MCP-3 (CCL7) and MCP-4 (CCL13) consistent with their structural similarity to CCL2. This flexibility in chemokine-receptor interaction may lead to different biological outcomes, depending on the particular chemokine and receptor pair engaged [19], or may produce similar effects, suggesting redundancies in chemokine function. For example, binding of either CCL7 or CCL2 to CCR2 can stimulate monocyte emigration from the bone marrow to sites of metastasis [20]. Likewise, binding or either CCL8 or CCL2 to CCR2 on colorectal cancer cells can provoke a similar increase in migration and invasion [21]. However, distinct effects have also been noted; binding of CCL8 to CCR8 promotes the recruitment of inflammatory Th2 cells whereas its binding to CCR2 did not generate the same response [22].
Because CCL2 and CCR2 bind other receptors and ligands respectively, understanding the specific effects and implications of CCL2-CCR2 signaling has been challenging. However, the CCL2-CCR2 pairing appears to be the prevalent interaction in vivo as mice deficient in CCL2 share similar phenotypes to those deficient in CCR2 [23, 24].
The role of CCL2-CCR2 signaling in the metastatic cascade
Metastasis is a multistep process that involves invasion of cancer cells into surrounding tissues, followed by intravasation into lymph or blood vessels whereupon tumor cells disseminate until their arrest and extravasation into secondary sites. At secondary sites, cancer cells must adapt to their new microenvironment in order to proliferate and form metastatic outgrowths; this latter stage is often referred to as metastatic colonization [25-27]. Cancer cells are vulnerable to death at any of these steps and will require intrinsic and extrinsic responses to enable their survival. CCL2-CCR2 signaling is especially important for successful metastasis, and has been shown to be involved in both the early and late steps of the metastatic process in experimental models. CCL2 can be produced by both cancer and stromal cells in the tumor microenvironment, exerting direct effects on cancer cells and functioning indirectly by recruiting host stromal cells with pro-tumorigenic activities during metastasis (Figure 1).
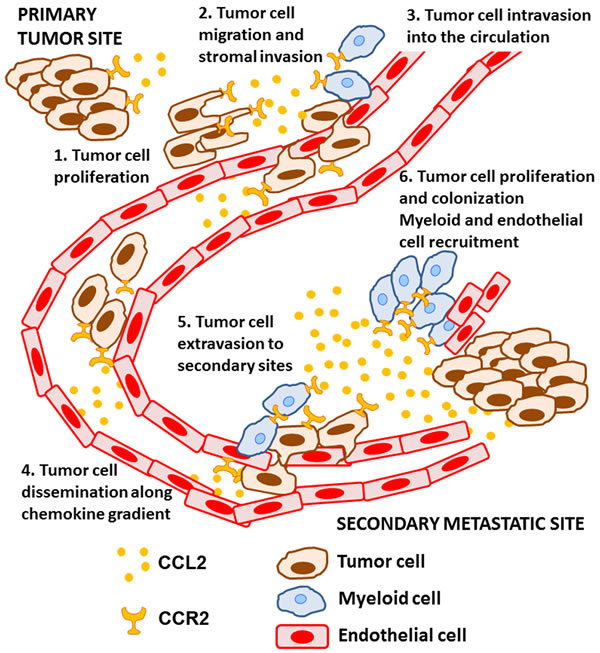
Figure 1: The role of CCL2-CCR2 signaling during the metastatic process. CCL2 is expressed by cancer and stromal cells in the tumor microenvironment and, 1) induces tumor cell proliferation at the primary tumor site and 2) stimulates tumor cell migration and invasion into the surrounding extracellular matrix. CCL2 subsequently 3) promotes tumor cell intravasation into the circulation, likely by recruiting host myeloid cells to facilitate this process. Once in the circulation, CCL2 may 4) direct the dissemination of cancer cells along a chemotactic gradient towards the metastatic site. Trapping of tumor cells in small capillaries initiates 5) tumor cell extravasation, which is further supported by CCR2+ myeloid cells and the CCR2+ endothelium. Finally, CCL2 6) promotes tumor growth at the metastatic site, and tumor colonization by recruiting additional myeloid and endothelial cells.
During the early stages of metastasis, cancer cells acquire a migratory and invasive phenotype, which allows them to break down surrounding extracellular matrix (ECM), invade neighboring tissues and move towards the blood or lymph vessels. CCL2 contributes towards this initial stage, guiding cancer cell migration by interacting with the CCR2 receptor expressed on tumor cells [28, 29]. Additionally, CCL2 induces expression of metalloproteinases MMP2 and MMP9 in cancer cells, leading to increased invasion [30, 31]. Following migration and invasion, intravasation of cancer cells into the circulation is required to allow for metastatic dissemination. Wyckoff et al. demonstrated that this process may necessitate cancer cell interaction with tumor associated macrophages (TAMs) [32]. Extravasation of cancer cells out of the circulation to secondary sites is also dependent on association with host stromal cells including TAMs [33] and bone marrow endothelial cells [34]. Because CCL2 is a potent chemoattractant for TAMs, it may indirectly promote the intravasation and extravasation process of cancer cells.
Arguably, the most challenging step of the metastatic cascade occurs after cancer cells have successfully extravasated from the circulation. At secondary sites, most cancer cells die, enter a homeostatic balance of cell proliferation and apoptosis that prevent further progression, or remain quiescent [35]. This state of dormancy is in keeping with clinical observations that many patients with melanoma, or breast and prostate carcinomas develop metastatic relapse years after the initial diagnosis or treatment. Breast cancer recurrences in particular are at times detected decades after remission [36, 37]. Tumor dormancy occurs in response to delayed adaptation to the new microenvironment and may be a coping mechanism whilst cancer cells accumulate sufficient resources and/or properties for successful proliferation and colonization. For example, cancer cells may need to develop means of evading immunosurveillance (to prevent immune dormancy), circumventing cell cycle arrest (to prevent cellular dormancy) and/or triggering an angiogenic switch (to prevent mass dormancy) for successful metastatic formation [38-40]. Although there has been little evidence linking CCL2 or CCR2 to cell cycle arrest, CCL2-CCR2 signaling has been shown to recruit myeloid cells such as TAMs to incite an angiogenic switch [41, 42], and myeloid-derived suppressor cells (MDSCs) to suppress and evade immune-mediated killing [43].
CCL2 has additional roles during the later stages of metastasis. Cancer cells can usurp the leukocyte trafficking mechanism to preferentially home to specific organs [44, 45]. Hence, in addition to leukocytes, CCL2 can also attract cancer cells to secondary sites, although this process is more complex and may require interactions with other hematopoietic cell types [46]. CCL2 can further stimulate cell proliferation and enhance survival once cancer cells have been recruited to metastatic sites [47, 48].
CCL2, along with other CXC chemokines such as CXCL1, CXCL2, CXCL3, CXCL5, CXCL6, CXCL7, and CXCL8 promote recruitment, migration and proliferation of endothelial cells. Both mouse and human endothelial cells express CCR2 and can be recruited to the metastatic microenvironment in response to CCL2. Additionally, CCL2-CCR2 signaling as a mediator of neovascularization has been previously demonstrated in several in vitro and in vivo models of angiogenesis [49-52].
Apart from direct effects on tumor and endothelial cells, CCL2 also recruits various immune cell subsets including monocytes [53] and macrophages [54] to site of metastasis, and mediates differentiation and polarization of these cell types [55, 56]. Immune cell polarization can result in distinct, if not opposing, functional properties, and may ultimately tip the balance between tumor promotion or inhibition. For example, macrophages polarized towards an M2 phenotype are immunosuppressive, and may enhance tumor cell survival by dampening immune surveillance and attack [55]. Similarly, CCL2 is involved in T cell differentiation and polarization [57], and has been shown to regulate Th2 polarization towards a more immunosuppressive T regulatory phenotype in vitro and in mouse models [58-61].
It is evident that CCL2-CCR2 signaling has multiple key functions, on both cancer and stromal cells (summarized in Table 1), altogether of which appear to predominately favor metastatic development and progression. In the following sections, we highlight effects of CCL2/CCR2 signaling in several experimental studies on metastatic cancers, with a particular focus on prostate, breast and colorectal cancers.
