
Introduction
Major molecular pathways in colorectal carcinogenesis include the chromosomal instability (CIN) pathway, the microsatellite instability (MSI) pathway, and the CpG island methylator phenotype (CIMP) pathway [1-3]. MSI and CIMP are unique molecular phenotypes found in subsets of colorectal carcinoma (CRC) and have been revealed to be associated with characteristic clinicopathologic features and serrated precursor lesions [1-3]. MSI is caused by a defect in the DNA mismatch repair (MMR) system induced by a germline mutation or promoter methylation in one of the MMR genes (MLH1, MSH2, MSH6, and PMS2) or by a germline deletion at the 3’ end of the EPCAM gene [4-7]. MSI is characterized by vulnerability to length alterations in microsatellite DNA repeats throughout the genome, and therefore, it frequently results in mutations in many tumor-related genes [4-7]. CRC with MSI-high (MSI-H), which is considered the MSI-positive status, can occur in a hereditary (Lynch syndrome) or sporadic (CIMP-associated or serrated pathway-associated) setting [4-7]. By contrast to MSI, which is associated with genetic alterations, the CIMP is associated with epigenetic alterations in CRC and is characterized by extensive promoter CpG island hypermethylation and the subsequent transcriptional silencing of many tumor-related genes [8-10]. The majority of CRCs with CIMP-high (CIMP-H), which is considered the CIMP-positive status, are thought to develop sporadically from serrated precursor lesions [11, 12].
MSI-H has been reported to be associated with unique clinicopathologic and molecular features in CRC, including proximal colonic tumor location, mucinous histology, medullary histology, tumor-infiltrating lymphocytes, a peritumoral lymphoid reaction, a Crohn-like lymphoid reaction, the BRAF V600E mutation, resistance to 5-fluorouracil-based adjuvant chemotherapy, and a favorable prognosis [6]. The characteristics of CIMP-H CRC have also been investigated and are similar to those of MSI-H CRC. Representative CIMP-H-associated features in CRC are old age, poor prognosis, female sex, a proximal colonic tumor location, poorly differentiated histology, signet ring cell histology, serrated histology, the BRAF V600E mutation, and MSI-H status [8]. In fact, the significant overlap between MSI-H and CIMP-H in CRC is not surprising because nearly all sporadic MSI-H CRCs are molecularly based on the promoter CpG island hypermethylation-induced silencing of the MLH1 gene, which is also found in a considerable number of CIMP-H CRCs. In this context, clear discrimination of CIMP-H-associated features from MSI-H-associated features in CRC might be difficult. Moreover, although MLH1 methylation is regarded as one of the major molecular determinants of CIMP-H CRC, detailed features associated with MLH1 methylation in CIMP-H CRC have not been fully elucidated.
Therefore, we aimed to comparatively and comprehensively investigate the differential clinicopathologic and molecular characteristics between MLH1-methylated tumors and MLH1-unmethylated tumors in CIMP-H CRC. In addition to analysis of CRC samples, independent statistical analysis of MLH1 silencing-associated features in CIMP-H CRC was conducted using The Cancer Genome Atlas (TCGA) data. Through these analyses, we expected to more precisely determine the MLH1 methylation-dependent clinicopathologic and molecular heterogeneity of CIMP-H CRC and to obtain a deeper understanding of the potential connection between CIMP-H CRC and serrated precursor pathways.
Results
Distinct clinicopathologic features according to MLH1 methylation status in CIMP-H CRCs
The differential clinicopathologic features of our study samples (65 CIMP-H CRCs) according to MLH1 promoter methylation status are summarized in Table 1. CIMP-H CRC with MLH1 methylation (n = 33) was significantly associated with old age (66 years or older; P < 0.001), a proximal colonic tumor location (82%; P = 0.026), extracellular mucinous histology (73%; P < 0.001), high-density tumor-infiltrating lymphocytes (TILs) (58%; P < 0.001), moderate to marked peritumoral lymphoid reaction (53%; P = 0.002), and active (34%; P = 0.005) and high-density (69%; P = 0.024) Crohn-like lymphoid reaction (Table 1). By contrast, CIMP-H CRC without MLH1 methylation (n = 32) demonstrated significantly higher frequencies of distant metastasis (34%; P = 0.033), vascular invasion (28%; P = 0.006), perineural invasion (47%; P = 0.006), and tumor budding (81%; P = 0.011) (Table 1).
Table 1: Differential clinicopathologic features of CIMP-H CRC according to MLH1 promoter methylation status (original study samples; n = 65)
Variables |
Case No. |
MLH1-methylated (n = 33) |
MLH1-unmethylated (n = 32) |
P-value |
|
Agea |
< 66 years |
27 |
5 (15%) |
22 (69%) |
< 0.001 |
≥ 66 years |
38 |
28 (85%) |
10 (31%) |
||
Sex |
Male |
32 |
15 (45%) |
17 (53%) |
0.536 |
Female |
33 |
18 (55%) |
15 (47%) |
||
Tumor location |
Proximal |
45 |
27 (82%) |
18 (56%) |
0.026 |
Distal |
20 |
6 (18%) |
14 (44%) |
||
Gross tumor type |
Fungating |
39 |
20 (61%) |
19 (59%) |
0.919 |
Infiltrative |
26 |
13 (39%) |
13 (41%) |
||
AJCC TNM stage |
Stage I/II |
20 |
12 (36%) |
8 (25%) |
0.321 |
Stage III/IV |
45 |
21 (64%) |
24 (75%) |
||
Depth of primary tumor invasion (pT category) |
pT1/pT2 |
2 |
1 (3%) |
1 (3%) |
1 |
pT3/pT4 |
63 |
32 (97%) |
31 (97%) |
||
Lymph node metastasis (pN category) |
Absent (pN0) |
20 |
12 (36%) |
8 (25%) |
0.321 |
Present (pN1/pN2) |
45 |
21 (64%) |
24 (75%) |
||
Distant metastasis (M category) |
Absent (M0) |
50 |
29 (88%) |
21 (66%) |
0.033 |
Present (M1) |
15 |
4 (12%) |
11 (34%) |
||
Tumor differentiation |
WD/MD |
48 |
23 (70%) |
25 (78%) |
0.44 |
PD |
17 |
10 (30%) |
7 (22%) |
||
Mucinous histology |
Absent |
34 |
9 (27%) |
25 (78%) |
< 0.001 |
Present |
31 |
24 (73%) |
7 (22%) |
||
Signet ring cell histology |
Absent |
58 |
27 (82%) |
31 (97%) |
0.105 |
Present |
7 |
6 (18%) |
1 (3%) |
||
Medullary histology |
Absent |
60 |
29 (88%) |
31 (97%) |
0.355 |
Present |
5 |
4 (12%) |
1 (3%) |
||
Serrated histology |
Absent |
50 |
26 (79%) |
24 (75%) |
0.717 |
Present |
15 |
7 (21%) |
8 (25%) |
||
Lymphatic invasion |
Absent |
22 |
12 (36%) |
10 (31%) |
0.663 |
Present |
43 |
21 (64%) |
22 (69%) |
||
Venous invasion |
Absent |
55 |
32 (97%) |
23 (72%) |
0.006 |
Present |
10 |
1 (3%) |
9 (28%) |
||
Perineural invasion |
Absent |
45 |
28 (85%) |
17 (53%) |
0.006 |
Present |
20 |
5 (15%) |
15 (47%) |
||
Tumor budding |
Absent |
22 |
16 (48%) |
6 (19%) |
0.011 |
Present |
43 |
17 (52%) |
26 (81%) |
||
Tumor-infiltrating lymphocytes (TILs)b |
Low-density (< 21 TILs/HPF) |
46 |
14 (42%) |
32 (100%) |
< 0.001 |
High-density (≥ 21 TILs/HPF) |
19 |
19 (58%) |
0 (0%) |
||
Peritumoral lymphoid reactionb |
Absent or mild |
42 |
15 (47%) |
27 (84%) |
0.002 |
Moderate to marked |
22 |
17 (53%) |
5 (16%) |
||
Crohn-like lymphoid reaction (Ueno criteria)c |
Inactive (largest LA < 1 mm) |
51 |
21 (66%) |
30 (94%) |
0.005 |
Active (largest LA ≥ 1 mm) |
13 |
11 (34%) |
2 (6%) |
||
Crohn-like lymphoid reaction (Väyrynen-Mäkinen criteria)c |
Low-density (< 0.38/mm) |
29 |
10 (31%) |
19 (59%) |
0.024 |
High-density (≥ 0.38/mm) |
35 |
22 (69%) |
13 (41%) |
Abbreviations: CIMP-H, CpG island methylator phenotype-high; AJCC, American Joint Committee on Cancer; TNM, tumor-node-metastasis; WD, well differentiated; MD, moderately differentiated; PD, poorly differentiated; HPF, high power field; LA, lymphoid aggregate.
