
Introduction
B-lineage acute lymphoblastic leukemia (B-ALL) represents 75-85% of all ALLs and is characterized by the accumulation of immature B cells, which results in the suppression of normal hematopoiesis. B-ALL is the most frequent cancer in children, with the highest incidence between 2 and 5 years, while it is relatively rare in adults, with a peak after the age of 50 [1-3]. While in childhood the survival rates are in the order of 80%, the outcome in adults is still unsatisfactory, with long-term survival rates of about 40-50% of cases [4-8]. An intermediate group is represented by the adolescents/young adults (AYA), who show an improved outcome when treated with pediatric-like protocols [9-11].
Besides the different prognosis, children and adults are characterized by a variable distribution of recurrent chromosomal aberrations with prognostic impact, such as high hyperdiploidy and ETV6-RUNX1 transcript, common in children, BCR-ABL1 and MLL rearrangements, more frequent in adults [3, 12-20]. While these lesions characterize about 50% of B-ALL, a considerable proportion of cases proves negative for gold-standard molecular screening (B-NEG ALL). The definition of the genetic landscape of this “genetically-orphan” subgroup has partially improved by the introduction of genome-wide technologies, such as gene expression profiling (GEP), single nucleotide polymorphism (SNP) arrays and next generation sequencing (NGS) [21-26]. The latter has clarified the molecular background of specific subgroups, such as “BCR-ABL1-like” and hypodiploid ALL, while a comprehensive description of B-NEG has so far not been provided [25-26].
We analyzed 168 B-NEG ALLs (50 children, 61 AYA, 57 adults) - distributed in a discovery panel (N = 13), screening panel 1 (N = 68) and screening panel 2 (N = 87) - by NGS and copy number aberration (CNA) analysis to refine the molecular scenario of B-NEG ALL cases and assess if different lesions might account for the different outcome between children, AYA and adults, with the ultimate goal of investigating the potential role of targeted therapeutic approaches.
Results
Incidence and outcome of B-NEG ALLs stratified for age cohorts
The meta-analysis of the incidence of known molecular aberrations in a cohort of over 5,000 ALL [3] revealed that within B-ALL, B-NEG cases represent 70.5% of childhood, 72.7% of AYA and 42.7% of adults. Furthermore, within the population included in the current study, we evaluated the outcome of the above mentioned age cohorts. Clinical outcome data were available for 142 patients (48 children, 50 AYA and 44 adults) with a median follow-up of 65 months. Overall survival (OS) was significantly different among children (83%), AYA (55%) and adults (29%), resembling that of ALL in general (Figure 1) [3].
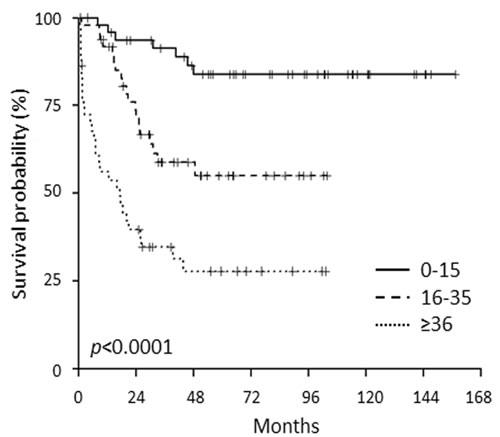
Figure 1: OS of B-NEG ALL patients, included in the present study, stratified for age cohorts.
Genomic overview of B-NEG ALL
Whole exome sequencing (WES) of the discovery panel cases revealed 136 mutated genes (10.5/case), with a similar load across the 3 age cohorts (Figure 2A). Mutated genes were highly heterogeneous across B-NEG ALL samples with only FLT3, PREX2, PAX5 - recurrently mutated, being found in 2 of the 13 samples (Table S1). In spite of this, in silico analyses revealed a significant enrichment of the following gene categories: focal adhesion/ECM interaction, small GTPase mediated signal transduction, ion transport and protein kinase activity, as described in Supplemental Material and Table S2.
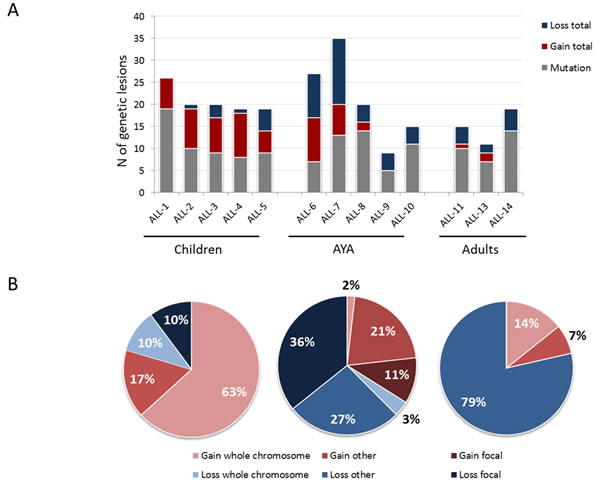
Figure 2: Overall load of genetic lesions of B-NEG ALL. A. Load of somatic mutations and CNAs across different age cohorts. B. CNAs distribution and type across different age cohorts.
By SNP arrays we detected 119 CNAs (9.1 CNAs/sample, range 4-22). The load and type of CNAs was heterogeneous across the age cohorts. Indeed, whilst childhood ALLs were affected by 9.8 CNAs, adult ALLs carried 4.6 CNAs on average. AYA displayed an intermediate behavior, with 2/5 samples being genetically complex and 3 harboring a number of CNAs similar to the adult cohort (Figure 2A). Pediatric samples were mostly characterized by gains of entire chromosomes. Contrariwise, AYA and adult patients were mainly affected by losses of variable size, ranging from deletions of chromosome arms to single exons (Figure 2B). The most common minimally deleted regions were those encompassing known target genes [21, 22]. Indeed, the screening of these known targets confirmed that the most frequently deleted genes were IKZF1 (41.4%), CDKN2A (36.9%), PAX5 (25.5%) and ETV6 (17.8%) (Figure 3A).
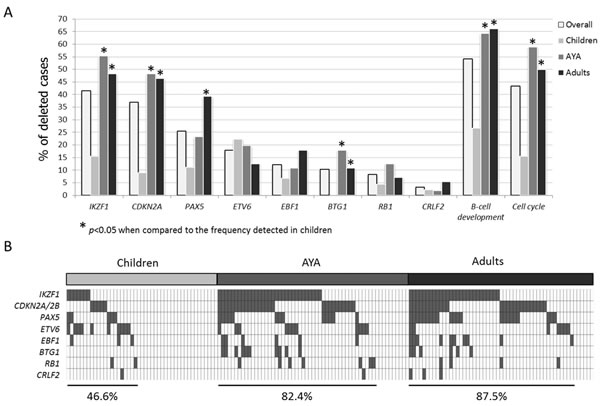
Figure 3: Incidence of the most common deletions in B-NEG ALL. A. Frequency of deletions targeting IKZF1, CDKN2A, PAX5, ETV6, EBF1, BTG1, RB1, CRLF2 - and grouped in functional categories - in the whole population and in age cohorts. Asterisks indicate deletions with a statistically significant different incidence when comparing children and AYA or children and adults. B. Distribution of the abovementioned deletions in children, AYA and adult populations.
Identification and screening of the recurrently mutated genes
Given the low number of recurrently mutated genes detected in the discovery panel, WES was broadened to 68 additional B-NEG cases (screening panel 1) and the bioinformatic analysis focused on the genes mutated in the discovery panel and/or reported as affected by mutations or CNAs in published ALL series [25-29]. This approach yielded a shorter list of genes frequently targeted by mutations, which overall clustered in the RAS/RTK and JAK/STAT cascades, or affected B-lineage differentiation, and also highlighted intriguing differences amongst the age cohorts (Table S3).
To validate the latter findings we sequenced the most frequently mutated genes within the abovementioned pathways - namely FLT3, KRAS, NRAS, IL7R, JAK2, CRLF2, JAK1 and PAX5 - in 87 additional B-NEG ALLs (screening panel 2). The results described below refer to a total population of 168 cases (50 children, 61 AYA and 57 adults).
Age-related genetic lesions in B-NEG ALL
Genetic lesions of PAX5
Among the mutations of B-lineage development genes, those affecting PAX5 were the most common, occurring in 20 patients (11.9%). Mutations clustered in the paired-box domain with roughly 50% being represented by the hotspot P80R (Figure S1A). The comparison among age groups highlighted a significant difference between the percentage of mutations detected in children and adults (2% vs 19.3%, p = 0.005, Figure S1B); AYA displayed an intermediate behavior, with mutations observed in 13.1% of cases.