Table 1: Effects of CCL2-CCR2 signaling on cancer and stromal cells in the tumor microenvironment
CCL2-CCR2 signaling on cancer cells |
||
Cancer cell types |
Effects |
References |
Breast cancer cells MCF-7 |
Stimulates cell migration |
[102] |
Breast cancer cells PyVmT, 4T1, MCF-7, MDA-MB-231 |
Stimulates cell survival and motility |
[103] |
Breast cancer cells MDA-MB-231 and MCF-7 |
Stimulates cell attachment to lymphatic endothelial cells |
[104] |
Prostate cancer cells PC-3, LNCaP, DU145 |
Stimulates cell adhesion and invasion |
[105] |
Prostate cancer cells PC-3 |
Stimulates cell proliferation and invasion |
[52] |
Prostate cancer cells PC3, DU145, and LNCaP |
Stimulates cell migration and invasion |
[48] |
Prostate cancer cells PC-3 and VCaP |
Stimulates proliferation and migration |
[47] |
Prostate cancer cells PC-3 |
Stimulates invasion and transendothelial cell migration |
[30] |
Glioblastoma cells T98G, and U87MG |
Simulates cell migration and invasion |
[106] |
Ovarian cancer cells SKOV-3 |
Stimulates cell invasion and adhesion |
[107] |
Bladder cancer cells SV-HUC-1, RT4, TSGH8301, and J82 |
Stimulates cell migration and tumorigenicity |
[24] |
Chondrosarcoma cells JJ012 |
Stimulates cell migration |
[27] |
CCL2-CCR2 signaling on stromal cells |
||
Stromal cell types |
Effects |
References |
Fibroblasts |
Stimulates anti-fibrotic effects, survival, and adhesion |
|
Myeloid-derived suppressor cells |
Stimulates accumulation in tumors and affects immunosuppressive features |
|
Macrophages |
Stimulates recruitment and infiltration; promotes normal peritoneal macrophages to acquire features of TAMs |
|
Monocytes |
Stimulates maturation into macrophages |
|
Neutrophils |
Stimulates recruitment |
|
Osteoclasts |
Stimulates osteoclast formation and differentiation |
|
Stem cells |
Stimulates migration and enhances pluripotency |
|
NK cells |
Stimulates migration |
|
T cells |
Stimulates migration, promotes Th2 polarization and negatively regulates Th1 response |
|
CD4+ Th17 T cells |
Inhibits proliferation and activity |
|
Effects of CCL2-CCR2 signaling in metastatic cancers: experimental evidence
Prostate cancer
CCL2 is predominantly expressed by endothelial cells in the prostate tumor microenvironment and by prostate cancer cell lines such as PC3 and LnCaP. CCL2 directly stimulates PC3 and VCaP prostate cancer cell proliferation and migration via activation of the PI3K/Akt signaling pathway and activation of Rac GTPase, respectively [62]. A separate, but similar study reported that CCL2 promoted PC3, LnCaP and DU145 prostate cancer cell migration in a PKC-dependent manner by upregulation of αvβ3 integrin expression on cancer cells [63]. In addition to proliferation and migration, CCL2 protects prostate cancer cells from autophagic death by activating the PI3K/Akt/survivin pathway [47].
In addition to direct effects on cancer cells, CCL2 also mediates stromal cell responses in the prostate tumor microenvironment. CCL2 blockade using CCL2 neutralizing antibodies (anti-mouse CCL2/JE C1142) in SCID mice after subcutaneous injection of VCaP cells suppressed tumor growth, decreased CD68+ macrophage infiltration, and led to reduced tumor-associated microvasculature [64]. However, although CCL2 has been shown to have direct angiogenic effects on HUVEC and HDMVEC, in this context, CCL2 was shown to stimulate tumor cells to upregulate expression of angiogenic factors such as VEGF [65].
Both tumor- and stromal-derived CCL2 contribute towards prostate tumor progression. Inhibition of tumor-derived CCL2 via administration of anti-human CCL2 blocking antibody (CNTO888) resulted in reduced tumor burden, albeit not to the extent as inhibition of stromal-derived CCL2 using the anti-mouse CCL2/JE antibodies. Additionally, use of both tumor and stromal-specific inhibitory CCL2 antibodies were not as effective compared to single-agent docetaxel, the standard treatment for hormone-refractory metastatic prostate cancer. However, when used in combination with docetaxel, a dramatic regression in tumor burden was observed; tumor regression was only maintained with repeated antibody administration as discontinuation of treatment resulted in tumor re-growth [66].
Prostate cancers typically metastasize to the bone, and inhibition of both tumor and stromal-derived CCL2 had striking effects in decreasing metastatic bone lesions [65, 66]. In accordance with this finding, CCL2 overexpression in PC3 cells increased metastatic bone lesions, with concurrent increases in the number of activated osteoclasts and infiltrating macrophages [54]. Lu et al. further showed that CCL2 may mediate bone resorption by inducing differentiation of osteoclast-like cells [67]. Targeting CCL2 using the anti-human CCL2 (CNTO888) antibodies, either alone or in combination with docetaxel, has also been shown to inhibit PCa prostate cancer cell growth in the bone [68]. Overall, these studies highlight the multifaceted roles of CCL2 in prostate cancer and suggest that both tumor and stromal derived CCL2 have direct and indirect effects in promoting prostate tumor growth and associated bone metastasis.
Breast cancer
CCL2 is highly expressed by various breast cancer cell lines including 4T1, 4T07 and 67NR, as well as by both the hematopoietic and non-hematopoietic cells in the tumor stroma [69, 70]. CCL2 was shown to promote the migration of mammary carcinoma cell lines MCF-7, T47D and ZR-75-1 [71], and enhanced the migration and survival of 4T1, PyVmT, MDA-MD-231 and MCF-7 cells via activation of Smad3 and p42/44 MAPK signaling [72].
In addition to its direct effects on tumor cells, tumor and stromal-derived CCL2 also stimulated metastatic progression by recruiting different subsets of myeloid cells including TAMs [73], metastasis associated macrophages (MAMs) [74] and inflammatory monocytes [75]. Stromal CCL2 prompted macrophage infiltration to mammary tumor xenografts and its blockade using neutralizing antibody significantly reduced macrophage recruitment and tumor growth [76]. Similarly, CCL2 deficiency in the tumor stroma led to decreased macrophage recruitment and angiogenesis, consequently reducing incidence of lung metastases [69]. The contribution of stromal CCL2 may outweigh that of tumor-derived CCL2 as inhibition of stromal CCL2 alone significantly reduced primary tumor growth and distant lung metastases in orthotopic breast cancer models [77].
Qian et al. demonstrated that total blockade of CCL2 (both stromal- and tumor-derived) inhibited recruitment of CCR2+ inflammatory monocytes and prolonged survival of tumor-bearing mice; depletion of tumor-derived CCL2 alone was sufficient to inhibit metastatic seeding in the lung. The authors further implicated CCL2-mediated recruitment of CCR2+ monocytes as essential in enhancing tumor cell extravasation [53]. CCR2+ monocytes differentiate into CCR2+ MAMs, and in a subsequent study, the authors showed that CCL3 secretion by CCR2+ MAMs prolonged their retention in metastatic sites [74]. Overall these studies indicate that CCL2-CCR2 signaling is an important initial event leading to pulmonary metastasis of breast cancer.
In breast cancers, the recruitment of macrophages by CCL2 may also lead to extensive neovascularization [78] suggesting that tumor infiltrating myeloid cells may directly contribute to the angiogenic process although the mechanisms for this are not yet well defined. CCL2 from immortalized human mammary epithelial cells was recently shown to induce angiogenesis via upregulation of the transcription factor Twist1 [41] and presence of both CCL2 and macrophage infiltration was necessary for Twist1-driven angiogenesis, suggesting that CCL2 may recruit macrophages and thus, indirectly upregulate Twist1.
Another interesting study by Tsuyada et al. [79] revealed that human cancer associated fibroblasts (CAFs) co-cultured with human breast cancer cell lines BT474, MDA-MB-361 and MCF7 upregulated CCL2 in the cancer cells. Upregulation of CCL2 provoked a stem cell-like mammosphere-forming phenotype in the breast cancer cells. Furthermore, CCL2 enhanced self-renewal and expansion of the sphere-initiating cancer stem cells through STAT3 and NOTCH1 signaling pathways, suggesting that CCL2 may confer a more aggressive stem cell-like tumor phenotype. Overall, these findings strongly indicate that CCL2-CCR2 signaling in cancer and stromal cells is a major factor in the promotion of breast cancer growth and metastasis.