aDichotomous age groups were classified using a cutoff value of the average age (66 years).
bDichotomous TIL groups were classified using a cutoff value of the average TIL density (21/HPF).
cPeritumoral lymphoid reaction and Crohn-like lymphoid reaction were assessed in 64 out of the 65 CIMP-H CRCs due to the inadequate quality of tissue sections for lymphoid reaction evaluations in one case.
Further detailed analyses taking into account age and tumor location were performed. The age distribution of the MLH1-unmethylated tumors (mean age ± standard deviation = 61.1 ± 10.3 years; age range = 36 to 79 years) was shifted more toward middle age compared with that of the MLH1-methylated tumors (mean age ± standard deviation = 70.2 ± 9.2 years; age range = 49 to 91 years) (P < 0.001; Figure 1A). A comparison of the bowel subsite distribution of tumor location between the MLH1-methylated and MLH1-unmethylated tumors is shown in Figure 1B. The most prevalent bowel subsite of the MLH1-methylated tumors was the ascending colon (55%), followed by the cecum (18%) (Figure 1B). By contrast, although the MLH1-unmethylated tumors were frequently located in the ascending colon (25%), the sigmoid colon and rectum were also major bowel subsites (19% and 16%, respectively) (Figure 1B).
Next, quantitatively measured raw data on TILs and Crohn-like lymphoid reactions were further analyzed. The mean value of the density of TILs in the MLH1-methylated tumors (mean density ± standard deviation = 34.4 ± 29.3 TILs/high power field) was significantly higher than that in the MLH1-unmethylated tumors (mean density ± standard deviation = 6.3 ± 5.8 TILs/high power field) (P < 0.001; Figure 1C). The mean value of the diameter of the largest lymphoid aggregate in the MLH1-methylated tumors (mean diameter ± standard deviation = 830.6 ± 295.3 μm) was also significantly higher than that in the MLH1-unmethylated tumors (mean diameter ± standard deviation = 503.5 ± 288.1 μm) (P < 0.001; Figure 1D). A comparison of lymphoid aggregate density between the MLH1-methylated (mean density ± standard deviation = 0.66 ± 0.51 lymphoid aggregates/mm) and MLH1-unmethylated tumors (mean density ± standard deviation = 0.46 ± 0.45 lymphoid aggregates/mm) is shown in Figure 1E.
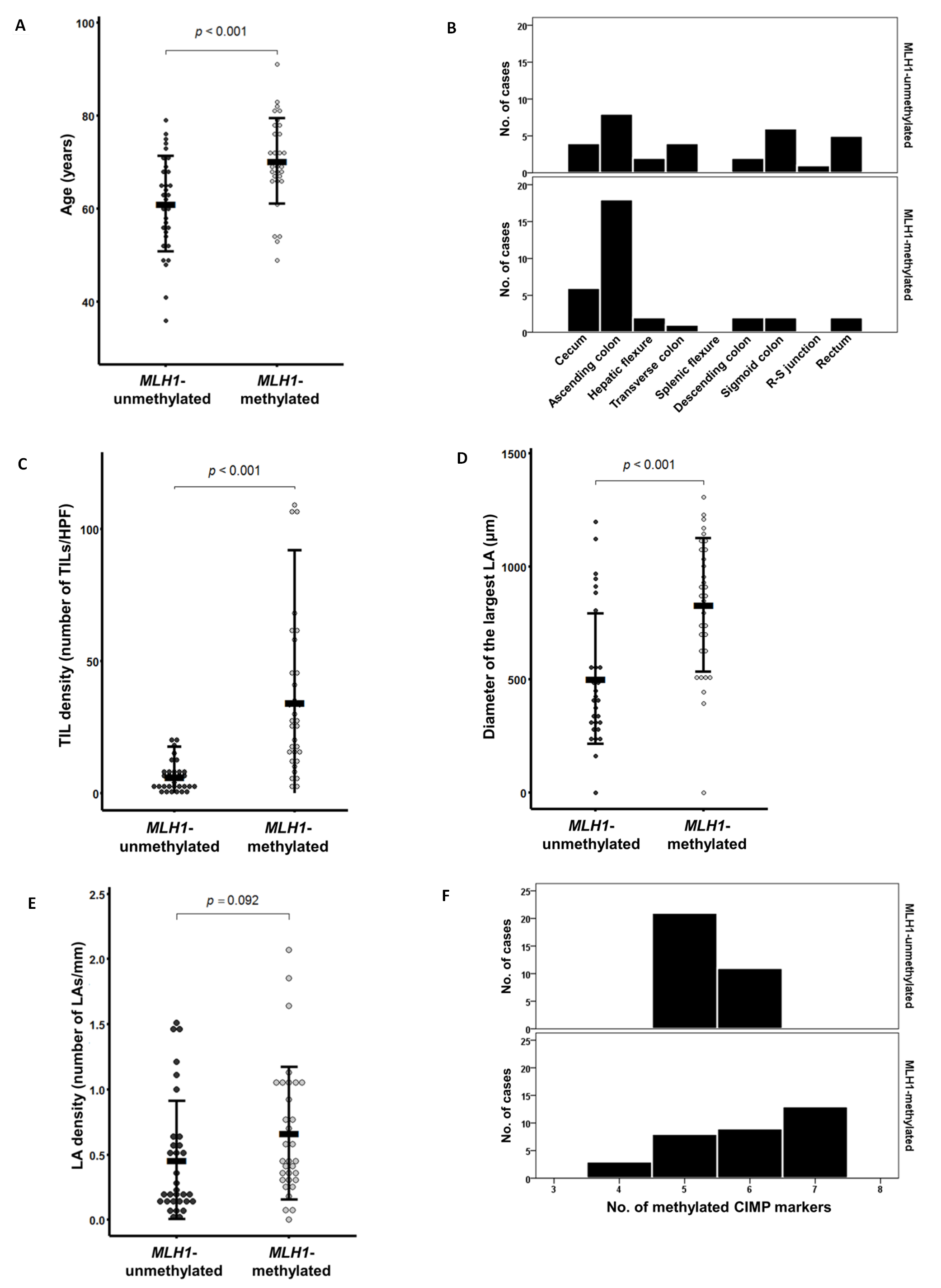
Figure 1: Comparison of quantitative clinicopathologic parameters according to MLH1 promoter methylation status in CIMP-H CRC (n = 65). A. Age at diagnosis. B. Tumor location bowel subsite. C. Tumor-infiltrating lymphocyte density (number of tumor-infiltrating lymphocytes/high power field). D. Size of the largest Crohn-like lymphoid aggregate (μm). E. Crohn-like lymphoid reaction density (number of lymphoid aggregates/mm). F. Number of methylated CIMP markers (except for MLH1). Abbreviations: TIL, tumor-infiltrating lymphocyte; HPF, high power field; LA, lymphoid aggregate.
Representative photomicrographs of the MLH1 methylation-dependent differential histopathologic features in our CIMP-H CRC samples are shown in Figure 2. The MLH1-methylated tumors frequently exhibited abundant mucin pools, intense peritumoral lymphoid reactions, and active Crohn-like lymphoid reactions, whereas the MLH1-unmethylated tumors were associated with non-mucinous-type adenocarcinoma and the absence of peritumoral lymphoid reaction and Crohn-like lymphoid reaction (Figure 2).
