
Introduction
Metastatic prostate cancer lacks any effective therapy, although clinical efficacy of the first anti-metastatic drug cabozantinib has shown some promise [1]. An impediment to improve outcome is lack of thorough understanding of the underlying molecular mechanisms. Deciphering key molecular mechanisms and drivers of prostate cancer metastasis will enable identification of new therapeutic targets and avenues, providing for critical advancement in prostate cancer treatment. The discovery and functional validation of recurrent gene fusions of 5 Ets factor genes (ETV1, ETV4, ETV5, ERG, Fli1) in prostate cancer highlights the relevance of Ets transcription factors as oncogenes in prostate cancer [2-4]. The potential role of the non-fusion epithelial-specific Ets factor, SPDEF, as prostate cancer tumor and metastasis suppressor recently emerged, demonstrating that SPDEF expression loss in advanced prostate cancer correlates with poor outcome [5-11]. Thus, downregulation of SPDEF expression may be critical for prostate cancer progression. Indeed, we and others have shown that inhibition of SPDEF expression in prostate cancer cells reduces adhesion and increases migration, invasion and EMT, while overexpression prevents these functions indicating a role of SPDEF in prostate cancer metastasis, but with apparent opposite activity to ERG [5, 6, 12]. The role of SPDEF in metastasis is further supported by the recent study showing that SPDEF knockdown in prostate cancer cells enhances and overexpression reduces survival of prostate cancer cells at distal sites and metastasis formation [11]. SPDEF acts as a tumor suppressor in colorectal cancer through the E-cadherin and β-catenin pathway and may play similar roles in a number of other epithelial cancers [5, 13-15]. While we have demonstrated interaction of SPDEF with the androgen receptor (AR) and various other proteins relevant for cancer signalling [5, 16], no studies have been reported as of yet about the precise regulatory mechanisms of SPDEF protein expression and activity in the context of metastasis.
In this study we sought to gain new insights into the role phosphorylation and protein degradation plays in SPDEF-dependent effects on prostate cancer cell migration and invasion. Our hypothesis is that a shift in SPDEF protein turnover is a key step in prostate cancer metastasis and that degradation of SPDEF initiates a cascade of events associated with metastatic spread of prostate cancer.
Results
GADD45α, like SPDEF, blocks migration and invasion of prostate cancer cells
We previously demonstrated that SPDEF effectively inhibits prostate cancer migration and invasion and may, thus, function as a tumor and metastasis suppressor [5]. Likewise, our data provided strong evidence that reduced SPDEF expression induces migration, invasion and EMT. We also showed that GADD45α and γ induce cell cycle arrest and apoptosis in prostate cancer cells, and NF-κB- as well as JunD-mediated repression of GADD45α and γ are essential for prostate cancer cells to escape cell death [17, 18]. GADD45α expression is repressed due to methylation [17], and reduced SPDEF expression directly correlates with poor outcome in prostate cancer [7, 9]. A review of published gene expression data, moreover, suggests that regulation of SPDEF expression correlates with GADD45α in LNCaP prostate cancer and MCF-7 breast cancer cells. Based on these findings we tested the hypothesis that SPDEF and GADD45α and γ are functionally interconnected and overlap in their tumor suppressive roles in prostate cancer.
We evaluated the impact of SPDEF and GADD45α on migration and invasion of DU145 prostate cancer cells. As previously reported [5], SPDEF reduced both migration and invasion and GADD45α likewise inhibited migration and invasion at a similar rate as SPDEF (Figure 1A-1B and see Additional file 1: Figure S1).