By integrating the mutational with the CN status, we confirmed that PAX5 lesions increase with age progression, being detected in 13.3% of children, 28.6% of AYA and 44.6% of adults: the difference was statistically significant between children and adults, as well as by combining AYA and adults (p = 0.001, p = 0.004, Figure S1C).
Recurrent deletions of B-cell differentiation and cell-cycle genes
Beside PAX5 deletions, the screening of CNAs of selected genes involved in B-cell development (i.e. IKZF1, EBF1, BTG1) as well as the cell-cycle related genes (i.e. CDKN2A/B and RB1) revealed that deletions were significantly more frequent in AYA and adult patients than in children. Overall, B-cell development genes were deleted in 26.6% of pediatric, 64.3% of AYA and 66.1% of adult cases. Similarly, 15.5% of pediatric, 58.9% of AYA and 50% of adult patients harbored deletions targeting cell-cycle genes. In particular, these differences achieved a statistical significance for IKZF1, BTG1 and CDKN2A, as detailed in Figure 3A.
As a result, these selected CNAs tended to accumulate with age, with 46.6% of children, 82.4% of AYA and 87.5% carrying at least one CNA (Figure 3B).
Distribution and features of RAS/RTK pathway mutations
Mutations of FLT3, KRAS and NRAS, grouped in the RAS/RTK pathway, were detected in 26.8% (45/168) of cases. In particular, KRAS was mutated in 11.3% (19/168) of cases, NRAS in 9.5% (16/168) and FLT3 in 8.3% (14/168) (Figure 4A). The latter was prevalently mutated in adults (12.3%) and AYA (9.8%), while only 2% of children carried FLT3 mutations (Figure 4A). This difference reached statistical significance when comparing adults vs children, as well as by combining AYA and adults (p = 0.046 and p = 0.043, Figure S2A). FLT3 mutations mainly targeted the tyrosine kinase catalytic domain (TKD, 50%) or were represented by internal tandem duplications (ITD) in the juxtamembrane domain (36%) (Table 1, Figure S2B).
KRAS and NRAS mutations accumulated mostly in childhood: in fact, collectively RAS mutations were detected in 30% of children and in 14.7% of AYA and 14% of adults (p = 0.052, p = 0.045).
Mutations targeted almost exclusively (92%) the known hotspot G12 and G13, and in the remaining cases the aminoacidic residues 60-63 (Table 1, Figure S3).
Finally, we found no overlap between FLT3 mutations and NRAS, KRAS G12-13 mutations; nonetheless, 1 sample carrying a FLT3 ITD and KRAS G60D mutations was detected. Moreover, 3 cases harbored both NRAS and KRAS mutations. Notably, concomitant NRAS and KRAS mutations were only observed in AYA and children.
Of interest, cytogenetic analyses - successful in 109/168 cases - revealed a hyperdiploid karyotype in 17, with only 5 of them harboring RAS/RTK mutations. Given the known association between hyperdiploidy and RAS/RTK mutations [30-36], the analysis was repeated excluding hyperdiploid cases and samples lacking a cytogenetic evaluation, a similar incidence of RAS/RTK-mutated cases was observed: KRAS/NRAS mutations were detected in 32% of children, 15.6% of AYA and 15.3% of adults, while FLT3 mutations were detected in 12.5% of AYA and 15.3% of adults.
Table 1: Features of the mutations affecting RAS/RTK pathway
ID |
Age |
Cohort |
Panel |
Gene |
Exon |
AA change |
Type of mutation |
Domain (Uniprot) |
GQW_63 |
4 |
Children |
Discovery |
FLT3 |
20 |
M837- |
In frame deletion |
TKD |
GQW_71 |
19 |
AYA |
Discovery |
FLT3 |
20 |
Y842N |
Missense |
TKD |
GQW_220 |
16 |
AYA |
Screening 1 |
FLT3 |
14 |
Y572C |
Missense |
JM |
GQW_210 |
22 |
AYA |
Screening 1 |
FLT3 |
14 |
ITD |
ITD |
JM |
GQW_222 |
23 |
AYA |
Screening 1 |
FLT3 |
14 |
ITD |
ITD |
JM |
GQW_236 |
30 |
AYA |
Screening 1 |
FLT3 |
16 |
N676K |
Missense |
TKD |
ALL_132 |
20 |
AYA |
Screening 2 |
FLT3 |
20 |
D835H |
Missense |
TKD |
GQW_187 |
56 |
Adults |
Screening 1 |
FLT3 |
14 |
ITD |
ITD |
JM |
GQW_199 |
59 |
Adults |
Screening 1 |
FLT3 |
20 |
D835Y |
Missense |
TKD |
GQW_230 |
39 |
Adults |
Screening 1 |
FLT3 |
11 |
S446L |
Missense |
Extracellular |
ALL_14 |
78 |
Adults |
Screening 2 |
FLT3 |
14 |
ITD |
ITD |
JM |
ALL_19 |
40 |
Adults |
Screening 2 |
FLT3 |
20 |
p.837_838delMSinsVQG |
In frame indel |
TKD |
ALL_104 |
65 |
Adults |
Screening 2 |
FLT3 |
14 |
ITD |
ITD |
JM |
ALL_125 |
40 |
Adults |
Screening 2 |
FLT3 |
21 |
P857S |
Missense |
TKD |
GQW_249 |
12 |
Children |
Screening 1 |
KRAS |
2 |
G12D |
Missense |
GTPase |
GQW_245 |
3 |
Children |
Screening 1 |
KRAS |
2 |
G13D |
Missense |
GTPase |
GQW_248 |
9 |
Children |
Screening 1 |
KRAS |
2 |
G13D |
Missense |
GTPase |
ALL_142 |
3 |
Children |
Screening 2 |
KRAS |
2 |
G13D |
Missense |
GTPase |
WB_6_PD |
3 |
Children |
Screening 2 |
KRAS |
2 |
G12D |
Missense |
GTPase |
WB_8_PD |
1 |
Children |
Screening 2 |
KRAS |
2 |
G12R |
Missense |
GTPase |
WB_10_PD |
3 |
Children |
Screening 2 |
KRAS |
2 |
G12S |
Missense |
GTPase |
WB_11_PD |
14 |
Children |
Screening 2 |
KRAS |
2 |
G12D |
Missense |
GTPase |
WB_14_PD |
5 |
Children |
Screening 2 |
KRAS |
2 |
G12D |
Missense |
GTPase |
GQW_208 |
19 |
AYA |
Screening 1 |
KRAS |
3 |
E63K |
Missense |
GTPase |
GQW_210 |
22 |
AYA |
Screening 1 |
KRAS |
3 |
G60D |
Missense |
GTPase |
GQW_221 |
16 |
AYA |
Screening 1 |
KRAS |
2 |
G12D |
Missense |
GTPase |
GQW_225 |
28 |
AYA |
Screening 1 |
KRAS |
2 |
G12D |
Missense |
GTPase |
ALL_138 |
21 |
AYA |
Screening 2 |
KRAS |
2 |
G12V |
Missense |
GTPase |
ALL_106 |
20 |
AYA |
Screening 2 |
KRAS |
2 |
G12D |
Missense |
GTPase |
GQW_81 |
53 |
Adults |
Discovery |
KRAS |
4 |
A146T |
Missense |
GTPase |
GQW_196 |
58 |
Adults |
Screening 1 |
KRAS |
2 |
G12S |
Missense |
GTPase |
ALL_91 |
48 |
Adults |
Screening 2 |
KRAS |
2 |
G13D |
Missense |
GTPase |
ALL_120 |
47 |
Adults |
Screening 2 |
KRAS |
2 |
G12V |
Missense |
GTPase |
GQW_247 |
2 |
Children |
Screening 1 |
NRAS |
2 |
G12A |
Missense |
GTPase |
GQW_242 |
14 |
Children |
Screening 1 |
NRAS |
2 |
G13D |
Missense |
GTPase |
GQW_254 |
9 |
Children |
Screening 1 |
NRAS |
2 |
G13D |
Missense |
GTPase |
ALL_111 |
9 |
Children |
Screening 2 |
NRAS |
2 |
G13D |
Missense |
GTPase |
ALL_146 |
14 |
Children |
Screening 2 |
NRAS |
3 |
Q61R |
Missense |
GTPase |
WB_2_PD |
11 |
Children |
Screening 2 |
NRAS |
2 |
G13D |
Missense |
GTPase |
WB_6_PD |
3 |
Children |
Screening 2 |
NRAS |
2 |
G12S |
Missense |
GTPase |
WB_14_PD |
5 |
Children |
Screening 2 |
NRAS |
2 |
G13D |
Missense |
GTPase |
GQW_225 |
28 |
AYA |
Screening 1 |
NRAS |
2 |
G13D |
Missense |
GTPase |
GQW_229 |
30 |
AYA |
Screening 1 |
NRAS |
2 |
G12D |
Missense |
GTPase |
ALL_88 |
23 |
AYA |
Screening 2 |
NRAS |
2 |
G12D |
Missense |
GTPase |
ALL_116 |
16 |
AYA |
Screening 2 |
NRAS |
2 |
G12D |
Missense |
GTPase |
GQW_181 |
44 |
Adults |
Screening 1 |
NRAS |
2 |
G12A |
Missense |
GTPase |
GQW_198 |
42 |
Adults |
Screening 1 |
NRAS |
2 |
G13D |
Missense |
GTPase |
ALL_29 |
42 |
Adults |
Screening 2 |
NRAS |
2 |
G12S |
Missense |
GTPase |
ALL_134 |
78 |
Adults |
Screening 2 |
NRAS |
2 |
G12D |
Missense |
GTPase |
Distribution and features of JAK/STAT pathway mutations
Mutations in genes converging into the JAK/STAT pathway - JAK2, IL7R, CRLF2 and JAK1 - accounted for 12.5% of cases (21/168). Missense mutations targeting arginine 863 of JAK2 and indels in exon 6 of IL7R were the most frequent, detected in 5.3% and 4.8% of cases, respectively. Less than 2% of cases harbored CRLF2 F232C mutations (N = 3) or JAK1 mutations (N = 2) (Table 2). JAK/STAT mutations were uniquely detected in patients >15 years (p < 0.001): none was in fact found in children, whereas 13% of AYA and 17.5% of adult patients were affected (Figure 4B). Furthermore, JAK/STAT mutations were associated with male gender (p = 0.01) and higher white blood cell (WBC) counts (90.6 vs 43.5 x109/L in mutated vs WT cases, p = 0.002). GEP analysis of JAK/STAT-mutated samples, revealed that 10 of the 21 mutated samples (47.6%) displayed a BCR-ABL1-like profile, thus indicating that JAK/STAT mutations, though characterizing the BCR-ABL1-like cases, are not exclusive of this cohort (Figure S4).