The MLH1-unmethylated CIMP-H CRCs showed a tendency toward worse disease-free survival compared with the MLH1-methylated CIMP-H CRCs, as shown by Kaplan-Meier survival analysis; however, this survival difference was not statistically significant (log-rank P = 0.438; Supplementary Figure S1A). In addition, prognostic significance of MLH1 methylation was not observed in 45 CIMP-H CRCs treated with 5-fluorouracil-based adjuvant chemotherapy (log-rank P = 0.157; Supplementary Figure S1B).
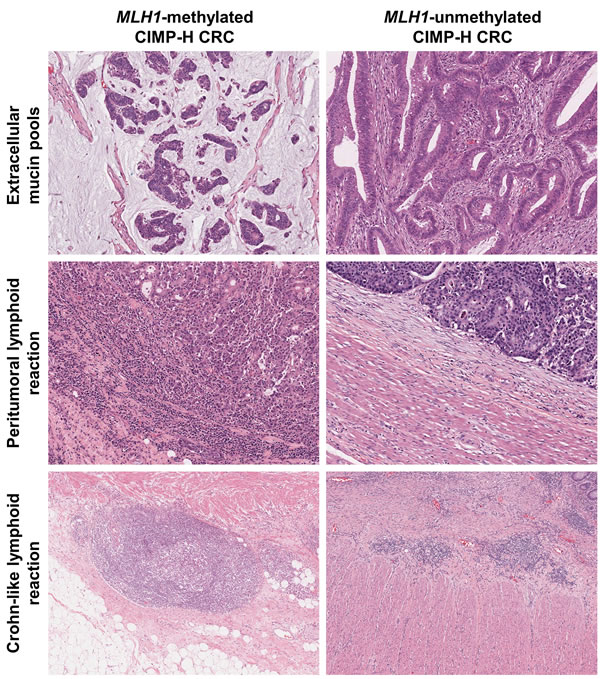
Figure 2: Differential histopathologic features according to MLH1 promoter methylation status in CIMP-H CRC. (Upper left) Typical mucinous histology in a case of MLH1-methylated CIMP-H CRC (H&E, ×100). (Upper right) Non-mucinous adenocarcinoma in a case of MLH1-unmethylated CIMP-H CRC (H&E, ×100). (Middle left) Intense peritumoral lymphoid reaction in a case of MLH1-methylated CIMP-H CRC (H&E, ×100). (Middle right) Absent peritumoral lymphoid reaction in a case of MLH1-unmethylated CIMP-H CRC (H&E, ×100). (Lower left) Active Crohn-like lymphoid reaction in a case of MLH1-methylated CIMP-H CRC. Note the large ( > 1 mm) lymphoid aggregate (H&E, ×50). (Lower right) Inactive Crohn-like lymphoid reaction in a case of MLH1-unmethylated CIMP-H CRC. Note the small ( < 1 mm) lymphoid aggregates (H&E, ×50).
Distinct molecular features according to MLH1 methylation status in CIMP-H CRCs
The differential molecular features of 65 CIMP-H CRCs according to MLH1 promoter methylation status are summarized in Table 2. As expected, most of the MLH1-methylated CIMP-H CRCs demonstrated MSI-H status (94%; P < 0.001; Table 2). The MLH1-methylated tumors were also associated with a relatively high frequency of the BRAF V600E mutation (36%; P = 0.03), whereas the MLH1-unmethylated tumors were significantly correlated with KRAS mutations (38%; P = 0.001) (Table 2).
Notably, the MLH1-methylated tumors were associated with more extensive CpG island methylation compared with the MLH1-unmethylated tumors. Twenty-two of the 33 MLH1-methylated tumors (67%) had six or seven methylated markers among the seven CIMP markers (except for MLH1) (Table 2 and Figure 1F). By contrast, 21 of the 32 MLH1-unmethylated tumors (66%) demonstrated five methylated CIMP markers (Table 2 and Figure 1F). This difference in the number of methylated CIMP markers between the MLH1-methylated and MLH1-unmethylated tumors was based primarily on differences in the methylation frequencies of two CIMP markers, RUNX3 and SOCS1. The MLH1-methylated tumors showed significantly higher frequencies of promoter methylation of RUNX3 (67%; P = 0.019) and SOCS1 (55%; P < 0.001) (Table 2).
Table 2: Differential molecular features of CIMP-H CRC according to MLH1 promoter methylation status (original study samples; n = 65)
Variables |
Case No. |
MLH1-methylated (n = 33) |
MLH1-unmethylated (n = 32) |
P-value |
|
MSI status |
MSI-L/MSS |
29 |
2 (6%) |
27 (84%) |
< 0.001 |
MSI-H |
36 |
31 (94%) |
5 (16%) |
||
No. of methylated CIMP markers (except for MLH1) |
< 6 |
32 |
11 (33%) |
21 (66%) |
0.009 |
≥ 6 |
33 |
22 (67%) |
11 (34%) |
||
CRABP1 methylation |
Methylated |
64 |
32 (97%) |
32 (100%) |
1 |
Unmethylated |
1 |
1 (3%) |
0 (0%) |
||
CDKN2A (p16) methylation |
Methylated |
62 |
31 (94%) |
31 (97%) |
1 |
Unmethylated |
3 |
2 (6%) |
1 (3%) |
||
CACNA1G methylation |
Methylated |
64 |
32 (97%) |
32 (100%) |
1 |
Unmethylated |
1 |
1 (3%) |
0 (0%) |
||
NEUROG1 methylation |
Methylated |
60 |
30 (91%) |
30 (94%) |
1 |
Unmethylated |
5 |
3 (9%) |
2 (6%) |
||
IGF2 methylation |
Methylated |
62 |
32 (97%) |
30 (94%) |
0.613 |
Unmethylated |
3 |
1 (3%) |
2 (6%) |
||
RUNX3 methylation |
Methylated |
34 |
22 (67%) |
12 (38%) |
0.019 |
Unmethylated |
31 |
11 (33%) |
20 (63%) |
||
SOCS1 methylation |
Methylated |
22 |
18 (55%) |
4 (13%) |
< 0.001 |
Unmethylated |
43 |
15 (45%) |
28 (88%) |
||
KRAS mutationa |
Mutant |
12 |
1 (3%) |
11 (38%) |
0.001 |
Wild type |
49 |
31 (97%) |
18 (62%) |
||
BRAF mutationb |
Mutant |
16 |
12 (36%) |
4 (13%) |
0.03 |
Wild type |
48 |
21 (64%) |
27 (87%) |
Abbreviations: CIMP-H, CpG island methylator phenotype-high; MSI, microsatellite instability; MSI-L, MSI-low; MSI-H, MSI-high; MSS, microsatellite stable.
aKRAS mutation analysis was performed on 61 out of the 65 CIMP-H CRCs due to a limited amount or suboptimal quality of extracted tumor DNA in four cases.
bBRAF mutation analysis was performed on 64 out of the 65 CIMP-H CRCs due to a limited amount or suboptimal quality of extracted tumor DNA in one case.