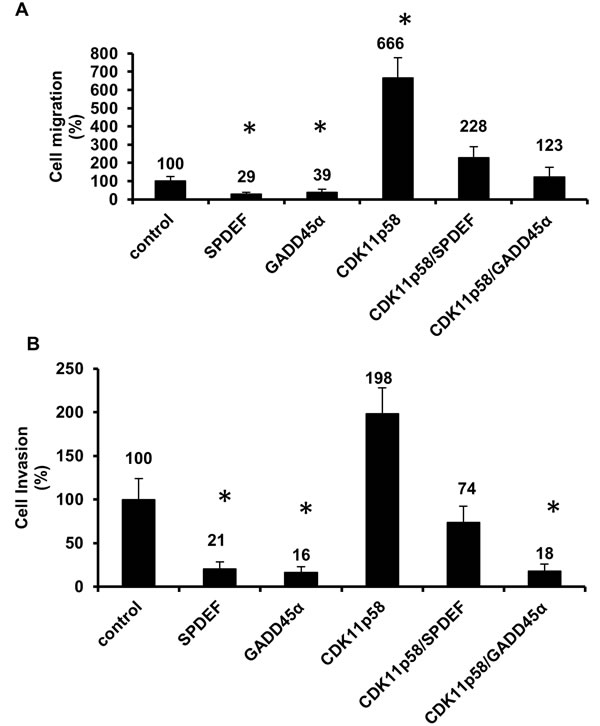
Figure 1: CDK11p58 and GADD45α affect prostate cancer cell migration and invasion. DU145 were transfected with SPDEF-Flag, GADD45α-Flag, CDK11p58-HA, CDK11p58-HA/SPDEF-Flag, CDK11p58-HA/GADD45 α-Flag or mock transfected. A. Migration and B. invasion were measured using transwells 24h post-transfection. Cells were fixed and stained, and 8-10 random microscopic fields were counted and the mean number of the control cells was considered as 100%, and the experimental groups are shown as relative to the control. Student-t test was used to evaluate the statistical difference among the groups compared to the control * indicates statistical significance with p-value < 0.0001.
All three GADD45 family members interact with the cell cycle kinase CDK11p58
Our previous mass spectrometric approach identified interaction partners of GADD45 proteins in vivo using HEK 293T cells, resulting in identification of MEKK4, the upstream kinase of JNK, as an in vivo interactor of all three members of the GADD45 family (α, β and γ) [18]. Here, we further explored these mass spectrometric data from the pull-down experiment and identified the CDK11 protein family member, CDK11p58, as a new in vivo interactor of GADD45γ (see Additional file 2: Figure S2). To validate this result, DU145 cells were transiently transfected with the GADD45γ-Flag expression vector or parental Flag vector as control. Immunoprecipitation was performed to pull down GADD45-interacting proteins using anti-Flag antibody. Western blot analysis of the immunoprecipitated proteins using anti-CDK11p58 antibody showed that CDK11p58 specifically interacts with GADD45γ (Figure 2A). To further confirm this interaction, the proteins were expressed in vitro in a cell-free system and the translated GADD45γ-Flag and CDK11p58-HA proteins were either combined or kept separate. Immunoprecipitation using anti-Flag-agarose beads followed by Western blot analysis using anti-HA antibody confirmed that CDK11p58 directly binds to GADD45γ, since CDK11p58 was only pulled down in the presence of GADD45γ (Figure 2B).
CDK11p58directly interacts with SPDEF
Since reduced SPDEF expression/activity is a key driver of metastasis in prostate cancer, we tested our hypothesis that the interplay between GADD45α and CDK11p58 elicits its effects on prostate cancer migration and invasion through modulation of SPDEF expression or activity. We tested whether SPDEF interacts with CDK11p58 and/or GADD45α. We co-transfected DU145 cells with CDK11p58-HA and SPDEF-Flag or Flag expression vectors and performed immunoprecipitation using anti-Flag antibody followed by Western blot analysis with anti-HA antibody. As shown in Figure 2C, CDK11p58 co-immunoprecipitates with SPDEF, but not with Flag. To evaluate whether CDK11p58-SPDEF interaction is direct, we expressed the SPDEF-GST fusion protein in a bacterial system. GST-pull down confirmed that CDK11p58-HA directly interacts with SPDEF (Figure 2D).