Colorectal cancer
CCL2 signaling was shown to be an important precursor to colorectal cancer development as its absence in CCL2-/- mice blocked neoplastic progression from dysplasia to adenocarcinoma. At later stages, CCL2 stimulated the accumulation of myeloid-derived suppressor cells (MDSCs) in colonic tumors, and enhanced MDSC-mediated suppression of T cells in a STAT3-dependent manner. Furthermore, CCL2 blocking antibodies decreased tumor and MDSC numbers, suggesting dual functions of CCL2 on both cancer and stromal cells [80].
Colorectal cancers typically metastasize to the liver although spread to the lung, peritoneum and other organs are also observed. Our group recently demonstrated the importance of CCL2-CCR2 signaling in an experimental mouse model of liver metastasis. CCL2, highly expressed by colorectal cancer cells, promoted the recruitment of a CD11b+ myeloid population expressing CCR2 from the bone marrow to the metastatic site in the liver. Depletion of CD11b+CCR2+ myeloid cells in the transgenic CD11b-DTR mouse model by diphtheria toxin administration markedly decreased metastatic growth and incidence [81].
Interestingly, in this system, attempts to inhibit CCL2 or CCR2 led to unexpected consequences. Inhibition of CCL2 using a functional blocking antibody, which targets both stromal and tumor cell-derived CCL2, did not alter myeloid cell recruitment and had little effect on metastatic tumor burden. This is in contrast to CCL2 inhibition in a breast cancer model, where Qian et al. showed decreased recruitment of CCR2+ inflammatory monocytes and reduced metastatic burden [53]. These differences may be model or organ specific but it is also likely that dosage of the CCL2 blocking antibody was not sufficient to inhibit CCL2-mediated effects. In support of this, serum CCL2 levels in mice were higher after treatment with the blocking antibody [81], suggesting a compensatory increase in CCL2 following its blockade.
In the experimental liver metastasis mouse model, knockdown of CCL2 expression in cancer cells caused a transient reduction in myeloid cell recruitment and a temporary delay in metastatic tumor growth, but these differences were not apparent 2 weeks after tumor cell injection [81]. The effect of CCR2 blockade was also disappointing; myeloid cell recruitment and metastatic tumor growth in the liver were reduced only to a small extent in CCR2 knockout mice, suggesting that CCL2 may bind other chemokine receptors apart from CCR2 to favor metastatic progression [81].
In addition to myeloid cell recruitment, tumor derived CCL2 has been shown to have direct effects on the tumor vasculature. CCL2 binding to CCR2+ endothelium enhanced vascular permeability in a p38/MAPK dependent manner, which in turn increased colon cancer cell extravasation and metastasis to the lungs [82].
Other cancers
Blockade of CCL2 using inhibitory antibodies suppressed growth of pancreatic cancer cells PANC-1 and BXPC3 in culture and in subcutaneous tumor models [83]. Blockade of the CCR2 receptor, on the other hand, inhibited infiltration of immunosuppressive CCR2+ myeloid cells in a pancreatic cancer mouse model, resulting in significant reduction in subcutaneous tumor growth, as well as incidence of liver metastases [84]. Inhibition of both tumor and stromal derived CCL2 in a mouse glioma and human glioma xenograft model prolonged survival of mice with a corresponding decrease in TAMs and MDSCs [85].
CCL2-CCR2 signaling has also been implicated in metastasis in other cancers. In chondrosarcoma, which preferentially metastasizes to the lungs, CCL2 promoted migration and invasion of chondrosarcoma cells, likely by inducing MMP9 expression via Ras/Raf-1 and NF-κB activation [31]. In ectopic and orthotopic xenograft models of gastric cancer, CCL2 overexpression in the gastric cancer cell line TMK-1 significantly increased tumorigenicity and induced metastasis to lymph nodes. Tumors formed by CCL2 overexpressing cancer cells were also more angiogenic compared to controls [86]. Similar findings were reported for bladder cancer whereby functional blockade of CCL2 or CCR2 inhibited migration of T24, J82 and SV-HUC-1 cancer cells, reduced tumorigenicity [28] and suppressed development of lung metastases through the inhibition of versican [87]. CCL2 inhibition also reduced macrophage infiltration to the lung, which may additionally contribute to inhibition of metastasis formation [87].
CCL2 was also shown to mediate the growth and proinvasive capacity of metastatic melanoma cells [88]. Targeting CCL2 using neutralizing antibodies or with BRAF inhibitors, which suppressed CCL2 gene expression, resulted in a marked inhibition of tumor growth in mouse models of metastatic melanoma [89]. In addition to having direct effects, CCL2 expressed by fibrocytes was implicated in recruitment of Ly6C monocytes, predisposing B16F10 cells to metastasize to the lung [90]. In agreement, other studies have shown that overexpression of CCL2 in melanoma cells enhanced vascular permeability and angiogenesis, and increased recruitment of mononuclear cells [91] and M2 polarized macrophages [92]. However, CCL2 may also have opposing anti-tumorigenic functions in melanoma progression. CCL2 expressed by melanoma cells has been shown to recruit cytotoxic T lymphocytes via binding to the CCR4 receptor on the lymphocytes [15]. Indeed, CCL2 overexpression in B16 melanoma cells enhanced Th2 cytokine production and reduced metastatic pulmonary tumor growth in wildtype C57BL/6 mice but not in nude mice with deficient adaptive immune responses [93], implying that T cells may be involved in CCL2-mediated anti-metastatic effects. Hence, effects of CCL2 may be dependent on specific receptor binding (CCR2 compared to CCR4) and also dependent on the type of effector cells involved.
Targeting CCL2-CCR2 signaling in the clinical setting
Elevated levels of CCL2 have been reported in patients with breast, colorectal, prostate, melanoma, gastric and ovarian cancers, and was often correlated with disease progression [94].
CCL2 was identified from meta analyses of gene expression datasets on prostate tumorigenesis as a primary driver of prostate cancer development [95] consistent with the experimental findings summarized above. Indeed, CCL2 appears to be a promising biomarker as it was overexpressed in tissue and serum samples of prostate cancer patients [96], and its expression correlated with Gleason score and pathologic state [97], and a more aggressive phenotype [48]. In ovarian cancer, CCL2 levels significantly correlated with histological grade and CCL2 was suggested as a differentiation marker between benign ovarian cysts and ovarian cancer [98].
CCL2 is overexpressed at tumor sites, malignant pleural effusions, and sera of breast cancer patients, and its increased levels in the tumor microenvironment and circulation were associated with disease progression, early relapse, tumor grade and aggressiveness, and poor prognosis [99]. Elevated CCL2 levels also inversely correlated with relapse-free survival, and predicted advanced tumor stage and lymph node involvement in breast cancer patients [100].
In patients with gastric cancer, elevated serum and intratumoral CCL2 levels significantly correlated with lymph node metastasis [101, 102]. Gastric cancer patients with high CCL2 expression also had a lower overall survival rate, suggesting CCL2 to be a prognostic marker for gastric cancer [103]. Similarly, in patients with primary and metastatic colorectal cancer, CCL2 levels were elevated in serum, and increased with progressive Dukes’ stages [81] and neoplastic progression [80]. CCL2 was further implicated as a prognostic marker and an independent predictor of liver metastasis in colorectal cancer patients [104].
Taken together with the many experimental studies supporting an important pro-tumorigenic role for CCL2-CCR2 signaling, several clinical trials have been established to investigate the safety and efficacy of CCL2 or CCR2 blockade as means of therapeutic intervention.
CCL2 inhibition (CNTO888) in solid tumors
Carlumab, previously known as CNTO888, is a human IgG1k monoclonal antibody developed by Centocor, that binds CCL2 with high affinity (disassociation constant of 15 pM), consequently inhibiting CCL2 binding to the CCR2 receptor [105]. CNTO888 interaction is also highly specific as it does not bind other mouse or human CC chemokines despite sharing high sequence homology. Analysis of the crystal structure of CNTO888 in complex with CCL2 [105] revealed residues 18-24 and 45-51 on CCL2 to be important for this interaction (Figure 2). Furthermore, comparison of the CCR2 receptor binding site indicated common epitopes shared by the inhibitory antibody and the CCR2 receptor, particularly Arg24 and Lys49 (Figure 2). Hence, the mode of action of CNTO888 is by direct competition with the CCR2 receptor binding site.