TCGA data analysis
From the TCGA dataset, a total of 36 CIMP-H CRCs were identified (24 MLH1-silenced and 12 MLH1-non-silenced tumors). Of these, the MLH1-silenced tumors were significantly associated with female sex (79%; P = 0.003), proximal colonic tumor location (100%; P = 0.031), MSI-H status (96%; P < 0.001), an exome-wide hypermutated phenotype (95%; P < 0.001), wild-type KRAS (95%; P < 0.001), and mutated BRAF (75%; P < 0.001) (Table 3). Characteristically, KRAS and APC mutations were predominantly observed in the MLH1-non-silenced tumors (each 83%; each P < 0.001; Table 3). Compared with the MLH1-non-silenced tumors, the MLH1-silenced tumors showed a tendency toward old age at diagnosis (mean age ± standard deviation = 78.3 ± 8.5 years; age range = 60 to 90 years; Figure 3A and Table 3) and mucinous histology (43%; Table 3), although these findings were not statistically significant. Two of the MLH1-non-silenced tumors were distant metastatic tumors (17%; Table 3) and three were rectal tumors (25%; Table 3 and Figure 3B), whereas the MLH1-silenced tumors showed no distant metastasis or distal colorectal localization (Table 3 and Figure 3B). Detailed analysis of the genomic mutation rate revealed that the MLH1-silenced tumors had significantly higher rates of both silent mutations (mean mutation rate ± standard deviation = 9.6 ± 4.4 mutations/Mb; range = 1.5 to 18.1 mutations/Mb; P < 0.001; Figure 3C) and non-silent mutations (mean mutation rate ± standard deviation = 26.9 ± 12.7 mutations/Mb; range = 4.5 to 54.2 mutations/Mb; P < 0.001; Figure 3D).
Table 3: Differential features of CIMP-H CRC according to MLH1 silencing status (TCGA dataset; n = 36)
Variables |
Case No. |
MLH1-silenced (n = 24) |
MLH1-non-silenced (n = 12) |
P-value |
|
Agea |
< 76 years |
15 |
8 (33%) |
7 (58%) |
0.151 |
≥ 76 years |
21 |
16 (67%) |
5 (42%) |
||
Sex |
Male |
14 |
5 (21%) |
9 (75%) |
0.003 |
Female |
22 |
19 (79%) |
3 (25%) |
||
Tumor location |
Proximal |
33 |
24 (100%) |
9 (75%) |
0.031 |
Distal |
3 |
0 (0%) |
3 (25%) |
||
AJCC TNM stageb |
Stage I/II |
23 |
16 (70%) |
7 (58%) |
0.709 |
Stage III/IV |
12 |
7 (30%) |
5 (42%) |
||
Depth of primary tumor invasion (pT category)c |
pT1/pT2 |
5 |
3 (16%) |
2 (18%) |
1 |
pT3/pT4 |
25 |
16 (84%) |
9 (82%) |
||
Lymph node metastasis (pN category) |
Absent (pN0) |
24 |
17 (71%) |
7 (58%) |
0.479 |
Present (pN1/pN2) |
12 |
7 (29%) |
5 (42%) |
||
Distant metastasis (M category) |
Absent (M0) |
34 |
24 (100%) |
10 (83%) |
0.105 |
Present (M1) |
2 |
0 (0%) |
2 (17%) |
||
Histologic subtyped |
Non-mucinous |
22 |
13 (57%) |
9 (75%) |
0.463 |
Mucinous |
13 |
10 (43%) |
3 (25%) |
||
Lymphatic invasione |
Absent |
17 |
13 (57%) |
4 (40%) |
0.465 |
Present |
16 |
10 (43%) |
6 (60%) |
||
Vascular invasionf |
Absent |
22 |
14 (78%) |
8 (89%) |
0.636 |
Present |
5 |
4 (22%) |
1 (11%) |
||
MSI status |
MSI-L/MSS |
12 |
1 (4%) |
11 (92%) |
< 0.001 |
MSI-H |
24 |
23 (96%) |
1 (8%) |
||
Mutational phenotypeg |
Non-hypermutated |
12 |
1 (5%) |
11 (92%) |
< 0.001 |
Hypermutated |
20 |
19 (95%) |
1 (8%) |
||
KRAS mutationh |
Mutant |
11 |
1 (5%) |
10 (83%) |
< 0.001 |
Wild type |
21 |
19 (95%) |
2 (17%) |
||
BRAF mutationh |
Mutant |
16 |
15 (75%) |
1 (8%) |
< 0.001 |
Wild type |
16 |
5 (25%) |
11 (92%) |
||
PIK3CA mutationh |
Mutant |
9 |
4 (20%) |
5 (42%) |
0.24 |
Wild type |
23 |
16 (80%) |
7 (58%) |
||
APC mutationh |
Mutant |
14 |
4 (20%) |
10 (83%) |
< 0.001 |
Wild type |
18 |
16 (80%) |
2 (17%) |
||
CTNNB1 mutationh |
Mutant |
1 |
0 (0%) |
1 (8%) |
0.375 |
Wild type |
31 |
20 (100%) |
11 (92%) |
||
TP53 mutationh |
Mutant |
7 |
3 (15%) |
4 (33%) |
0.379 |
Wild type |
25 |
17 (85%) |
8 (67%) |
Abbreviations: CIMP-H, CpG island methylator phenotype-high; AJCC, American Joint Committee on Cancer; TNM, tumor-node-metastasis; MSI, microsatellite instability; MSI-L, MSI-low; MSI-H, MSI-high; MSS, microsatellite stable.
aDichotomous age groups were classified using a cutoff value of the average age (76 years).
bAJCC TNM stage data were not available in one case among the 36 CIMP-H CRCs from the TCGA dataset.
cDepth of primary tumor invasion (pT) data were not available in six cases among the 36 CIMP-H CRCs from the TCGA dataset.
dHistologic subtype (mucinous adenocarcinoma) data were not available in one case among the 36 CIMP-H CRCs from the TCGA dataset.
eLymphatic invasion data were not available in three cases among the 36 CIMP-H CRCs from the TCGA dataset.
fVascular invasion data were not available in nine cases among the 36 CIMP-H CRCs from the TCGA dataset.
gMutation rate data were not available in four cases among the 36 CIMP-H CRCs from the TCGA dataset.
hKRAS/BRAF/PIK3CA/APC/CTNNB1/TP53 mutations data were not available in four cases among the 36 CIMP-H CRCs from the TCGA dataset.
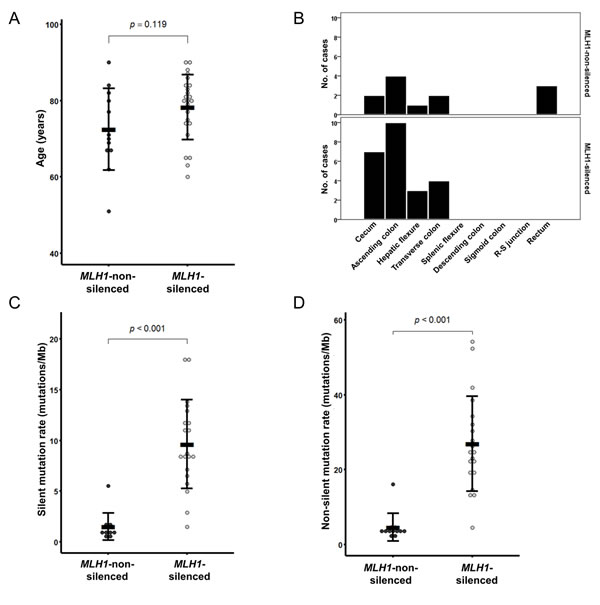
Figure 3: MLH1 silencing-associated features in CIMP-H CRC from the TCGA dataset (n = 36). A. Age at diagnosis. B. Tumor location bowel subsite. C. Silent mutation rate. D. Non-silent mutation rate.
Discussion
The presence or absence of MLH1 methylation is a critical determinant of two distinct molecular subtypes of CIMP-H CRC: MSI-positive (MSI-H) and MSI-negative (MSS or MSI-L) CIMP-H CRCs. According to previous findings, the development of MSI-H status in a tumor genome is thought to be a late event that occurs in the serrated neoplasia pathway: although it is frequently found in combination with CIMP-H CRC, the majority of serrated precursor lesions, including sessile serrated adenomas/polyps (SSA/Ps) and traditional serrated adenomas (TSAs), have an MSS or MSI-L status [13]. Thus, MLH1 promoter methylation and the subsequent acquisition of the MSI-H molecular phenotype may preferentially occur at the advanced dysplastic stage and/or the initiation of malignant transformation of serrated lesions. MSI-H status leads to genomic vulnerability associated with accumulation of mutations and neoplastic progression, finally inducing development of the unique clinicopathologic features of CRC. Representative MSI-H-associated features in CRC include frequent frameshift mutations, prominent intratumoral and peritumoral lymphocytic reactions, and favorable prognosis [6, 7, 14]. In our present study, to display the heterogeneous landscape of CIMP-H CRC according to MLH1 methylation status, which critically determines MSI status, we analyzed various clinicopathologic and molecular factors as well as known MSI-associated features in CIMP-H CRC. As a result, we successfully obtained comprehensive data regarding MLH1 methylation-dependent characteristics in CIMP-H CRC and their implications in different serrated precursor pathways.