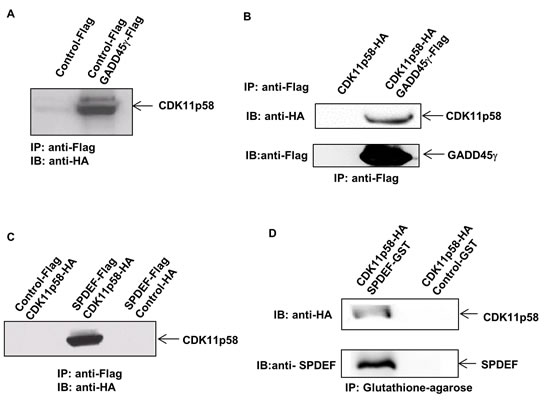
Figure 2: Interaction of SPDEF, CDK11p58 and GADD45γ. A. DU145 cells were transfected with GADD45γ-Flag and CDK11p58-HA. Protein extracts were immunoprecipitated using anti-FLAG mAb and interaction with CDK11p58 was detected by anti-HA antibody. B. CDK11p58 interacts directly with GADD45γ. GADD45γ-Flag and CDK11p58-HA proteins were in vitro translated and immunoprecipitated using anti-FLAG mAb. Interaction with CDK11p58 was detected by anti-HA antibody. C. CDK11p58 interacts with SPDEF. DU145 cells were transfected with SPDEF-Flag and CDK11p58-HA. Protein extracts were immunoprecipitated using anti-FLAG mAb. Interaction with CDK11p58 was detected by anti-HA antibody. D. Direct interaction between CDK11p58 and SPDEF. GST and SPDEF-GST proteins were incubated with in vitro translated CDK11p58 and immunoprecipitated using glutathione-agarose beads and after SDS-PAGE immunoblotted with anti-HA and anti-PDEF antibodies.
GADD45α inhibits CDK11p58-mediated increase in migration and invasion
We evaluated whether all three GADD45 family proteins, GADD45α, GADD45β and GADD45γ, bind to CDK11p58. HEK 293T cells were transfected with expression vectors for the GADD45-Flag family members and CDK11p58-HA. Immunoprecipitation using anti-Flag antibody followed by Western blot analysis using anti-HA antibody demonstrated that CDK11p58 interact with GADD45α, β and γ proteins (Figure 3A).
To investigate the impact of interaction between GADD45α and CDK11p58 on CDK11p58 function, we evaluated the effect of transiently transfected CDK11p58 on migration and invasion of DU145 cells. In contrast to SPDEF and GADD45α, CDK11p58 strongly increased prostate cancer migration and invasion (Figure 1A-1B). However, co-transfection with CDK11p58 and GADD45α significantly reduced CDK11p58-dependent migration and invasion (Figure 1A-1B), suggesting that GADD45α interaction with CDK11p58 attenuates the pro-metastatic activity of CDK11p58.
The impact of CDK11p58 on migration and invasion was confirmed in DU145 cells infected with a lentivirus encoding shRNA against CDK11p58 (DU145-CDK11p58-/-) or GFP (DU145GFP-/-). Analysis of a wound-healing assay in DU145GFP-/- and DU145CDK11p58-/- cell lines showed a slower rate of migration of DU145CDK11p58-/- cells as compared to the control DU145GFP-/- cells (see Additional file 1: Figure S1C).
In addition, GST pull down of SPDEF-GST and GADD45α-Flag did not shown any direct interaction (Figure 3B). This result was further corroborated by immunoprecipitation of in vitro translated SPDEF-myc and GADD45α-Flag proteins using anti-myc agarose or anti-Flag agarose (Figure 3C-3D). Thus, SPDEF,GADD45α and GADD45γ directly interact with CDK11p58, but not with each other.