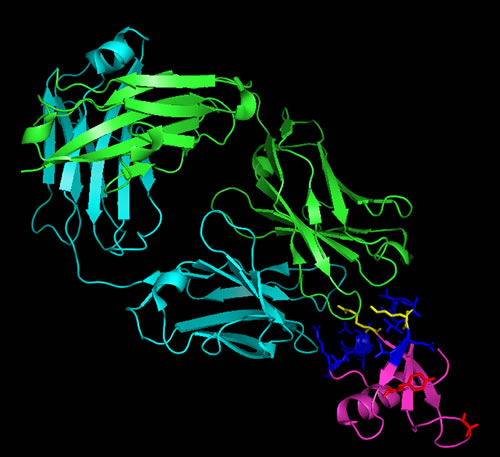
Figure 2: Ribbon representation of CNTO888 in complex with CCL2. CCL2, represented in magenta, comprises an anti-parallel 3-stranded β-sheet and a C-terminal α helix. The light chain of the CNTO888 antibody is shown in cyan whilst the heavy chain is shown in green. The epitope (on CCL2) recognized by CNTO888 (residues 18-24 and 45-51) is shown in blue whilst those important in CCR2 binding (Tyr13, Arg24, Lys35 and Lys49) is shown in red. The CNTO888 and CCR2 receptor epitope both include Arg24 and Lys49, shown in yellow. Figure was generated with PyMoL [116] using crystal structure of CNTO888 and CCL2 complex (PDB ID: 4DN4).
CCL2 inhibition was evaluated in solid tumors for safety and pharmacokinetic/pharmacodynamic parameters, in which CNTO888 was administered either alone or in combination with other standard of care chemotherapies. A phase 1 clinical trial (NCT00537368) was conducted in 44 patients with advanced solid tumors refractory to conventional treatments. As secondary measures, anti-tumor responses based on PSA and cancer antigen 125 (CA125) levels were also monitored. CNTO888 was administered intravenously at a starting dose of 0.3 mg/kg to a maximum planned dose of 15 mg/kg.
CNTO888 was well tolerated in these patients with less than 37% experiencing adverse events, all of which were mild in severity. These adverse events were likely related to CNTO888 administration as they were resolved following treatment discontinuation. However, CCL2 was only transiently suppressed following the first CNTO888 administration and subsequent dosing saw increased free CCL2 concentration to levels beyond pretreatment baseline values. None of the patients had an objective anti-tumor response although 4 maintained stable disease for 10.5 (patient with ovarian cancer), 5 (patient with prostate cancer), 7.2 (patient with ocular melanoma) and 15.7 (patient with neuroendocrine tumor) months [106].
In another phase 1 clinical trial (NCT01204996), the safety and pharmacokinetic/pharmacodynamics profile of CNTO888 in combination with 4 standard of care chemotherapies was assessed in 53 patients with solid tumors. These patients were divided into 4 treatment arms receiving CNTO888 with either docetaxel, gemcitabine, paclitaxel-carboplatin, or pegylated liposomal doxorubicin hydrochloride. Not unexpectedly given the combination treatments, more adverse events were observed with up to 93% of patients from each group experiencing hematological (ie neutropenia, thrombocytopenia, anemia) and non-hematological (ie nausea, stomatitis, fatigue) complications. Addition of CNTO888 to the treatment regime did not affect the pharmacokinetics of standard chemotherapies but the treatments also did not cause prolonged inhibition of CCL2; serum CCL2 initially declined but gradually increased. Despite the lack of CCL2 inhibition, 3 patients showed a 30% decrease in urinary cross-linked N-telopeptide of type I collagen (uNTX) values, used as a biomarker of bone resorption. Furthermore, 18 patients from all treatment arms showed stable disease ranging from 2 to 10.8 months and one patient achieved partial response. However, these objective responses were attributed to effects of the standard chemotherapies [107].
Follow-up pharmacodynamic analysis from these trials indicated that, contrary to its high binding affinity reported from in vitro studies, CNTO888 disassociation constant was much higher (2.4 nM) in patients, suggesting that it bound free CCL2 with lesser affinity, and as such, may not be as efficient in inhibiting CCL2 in humans [106].
CCL2 inhibition (CNTO888) in metastatic resistant prostate cancer
Administration of CNTO888 has previously demonstrated remarkable anti-tumor effects, both alone and in combination with docetaxel, in murine models of metastatic prostate cancer [66]. Based on these encouraging results, a phase 2 study (NTC00992186) was performed to assess the efficacy of CNTO888 in 46 patients with metastatic resistant prostate cancer; these patients had previously received docetaxel-based chemotherapy and the majority had additional radiotherapy, hormonal therapy or surgery. CNTO888 (15 mg/kg) was administered intravenously every two weeks for the duration of the study. CNTO888 treatment did not result in any complete or partial responses, and only 34% of patients maintained stable disease for more than 3 months. Furthermore, none of the patients showed more than 50% reduction in PSA value. Although CCL2 levels were initially suppressed following treatment, CCL2 concentration quickly increased, surpassing pre-treatment serum concentration, and elevated levels were sustained even after subsequent CNTO888 administration [108].
Taken together, CNTO888 was generally well tolerated with few mild-to-moderate adverse events. Nevertheless, these trials have consistently indicated that CNTO888 is ineffective at sustaining CCL2 blockade in humans. It is important to point out, however, that CCL2 inhibition using the CNTO888 antibody has shown robust anti-tumor responses in several pre-clinical cancer models. Indeed, pre-clinical results were so promising that a phase 2 study was initially planned in ovarian cancer [109] but given the current findings, further development of CNTO888 in oncology needs to be reviewed.
CCR2 inhibition (MLN1202) in bone metastases
A different approach to CCL2/CCR2 interference was blocking the cognate CCR2 receptor. A humanized IgG1 antibody developed by Millennium Pharmaceuticals, MLN1202, was shown to be specific for the CCR2 receptor, and has been previously tested in several inflammation-related diseases including rheumatoid arthritis, multiple sclerosis, chronic obstructive pulmonary disorder and atherosclerosis with varying results; Phase 2 clinical trials of MLN1202 in multiple sclerosis [110] and in atherosclerosis [111] had moderate success but its use in rheumatoid arthritis showed no clinical improvement [112, 113].
A phase 2 clinical trial (NCT01015560) was conducted to establish the efficacy of MLN1202 in 44 patients with bone metastases resulting from unspecified solid tumors. MLN1202 (8mg/kg) was administered intravenously as a monotherapy on days 1, 15 and 29, and several primary and secondary measures were assessed including uNTX, anti-tumor and immune responses, the latter two which were defined by tumor cell proliferation and myeloid cell infiltration. Similar to CCL2 inhibition by CNTO888, administration of MLN1202 was well tolerated in patients with < 8% reporting serious adverse events although only 14 patients (~32%) had a considerable reduction in uNTX values after 43 days of treatment. Effects of MLN1202 on anti-tumor and immune responses were not disclosed [114].
Conclusions
CCL2/CCR2 signaling was demonstrated to play crucial roles in the metastatic process, stimulating cancer cell proliferation, invasion and migration, and promoting metastatic outgrowth and colonization. Experimental evidence from various pre-clinical cancer models have been encouraging and indicate that CCL2 and CCR2 are attractive targets for intervention of metastatic diseases. However, thus far, therapeutics aimed at interference of this chemokine receptor pair have had disappointing results in the clinic.
In depth analysis of the data from clinical trials could shed light into how we can tailor CCL2 or CCR2 blockade to be more effective. Pharmacodynamic studies of the CCL2 inhibitory antibody CNTO888 showed that CCL2 expression was augmented in response to the initial CCL2 inhibition, likely due to a homeostatic feedback mechanism. Indeed, the fact that targeting this pathway seems to have no considerable or long term benefits either in solid tumors or metastatic cancers suggest that other compensatory mechanisms may be involved; whether this is due to increased CCL2, upregulation of CCR2, or contribution by other functionally analogous chemokines and/or chemokine receptors is yet to be delineated. Moreover, it is also important to establish whether inhibition of CCL2 had distinct effects compared to CCR2 inhibition, and if interference of both can lead to better anti-tumor responses in clinical trials.