Moreover, when analyzed in light of other features bearing a negative prognostic impact [23-25], 57.1% (12/21) carried an IKZF1 deletion and 3 harbored also a CRLF2-P2RY8 rearrangement (Figure S4). The latter aberration was overall detected in 5 (3 adults, 1 AYA and 1 child) out of 157 samples analyzed by MLPA.
Mutations targeting the JAK/STAT genes were mainly mutually exclusive, with only 1 sample carrying 2 mutations: one targeting JAK2 and the other CRLF2, the latter detected at the subclonal level.
Moreover, only 4 cases displayed concomitant RAS/RTK and JAK/STAT pathway mutations: in 2 cases we found IL7R and NRAS mutations, in 1 sample IL7R and KRAS mutations, and, lastly, 1 case harbored JAK1 and KRAS mutations (Figure 4C).
Contrariwise, PAX5 mutations frequently overlapped with RAS/RTK (8/45) and JAK/STAT (7/21) mutations.
Table 2: Features of the mutations affecting JAK/STAT pathway
ID |
Age |
Cohort |
Panel |
Gene |
Ex on |
AA change |
Type of mutation |
Domain (Uniprot) |
GQW_211 |
22 |
AYA |
Screening 1 |
JAK2 |
16 |
I682F |
Missense |
TK1 |
GQW_239 |
19 |
AYA |
Screening 1 |
JAK2 |
16 |
R683S |
Missense |
TK1^ |
B-ALL_045 |
32 |
AYA |
Screening 2 |
JAK2 |
16 |
R683S |
Missense |
TK1 |
B-ALL_092 |
16 |
AYA |
Screening 2 |
JAK2 |
16 |
R683G |
Missense |
TK1 |
GQW_182 |
48 |
Adults |
Screening 1 |
JAK2 |
16 |
R683G |
Missense |
TK1 |
GQW_235 |
37 |
Adults |
Screening 1 |
JAK2 |
16 |
R683S |
Missense |
TK1 |
GQW_184 |
51 |
Adults |
Screening 1 |
JAK2 |
16 |
R683G |
Missense |
TK1^ |
GQW_185 |
49 |
Adults |
Screening 1 |
JAK2 |
16 |
R683G |
Missense |
TK1^ |
B-ALL_028 |
47 |
Adults |
Screening 2 |
JAK2 |
16 |
R683S |
Missense |
TK1 |
GQW_255 |
31 |
AYA |
Screening 1 |
IL7R |
5 |
S185C |
Missense |
Extracellular |
B-ALL_073 |
27 |
AYA |
Screening 2 |
IL7R |
6 |
LTIS -> PRC (243..246) |
Inframe indel |
TM^ |
GQW_188 |
44 |
Adults |
Screening 1 |
IL7R |
5 |
S234N |
Missense |
Extracellular |
GQW_181 |
44 |
Adults |
Screening 1 |
IL7R |
6 |
DPILLTIS -> DRGC (239..246) |
Inframe indel |
TM |
GQW_186 |
66 |
Adults |
Screening 1 |
IL7R |
6 |
LLTI -> C (242..245) |
Inframe deletion |
TM |
GQW_198 |
42 |
Adults |
Screening 1 |
IL7R |
6 |
LT -> LMVKGSFNICG (244) |
Inframe insertion |
TM |
B-ALL_03 |
40 |
Adults |
Screening 2 |
IL7R |
6 |
ILLTIS -> MC (241..246) |
Inframe indel |
TM^ |
B-ALL_013 |
48 |
Adults |
Screening 2 |
IL7R |
6 |
LTIS -> QKGEC (243..246) |
Inframe indel |
TM |
B-ALL_07 |
31 |
AYA |
Screening 2 |
CRLF2 |
6 |
F232C |
Missense |
TM |
B-ALL_061 |
27 |
AYA |
Screening 2 |
CRLF2 |
6 |
F232C |
Missense |
TM^ |
B-ALL_028 |
47 |
Adults |
Screening 2 |
CRLF2 |
6 |
F232C |
Missense |
TM^ |
B-ALL_060 |
21 |
AYA |
Screening 1 |
JAK1 |
14 |
A634D |
Missense |
PK1 |
GQW-195 |
43 |
Adults |
Screening 1 |
JAK1 |
16 |
R724C |
Missense |
PK1 |
^Subclonal mutations: detected in <20% of tumor population.
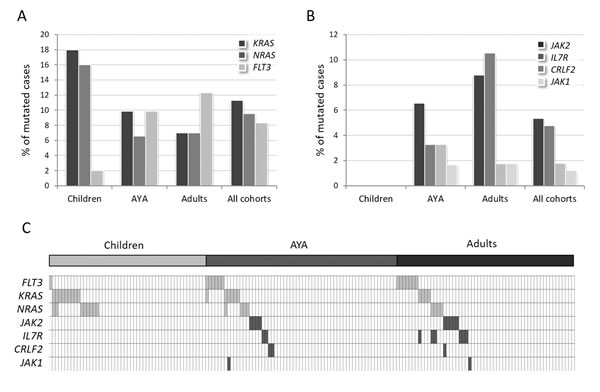
Figure 4: Incidence of RAS/RTK and JAK/STAT pathway mutations in B-NEG ALL. A. Frequency of mutations targeting KRAS, NRAS and FLT3. B. Frequency of mutations targeting JAK2, IL7R, CRLF2, JAK1. C. Distribution of the mutations targeting the RAS/RTK pathway (light grey cells) and JAK/STAT signaling (dark grey cells).
Outcome of B-NEG ALL stratified for age cohorts
To assess whether the mutations identified had a role on patients’ outcome, we evaluated the impact on OS and DFS of RAS/RTK and JAK/STAT pathway mutations. Provided the relatively low incidence of the single gene mutations, clonal mutations (detected in >20% of tumor population) were grouped into the JAK/STAT (IL7R, JAK2, JAK1, CRLF2 mutations and CRLF2-rearrangements) and RAS/RTK (FLT3, KRAS, NRAS) pathways. Among the latter, only the most common hot-spot mutations - namely, ITD and D835 FLT3 mutations and those targeting the G12-13 aminoacidic residues of KRAS and NRAS - were considered [37, 38]. When analyzing the impact of mutations targeting one pathway, samples harboring mutations of the other pathway were filtered out from the WT cohort.