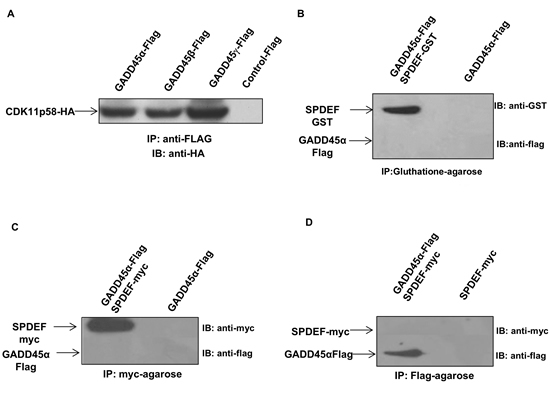
Figure 3: Interaction of SPDEF, GADD45 and CDK11p58 proteins. A. CDK11p58 binds to the GADD45 family genes. HEK293T cells were transfected with GADD45α-Flag, GADD45β-Flag, GADD45γ-Flag, and CDK11p58-HA constructs. Protein extracts were immunoprecipitated using anti-FLAG mAb antibody, and the presence of interaction with CDK11p58 were probed by anti-HA antibody. B. GADD45 does not interact with SPDEF. SPDEF-GST protein was expressed in BL21 bacteria cells followed by GST-pull down assay and GADD45 protein was in vitro translated using TNT® Quick Coupled transcription/translation system (Promega). GST and SPDEF-GST proteins were incubated with in vitro translated GADD45 protein for 16 hours and subjected to SDS-PAGE gel. Immunoblot was performed using anti-GST and anti-flag antibodies, (C and D)GADD45 does not interact with SPDEF. SPDEF-myc and GADD45 α-Flag proteins were in vitro translated using TNT ® Quick Coupled transcription/translation system (Promega), immunoprecipitated using anti Myc-agarose or anti Flag-agarose and subjected to SDS-PAGE gel. The detection of SPDEF-myc protein was performed using anti-myc antibodies, while GADD45α-Flag was detected using anti-FLAG antibodies.
CDK11p58 phosphorylates SPDEF
The nuclear Ser/Thr kinase CDK11p58 is involved in degradation of several transcriptional factors such as Estrogen receptor α (ERα) and AR [19-21]. We evaluated whether, as a consequence of CDK11p58 interaction with SPDEF, CDK11p58 phosphorylates SPDEF. An in vitro kinase assay with SPDEF in the presence or absence of CDK11p58 followed by Western blot analysis using monoclonal antibodies against phospho-Serine (pSer) demonstrated phosphorylation of SPDEF on serine residues only in the presence of CDK11p58 (see Additional file 3: Figure S3).
GADD45α inhibits CDK11p58 kinase activity
GADD45 family members interact with various signaling kinases [22, 23]. In some instances GADD45 proteins activate the interacting kinase such as MTK1/MEKK4, whereas interaction of all three GADD45 family members with CDK1 inhibits CDK1 kinase activity [23]. To determine whether GADD45α enhances or decreases CDK11p58 kinase activity, an in vitro CDK11p58 kinase assay with SPDEF as a substrate was performed. No significant serine phosphorylation on SPDEF occurred when only GADD45α was present. CDK11p58 induced SPDEF phosphorylation which was completely inhibited by GADD45α indicating that GADD45α blocks the kinase activity of CDK11p58 (Figure 4A).
To determine whether inhibition of CDK11p58 kinase activity by GADD45α prevents CDK11p58-mediated SPDEF degradation, the effect of GADD45α on SPDEF degradation was tested in HEK 293T cells in the presence of CDK11p58. Western blot analysis for SPDEF revealed that overexpression of GADD45α, β and γ prevents SPDEF degradation mediated by CDK11p58 (Figure 4B). These data confirm that CDK11p58 regulation of SPDEF protein stability is controlled by GADD45 and that shifts in the levels of GADD45 and CDK11p58 proteins may play an important role in prostate cancer migration and invasion.
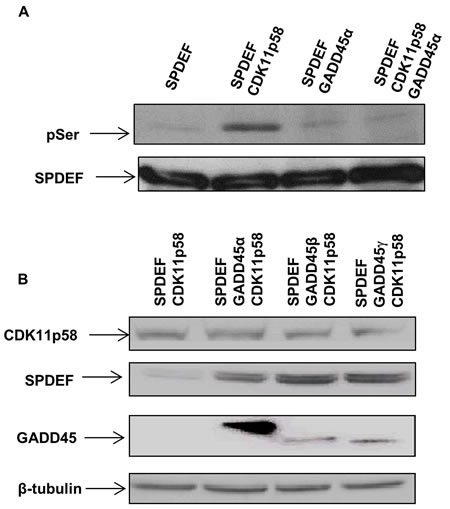
Figure 4: GADD45 inhibits CDK11p58 kinase activity and SPDEF degradation. A. Phosphorylation of SPDEF by CDK11p58 is inhibited by GADD45 proteins. SPDEF, GADD45 and CDK11p58 were produced in vitro by TNT® Quick Coupled transcription/translation system (Promega) and combinations of SPDEF and mock or SPDEF and GADD45 were subjected to in vitro kinase assay mediated by CDK11p58. SPDEF was detected using anti-flag and phosphorylated SPDEF was detected with anti-serine antibody. B. CDK11p58-mediated degradation of SPDEF is blocked by GADD45. Western blot of HEK 293T cells transfected with SPDEF-Flag and CDK11p58-HA in absence or presence of GADD45α-Flag, GADD45β-Flag or GADD45γ-Flag using anti-Flag and anti-HA antibodies.