Overall, this brings into question the translationability of previous pre-clinical data, and further highlights our incomplete understanding of the CCL2-CCR2 signaling network, which is undeniably far more complex than what we initially surmised. Interestingly, a recent study indicated that targeting CCL2-CCR2 signaling may provoke unexpected adverse effects. Cessation or interruption of CCL2 inhibition was shown to increase metastases and accelerate death in four mouse models of metastatic breast cancer. This effect was attributed to augmented cancer cell mobilization from the tumor, increased blood vessel formation and enhanced proliferation of metastatic cells [115]. This is in keeping with the observation in a prostate cancer metastasis model that CCL2 inhibition must be sustained to maintain tumor regression [66], and further questions the effects of CNTO 888 or MLN1202 treatment withdrawal in clinical trials. Taking all these findings into account, it is clear that we may still have a long way to go before we can successfully design chemokine or chemokine receptor-targeted therapies that will profoundly alleviate metastatic cancers.
Acknowledgments
The authors acknowledge CRUK/EPSRC Oxford Cancer Imaging Centre and CRUK Oxford Centre for providing funding support.
Conflicts of interest
The authors declare no conflicts of interest.
Grant support
CRUK/EPSRC Oxford Cancer Imaging Centre (RJM) and CRUK Oxford Centre grant number C38302/A1731 (SYL and RJM)
References
1. Statistics OfN. Cancer survival in England: patients diagnosed 2008-2012, followed up to 2013. In. London: ONS; 2013.
2. Zlotnik A, Yoshie O, Nomiyama H. The chemokine and chemokine receptor superfamilies and their molecular evolution. Genome Biol 2006; 7: 243.
3. Rollins BJ. Chemokines. Blood 1997; 90: 909-28.
4. Zlotnik A. Chemokines and cancer. Int J Cancer 2006; 119: 2026-9.
5. Balkwill F. Cancer and the chemokine network. Nat Rev Cancer 2004; 4: 540-50.
6. Balkwill FR. The chemokine system and cancer. J Pathol 2012; 226: 148-57.
7. Lazennec G, Richmond A. Chemokines and chemokine receptors: new insights into cancer-related inflammation. Trends Mol Med 2010; 16: 133-44.
8. Deshmane SL, Kremlev S, Amini S, Sawaya BE. Monocyte chemoattractant protein-1 (MCP-1): an overview. J Interferon Cytokine Res 2009; 29: 313-26.
9. Bachelerie F, Ben-Baruch A, Burkhardt AM, Combadiere C, Farber JM, Graham GJ, Horuk R, Sparre-Ulrich AH, Locati M, Luster AD, Mantovani A, Matsushima K, Murphy PM, et al. International Union of Pharmacology. LXXXIX. Update on the Extended Family of Chemokine Receptors and Introducing a New Nomenclature for Atypical Chemokine Receptors. Pharmacological Reviews 2014; 66: 1-79.
10. Charo IF, Myers SJ, Herman A, Franci C, Connolly AJ, Coughlin SR. Molecular cloning and functional expression of two monocyte chemoattractant protein 1 receptors reveals alternative splicing of the carboxyl-terminal tails. Proc Natl Acad Sci U S A 1994; 91: 2752-6.
11. Van Coillie E, Van Damme J, Opdenakker G. The MCP/eotaxin subfamily of CC chemokines. Cytokine Growth Factor Rev 1999; 10: 61-86.
12. Wong LM, Myers SJ, Tsou CL, Gosling J, Arai H, Charo IF. Organization and differential expression of the human monocyte chemoattractant protein 1 receptor gene. Evidence for the role of the carboxyl-terminal tail in receptor trafficking. J Biol Chem 1997; 272: 1038-45.
13. Bartoli C, Civatte M, Pellissier JF, Figarella-Branger D. CCR2A and CCR2B, the two isoforms of the monocyte chemoattractant protein-1 receptor are up-regulated and expressed by different cell subsets in idiopathic inflammatory myopathies. Acta Neuropathol 2001; 102: 385-92.
14. Sanders SK, Crean SM, Boxer PA, Kellner D, LaRosa GJ, Hunt SW. Functional differences between monocyte chemotactic protein-1 receptor A and monocyte chemotactic protein-1 receptor B expressed in a Jurkat T cell. Journal of Immunology 2000; 165: 4877-83.
15. Zhang T, Somasundaram R, Berencsi K, Caputo L, Gimotty P, Rani P, Guerry D, Swoboda R, Herlyn D. Migration of cytotoxic T lymphocytes toward melanoma cells in three-dimensional organotypic culture is dependent on CCL2 and CCR4. Eur J Immunol 2006; 36: 457-67.
16. Nishikawa H, Sakaguchi S. Regulatory T cells in tumor immunity. Int J Cancer 2010; 127: 759-67.
17. Bachelerie F, Graham GJ, Locati M, Mantovani A, Murphy PM, Nibbs R, Rot A, Sozzani S, Thelen M. New nomenclature for atypical chemokine receptors. Nature Immunology 2014; 15: 207-08.
18. Lee KM, Nibbs RJ, Graham GJ. D6: the ‘crowd controller’ at the immune gateway. Trends Immunol 2013; 34: 7-12.
19. Rossi D, Zlotnik A. The biology of chemokines and their receptors. Annu Rev Immunol 2000; 18: 217-42.
20. Shi C, Pamer EG. Monocyte recruitment during infection and inflammation. Nat Rev Immunol 2011; 11: 762-74.
21. Torres S, Bartolome RA, Mendes M, Barderas R, Fernandez-Acenero MJ, Pelaez-Garcia A, Pena C, Lopez-Lucendo M, Villar-Vazquez R, de Herreros AG, Bonilla F, Casal JI. Proteome profiling of cancer-associated fibroblasts identifies novel proinflammatory signatures and prognostic markers for colorectal cancer. Clin Cancer Res 2013; 19: 6006-19.
22. Islam SA, Chang DS, Colvin RA, Byrne MH, McCully ML, Moser B, Lira SA, Charo IF, Luster AD. Mouse CCL8, a CCR8 agonist, promotes atopic dermatitis by recruiting IL-5+ T(H)2 cells. Nat Immunol 2011; 12: 167-77.
23. Lu B, Rutledge BJ, Gu L, Fiorillo J, Lukacs NW, Kunkel SL, North R, Gerard C, Rollins BJ. Abnormalities in monocyte recruitment and cytokine expression in monocyte chemoattractant protein 1-deficient mice. J Exp Med 1998; 187: 601-8.
24. Boring L, Gosling J, Chensue SW, Kunkel SL, Farese RV, Jr., Broxmeyer HE, Charo IF. Impaired monocyte migration and reduced type 1 (Th1) cytokine responses in C-C chemokine receptor 2 knockout mice. J Clin Invest 1997; 100: 2552-61.
25. Chaffer CL, Weinberg RA. A perspective on cancer cell metastasis. Science 2011; 331: 1559-64.
26. Pantel K, Brakenhoff RH. Dissecting the metastatic cascade. Nat Rev Cancer 2004; 4: 448-56.
27. Gupta GP, Massague J. Cancer metastasis: building a framework. Cell 2006; 127: 679-95.
28. Chiu HY, Sun KH, Chen SY, Wang HH, Lee MY, Tsou YC, Jwo SC, Sun GH, Tang SJ. Autocrine CCL2 promotes cell migration and invasion via PKC activation and tyrosine phosphorylation of paxillin in bladder cancer cells. Cytokine 2012; 59: 423-32.
29. Monti P, Leone BE, Marchesi F, Balzano G, Zerbi A, Scaltrini F, Pasquali C, Calori G, Pessi F, Sperti C, Di Carlo V, Allavena P, Piemonti L. The CC chemokine MCP-1/CCL2 in pancreatic cancer progression: regulation of expression and potential mechanisms of antimalignant activity. Cancer Res 2003; 63: 7451-61.