Both histopathologic and prognostic features associated with MLH1 methylation can be regarded as being directly related to MSI status in CIMP-H CRC. In the current study, MLH1-methylated tumors were found to be significantly associated with dense TILs, intense peritumoral lymphoid reactions and active/dense Crohn-like lymphoid reactions (Table 1 and Figures 1 and 2). By contrast, most histopathologic factors that are known to be associated with poor prognosis, including vascular invasion, perineural invasion, tumor budding, and distant metastasis, were observed more frequently in MLH1-unmethylated tumors than in MLH1-methylated tumors (Table 1). Moreover, although statistical significance was not found, a tendency toward better survival was observed in the MLH1-methylated tumors (Supplementary Figures S1A and S1B). All of these findings are consistent with the expected effects of MSI-H status in CRC. Because a significant association between anti-tumor immune reactions and improved patient survival has been demonstrated in diverse human malignancies, including CRC [15], it is reasonable to infer that the favorable prognostic impact of MSI-H in CRC may be mainly due to accompanying prominent lymphoid reactions [14]. Several previous studies have explained the potential mechanism underlying MSI-induced lymphoid reactions in CRC. Schwitalle et al. demonstrated that many tumor-specific frameshift peptides are generated in MSI-H CRC and that these neoantigens cause increased anti-tumor immune responses [16]. A recent investigation also revealed that the number of frameshift mutations is positively correlated with the density of T cell infiltrates in MSI-H CRC [17].
Mucinous differentiation has been shown to be associated with MSI-H, CIMP-H, BRAF mutation, and proximal colonic tumor location in CRC [18]. In the present study, MLH1-methylated CIMP-H CRCs were significantly correlated with a high frequency of mucinous histology (Table 1 and Figure 2). This finding suggests that extracellular mucin production might be molecularly related to MSI-H rather than to CIMP-H in CRC. Because the MLH1-methylated CIMP-H CRCs were concentrated in the proximal colon, to exclude the possibility of a tumor location effect on mucinous histology, we additionally performed subgroup analyses according to tumor location. The results demonstrated that in both the proximal colon and distal colorectum, the MLH1-methylated CIMP-H CRCs showed significantly more mucinous differentiation than the MLH1-unmethylated CIMP-H CRCs (Supplementary Tables S1 and S2). Furthermore, in an independent analysis using our extended CRC cohort (n = 687), we found that mucinous differentiation was more frequent in MSI-H, CIMP-low/negative (CIMP-L/0) CRC than in MSS/MSI-L, CIMP-L/0 CRC (36% and 9%, respectively; P < 0.001; Supplementary Table S3). Therefore, regardless of the proximal or distal tumor location and CIMP status, MSI-H can be regarded as a significant molecular predisposing factor for extracellular mucinous histology in CRC. In addition, because the frequency of mucinous histology was remarkably higher in MLH1-methylated CIMP-H CRC (73%; Table 1) than in MSI-H, CIMP-L/0 CRC (36%; Supplementary Table S3), there may be a synergistic impact of MSI-H and CIMP-H on mucinous differentiation in CRC.
The differential clinicopathologic and molecular features between MLH1-methylated and MLH1-unmethylated CIMP-H CRCs provide important clues regarding the major precursor lesions of CIMP-H CRC. Intensive investigations conducted in the past decade have revealed that the majority of CIMP-H CRCs develop from serrated precursor lesions, especially from SSA/Ps [12]. However, our results indicate that substantial proportions of MLH1-methylated and MLH1-unmethylated CIMP-H CRCs may be derived from different subtypes of serrated lesions. Fundamentally, although the data are variable across studies, previous investigations have reported that the MLH1 methylation frequency is higher in SSA/Ps (16% to 75%) than in TSAs (3% to 48%) [19]. Previous studies have also revealed that SSA/Ps are significantly associated with a proximal colonic tumor location, BRAF mutations, and CIMP-H status, whereas TSAs show relatively heterogeneous clinicopathologic and molecular features [12]. Therefore, the close relationship between SSA/Ps and MLH1-methylated CIMP-H CRC can be easily inferred, but a detailed approach is needed to identify the link between TSA and CIMP-H CRC. Fortunately, recent advances in the characterization of TSA suggest that it can be classified into three distinct molecular subtypes: KRAS-mutated, BRAF-mutated, and KRAS/BRAF-wild type [20-23]. Importantly, compared with KRAS-mutated TSA, BRAF-mutated TSA is significantly associated with a proximal colonic tumor location and more widespread CpG island methylation [20, 21]. According to a study by Bettington et al., MLH1 methylation occurs in BRAF-mutated TSA but not in KRAS-mutated TSA, although the frequency is low (7% of BRAF-mutated TSAs) [20]. In addition, Wiland et al. detected RUNX3 and SOCS1 methylation in BRAF-mutated TSA but not in KRAS-mutated TSA [21]. In our study cohort, BRAF-mutated, MLH1-unmethylated CIMP-H CRCs generally showed more extensive methylation of CIMP markers than KRAS-mutated, MLH1-unmethylated CIMP-H CRCs (Supplementary Figure S2). These features imply that BRAF mutation is tightly associated with extensive CpG island methylation in colorectal carcinogenesis, regardless of whether the carcinogenesis pathway is the SSA/P pathway or the TSA pathway (Figure 4A and 4B).
According to our findings, MLH1-methylated CIMP-H CRC was significantly associated with high frequencies of a proximal colonic tumor location and RUNX3/SOCS1 promoter methylation compared with MLH1-unmethylated CIMP-H CRC (Tables 1, 2). On the basis of comprehensive profiles of proximal/distal tumor locations, KRAS/BRAF mutations, and MLH1/RUNX3/SOCS1 methylation of serrated lesions, the major precursor of MLH1-methylated CIMP-H CRC may be SSA/P, whereas the major precursors of MLH1-unmethylated CIMP-H CRC may be KRAS-mutated TSA, followed by BRAF-mutated TSA and SSA/P (Figure 4A and 4B).
According to our data, although KRAS mutations were the dominant form of RAS/RAF pathway mutations in MLH1-unmethylated CIMP-H CRC, a small portion of MLH1-unmethylated CIMP-H CRCs displayed BRAF mutations (8% to 13%; Tables 2, 3). Therefore, the minority of MLH1-unmethylated CIMP-H CRCs, which have BRAF mutations, may develop from SSA/P and/or BRAF-mutated TSA (Figure 4B). In our samples, BRAF-mutated, MLH1-unmethylated CIMP-H CRC (n = 4) was associated with aggressive clinicopathologic features (Supplementary Table S4). All four cases of BRAF-mutated, MLH1-unmethylated tumor histologically showed perineural invasion and tumor budding-positivity. Of the four cases, two patients were stage IV and died within 4 months after surgical treatment. Another patient was stage III, and cancer recurred at 19.5 months after surgical treatment. These findings were consistent with the results of a study by Pai et al., who demonstrated that BRAF-mutated, MSS-type proximal colonic adenocarcinoma was associated with adverse histologic features and poor survival [24]. Collectively, a small subset of MLH1-unmethylated CIMP-H CRCs may be derived from BRAF-mutated TSA and/or SSA/P (Figure 4B), and these BRAF-mutated, MLH1-unmethylated tumors constitute a highly aggressive phenotype among CRCs.