CDK11p58 induces degradation of SPDEF protein
CDK11p58 induces ubiquitin/proteasome dependent protein degradation of several transcription factors [20, 21, 24]. To gain insights into the functional consequences of CDK11p58-mediated phosphorylation of SPDEF, we applied gain-of-function and loss-of-function of CDK11p58 assays. We developed DU145 cell lines either overexpressing CDK11p58 protein (DU145-CDK11p58+/+) or knocking down CDK11p58 protein expression (DU145-CDK11p58-/-). DU145-CDK11p58+/+ cells showed a 13 fold increase in CDK1158 expression and DU145-CDK11p58-/- cells had a 55% decrease in CDK11p58 expression (Figure 5A). Western blot analysis of endogenous SPDEF of DU145-CDK11p58-/- and DU145-CDK11p58+/+ cells revealed a 43% decrease of SPDEF protein in DU145-CDK11p58+/+ cells and a 1.8 fold increase in DU145-CDK11p58-/- cells when compared to parental DU145 cells (Figure 5B), corroborating that CDK11p58-mediated phosphorylation of SPDEF induces SPDEF degradation. Immunoprecipitation of SPDEF-Flag in DU145 and DU145-CDK11p58-/- in the presence of ubiquitin-myc with and without the proteasome inhibitor MG132, which inhibits proteasome mediated degradation of ubiquitinated proteins, followed by Western blot analysis with anti-myc antibody demonstrates that SPDEF is poly-ubiquitinated and degraded by the proteasome pathway (Figure 5C), since MG132 increase poly-ubiquinated SPDEF protein levels. In contrast, in CDK11p58 knockdown cells, the increase in SPDEF ubiquitination upon MG132 treatment is reduced (Figure 5C). These findings indicate that CDK11p58 induces SPDEF degradation through the ubiquitination pathway.
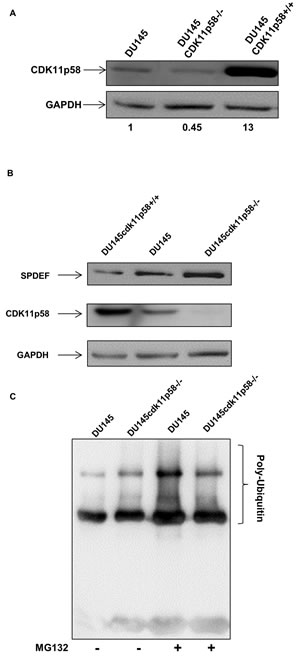
Figure 5: Degradation of SPDEF mediated by CDK11p58. A. Differential expression of CDK11p58 expression in DU145-CDK11p58-/-, DU145-CDK11p58+/+ and DU145 cell lines. Whole cell extract was subjected to SDS-Page and immunoblot performed using anti-HA and anti-GAPDH (Bio Rad) antibodies. B. Differential expression of SPDEF. DU145-CDK11p58-/-, DU145-CDK11p58+/+ and DU145 cell lines were transfected with CDK11p58-HA and SPDEF-flag vectors and protein were analysed by western-blot. Detection of CDK11p58 by anti HA-HRP antibody (Sigma), anti-SPDEF and anti-GAPDH (Bio Rad) antibodies. C. Ubiquitination of SPDEF is mediated by CDK11p58. DU145 and DU145-CDK11p58-/- cells were transfected with SPDEF-Flag and Ubiquitin-myc vectors. 24 hours after transfection, cells were treated with MG132 for 4 hours and 500μg of total protein were immunoprecipitated using anti-Flag antibody and ubiquitin was detected with anti-myc antibody.