30. Dagouassat M, Suffee N, Hlawaty H, Haddad O, Charni F, Laguillier C, Vassy R, Martin L, Schischmanoff PO, Gattegno L, Oudar O, Sutton A, Charnaux N. Monocyte chemoattractant protein-1 (MCP-1)/CCL2 secreted by hepatic myofibroblasts promotes migration and invasion of human hepatoma cells. Int J Cancer 2010; 126: 1095-108.
31. Tang CH, Tsai CC. CCL2 increases MMP-9 expression and cell motility in human chondrosarcoma cells via the Ras/Raf/MEK/ERK/NF-kappaB signaling pathway. Biochem Pharmacol 2012; 83: 335-44.
32. Wyckoff JB, Wang Y, Lin EY, Li JF, Goswami S, Stanley ER, Segall JE, Pollard JW, Condeelis J. Direct visualization of macrophage-assisted tumor cell intravasation in mammary tumors. Cancer Res 2007; 67: 2649-56.
33. Qian B, Deng Y, Im JH, Muschel RJ, Zou Y, Li J, Lang RA, Pollard JW. A distinct macrophage population mediates metastatic breast cancer cell extravasation, establishment and growth. PLoS One 2009; 4: e6562.
34. van Golen KL, Ying C, Sequeira L, Dubyk CW, Reisenberger T, Chinnaiyan AM, Pienta KJ, Loberg RD. CCL2 induces prostate cancer transendothelial cell migration via activation of the small GTPase Rac. J Cell Biochem 2008; 104: 1587-97.
35. Holmgren L, O’Reilly MS, Folkman J. Dormancy of micrometastases: balanced proliferation and apoptosis in the presence of angiogenesis suppression. Nat Med 1995; 1: 149-53.
36. Aguirre-Ghiso JA. Models, mechanisms and clinical evidence for cancer dormancy. Nat Rev Cancer 2007; 7: 834-46.
37. Schmidt-Kittler O, Ragg T, Daskalakis A, Granzow M, Ahr A, Blankenstein TJ, Kaufmann M, Diebold J, Arnholdt H, Muller P, Bischoff J, Harich D, Schlimok G, et al. From latent disseminated cells to overt metastasis: genetic analysis of systemic breast cancer progression. Proc Natl Acad Sci U S A 2003; 100: 7737-42.
38. Almog N. Molecular mechanisms underlying tumor dormancy. Cancer Letters 2010; 294: 139-46.
39. Giancotti FG. Mechanisms governing metastatic dormancy and reactivation. Cell 2013; 155: 750-64.
40. Quail DF, Joyce JA. Microenvironmental regulation of tumor progression and metastasis. Nat Med 2013; 19: 1423-37.
41. Low-Marchelli JM, Ardi VC, Vizcarra EA, van Rooijen N, Quigley JP, Yang J. Twist1 Induces CCL2 and Recruits Macrophages to Promote Angiogenesis. Cancer Research 2013; 73: 662-71.
42. Lin EY, Pollard JW. Tumor-associated macrophages press the angiogenic switch in breast cancer. Cancer Res 2007; 67: 5064-6.
43. Huang B, Lei Z, Zhao J, Gong W, Liu J, Chen Z, Liu Y, Li D, Yuan Y, Zhang GM, Feng ZH. CCL2/CCR2 pathway mediates recruitment of myeloid suppressor cells to cancers. Cancer Lett 2007; 252: 86-92.
44. Shields JD, Fleury ME, Yong C, Tomei AA, Randolph GJ, Swartz MA. Autologous chemotaxis as a mechanism of tumor cell homing to lymphatics via interstitial flow and autocrine CCR7 signaling. Cancer Cell 2007; 11: 526-38.
45. Geminder H, Sagi-Assif O, Goldberg L, Meshel T, Rechavi G, Witz IP, Ben-Baruch A. A possible role for CXCR4 and its ligand, the CXC chemokine stromal cell-derived factor-1, in the development of bone marrow metastases in neuroblastoma. J Immunol 2001; 167: 4747-57.
46. Witz IP. Tumor-microenvironment interactions: dangerous liaisons. Adv Cancer Res 2008; 100: 203-29.
47. Roca H, Varsos Z, Pienta KJ. CCL2 protects prostate cancer PC3 cells from autophagic death via phosphatidylinositol 3-kinase/AKT-dependent survivin up-regulation. J Biol Chem 2008; 283: 25057-73.
48. Zhang J, Patel L, Pienta KJ. CC chemokine ligand 2 (CCL2) promotes prostate cancer tumorigenesis and metastasis. Cytokine Growth Factor Rev 2010; 21: 41-8.
49. Salcedo R, Ponce ML, Young HA, Wasserman K, Ward JM, Kleinman HK, Oppenheim JJ, Murphy WJ. Human endothelial cells express CCR2 and respond to MCP-1: direct role of MCP-1 in angiogenesis and tumor progression. Blood 2000; 96: 34-40.
50. Stamatovic SM, Keep RF, Mostarica-Stojkovic M, Andjelkovic AV. CCL2 regulates angiogenesis via activation of Ets-1 transcription factor. J Immunol 2006; 177: 2651-61.
51. Weber KS, Nelson PJ, Grone HJ, Weber C. Expression of CCR2 by endothelial cells : implications for MCP-1 mediated wound injury repair and In vivo inflammatory activation of endothelium. Arterioscler Thromb Vasc Biol 1999; 19: 2085-93.
52. Goede V, Brogelli L, Ziche M, Augustin HG. Induction of inflammatory angiogenesis by monocyte chemoattractant protein-1. Int J Cancer 1999; 82: 765-70.
53. Qian BZ, Li J, Zhang H, Kitamura T, Zhang J, Campion LR, Kaiser EA, Snyder LA, Pollard JW. CCL2 recruits inflammatory monocytes to facilitate breast-tumour metastasis. Nature 2011; 475: 222-5.
54. Mizutani K, Sud S, McGregor NA, Martinovski G, Rice BT, Craig MJ, Varsos ZS, Roca H, Pienta KJ. The chemokine CCL2 increases prostate tumor growth and bone metastasis through macrophage and osteoclast recruitment. Neoplasia 2009; 11: 1235-42.
55. Mantovani A, Sozzani S, Locati M, Allavena P, Sica A. Macrophage polarization: tumor-associated macrophages as a paradigm for polarized M2 mononuclear phagocytes. Trends Immunol 2002; 23: 549-55.
56. Roca H, Varsos ZS, Sud S, Craig MJ, Ying C, Pienta KJ. CCL2 and interleukin-6 promote survival of human CD11b+ peripheral blood mononuclear cells and induce M2-type macrophage polarization. J Biol Chem 2009; 284: 34342-54.
57. Luther SA, Cyster JG. Chemokines as regulators of T cell differentiation. Nat Immunol 2001; 2: 102-7.
58. Gu L, Tseng S, Horner RM, Tam C, Loda M, Rollins BJ. Control of TH2 polarization by the chemokine monocyte chemoattractant protein-1. Nature 2000; 404: 407-11.
59. Karpus WJ, Lukacs NW, Kennedy KJ, Smith WS, Hurst SD, Barrett TA. Differential CC chemokine-induced enhancement of T helper cell cytokine production. J Immunol 1997; 158: 4129-36.
60. Matsukawa A, Lukacs NW, Standiford TJ, Chensue SW, Kunkel SL. Adenoviral-mediated overexpression of monocyte chemoattractant protein-1 differentially alters the development of Th1 and Th2 type responses in vivo. J Immunol 2000; 164: 1699-704.
61. Bonecchi R, Bianchi G, Bordignon PP, D’Ambrosio D, Lang R, Borsatti A, Sozzani S, Allavena P, Gray PA, Mantovani A, Sinigaglia F. Differential expression of chemokine receptors and chemotactic responsiveness of type 1 T helper cells (Th1s) and Th2s. J Exp Med 1998; 187: 129-34.
62. Loberg RD, Day LL, Harwood J, Ying C, St John LN, Giles R, Neeley CK, Pienta KJ. CCL2 is a potent regulator of prostate cancer cell migration and proliferation. Neoplasia 2006; 8: 578-86.
63. Lin TH, Liu HH, Tsai TH, Chen CC, Hsieh TF, Lee SS, Lee YJ, Chen WC, Tang CH. CCL2 increases alphavbeta3 integrin expression and subsequently promotes prostate cancer migration. Biochim Biophys Acta 2013; 1830: 4917-27.