In terms of the age distribution of CIMP-H CRCs, we found that older patients had MLH1-methylated tumors more frequently than they had MLH1-unmethylated tumors (Table 1 and Figure 1a). However, although advanced SSA/P is known to be prevalent in old females [25], there are no established data showing a significant age difference between individuals with SSA/P and TSA. Therefore, a hypothetical theory can be suggested that is based on different rates of dysplastic change and/or different chances of malignant transformation between SSA/P and TSA. SSA/P (4% to 23% of all serrated lesions) is known to be more frequent than TSA (1% to 2% of all serrated lesions) [19]. However, advanced SSA/P and its malignant counterpart, MLH1-methylated CIMP-H CRC, occur at an older age than advanced TSA and MLH1-unmethylated CIMP-H CRC. These features imply that dysplastic change and the malignant transformation of SSA/P may be more slow and rare than those of TSA. This presumed biological difference in neoplastic progression between SSA/P and TSA might be based on the underlying molecular differences. In fact, KRAS-mutated TSA has been regarded as an aggressive serrated lesion that is significantly associated with high-grade dysplasia and intramucosal adenocarcinoma and is frequently found in combination with MGMT and CDKN2A methylation, TP53 mutations, and Wnt pathway activation, suggestive of the combined activity of the CIMP and CIN pathways [23, 26]. Moreover, a recent study by Panarelli et al. reported a lack of evidence of Wnt pathway activation in sporadic MSI-H CRC [27]. Our results from TCGA data analysis also demonstrated that MLH1-non-silenced CIMP-H CRC showed frequent APC mutations, but MLH1-silenced CIMP-H CRC did not (83% vs. 20%; Table 3). These findings support that in contrast with the TSA pathway, which may partly depend on APC mutation-associated conventional adenoma-carcinoma pathway, the SSA/P pathway may be molecularly associated with the pure CIMP pathway (Figure 4B). Therefore, it can be hypothesized that the synergistic fusion of molecular pathways in TSA may contribute to the relatively rapid and frequent progression of low-grade dysplasia to high-grade dysplasia and carcinoma and may ultimately underlie the age difference between patients with MLH1-methyated and MLH1-unmethylated CIMP-H CRCs.
In this study, to identify distinct features between MLH1-methylated and unmethylated CIMP-H CRCs, we analyzed both our original cohort data and the TCGA data. We successfully confirmed that the majority of differential features between MLH1-methylated and unmethylated tumors identified from our cohort data was also maintained in results from the TCGA data analysis. However, underlying characteristics of our samples and TCGA samples were somewhat different, which representatively included BRAF mutation frequency. In our cohort, only 36% of MLH1-methylated CIMP-H CRCs harbor BRAF mutations, whereas BRAF mutation frequency of MLH1-silenced CIMP-H CRCs in the TCGA data was 75%. This discrepancy of BRAF mutation frequency may be due to the ethnic difference of molecular features in CRCs. According to our previous investigation, frequencies of BRAF mutation and CIMP-H in East Asian patients with CRC were significantly lower than those in Western country patients with CRC [6]. In fact, our cohort was comprised exclusively of East Asians, but the racial composition of the TCGA cohort was heterogeneous. This difference of study populations can induce differences in molecular features of CRCs, especially in BRAF mutation frequency.
In conclusion, MLH1 promoter methylation is a major determining factor for the clinicopathologic and molecular heterogeneity of CIMP-H CRC. In contrast with the typical features of MLH1-methylated CIMP-H CRC, which include an older age of onset, a proximal colonic tumor location, mucinous histology, intense lymphoid reactions, frequent BRAF mutations, frequent RUNX3/SOCS1 promoter methylation, MSI-H status, and a hypermutated phenotype, MLH1-unmethylated CIMP-H CRC is characterized by a middle to old age of onset, a relatively even distribution between the proximal and distal colorectum, less prominent mucinous and lymphoid histologic features, frequent KRAS and APC mutations, MSS/MSI-L status, and a non-hypermutated phenotype (Figure 4A). Although different serrated pathways to CRC remain to be further understood, our findings suggest that MLH1-methylated CIMP-H CRC may be derived typically from the SSA/P pathway but that MLH1-unmethylated CIMP-H CRC may develop mainly through the KRAS-mutated TSA pathway and also less commonly through the BRAF-mutated TSA pathway and/or the SSA/P pathway (Figure 4B).
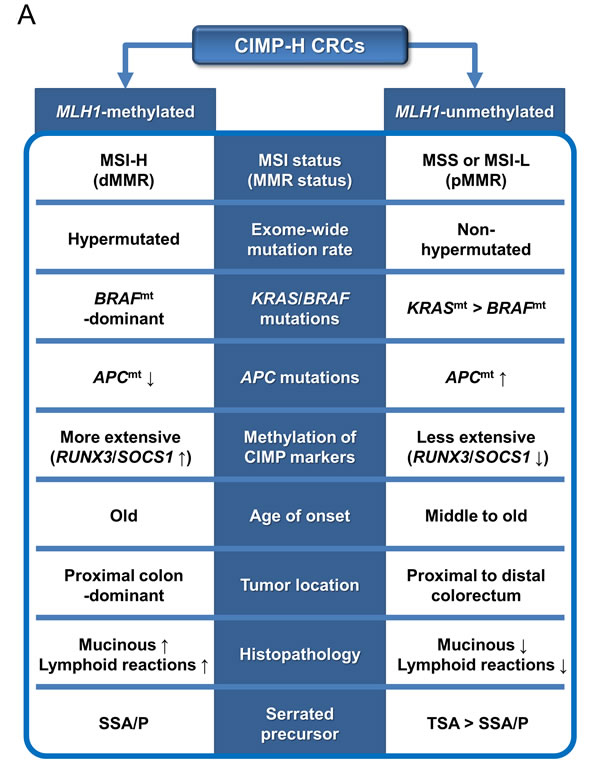
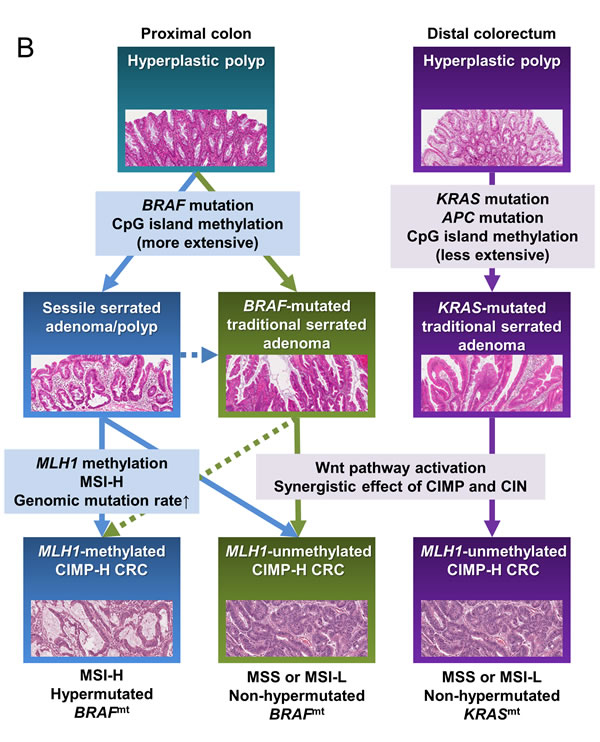
Figure 4: Schematic summary of this study. A. Distinct features and precursors of the MLH1-methylated and unmethylated CIMP-H CRCs. B. A molecular pathogenetic model of CIMP-H CRCs based on different serrated precursor pathways. Abbreviations: CIMP-H, CpG island methylator phenotype-high; MSI, microsatellite instability; MSI-H, MSI-high; MSI-L, MSI-low; MSS, microsatellite stable; MMR, DNA mismatch repair; dMMR, MMR-deficient; pMMR, MMR-proficient; BRAFmt, BRAF mutation or BRAF-mutated; KRASmt, KRAS mutation or KRAS-mutated; APCmt, APC mutation; SSA/P, sessile serrated adenoma/polyp; TSA, traditional serrated adenoma; CIN, chromosomal instability.