SPDEF, CDK11p58, and ubiquitin co-localize in the nucleus
To establish the cellular location of interaction between CDK11p58 and SPDEF, SPDEF-Flag and Ubiquitin-myc expression vectors were transfected into DU145 cells in the absence or presence of the CDK11p58-HA expression vector. Confocal laser microscopy of transfected cells revealed that SPDEF-Flag is localized in the nucleus, while Ubiquitin-myc is distributed in the cytoplasm and the nucleus and does not specifically co-localize with SPDEF (Figure 6A). Co-transfection of CDK11p58 results in a clear co-localization of CDK11p58, SPDEF and Ubiquitin-myc in clusters inside the nucleus (Figure 6B). Similar analysis indicates that GADD45α and CDK11p58 co-localize in both the nucleus and cytoplasm (see Additional file 4: Figure S4).
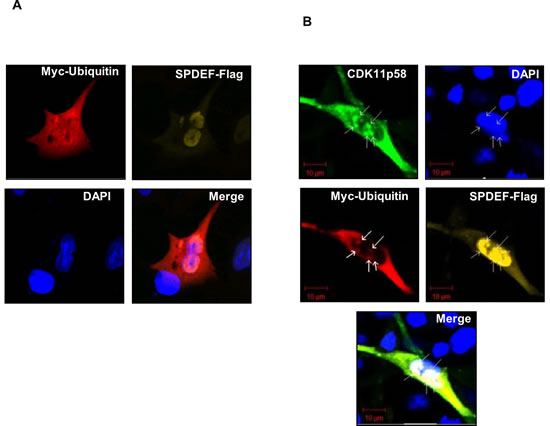
Figure 6: Co-localization of SPDEF, CDK11p58 and ubiquitin. DU145 cells were transfected with A. SPDEF-Flag and Ubiquitin-myc and B. CDK11p58-HA, Ubiquitin-myc and SPDEF-Flag vectors. 24 hours after transfection cells were treated with MG132 for 4 hours, fixed, and incubated for 1 hour with antibodies against HA-FITC, Myc-Cy3 and against Flag produced in rabbit and then incubated for 1 hour with antibody against rabbit conjugated with Cy5 and DAPI. Cells were analysed by Confocal Laser Scanning Microscope (Carl Zeiss). Magnification, X 200.
Mutation analysis of SPDEF identifies phosphorylation sites for CDK11p58 involved in protein degradation
Candidate SPDEF phosphorylation sites were predicted by the NetPhos2.0 server. To identify which sites are responsible for SPDEF phosphorylation and consequent degradation by CDK11p58 we replaced high scoring candidate serine phosphorylation sites of SPDEF with alanine. Some of these mutants contained more than one mutation (S44A/S46A, S61A, S61A/S62A, S220A, S231A/S234A, S238A/S242A/S243A, S308A). We co-transfected HEK 293T cells with CDK11p58-HA plasmid together with wild type or mutant SPDEF plasmids. Western blot analysis demonstrated that only one of the mutant constructs, consisting of mutations of serine 238, 242 and 243, resulted in increased levels of SPDEF protein expression as compared to wild type SPDEF (Figure 7A), indicating that alterations of the serine residues 238, 242 and/or 243 may increase protein stability and may be the targets of CDK11p58 kinase activity. An in vitro kinase assay comparing phosphorylation of SPDEFwt to mutated SPDEF-S238A/S242A/S243A (SPDEFmu) supported this conclusion, since levels of phosphorylation of this mutant were reduced in the presence of CDK11p58 as compared to wild type SPDEF (Figure 7B).