64. Loberg RD, Ying C, Craig M, Yan L, Snyder LA, Pienta KJ. CCL2 as an important mediator of prostate cancer growth in vivo through the regulation of macrophage infiltration. Neoplasia 2007; 9: 556-62.
65. Lim SY, Raftery MJ, Goyette J, Hsu K, Geczy CL. Oxidative modifications of S100 proteins: functional regulation by redox. J Leukoc Biol 2009; 86: 577-87.
66. Loberg RD, Ying C, Craig M, Day LL, Sargent E, Neeley C, Wojno K, Snyder LA, Yan L, Pienta KJ. Targeting CCL2 with systemic delivery of neutralizing antibodies induces prostate cancer tumor regression in vivo. Cancer Res 2007; 67: 9417-24.
67. Lu Y, Xiao G, Galson DL, Nishio Y, Mizokami A, Keller ET, Yao Z, Zhang J. PTHrP-induced MCP-1 production by human bone marrow endothelial cells and osteoblasts promotes osteoclast differentiation and prostate cancer cell proliferation and invasion in vitro. Int J Cancer 2007; 121: 724-33.
68. Kirk PS, Koreckij T, Nguyen HM, Brown LG, Snyder LA, Vessella RL, Corey E. Inhibition of CCL2 Signaling in Combination with Docetaxel Treatment Has Profound Inhibitory Effects on Prostate Cancer Growth in Bone. International Journal of Molecular Sciences 2013; 14: 10483-96.
69. Yoshimura T, Howard OMZ, Ito T, Kuwabara M, Matsukawa A, Chen KQ, Liu Y, Liu MY, Oppenheim JJ, Wang JM. Monocyte Chemoattractant Protein-1/CCL2 Produced by Stromal Cells Promotes Lung Metastasis of 4T1 Murine Breast Cancer Cells. PLoS One 2013; 8.
70. Rego SL, Swamydas M, Kidiyoor A, Helms R, De Piante A, Lance AL, Mukherjee P, Dreau D. Soluble Tumor Necrosis Factor Receptors Shed by Breast Tumor Cells Inhibit Macrophage Chemotaxis. Journal of Interferon and Cytokine Research 2013; 33: 672-81.
71. Youngs SJ, Ali SA, Taub DD, Rees RC. Chemokines induce migrational responses in human breast carcinoma cell lines. International Journal of Cancer 1997; 71: 257-66.
72. Fang WB, Jokar I, Zou A, Lambert D, Dendukuri P, Cheng N. CCL2/CCR2 Chemokine Signaling Coordinates Survival and Motility of Breast Cancer Cells through Smad3 Protein- and p42/44 Mitogen-activated Protein Kinase (MAPK)-dependent Mechanisms. Journal of Biological Chemistry 2012; 287: 36593-608.
73. Saji H, Koike M, Yamori T, Saji S, Seiki M, Matsushima K, Toi M. Significant correlation of monocyte chemoattractant protein-1 expression with neovascularization and progression of breast carcinoma. Cancer 2001; 92: 1085-91.
74. Kitamura T, Qian BZ, Soong D, Cassetta L, Noy R, Sugano G, Kato Y, Li J, Pollard JW. CCL2-induced chemokine cascade promotes breast cancer metastasis by enhancing retention of metastasis-associated macrophages. J Exp Med 2015; 212: 1043-59.
75. Silzle T, Kreutz M, Dobler MA, Brockhoff G, Knuechel R, Kunz-Schughart LA. Tumor-associated fibroblasts recruit blood monocytes into tumor tissue. European Journal of Immunology 2003; 33: 1311-20.
76. Fujimoto H, Sangai T, Ishii G, Ikehara A, Nagashima T, Miyazaki M, Ochiai A. Stromal MCP-1 in mammary tumors induces tumor-associated macrophage infiltration and contributes to tumor progression. International Journal of Cancer 2009; 125: 1276-84.
77. Campion L, Shi F, Kaiser E, Johns L, Egenolf D, Ferrante C, McCabe F, Millar H, Rafferty P, Rudnick K, Bugelski P, Snyder L. Neutralizing CCL2 Inhibits Breast Tumor Growth Via Impact on the Tumor/Stroma Microenvironment. Cancer Research 2009; 69: 855s-55s.
78. Arendt LM, McCready J, Keller PJ, Baker DD, Naber SP, Seewaldt V, Kuperwasser C. Obesity Promotes Breast Cancer by CCL2-Mediated Macrophage Recruitment and Angiogenesis. Cancer Research 2013; 73: 6080-93.
79. Tsuyada A, Wang SE. Fibroblast-Derived CCL2 Induces Cancer Stem Cells-Response. Cancer Research 2013; 73: 1032-33.
80. Chun E, Lavoie S, Michaud M, Gallini CA, Kim J, Soucy G, Odze R, Glickman JN, Garrett WS. CCL2 Promotes Colorectal Carcinogenesis by Enhancing Polymorphonuclear Myeloid-Derived Suppressor Cell Population and Function. Cell Rep 2015; 12: 244-57.
81. Zhao L, Lim SY, Gordon-Weeks AN, Tapmeier TT, Im JH, Cao YH, Beech J, Allen D, Smart S, Muschel RJ. Recruitment of a Myeloid Cell Subset (CD11b/Gr1(mid)) via CCL2/CCR2 Promotes the Development of Colorectal Cancer Liver Metastasis. Hepatology 2013; 57: 829-39.
82. Wolf MJ, Hoos A, Bauer J, Boettcher S, Knust M, Weber A, Simonavicius N, Schneider C, Lang M, Sturzl M, Croner RS, Konrad A, Manz MG, et al. Endothelial CCR2 Signaling Induced by Colon Carcinoma Cells Enables Extravasation via the JAK2-Stat5 and p38MAPK Pathway. Cancer Cell 2012; 22: 91-105.
83. Snyder LA, Kesavan P, Kaiser E, Rudnick KA, McCabe F, Millar H, Nakada MT, Yan L. Neutralization of CCL2 inhibits tumor angiogenesis and pancreatic tumor growth. Molecular Cancer Therapeutics 2007; 6: 3357s-57s.
84. Sanford DE, Belt BA, Panni RZ, Mayer A, Deshpande AD, Carpenter D, Mitchem JB, Plambeck-Suess SM, Worley LA, Goetz BD, Wang-Gillam A, Eberlein TJ, Denardo DG, et al. Inflammatory Monocyte Mobilization Decreases Patient Survival in Pancreatic Cancer: A Role for Targeting the CCL2/CCR2 Axis. Clinical Cancer Research 2013; 19: 3404-15.
85. Zhu X, Fujita M, Snyder LA, Okada H. Systemic delivery of neutralizing antibody targeting CCL2 for glioma therapy. J Neurooncol 2011; 104: 83-92.
86. Kuroda T, Kitadai Y, Tanaka S, Yang X, Mukaida N, Yoshihara M, Chayama K. Monocyte chemoattractant protein-1 transfection induces angiogenesis and tumorigenesis of gastric carcinoma in nude mice via macrophage recruitment. Clin Cancer Res 2005; 11: 7629-36.
87. Said N, Sanchez-Carbayo M, Smith SC, Theodorescu D. RhoGDI2 suppresses lung metastasis in mice by reducing tumor versican expression and macrophage infiltration. J Clin Invest 2012; 122: 1503-18.
88. Ohanna M, Giuliano S, Bonet C, Imbert V, Hofman V, Zangari J, Bille K, Robert C, Bressac-de Paillerets B, Hofman P, Rocchi S, Peyron JF, Lacour JP, et al. Senescent cells develop a PARP-1 and nuclear factor-{kappa}B-associated secretome (PNAS). Genes Dev 2011; 25: 1245-61.
89. Knight DA, Ngiow SF, Li M, Parmenter T, Mok S, Cass A, Haynes NM, Kinross K, Yagita H, Koya RC, Graeber TG, Ribas A, McArthur GA, et al. Host immunity contributes to the anti-melanoma activity of BRAF inhibitors. J Clin Invest 2013; 123: 1371-81.