Firstly, the entire B-NEG cohort - comprising children, AYA and adults - was analyzed and we observed that the RAS/RTK pathway mutations did not impact on long-term survival. At variance, the OS of JAK/STAT-mutated cases was significantly shorter than that of WT cases (29.4% vs 62.8% at 4 years; 95% CI 14.1-61.4 and 53.3-73.9, p = 0.003) (Figure 5A).
Samples carrying mutations in both pathways (N = 4) experienced the worse OS when compared to the samples harboring only JAK/STAT or RAS/RTK (p = 0.006); in fact, all the events occurred within 30 months (Figure 5B).
Given the diverse distribution of mutations and the heterogeneous treatments across age cohorts, we repeated these analyses by grouping AYA and adults, whose mutational profile and therapeutic regimens were comparable. This approach led to the results described below.
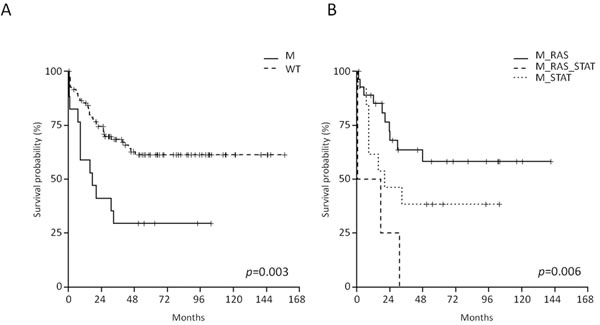
Figure 5: Impact of JAK/STAT pathway mutations on B-NEG ALL patients’ outcome. A. OS of JAK/STAT-mutated vs WT cases (analysis on the entire study population). B. OS of JAK/STAT-mutated vs RAS/RTK-mutated vs double mutant cases (analysis on the entire study population).
Impact of RAS/RTK pathway mutations on B-NEG ALL patients’ outcome
Among the RAS/RTK pathway members, the mutations targeting KRAS/NRAS hotspot exerted the strongest effect on outcome. AYA and adult B-NEG ALL patients harboring RAS mutations displayed a shorter OS at 4 years than WT cases (11.4% vs 50%, CI 95% 1.8-70.8 and 37.8-66.2, p = 0.039) (Figure 6A). RAS-mutated cases had also a significantly shorter disease-free survival (DFS) (p = 0.006), being 0 at 4 years compared to 47.8% (95% CI 33.5-68.3) for WT cases (Figure 6B). On multivariate analysis, RAS mutations and age retained their significance on OS (p = 0.0171 and p = 0.0002, respectively) and DFS (p = 0.0458 and p = 0.0128, respectively).
Contrariwise, in the pediatric cohort the survival of RAS/RTK-mutated cases resembled that observed in WT cases.
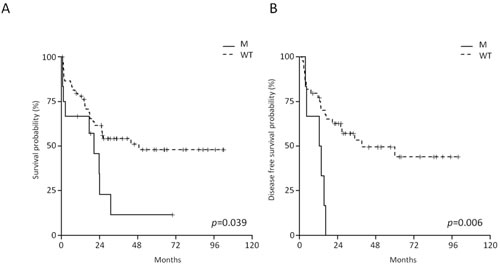
Figure 6: Impact of RAS mutations on the outcome of adults and AYA B-NEG ALL patients. A. OS of RAS-mutated vs WT cases. B. DFS of RAS-mutated vs WT cases.
Impact of JAK/STAT mutations on B-NEG ALL patient’s outcome
The evaluation of the impact of JAK/STAT mutations on B-NEG ALL patient’s outcome was confined to AYA and adult cases, since these mutations were absent in children. This analysis documented the negative impact of JAK/STAT mutations on the OS of adult patients only, with JAK/STAT-mutated cases having a 10% (95% CI 1.6-64.2) survival probability at 4 years, as opposed to 31.3% (95% CI 16.6-59.2) for WT patients (Figure 7A). JAK/STAT-mutated cases also had a DFS at 4 years of 16.7% (95% CI 2.8-99.7) compared to 46.9% (95% CI 25.6-86) for WT patients (p = 0.009, Figure 7B).
On multivariate analysis, JAK/STAT pathway mutations and WBC retained their significance on DFS (p = 0.0259 and p = 0.0273, respectively).
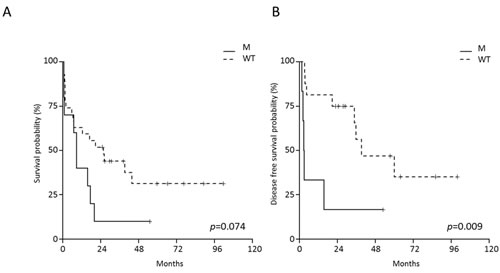
Figure 7: Impact of JAK/STAT mutations on the outcome of adult B-NEG ALL patients. A. OS of JAK/STAT-mutated vs WT. B. DFS of JAK/STAT-mutated vs WT cases.
In vitro sensitivity assays show the efficacy of target specific inhibition
Finally, in order to examine the potential of targeting the RAS/RTK and JAK/STAT pathways in B-NEG ALLs, we performed in vitro sensitivity experiments by treating primary cells from cases harboring known mutations with gene or pathway specific inhibitors.
The FLT3 inhibitors quizartinib and crenolanib and the pan-TK inhibitor ponatinib (0.1 μM) were able to reduce the proliferation rate of primary B-NEG ALL cells from 3 patients carrying FLT3 ITD mutations and 1 harboring a TKD mutation: indeed, after 72 hours of treatment, quizartinib reduced the percentage of proliferating cells to 28.3±9%, crenolanib to 29.5±21.1% and ponatinib to 20.7±7% (Figure 8A). Primary cells from 4 samples harboring RAS mutations - targeting the G12-13 hotspot - were sensitive to PI3K/mTOR inhibitors (0.1 μM): in particular, BEZ235 reduced the percentage of proliferating cells to 21.3±9% and rapamycin to 37.8±21.1%; in addition, cell proliferation decreased to 62.4±16.9% upon treatment with the MEK inhibitor selumetinib (0.1 μM) (Figure 8B). As expected, ruxolitinib and tofacitinib - not targeting FLT3 nor PI3K/mTOR or MEK signaling - proved ineffective in reducing the proliferation of FLT3 as well as KRAS/NRAS-mutated primary cells.
When the sensitivity of primary cells carrying JAK/STAT pathway mutations was tested, ruxolitinib (0.1 μM) proved effective in reducing to 59% the proliferation rate of JAK2-mutated cells from 1 case and rapamycin (0.1 μM) reduced to 35.4% the proliferation of IL7R-mutated primary cells from another patient, in line with a downstream PI3K/mTOR cascade activation.
Beside their anti-proliferative activity, the above-mentioned drugs also exerted a pro-apoptotic effect, particularly evident for FLT3 inhibitors.
To assess whether the response to the selected inhibitors was specifically associated to the mutational status of B-ALL primary cells, we compared it with B-ALL cases that proved negative for FLT3, KRAS and NRAS mutations. The reduction of proliferation induced by crenolanib, quizartinib and ponatinib (0.1 μM) was significantly higher in FLT3-mutated than in FLT3-WT cases (N = 4, p = 0.002, p < 0.0001, p = 0.0004, respectively) (Figure 8C). Similarly, the proliferation rate of RAS-mutated cases was considerably reduced in response to rapamycin and selumetinib (0.1 μM) when compared to RAS-WT B-ALL samples (N = 3, p = 0.024, p = 0.037) (Figure 8D). At variance, BEZ235, which simultaneously targets mTOR and PI3K, was also partly active in reducing the proliferation (50.5±21.4%, 0.1 μM) of RAS-WT primary cells.