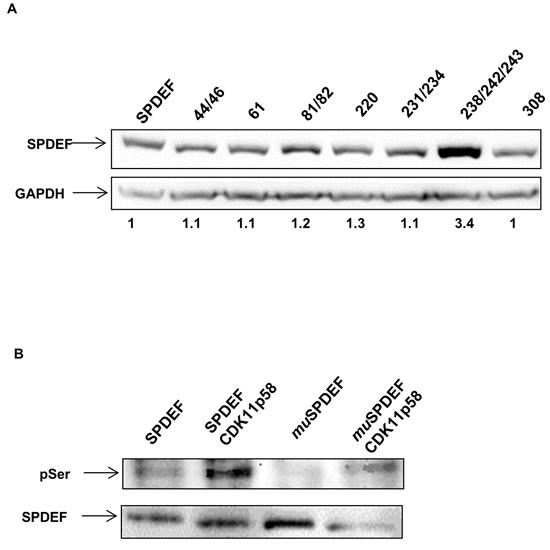
Figure 7: SPDEF mutational analysis. A. Western-blot analysis of SPDEF and SPDEF mutant proteins. SPDEF and SPDEF mutant plasmids were transfected in HEK 293T and immunoblotted using anti-SPDEF antibody. B. In vitro kinase assay of SPDEF and SPDEF mutated in the residues 238/242/243 by CDK11p58 and detection using anti p-serine antibody.
CDK11p58 counteracts the metastasis suppressor activity of SPDEF
To further confirm that CDK11p58 enhances migration and invasion of prostate cancer cells by inducing degradation of SPDEF, a critical step in prostate cancer metastasis, we measured migration and invasion of DU145 cells overexpressing SPDEF in the absence or presence of CDK11p58. While SPDEF suppressed migration and invasion, CDK11p58 completely reversed the metastasis suppressive function of SPDEF, most likely as a consequence of CDK11p58-mediated SPDEF degradation (Figure 1A-1B).
Deregulation of various Ets factors leads to altered expression of key genes required for cancer progression. SPDEF has been previously shown to act as a negative regulator of tumor progression and to elicit its metastasis suppressor function by regulating a gene expression program linked to maintaining the epithelial phenotype, while knock down of SPDEF induces EMT by reducing expression of epithelial-specific genes such as E-cadherin and cytokeratin 18 and enhancing expression of mesenchymal genes that promote prostate cancer migration, invasion and metastasis [5, 13-15, 25]. Indeed SPDEF directly transactivates the E-cadherin gene in prostate cancer [10]. We measured expression of two SPDEF target genes, cytokeratin 18 and E-cadherin, by real-time PCR in DU145 knockdown CDK11p58 (DU145-CDK11p58-/-) and SPDEF-overexpressing DU145 cells as compared to DU145 cells expressing siRNA against GFP (DU145-GFP-/-). Compared to control cells (DU145-GFP-/-), overexpression of SPDEF induced cytokeratin 18 and E-cadherin expression (Figure 8A). The same upregulation of cytokeratin 18 and E-cadherin expression was obtained when CDK11p58 expression was knocked down (Figure 8B), further supporting our conclusions that CDK11p58 impacts prostate cancer cell migration and invasion through regulation of SPDEF protein stability. Similar effects of CDK11p58 and SPDEF on Cytokeratin 18 and E-cadherin expression were seen in another prostate cancer cell line, PC-3 (data not shown).
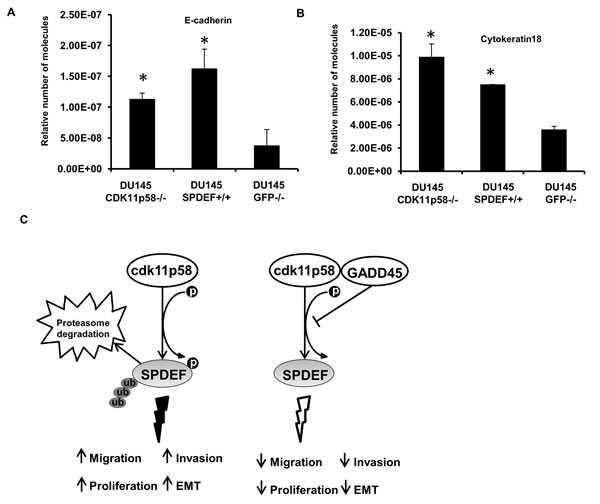
Figure 8: SPDEF induces expression of genes associated with epithelial phenotype. Real-time PCR for Cytokeratin-18 A. and E-cadherin B. in DU145GFP-/-, DU145-CDK11p58-/- and DU145-SPDEF+/+ cells. Student-t test was used to evaluate the statistical difference among the groups compared to the control. * indicates statistical significance with p-value < 0.01. C. Schematic representation of SPDEF regulation by CDK11p58 in prostate cancer cells.