Materials and Methods
Case selection
For the present study, 65 CIMP-H CRC samples were selected from our previously established CRC sample cohorts, which consisted of 814 CRC samples in total [28, 29]. All samples from our CRC cohorts were retrospectively collected formalin-fixed paraffin-embedded (FFPE) tissues from 814 patients who underwent surgical resection for CRC at Seoul National University Hospital, Seoul, Korea, between 2004 and 2008. Of the 65 CIMP-H CRCs, 33 were MLH1-methylated CIMP-H CRCs, and 32 were MLH1-unmethylated CIMP-H CRCs. Based on previous studies [28, 29], by immunohistochemical staining, all 33 of the MLH1-methylated tumors were confirmed to have lost MLH1 expression, whereas all 32 of the MLH1-unmethylated tumors were confirmed to have retained MLH1 expression. A detailed procedure for CIMP determination, including MLH1 methylation analysis, is described in the section below. This study was approved by the institutional review board.
Clinicopathologic data
The clinical and pathologic features of the 65 CIMP-H CRCs were investigated by a review of medical records or by microscopic examination of hematoxylin and eosin-stained FFPE tissue slides. The clinicopathologic parameters included age, sex, tumor location, gross tumor type, American Joint Committee on Cancer (AJCC) tumor-node-metastasis (TNM) stage, disease-free survival, tumor differentiation, mucinous histology, signet ring cell histology, medullary histology, serrated histology, lymphatic invasion, vascular invasion, perineural invasion, tumor budding, tumor-infiltrating lymphocytes (TILs), peritumoral lymphoid reaction, and Crohn-like lymphoid reaction. The mean follow-up time for disease-free survival data of our original cohort (65 patients with CIMP-H CRC) was 36 months (ranging from 1 to 89 months). The detailed assessment criteria for the histopathologic parameters used in this study have been previously described [29, 30]. The number of TILs was counted in one high power field (×400) of the most TIL-intense area. All of the histopathologic parameters were independently evaluated by two pathologists (J.H.K. and J.M.B.). Conflicting assessment results between the pathologists were reviewed and discussed until consensus was reached.
CIMP analysis
Genomic DNA extraction from 814 CRC FFPE tissues and subsequent bisulfite modification and CIMP analysis of these DNA samples were conducted as previously described [28, 31]. For CIMP determination, methylation-specific quantitative PCR analysis (MethyLight assay) was performed using eight CIMP markers (MLH1, CACNA1G, CDKN2A (p16), CRABP1, IGF2, NEUROG1, RUNX3, and SOCS1). A CpG island locus was defined as hypermethylated when the percentage of the methylated reference value exceeded 4. CIMP-H was defined when a tumor showed promoter CpG island hypermethylation in five or more CIMP markers [32]. CIMP-low (CIMP-L) was defined when a tumor showed promoter CpG island hypermethylation in one to four CIMP markers. CIMP-negative (CIMP-0) was defined when all CIMP markers in a tumor were unmethylated. CIMP-H generally represents the true CIMP-positive status. Of the 814 CRCs subjected to CIMP analysis, 65 were determined to be CIMP-H CRCs and were finally included in this study. Of the remaining 749 cases, 445 were CIMP-L tumors, and 304 were CIMP-0 tumors. Reproducibility of MethyLight assay for quantification of promoter methylation of CIMP markers had been validated by previous data [33]. Performance of the eight-marker panel with a cutoff value of five for determination of CIMP-H in CRC had been validated by our previous study [32].
MSI analysis
MSI analysis was performed as previously described [28, 31]. In detail, the instability statuses of 65 paired CIMP-H CRC tumor and normal DNA samples were analyzed for the five NCI-recommended microsatellite markers (the Bethesda panel; BAT-25, BAT-26, D5S346, D17S250, and D2S123) using fluorescent multiplex PCR [34]. MSI-H was defined when tumor DNA showed instability (an amplicon length alteration) in two or more microsatellite markers compared with normal DNA. MSI-low (MSI-L) was defined when tumor DNA showed instability in one microsatellite marker. Microsatellite stable (MSS) status was considered when tumor DNA showed no instability in any of the five microsatellite markers. MSI-H generally represents the true MSI-positive status, whereas MSI-L and MSS are considered MSI-negative statuses.
KRAS/BRAF mutation analysis
The mutational statuses of KRAS codons 12 and 13 and BRAF codon 600 were assessed as previously described [28, 31]. Allele-specific PCR and direct sequencing methods were used for these mutation analyses. Of the 65 CIMP-H CRCs, due to the suboptimal quantity or quality of extracted DNA, four samples and one sample were finally excluded from KRAS and BRAF mutation analyses, respectively.
TCGA data analysis
Using the publically accessible TCGA data portal (https://tcga-data.nci.nih.gov/tcga/) and TCGA-based publications [35, 36], clinical and molecular data on 36 CIMP-H CRCs were collected and analyzed. The assessed parameters included age, sex, tumor location, TNM stage, histologic subtype (mucinous and non-mucinous adenocarcinomas), lymphatic invasion, vascular invasion, MSI status, mutational phenotype (hypermutated and non-hypermutated; using a cutoff value of 12 mutations per 106 exonic bases) [35, 36], exome-wide mutation rate (including silent and non-silent mutations) [35], and KRAS/BRAF/PIK3CA/APC/CTNNB1/TP53 mutations. The differential features according to MLH1 epigenetic silencing status (silenced versus non-silenced) of the CIMP-H CRCs were statistically analyzed.
Statistical analysis
Statistical analyses in this study were performed using IBM SPSS Statistics version 20 (IBM Co., Armonk, NY, USA) and the R software (http://www.r-project.org/). Comparisons of categorical variables between MLH1-methylated (-silenced) and MLH1-unmethylated (-non-silenced) tumors were conducted using the chi-square test or Fisher’s exact test. Comparisons of continuous variables between MLH1-methylated (-silenced) and MLH1-unmethylated (-non-silenced) tumors were conducted using the t-test. Survival analysis was performed using the Kaplan-Meier method with the log-rank test. All P-values were two-sided, and statistical significance was determined at P < 0.05.
Acknowledgments
The results in this study are in part based upon data generated by the TCGA Research Network: http://cancergenome.nih.gov/. This study was supported by a grant from Basic Science Research Program through the National Research Foundation (NRF) funded by the Korean Ministry of Education (2013R1A1A2059080), a grant from the Korea Health Technology R&D Project funded by the Korean Ministry of Health and Welfare (HI13C1804), the NRF grant funded by the Korean Ministry of Science, ICT and Future planning (2011-0030049), and a grant from the Korea Health Technology R&D Project through the Korea Health Industry Development Institute funded by the Korean Ministry of Health and Welfare (HI14C1277).
Conflicts of Interest
The authors declare no conflict of interest.
Editorial note
This paper has been accepted based in part on peer-review conducted by another journal and the authors' response and revisions as well as expedited peer-review in Oncotarget.
References
1. Grady WM, Carethers JM. Genomic and epigenetic instability in colorectal cancer pathogenesis. Gastroenterology. 2008; 135:1079-1099.
2. Ogino S, Goel A. Molecular classification and correlates in colorectal cancer. J Mol Diagn. 2008; 10:13-27.
3. Colussi D, Brandi G, Bazzoli F, Ricciardiello L. Molecular pathways involved in colorectal cancer: implications for disease behavior and prevention. Int J Mol Sci. 2013; 14:16365-16385.
4. Boland CR, Goel A. Microsatellite instability in colorectal cancer. Gastroenterology. 2010; 138:2073-2087 e2073.
5. Geiersbach KB, Samowitz WS. Microsatellite instability and colorectal cancer. Arch Pathol Lab Med. 2011; 135:1269-1277.
6. Kim JH, Kang GH. Molecular and prognostic heterogeneity of microsatellite-unstable colorectal cancer. World J Gastroenterol. 2014; 20:4230-4243.
7. Vilar E, Tabernero J. Molecular dissection of microsatellite instable colorectal cancer. Cancer Discov. 2013; 3:502-511.