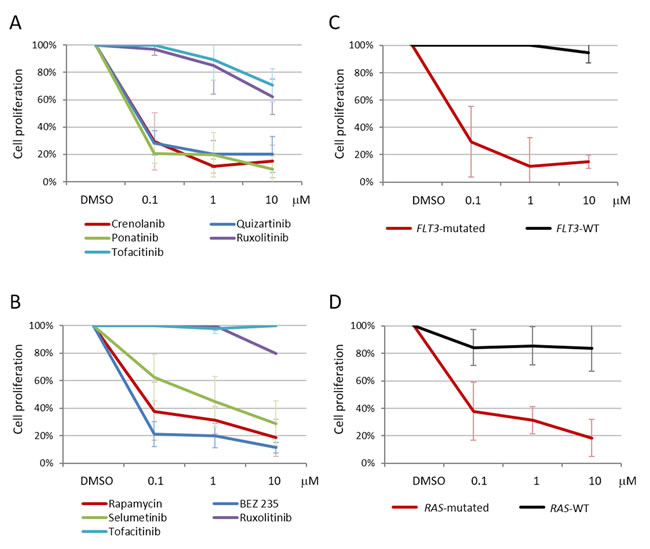
Figure 8: Target-specific inhibitors are effective in reducing B-NEG ALL primary cell proliferation. A. Average values of 3H-thymidine incorporation of primary B-NEG ALL cells from 4 FLT3-mutated samples after 72 hours of treatment; crenolanib (0.1 μM) reduced the percentage of proliferating cells to 29.5±21.1%, quizartinib (0.1 μM) to 28.3±9.0% and ponatinib (0.1 μM) to 20.7±7.0%, on average. Compounds not targeting FLT3 (ruxolitinib and tofacitinib) proved ineffective. B. Average values of 3H-thymidine incorporation of primary B-NEG ALL cells from 4 samples carrying KRAS or NRAS mutations after 72 hours of treatment; BEZ235 (0.1 μM) reduced the percentage of proliferating cells to 21.3±9.0%, rapamycin (0.1 μM) to 37.8±21.1% and selumetinib to 62.4±16.9%, on average. Compounds not targeting PI3K/mTOR or MEK signaling (ruxolitinib and tofacitinib) proved ineffective. Each condition was run in triplicate. C. Average values of 3H-thymidine incorporation of primary B-NEG ALL cells from 4 FLT3-mutated and 4 FLT3-WT samples after 72 hours of treatment with crenolanib (0.1 μM). D. Average values of 3H-thymidine incorporation of primary B-NEG ALL cells from 4 RAS-mutated and 3 RAS-WT samples after 72 hours of treatment with rapamycin (0.1 μM).
Discussion
We describe the first in-depth genomic analysis of a broad cohort of B-ALLs lacking known fusion transcripts, including pediatric, AYA and adult cases. This “genetically orphan” subgroup deserves special attention, since it constitutes 70.5% of childhood, 72.7% of AYA and 42.7% of adult ALL cases [3], represents a suitable setting to identify additional lesions sustaining the heterogeneous clinical scenario between children and adults, and reflects the clinical outcome observed in ALL, where the worse prognosis is observed in adults.
By WES of the discovery panel we found that the load of somatic mutations in B-NEG ALL resembles what observed in other leukemias, being 10.5 per sample [26, 28, 39]. Since few recurrent mutations were detected, the study was broadened to two screening panels, for a total of 168 B-NEG ALL samples. This analysis, focused on the genes commonly altered in ALL, revealed an enrichment of mutations targeting genes involved in B-cell development and protein kinases signaling [25-29].
Among the B-cell development genes, PAX5 mutations were the most common event - occurring in 19% of cases - and increased with age. When PAX5 deletions were also considered, we observed the same pattern, with 44.6% of adults and only 13.3% of children carrying PAX5 genetic lesions. Remarkably, similar differences were found when the analysis of CNAs was extended to other B-cell development genes and cell-cycle genes commonly affected by deletions in B-ALL that tend to accumulate with age. Taken together, these results indicate that the genetic background of children is considerably different from patients older than 15 years old.
When focusing on protein kinases cascades, we found that 26.8% of B-NEG ALL cases carry mutations in the RAS/RTK pathway. The deregulation of this pathway may be due to alterations of upstream signaling molecules, like the receptor tyrosine kinase FLT3, or of essential players of the pathway like NRAS and KRAS, as occurred in our series [37].
Within B-ALL, the role of FLT3, KRAS and NRAS mutations has been investigated mostly in the pediatric setting and has been linked to hyperdiploid, MLL-rearranged ALL, hypodiploid, as well as high-risk pediatric cases, as elucidated by NGS [26, 30-36]. Our findings encourage a thoughtful reconsideration of their role since we documented that FLT3, KRAS and NRAS mutations are also a common event in B-NEG ALL with a different distribution across age cohorts. In particular, FLT3 mutations were specific of AYA and adults, while NRAS and KRAS alterations were prominent in children: in fact, in the latter cohort they collectively accounted for 30% of cases, consistent with the previously reported association with younger age at presentation [36]. Importantly, RAS/RTK mutations were not confined to hyperdiploid cases, as previously reported: in fact, in our cohort mutations were detected in both hyperdiploid and non-hyperdiploid cases.
The clinical impact of RAS/RTK pathway mutations has been explored with contradictory results and seems to be mainly subgroup-dependent. In fact, while several studies, including the largest analysis performed in pediatric cases [36], failed to document any significant effect of RAS mutations on survival, a recent paper focusing on MLL-rearranged infant ALL documented an independent prognostic factor of NRAS/KRAS [33, 35, 36]. Similarly, while the impact of FLT3 mutations has been widely explored in acute myeloid leukemia [40], it is unclear whether FLT3 mutations affect the outcome of ALL patients [26, 30, 31]. In our series, given the low number of mutations affecting each single gene we grouped them according to their pathway. When we evaluated the outcome of patients carrying RAS/RTK mutations, we observed a peculiar pattern: AYA and adult mutated cases - all enrolled in clinical trials for adults - had a significantly reduced OS and DFS when compared to WT patients, while the outcome of pediatric cases was not influenced by these mutations, in line with previous reports, most likely because of the effect of more aggressive regimens.
Overall, these results prompted us to investigate the efficacy of specific inhibitors, in light of their availability. Among the FLT3 inhibitors, the newly introduced quizartinib and crenolanib are promising against ITD and D835 mutations [41]. In our in vitro assays, both compounds, as well as the third-generation pan-TKI ponatinib were effective in reducing the proliferation of FLT3-mutated primary cells.
Since RAS proteins have proven challenging to inhibit, an alternative strategy is to interfere with the RAS signaling at different levels of the pathway [42, 43]. Encouraging results have been obtained with the inhibitors of PI3K/mTOR and MEK1/2 [26, 44]. Consistently, in the current study, samples carrying NRAS or KRAS mutations proved highly sensitive to rapamycin and to the dual PI3K/mTOR inhibitor BEZ235; the proliferation rate was also inhibited by the MEK1/2 inhibitor selumetinib, though at a lower extent.
These findings qualify FLT3, PI3K/mTOR and MEK1/2 inhibitors as alternative therapeutic agents for B-NEG ALL patients, especially in adults/elderly who frequently suffer of comorbidities and often cannot undergo intensive chemotherapeutic approaches.
The other set of mutations that captured our attention were those leading to the JAK/STAT signaling activation, being druggable and frequently altered in other poor prognosis ALL subsets [23-25, 34, 45-47]. Overall, we detected JAK/STAT mutations in 12.5% of B-NEG cases and, importantly, only AYA and adults were affected, with an incidence of 14.7% and 21%, respectively. This skewed distribution of lesions might be ascribed to the characteristics of our study population: in fact, unlike previously published papers describing high-risk and BCR-ABL1-like pediatric ALL cases [23-25, 34, 45-47], our cohort was not chosen for unfavorable clinical features nor for a peculiar GEP, and included all age cohorts. Therefore, our data indicate that unselected pediatric B-ALLs are rarely affected by these mutations, whereas their incidence increases with age. We provide direct evidence that considering only the mutational status of JAK/STAT members, regardless of GEP and concomitant rearrangements, it is possible to single out adult B-NEG ALLs experiencing a worse OS and DFS. As a matter of fact, the cohort of JAK/STAT-mutated only partly overlap with the BCR-ABL1-like profile, thus indicating that the mutational characterization may integrate the identification of BCR-ABL1-like cases, presently not completely elucidated nor straightforward.
The identification of JAK/STAT lesions bears also important therapeutic implications given the possibility of interfering with JAK members or downstream pathways as shown by previous experimental models [42, 43, 48] and our pilot experiments on primary cells.