90. van Deventer HW, Palmieri DA, Wu QP, McCook EC, Serody JS. Circulating Fibrocytes Prepare the Lung for Cancer Metastasis by Recruiting Ly-6C(+) Monocytes Via CCL2. Journal of Immunology 2013; 190: 4861-67.
91. Stathopoulos GT, Psallidas I, Moustaki A, Moschos C, Kollintza A, Karabela S, Porfyridis I, Vassiliou S, Karatza M, Zhou Z, Joo M, Blackwell TS, Roussos C, et al. A central role for tumor-derived monocyte chemoattractant protein-1 in malignant pleural effusion. J Natl Cancer Inst 2008; 100: 1464-76.
92. Gazzaniga S, Bravo AI, Guglielmotti A, van Rooijen N, Maschi F, Vecchi A, Mantovani A, Mordoh J, Wainstok R. Targeting tumor-associated macrophages and inhibition of MCP-1 reduce angiogenesis and tumor growth in a human melanoma xenograft. J Invest Dermatol 2007; 127: 2031-41.
93. Hu K, Xiong J, Ji K, Sun H, Wang J, Liu H. Recombined CC chemokine ligand 2 into B16 cells induces production of Th2-dominant [correction of dominanted] cytokines and inhibits melanoma metastasis. Immunology Letters 2007; 113: 19-28.
94. Craig MJ, Loberg RD. CCL2 (Monocyte Chemoattractant Protein-1) in cancer bone metastases. Cancer and Metastasis Reviews 2006; 25: 611-19.
95. Gorlov IP, Sircar K, Zhao H, Maity SN, Navone NM, Gorlova OY, Troncoso P, Pettaway CA, Byun JY, Logothetis CJ. Prioritizing genes associated with prostate cancer development. BMC Cancer 2010; 10: 599.
96. Tsaur I, Noack A, Makarevic J, Oppermann E, Waaga-Gasser AM, Gasser M, Borgmann H, Huesch T, Gust KM, Reiter M, Schilling D, Bartsch G, Haferkamp A, et al. CCL2 Chemokine as a Potential Biomarker for Prostate Cancer: A Pilot Study. Cancer Res Treat 2015; 47: 306-12.
97. Lu Y, Cai Z, Galson DL, Xiao G, Liu Y, George DE, Melhem MF, Yao Z, Zhang J. Monocyte chemotactic protein-1 (MCP-1) acts as a paracrine and autocrine factor for prostate cancer growth and invasion. Prostate 2006; 66: 1311-8.
98. Hefler L, Tempfer C, Heinze G, Mayerhofer K, Breitenecker G, Leodolter S, Reinthaller A, Kainz C. Monocyte chemoattractant protein-1 serum levels in ovarian cancer patients. Br J Cancer 1999; 81: 855-9.
99. Soria G, Ben-Baruch A. The inflammatory chemokines CCL2 and CCL5 in breast cancer. Cancer Letters 2008; 267: 271-85.
100. Ueno T, Toi M, Saji H, Muta M, Bando H, Kuroi K, Koike M, Inadera H, Matsushima K. Significance of macrophage chemoattractant protein-1 in macrophage recruitment, angiogenesis, and survival in human breast cancer. Clinical Cancer Research 2000; 6: 3282-89.
101. Tonouchi H, Miki C, Tanaka K, Kusunoki M. Profile of monocyte chemoattractant protein-1 circulating levels in gastric cancer patients. Scand J Gastroenterol 2002; 37: 830-3.
102. Futagami S, Tatsuguchi A, Hiratsuka T, Shindo T, Horie A, Hamamoto T, Ueki N, Kusunoki M, Miyake K, Gudis K, Tsukui T, Sakamoto C. Monocyte chemoattractant protein 1 and CD40 ligation have a synergistic effect on vascular endothelial growth factor production through cyclooxygenase 2 upregulation in gastric cancer. J Gastroenterol 2008; 43: 216-24.
103. Tao LL, Shi SJ, Chen LB, Huang GC. Expression of monocyte chemotactic protein-1/CCL2 in gastric cancer and its relationship with tumor hypoxia. World J Gastroenterol 2014; 20: 4421-7.
104. Hu H, Sun L, Guo C, Liu Q, Zhou Z, Peng L, Pan J, Yu L, Lou J, Yang Z, Zhao P, Ran Y. Tumor cell-microenvironment interaction models coupled with clinical validation reveal CCL2 and SNCG as two predictors of colorectal cancer hepatic metastasis. Clin Cancer Res 2009; 15: 5485-93.
105. Obmolova G, Teplyakov A, Malia TJ, Grygiel TL, Sweet R, Snyder LA, Gilliland GL. Structural basis for high selectivity of anti-CCL2 neutralizing antibody CNTO 888. Mol Immunol 2012; 51: 227-33.
106. Sandhu SK, Papadopoulos K, Fong PC, Patnaik A, Messiou C, Olmos D, Wang G, Tromp BJ, Puchalski TA, Balkwill F, Berns B, Seetharam S, de Bono JS, et al. A first-in-human, first-in-class, phase I study of carlumab (CNTO 888), a human monoclonal antibody against CC-chemokine ligand 2 in patients with solid tumors. Cancer Chemotherapy and Pharmacology 2013; 71: 1041-50.
107. Brana I, Calles A, LoRusso PM, Yee LK, Puchalski TA, Seetharam S, Zhong B, de Boer CJ, Tabernero J, Calvo E. Carlumab, an anti-C-C chemokine ligand 2 monoclonal antibody, in combination with four chemotherapy regimens for the treatment of patients with solid tumors: an open-label, multicenter phase 1b study. Target Oncol 2014.
108. Pienta KJ, Machiels JP, Schrijvers D, Alekseev B, Shkolnik M, Crabb SJ, Li S, Seetharam S, Puchalski TA, Takimoto C, Elsayed Y, Dawkins F, de Bono JS. Phase 2 study of carlumab (CNTO 888), a human monoclonal antibody against CC-chemokine ligand 2 (CCL2), in metastatic castration-resistant prostate cancer. Invest New Drugs 2013; 31: 760-8.
109. Forster M. D. AP, S. K. Sandhu, K. Papadopoulos, B. J. Tromp, C. Messiou, F. Balkwill, B. Berns, J. S. De Bono and A. W. Tolcher. Pre-final analysis of first-in-human, first-in-class, phase I clinical trial of CNTO 888, a human monoclonal antibody to the CC-chemokine ligand 2 (CCL2) in patients (pts) with advanced solid tumors. In: ASCO Annual Meeting; 2010; 2010. p. 2548.
110. Sharrack B, Leach T, Jacobson E, Donaldson DD, Xu X, Hu M. Frequent MRI study of a novel CCR2 antagonist in relapsing-remitting multiple sclerosis. Annals of Neurology 2007; 62: S74-S75.
111. Gilbert J, Lekstrom-Himes J, Donaldson D, Lee Y, Hu MX, Xu J, Wyant T, Davidson M, Grp MS. Effect of CC Chemokine Receptor 2 CCR2 Blockade on Serum C-Reactive Protein in Individuals at Atherosclerotic Risk and With a Single Nucleotide Polymorphism of the Monocyte Chemoattractant Protein-1 Promoter Region. American Journal of Cardiology 2011; 107: 906-11.
112. Xia M, Sui Z. Recent developments in CCR2 antagonists. Expert Opin Ther Pat 2009; 19: 295-303.
113. Vergunst CE, Gerlag DM, Lopatinskaya L, Klareskog L, Smith MD, van den Bosch F, Dinant HJ, Lee Y, Wyant T, Jacobson EW, Baeten D, Tak PP. Modulation of CCR2 in rheumatoid arthritis - A double-blind, randomized, placebo-controlled clinical trial. Arthritis and Rheumatism 2008; 58: 1931-39.
114. Vela M, Aris M, Llorente M, Garcia-Sanz JA, Kremer L. Chemokine receptor-specific antibodies in cancer immunotherapy: achievements and challenges. Front Immunol 2015; 6: 12.
115. Bonapace L, Coissieux MM, Wyckoff J, Mertz KD, Varga Z, Junt T, Bentires-Alj M. Cessation of CCL2 inhibition accelerates breast cancer metastasis by promoting angiogenesis. Nature 2014; 515: 130-3.
116. Schrodinger, LLC. The PyMOL Molecular Graphics System, Version 1.3r1. In; 2010.