8. Bae JM, Kim JH, Kang GH. Epigenetic alterations in colorectal cancer: the CpG island methylator phenotype. Histol Histopathol. 2013; 28:585-595.
9. Bardhan K, Liu K. Epigenetics and colorectal cancer pathogenesis. Cancers (Basel). 2013; 5:676-713.
10. Ng JM, Yu J. Promoter hypermethylation of tumour suppressor genes as potential biomarkers in colorectal cancer. Int J Mol Sci. 2015; 16:2472-2496.
11. Bettington M, Walker N, Clouston A, Brown I, Leggett B, Whitehall V. The serrated pathway to colorectal carcinoma: current concepts and challenges. Histopathology. 2013; 62:367-386.
12. Rex DK, Ahnen DJ, Baron JA, Batts KP, Burke CA, Burt RW, Goldblum JR, Guillem JG, Kahi CJ, Kalady MF, O’Brien MJ, Odze RD, Ogino S, et al. Serrated lesions of the colorectum: review and recommendations from an expert panel. Am J Gastroenterol. 2012; 107:1315-1329; quiz 1314, 1330.
13. Yamane L, Scapulatempo-Neto C, Reis RM, Guimaraes DP. Serrated pathway in colorectal carcinogenesis. World J Gastroenterol. 2014; 20:2634-2640.
14. Deschoolmeester V, Baay M, Lardon F, Pauwels P, Peeters M. Immune Cells in Colorectal Cancer: Prognostic Relevance and Role of MSI. Cancer Microenviron. 2011; 4:377-392.
15. Ogino S, Nosho K, Irahara N, Meyerhardt JA, Baba Y, Shima K, Glickman JN, Ferrone CR, Mino-Kenudson M, Tanaka N, Dranoff G, Giovannucci EL, Fuchs CS. Lymphocytic reaction to colorectal cancer is associated with longer survival, independent of lymph node count, microsatellite instability, and CpG island methylator phenotype. Clin Cancer Res. 2009; 15:6412-6420.
16. Schwitalle Y, Kloor M, Eiermann S, Linnebacher M, Kienle P, Knaebel HP, Tariverdian M, Benner A, von Knebel Doeberitz M. Immune response against frameshift-induced neopeptides in HNPCC patients and healthy HNPCC mutation carriers. Gastroenterology. 2008; 134:988-997.
17. Maby P, Tougeron D, Hamieh M, Mlecnik B, Kora H, Bindea G, Angell HK, Fredriksen T, Elie N, Fauquembergue E, Drouet A, Leprince J, Benichou J, et al. Correlation between density of CD8+ T cell infiltrates in microsatellite unstable colorectal cancers and frameshift mutations: a rationale for personalized immunotherapy. Cancer Res. 2015; 75:3446-55. doi: 10.1158/0008-5472.CAN-14-3051.
18. Tanaka H, Deng G, Matsuzaki K, Kakar S, Kim GE, Miura S, Sleisenger MH, Kim YS. BRAF mutation, CpG island methylator phenotype and microsatellite instability occur more frequently and concordantly in mucinous than non-mucinous colorectal cancer. Int J Cancer. 2006; 118:2765-2771.
19. Yang HM, Mitchell JM, Sepulveda JL, Sepulveda AR. Molecular and histologic considerations in the assessment of serrated polyps. Arch Pathol Lab Med. 2015; 139:730-741.
20. Bettington ML, Walker NI, Rosty C, Brown IS, Clouston AD, McKeone DM, Pearson SA, Klein K, Leggett BA, Whitehall VL. A clinicopathological and molecular analysis of 200 traditional serrated adenomas. Mod Pathol. 2015; 28:414-427.
21. Wiland HOt, Shadrach B, Allende D, Carver P, Goldblum JR, Liu X, Patil DT, Rybicki LA, Pai RK. Morphologic and molecular characterization of traditional serrated adenomas of the distal colon and rectum. Am J Surg Pathol. 2014; 38:1290-1297.
22. Tsai JH, Liau JY, Lin YL, Lin LI, Cheng YC, Cheng ML, Jeng YM. Traditional serrated adenoma has two pathways of neoplastic progression that are distinct from the sessile serrated pathway of colorectal carcinogenesis. Mod Pathol. 2014; 27:1375-1385.
23. Bettington ML, Chetty R. Traditional serrated adenoma: an update. Hum Pathol. 2015; 46:933-938.
24. Pai RK, Jayachandran P, Koong AC, Chang DT, Kwok S, Ma L, Arber DA, Balise RR, Tubbs RR, Shadrach B, Pai RK. BRAF-mutated, microsatellite-stable adenocarcinoma of the proximal colon: an aggressive adenocarcinoma with poor survival, mucinous differentiation, and adverse morphologic features. Am J Surg Pathol. 2012; 36:744-752.
25. Yang JF, Tang SJ, Lash RH, Wu R, Yang Q. Anatomic distribution of sessile serrated adenoma/polyp with and without cytologic dysplasia. Arch Pathol Lab Med. 2015; 139:388-393.
26. Kim KM, Lee EJ, Kim YH, Chang DK, Odze RD. KRAS mutations in traditional serrated adenomas from Korea herald an aggressive phenotype. Am J Surg Pathol. 2010; 34:667-675.
27. Panarelli NC, Vaughn CP, Samowitz WS, Yantiss RK. Sporadic microsatellite instability-high colon cancers rarely display immunohistochemical evidence of Wnt signaling activation. Am J Surg Pathol. 2015; 39:313-317.
28. Bae JM, Kim JH, Cho NY, Kim TY, Kang GH. Prognostic implication of the CpG island methylator phenotype in colorectal cancers depends on tumour location. Br J Cancer. 2013; 109:1004-1012.
29. Kim JH, Kim KJ, Bae JM, Rhee YY, Cho NY, Lee HS, Kang GH. Comparative validation of assessment criteria for Crohn-like lymphoid reaction in colorectal carcinoma. J Clin Pathol. 2015; 68:22-28.
30. Kim JH, Bae JM, Oh HJ, Lee HS, Kang GH. Pathologic Factors Associated with Prognosis after Adjuvant Chemotherapy in Stage II/III Microsatellite-Unstable Colorectal Cancers. J Pathol Transl Med. 2015; 49:118-128.
31. Kim JH, Rhee YY, Bae JM, Kwon HJ, Cho NY, Kim MJ, Kang GH. Subsets of microsatellite-unstable colorectal cancers exhibit discordance between the CpG island methylator phenotype and MLH1 methylation status. Mod Pathol. 2013; 26:1013-1022.
32. Kim JH, Shin SH, Kwon HJ, Cho NY, Kang GH. Prognostic implications of CpG island hypermethylator phenotype in colorectal cancers. Virchows Arch. 2009; 455:485-494.
33. Ogino S, Kawasaki T, Brahmandam M, Cantor M, Kirkner GJ, Spiegelman D, Makrigiorgos GM, Weisenberger DJ, Laird PW, Loda M, Fuchs CS. Precision and performance characteristics of bisulfite conversion and real-time PCR (MethyLight) for quantitative DNA methylation analysis. J Mol Diagn. 2006; 8:209-217.
34. Boland CR, Thibodeau SN, Hamilton SR, Sidransky D, Eshleman JR, Burt RW, Meltzer SJ, Rodriguez-Bigas MA, Fodde R, Ranzani GN, Srivastava S. A National Cancer Institute Workshop on Microsatellite Instability for cancer detection and familial predisposition: development of international criteria for the determination of microsatellite instability in colorectal cancer. Cancer Res. 1998; 58:5248-5257.
35. Cancer Genome Atlas N. Comprehensive molecular characterization of human colon and rectal cancer. Nature. 2012; 487:330-337.
36. Donehower LA, Creighton CJ, Schultz N, Shinbrot E, Chang K, Gunaratne PH, Muzny D, Sander C, Hamilton SR, Gibbs RA, Wheeler D. MLH1-silenced and non-silenced subgroups of hypermutated colorectal carcinomas have distinct mutational landscapes. J Pathol. 2013; 229:99-110.