In conclusion, our study, that was carried out in B-ALL cases without major molecular rearrangements of all ages, implements the current knowledge on ALL. Firstly, we hereby show that the genetic background is significantly different between children, AYA and adults, with the number of deleterious lesions incrementing with age progression; this phenomenon might, at least in part, account for the different outcome between age cohorts. Second we show that RAS/RTK pathway mutations negatively impact on OS and DFS while they do not affect children’s outcome. Along the same line, we show that JAK/STAT mutations confer a worse prognosis within the adult population. Finally, we show that by targeting these lesions we can reduce the in vitro leukemic cell proliferation, thus indicating that specific inhibitors might be integrated in the therapeutic backbone of ALL.
Materials and Methods
Experimental strategy
Bone marrow samples from 168 patients (median age: 23 years, range 1.2-78) with newly diagnosed B-ALL, containing >70% of leukemic blasts and negative for recurrent fusion genes (BCR-ABL1, ETV6-RUNX1, E2A-PBX1, MLL rearrangements, defined B-NEG), were included in the present research. Three age cohorts were considered: 1-15 years (children, 50 patients); 16-35 years (AYA, 61 patients); >36 years (adults, 57 patients). The study involved 3 phases (Figure S5): 1) WES and CNA analysis of 13 samples and their paired germline DNA (discovery panel, Table S4); 2) WES of 68 additional cases (screening panel 1, Table S5); 3) sequencing of selected genes by Sanger or NGS in 87 patients (screening panel 2, Table S5). Further details are provided in Supplemental Material and Table S6.
Sequencing analyses
Tumor and germline genomic DNA (gDNA, 3 µg) from the discovery panel patients and gDNA (1 µg) from the screening panel 1 tumor samples were enriched in protein coding sequences by the SureSelect Human All Exon 50Mb and the SureSelectXT2 Human All Exon kits, respectively (Agilent Technologies, Santa Clara, CA). Captured targets were subjected to massively parallel sequencing (HiSeq 2000 analyzer, Illumina, San Diego, CA) with the paired-end 2 × 100 bp read option (Tables S7 and S8).
Mapping to the hg19/NCBI GRCh37 and identification of tumor-specific variants of the discovery panel was performed as described [39, 49], while the analysis of screening panel 1 required more stringent criteria (Supplemental Material).
After validation by Sanger, frequently mutated genes (PAX5, FLT3, KRAS, NRAS, JAK2, JAK1, IL7R, CRLF2) were sequenced in the screening panel 2 by Sanger sequencing or NGS (Genome Sequencer Junior 454, Roche Applied Science®) (Supplemental Material, Table S9).
CNA and GEP analyses
CNA analysis of the discovery panel was performed with the Genome-Wide Human SNP Array 6.0 (Affymetrix, Santa Clara, CA; GSE67405) and processed as previously described [39]. Recurrent deletions and CRLF2 rearrangements were screened by the Salsa MLPA P335-A3 ALL-IKZF1 kit (MRC-Holland, Amsterdam, the Netherlands) and analyzed as reported [50, 51].
GEP of B-NEG ALLs carrying JAK/STAT mutations was performed by using the HGU133 Plus 2.0 gene chips (Affymetrix) according to manufacturer’s instructions and GEP analyses were carried out by dChip software (http://www.hsph.harvard.edu/cli/complab/dchip/). In order to recognize BCR-ABL1-like cases, gene expression levels of a set of genes specific of the BCR-ABL1-like profile, selected in the context of a previously described dataset [52], were evaluated in JAK/STAT-mutated cases.
Statistical analyses
Patients’ characteristics were compared by chi-squared or Fisher’s exact test for categorical variables. Differences in distributions were assessed by Wilcoxon test for continuous data. OS and DFS curves were estimated by Kaplan-Meier product-limit method and compared using the log-rank test. Multivariate analysis was performed with Cox proportional hazards regression model to adjust the effect of RAS and JAK/STAT pathway mutations for WBC and platelets counts, hemoglobin levels, age and gender, on OS and DFS. All tests were 2-sided, accepting p < 0.05 as index of statistical significance. All analyses relied on SAS v9.4 software.
In vitro assays
Annexin V/7AAD apoptotic test (BD Bioscience, San Josè, CA), MTT (TOX1 assay kit, Sigma Aldrich, St Louis, MO) and 3H-thymidine (Perkin Elmer, Waltham, MA) proliferation assays were performed to assess the sensitivity of primary B-NEG ALL cells carrying mutations of FLT3, KRAS, NRAS, IL7R, JAK2 or WT for the same mutations to increasing doses (0.1-10 μM) of selected inhibitors (Selleck Chemicals Houston, TX) - of tyrosine kinases (ponatinib), FLT3 (quizartinib, crenolanib), PI3K/mTOR/MEK pathway (rapamycin, BEZ235, selumetinib) and JAK1/2 (tofacitinib, ruxolitinib). Drugs were added at time 0 and viability was measured after 48 and 72 hours. Cells were plated at 1x106/ml and each condition was run in triplicate.
Acknowledgments
The authors would like to thank Laura Pasqualucci (Institute for Cancer Genetics and the Herbert Irving Comprehensive Cancer Center, Columbia University, New York, NY) for fruitful discussions and precious suggestions; Genewiz, Inc (South Plainfield, NJ) where the automated DNA sequencing of the validation phase was carried out; Fasteris SA HiSeq Service (Plan-les-Ouates, Switzerland) that performed exome capture and massively parallel sequencing; Roche diagnostics for helpful discussion and support.
Conflicts of Interest
The authors have no conflicts of interest to disclose.
Grant support
This work was supported by Associazione Italiana per la Ricerca sul Cancro (AIRC) Special Program Molecular Clinical Oncology, 5 x 1000, Milan (MCO-10007), Italy, Ministero dell’Istruzione, dell’Università e della Ricerca (MIUR), Fondo per gli investimenti della ricerca di base (FIRB, RBAP11TF7Z_003), Progetto Giovani Ricercatori 2010, Policlinico di Modena (GR-2010-2313609). MM was partly supported by Associazione Cristina Bassi Onlus (Genova). GteK received funding from the EU FP7 under the project ENCCA grant agreement n° 261743. NCI 1 U54 CA193313-01 to RR; Precision Medicine Fellowship (UL1 TR000040) to JW.
Authorship
MM performed experiments, interpreted data and wrote the manuscript; SC designed the study, interpreted data and revised the manuscript; JW and RR developed bioinformatics tools, performed bioinformatic analysis and revised the manuscript; ALF performed the screening of recurrently mutated genes and MLPA experiments; NP performed in vitro assays and analyzed data; FDG and SP performed in vitro assays; VG, FB and GC performed the screening of recurrently mutated genes and analysis of sequencing data; AP and MV performed the statistical analysis of mutations clinical impact; ABH contributed to the copy number experiment analysis; MCP contributed to MLPA experiment and analysis; VA provided biological material; M Mancini, GteK, AT and AV provided clinical samples and data; AG and RF designed the study, analyzed data and critically revised the manuscript.
Editorial note
This paper has been accepted based in part on peer-review conducted by another journal and the authors’ response and revisions as well as expedited peer-review in Oncotarget.
References
1. Redaelli A, Laskin BL, Stephens JM, Botteman MF, Pashos CL. A systematic literature review of the clinical and epidemiological burden of acute lymphoblastic leukaemia (ALL). Eur J Cancer Care. 2005; 14: 53-62.
2. Inaba H, Greaves M, Mullighan CG. Acute lymphoblastic leukaemia. Lancet. 2013; 381: 1943-55.
3. Chiaretti S, Vitale A, Cazzaniga G, Orlando SM, Silvestri D, Fazi P, Valsecchi MG, Elia L, Testi AM, Mancini F, Conter V, te Kronnie G, Ferrara F, et al. Clinico-biological features of 5202 patients with acute lymphoblastic leukemia enrolled in the Italian AIEOP and GIMEMA protocols and stratified in age cohorts. Haematologica. 2013; 98: 1702-10.
4. Silverman LB, Gelber RD, Dalton VK, Asselin BL, Barr RD, Clavell LA, Hurwitz CA, Moghrabi A, Samson Y, Schorin MA, Arkin S, Declerck L, Cohen HJ, Sallan SE. Improved outcome for children with acute lymphoblastic leukemia: results of Dana-Farber Consortium Protocol 91-01. Blood. 2001; 97: 1211-8.
5. Annino L, Vegna ML, Camera A, Specchia G, Visani G, Fioritoni G, Ferrara F, Peta A, Ciolli S, Deplano W, Fabbiano F, Sica S, Di Raimondo F, et al. Treatment of adult acute lymphoblastic leukemia (ALL): long-term follow-up of the GIMEMA ALL 0288 randomized study. Blood. 2002; 99: 863-71.
6. Thomas X, Boiron JM, Huguet F, Dombret H, Bradstock K, Vey N, Kovacsovics T, Delannoy A, Fegueux N, Fenaux P, Stamatoullas A, Vernant JP, Tournilhac O, et al. Outcome of treatment in adults with acute lymphoblastic leukemia: analysis of the LALA-94 trial. J Clin Oncol. 2004; 22: 4075-86.
7. Rowe JM, Buck G, Burnett AK, Chopra R, Wiernik PH, Richards SM, Lazarus HM, Franklin IM, Litzow MR, Ciobanu N, Prentice HG, Durrant J, Tallman MS, et al. Induction therapy for adults with acute lymphoblastic leukemia: results of more than 1500 patients from the international ALL trial: MRC UKALL XII/ECOG E2993. Blood. 2005;106:3760-7.
8. Bassan R, Hoelzer D. Modern therapy of acute lymphoblastic leukemia. J Clin Oncol. 2011; 29: 532-43.
9. Stock W, La M, Sanford B, Bloomfield CD, Vardiman JW, Gaynon P, Larson RA, Nachman J; Children’s Cancer Group; Cancer and Leukemia Group B studies. What determines the outcomes for adolescents and young adults with acute lymphoblastic leukemia treated on cooperative group protocols? A comparison of Children’s Cancer Group and Cancer and Leukemia Group B studies. Blood. 2008; 112: 1646-54.
10. Ribera JM, Oriol A, Sanz MA, Tormo M, Fernández-Abellán P, del Potro E, Abella E, Bueno J, Parody R, Bastida P, Grande C, Heras I, Bethencourt C, Feliu E, Ortega JJ. Comparison of the results of the treatment of adolescents and young adults with standard-risk acute lymphoblastic leukemia with the Programa Español de Tratamiento en Hematología pediatric-based protocol ALL-96. J Clin Oncol. 2008; 26: 1843-9.
11. Nachman JB, La MK, Hunger SP, Heerema NA, Gaynon PS, Hastings C, Mattano LA Jr, Sather H, Devidas M, Freyer DR, Steinherz PG, Seibel NL. Young adults with acute lymphoblastic leukemia have an excellent outcome with chemotherapy alone and benefit from intensive postinduction treatment: a report from the children’s oncology group. J Clin Oncol. 2009; 27: 5189-94.
12. Jabber Al-Obaidi MS, Martineau M, Bennett CF, Franklin IM, Goldstone AH, Harewood L, Jalali GR, Prentice HG, Richards SM, Roberts K, Harrison CJ; Medical Research Council Adult Leukaemia Working Party. ETV6/AML1 fusion by FISH in adult acute lymphoblastic leukemia. Leukemia. 2002; 16: 669-74.
13. Cimino G, Elia L, Mancini M, Annino L, Anaclerico B, Fazi P, Vitale A, Specchia G, Di Raimondo F, Recchia A, Cuneo A, Mecucci C, Pane F, et al. Clinico-biologic features and treatment outcome of adult pro-B-ALL patients enrolled in the GIMEMA 0496 study: absence of the ALL1/AF4 and of the BCR/ABL fusion genes correlates with a significantly better clinical outcome. Blood. 2003; 102: 2014-20.
14. Mancini M, Scappaticci D, Cimino G, Nanni M, Derme V, Elia L, Tafuri A, Vignetti M, Vitale A, Cuneo A, Castoldi G, Saglio G, Pane F, et al. A comprehensive genetic classification of adult acute lymphoblastic leukemia (ALL): analysis of the GIMEMA 0496 protocol. Blood. 2005; 105: 3434-41.
15. Harrison CJ, Moorman AV, Barber KE, Broadfield ZJ, Cheung KL, Harris RL, Jalali GR, Robinson HM, Strefford JC, Stewart A, Wright S, Griffiths M, Ross FM, et al. Interphase molecular cytogenetic screening for chromosomal abnormalities of prognostic significance in childhood acute lymphoblastic leukaemia: a UK Cancer Cytogenetics Group Study. Br J Haematol. 2005; 129: 520-30.
16. Moorman AV, Harrison CJ, Buck GA, Richards SM, Secker-Walker LM, Martineau M, Vance GH, Cherry AM, Higgins RR, Fielding AK, Foroni L, Paietta E, Tallman MS, et al. Karyotype is an independent prognostic factor in adult acute lymphoblastic leukemia (ALL): analysis of cytogenetic data from patients treated on the Medical Research Council (MRC) UKALLXII/Eastern Cooperative Oncology Group (ECOG) 2993 trial. Blood. 2007; 109: 3189-97.
17. Rubnitz JE, Wichlan D, Devidas M, Shuster J, Linda SB, Kurtzberg J, Bell B, Hunger SP, Chauvenet A, Pui CH, Camitta B, Pullen J; Children’s Oncology Group. Prospective analysis of TEL gene rearrangements in childhood acute lymphoblastic leukemia: a Children’s Oncology Group study. J Clin Oncol. 2008; 26: 2186-91.
18. Burmeister T, Schwartz S, Bartram CR, Gökbuget N, Hoelzer D, Thiel E; GMALL study group. Patients’ age and BCR-ABL frequency in adult B-precursor ALL: a retrospective analysis from the GMALL study group. Blood. 2008; 112: 918-9.
19. Moorman AV, Ensor HM, Richards SM, Chilton L, Schwab C, Kinsey SE, Vora A, Mitchell CD, Harrison CJ. Prognostic effect of chromosomal abnormalities in childhood B-cell precursor acute lymphoblastic leukaemia: results from the UKMedical Research Council ALL97/99 randomised trial. Lancet Oncol. 2010; 11:429-38.
20. Cimino G, Cenfra N, Elia L, Sica S, Luppi M, Meloni G, Vignetti M, Paoloni F, Foà R, Mandelli F. The therapeutic response and clinical outcome of adults with ALL1(MLL)/AF4 fusion positive acute lymphoblastic leukemia according to the GIMEMA experience. Haematologica. 2010; 95: 837-40.
21. Kuiper RP, Schoenmakers EF, van Reijmersdal SV, Hehir-Kwa JY, van Kessel AG, van Leeuwen FN, Hoogerbrugge PM. High-resolution genomic profiling of childhood ALL reveals novel recurrent genetic lesions affecting pathways involved in lymphocyte differentiation and cell cycle progression. Leukemia. 2007; 21: 1258-66.
22. Mullighan CG, Goorha S, Radtke I, Miller CB, Coustan-Smith E, Dalton JD, Girtman K, Mathew S, Ma J, Pounds SB, Su X, Pui CH, Relling MV, Evans WE, Shurtleff SA, Downing JR. Genome-wide analysis of genetic alterations in acute lymphoblastic leukaemia. Nature. 2007; 446: 758-64.
23. Den Boer ML, van Slegtenhorst M, De Menezes RX, Cheok MH, Buijs-Gladdines JG, Peters ST, Van Zutven LJ, Beverloo HB, Van der Spek PJ, Escherich G, Horstmann MA, Janka-Schaub GE, Kamps WA, et al. A subtype of childhood acute lymphoblastic leukaemia with poor treatment outcome: a genome-wide classification study. Lancet Oncol. 2009; 10: 125-34.
24. Harvey RC, Mullighan CG, Wang X, Dobbin KK, Davidson GS, Bedrick EJ, Chen IM, Atlas SR, Kang H, Ar K, Wilson CS, Wharton W, Murphy M, et al. Identification of novel cluster groups in pediatric high-risk B-precursor acute lymphoblastic leukemia with gene expression profiling: correlation with genome-wide DNA copy number alterations, clinical characteristics, and outcome. Blood. 2010; 116: 4874-84.
25. Roberts KG, Morin RD, Zhang J, Hirst M, Zhao Y, Su X, Chen SC, Payne-Turner D, Churchman ML, Harvey RC, Chen X, Kasap C, Yan C, et al. Genetic alterations activating kinase and cytokine receptor signaling in high-risk acute lymphoblastic leukemia. Cancer Cell. 2012; 22 :153-66.
26. Holmfeldt L, Wei L, Diaz-Flores E, Walsh M, Zhang J, Ding L, Payne-Turner D, Churchman M, Andersson A, Chen SC, McCastlain K, Becksfort J, Ma J, et al. The genomic landscape of hypodiploid acute lymphoblastic leukemia. Nat Genet. 2013; 45: 242-52.
27. Mullighan CG. Molecular genetics of B-precursor acute lymphoblastic leukemia. J Clin Invest. 2012; 122: 3407-15.
28. Zhang J, Ding L, Holmfeldt L, Wu G, Heatley SL, Payne-Turner D, Easton J, Chen X, Wang J, Rusch M, Lu C, Chen SC, Wei L, Collins-Underwood JR, Ma J, et al. The genetic basis of early T-cell precursor acute lymphoblastic leukaemia. Nature. 2012; 481: 157-63.
29. Tzoneva G, Perez-Garcia A, Carpenter Z, Khiabanian H, Tosello V, Allegretta M, Paietta E, Racevskis J, Rowe JM, Tallman MS, Paganin M, Basso G, Hof J, et al. Activating mutations in the NT5C2 nucleotidase gene drive chemotherapy resistance in relapsed ALL. Nat Med. 2013; 19: 368-71.
30. Taketani T, Taki T, Sugita K, Furuichi Y, Ishii E, Hanada R, Tsuchida M, Sugita K, Ida K, Hayashi Y. FLT3 mutations in the activation loop of tyrosine kinase domain are frequently found in infant ALL with MLL rearrangements and pediatric ALL with hyperdiploidy. Blood. 2004; 103: 1085-8.
31. Armstrong SA, Mabon ME, Silverman LB, Li A, Gribben JG, Fox EA, Sallan SE, Korsmeyer SJ. FLT3 mutations in childhood acute lymphoblastic leukemia. Blood. 2004; 103: 3544-6.
32. Liang DC, Shih LY, Fu JF, Li HY, Wang HI, Hung IJ, Yang CP, Jaing TH, Chen SH, Liu HC. K-Ras mutations and N-Ras mutations in childhood acute leukemias with or without mixed-lineage leukemia gene rearrangements. Cancer. 2006; 106: 950-6.
33. Paulsson K, Horvat A, Strömbeck B, Nilsson F, Heldrup J, Behrendtz M, Forestier E, Andersson A, Fioretos T, Johansson B. Mutations of FLT3, NRAS, KRAS, and PTPN11 are frequent and possibly mutually exclusive in high hyperdiploid childhood acute lymphoblastic leukemia. Genes Chromosomes Cancer. 2008; 47: 26-33.
34. Zhang J, Mullighan CG, Harvey RC, Wu G, Chen X, Edmonson M, Buetow KH, Carroll WL, Chen IM, Devidas M, Gerhard DS, Loh ML, Reaman GH, et al. Key pathways are frequently mutated in high-risk childhood acute lymphoblastic leukemia: a report from the Children’s Oncology Group. Blood. 2011; 118: 3080-7.
35. Driessen EM, van Roon EH, Spijkers-Hagelstein JA, Schneider P, de Lorenzo P, Valsecchi MG, Pieters R, Stam RW. Frequencies and prognostic impact of RAS mutations in MLL-rearranged acute lymphoblastic leukemia in infants. Haematologica. 2013; 98: 937-44.
36. Perentesis JP, Bhatia S, Boyle E, Shao Y, Shu XO, Steinbuch M, Sather HN, Gaynon P, Kiffmeyer W, Envall-Fox J, Robison LL. RAS oncogene mutations and outcome of therapy for childhood acute lymphoblastic leukemia. Leukemia. 2004; 18: 685-92.
37. Annesley CE, Brown P. The Biology and Targeting of FLT3 in Pediatric Leukemia. Front Oncol. 2014; 4: 263.
38. Knight T, Irving JA. Ras/Raf/MEK/ERK Pathway Activation in Childhood Acute Lymphoblastic Leukemia and Its Therapeutic Targeting. Front Oncol. 2014; 4: 160.
39. Messina M, Del Giudice I, Khiabanian H, Rossi D, Chiaretti S, Rasi S, Spina V, Holmes AB, Marinelli M, Fabbri G, Piciocchi A, Mauro FR, Guarini A, et al. Genetic lesions associated with chronic lymphocytic leukemia chemo-refractoriness. Blood. 2014; 123: 2378-88.
40. Swords R, Freeman C, Giles F. Targeting the FMS-like tyrosine kinase 3 in acute myeloid leukemia. Leukemia. 2012; 26: 2176-85.
41. Montalban-Bravo G, Garcia-Manero G. Novel drugs for older patients with acute myeloid leukemia. Leukemia. 2015; 29: 760-9.
42. Chappell WH, Steelman LS, Long JM, Kempf RC, Abrams SL, Franklin RA, Bäsecke J, Stivala F, Donia M, Fagone P, Malaponte G, Mazzarino MC, Nicoletti F, et al. Ras/Raf/MEK/ERK and PI3K/PTEN/Akt/mTOR inhibitors: rationale and importance to inhibiting these pathways in human health. Oncotarget. 2011; 2: 135-64. doi 10.18632/oncotarget.240.
43. Tasian SK, Teachey DT, Rheingold SR. Targeting the PI3K/mTOR Pathway in Pediatric Hematologic Malignancies. Front Oncol. 2014; 4: 108.
44. Irving J, Matheson E, Minto L, Blair H, Case M, Halsey C, Swidenbank I, Ponthan F, Kirschner-Schwabe R, Groeneveld-Krentz S, Hof J, Allan J, Harrison C, et al. Ras pathway mutations are highly prevalent in relapsed childhood acute lymphoblastic leukaemia, may act as relapse-drivers and confer sensitivity to MEK inhibition. Blood. 2014; 124: 3420-30.
45. Mullighan CG, Collins-Underwood JR, Phillips LA, Loudin MG, Liu W, Zhang J, Ma J, Coustan-Smith E, Harvey RC, Willman CL, Mikhail FM, Meyer J, Carroll AJ, et al. Rearrangement of CRLF2 in B-progenitor- and Down syndrome-associated acute lymphoblastic leukemia. Nat Genet. 2009;41: 1243-6.
46. Mullighan CG, Zhang J, Harvey RC, Collins-Underwood JR, Schulman BA, Phillips LA, Tasian SK, Loh ML, Su X, Liu W, Devidas M, Atlas SR, et al. JAK mutations in high-risk childhood acute lymphoblastic leukemia. Proc Natl Acad Sci U S A. 2009; 106: 9414-8.
47. Cario G, Zimmermann M, Romey R, Gesk S, Vater I, Harbott J, Schrauder A, Moericke A, Izraeli S, Akasaka T, Dyer MJ, Siebert R, Schrappe M, Stanulla M. Presence of the P2RY8-CRLF2 rearrangement is associated with a poor prognosis in non-high-risk precursor B-cell acute lymphoblastic leukemia in children treated according to the ALL-BFM 2000 protocol. Blood. 2010; 115: 5393-7.
48. Maude SL, Tasian SK, Vincent T, Hall JW, Sheen C, Roberts KG, Seif AE, Barrett DM, Chen IM, Collins JR, Mullighan CG, Hunger SP, Harvey RC, et al. Targeting JAK1/2 and mTOR in murine xenograft models of Ph-like acute lymphoblastic leukemia. Blood. 2012; 120: 3510-8.
49. Trifonov V, Pasqualucci L, Tiacci E, Falini B, Rabadan R. SAVI: a statistical algorithm for variant frequency identification. BMC Syst Biol. 2013;7 Suppl 2:S2.
50. Palmi C, Lana T, Silvestri D, Savino A, Kronnie GT, Conter V, Basso G, Biondi A, Valsecchi MG, Cazzaniga G. Impact of IKZF1 deletions on IKZF1 expression and outcome in Philadelphia chromosome negative childhood BCP-ALL. Reply to “incidence and biological significance of IKZF1/Ikaros gene deletions in pediatric Philadelphia chromosome negative and Philadelphia chromosome positive B-cell precursor acute lymphoblastic leukemia”. Haematologica. 2013; 98: e164-5.
51. Moorman AV, Enshaei A, Schwab C, Wade R, Chilton L, Elliott A, Richardson S, Hancock J, Kinsey SE, Mitchell CD, Goulden N, Vora A, Harrison CJ. A novel integrated cytogenetic and genomic classification refines risk stratification in pediatric acute lymphoblastic leukemia. Blood. 2014;124:1434-44.
52. Messina M, Chiaretti S, Tavolaro S, Peragine N, Vitale A, Elia L, Sica S, Levis A, Guarini A, Foà R. Protein kinase gene expression profiling and in vitro functional experiments identify novel potential therapeutic targets in adult acute lymphoblastic leukemia. Cancer. 2010;116:3426-37.