Discussion
We have identified an intricate pathway of SPDEF regulation that involves CDK11p58-dependent phosphorylation of SPDEF on amino acids residues 238, 242 and/or 243, resulting in SPDEF ubiquitination and degradation through the proteasome pathway, and inhibition of CDK11p58 activity by direct interaction with GADD45α and other GADD45 family members, which induce apoptosis in prostate cancer cell lines [18] and are repressed by promoter methylation in PCa [26] Thereby, the precise level of SPDEF protein and the actual metastasis-suppressive or -promoting activity can be tightly regulated by unique and dynamic signaling inputs.
We demonstrate that CDK11p58 directly interacts with SPDEF. The CDK11 protein family is encoded by two nearly identical genes, CDC2L1 and CDC2L2, and forms three main isoforms, CDK11p110, CDK11p58, and CDK11p46 [27]. CDK11p58 inhibits p21 activated kinase 1 (PAK1) [20], ERα [28], vitamin D receptor (VDR) [19] and AR [21] by inducing their phsophorylation and degradation through the proteasome pathway. Under normal physiological conditions CDK11p58 is primarily expressed during G2/M, is essential for cell cycle progression during mitosis and has been linked to tumorigenesis, although the precise role in cancer remains unknown [29, 30]. In osteosarcoma CDK11 is overexpressed and correlates with poor outcome [31]. CDK11 is critical for proliferation and escape from programmed cell death [31]. Overexpression of CDK11p58 is observed in pancreatic cancer cells and multiple myeloma, promoting proliferation and protecting cells from programed cell death [32]. With regard to prostate cancer, one report suggests that CDK11p58 expression is reduced in primary prostate cancer tissue; however, neither kinase activity nor level of activation of CDK11p58 in metastatic prostate cancer was tested [24].
Oncogenic activities of Ets fusion proteins in prostate cancer have been studied in detail; however, potential tumor and metastasis suppressor activities of Ets factors have been neglected until recently, when we and others showed that SPDEF is a tumor and metastasis suppressor in prostate cancer [5, 11]. Metastasis, the main cause of mortality in prostate cancer, involves changes in adhesion, migration and invasion and likely includes EMT-like properties, a switch from an epithelial to a mesenchymal gene expression program [33]. Although the EMT-like phenotype plays a critical role in progression and metastasis of various cancer types, EMT per se has not been typically observed in prostate cancer. Nevertheless, evidence of EMT-like changes exists during prostate cancer progression, particularly reduced expression of epithelial markers and increased expression of mesenchymal markers. We show that CDK11p58-induced SPDEF degradation reduces expression of the epithelial-specific genes E-cadherin and cytokeratin 18 indicating the CDK11p58-SPDEF axis may play an important role in cancer cell migration and invasion.
A key role for SPDEF in cancer cell migration, invasion and metastasis has emerged, although with opposite conclusions in different cancer types. Whereas, SPDEF overexpression has been shown to inhibit breast cancer cell growth and reduce invasion and migration [5-7, 13, 15], other studies indicate that SPDEF overexpression induces invasion and migration in breast cancer cells [34]. We and others previously demonstrated that loss of SPDEF expression induces morphologic changes, increased migration and invasiveness, and triggers a transcriptional program of genes involved in TGFβ signaling, migration, invasion, adhesion, and epithelial dedifferentiation, establishing SPDEF as a critical regulator of key aspects linked to metastasis in prostate, breast and colon cancer [5, 13-16, 35]. The relevance of SPDEF downregulation for prostate cancer metastasis has been confirmed in xenograft models, supporting our previous conclusions [5, 11, 12]. Most importantly, reduced SPDEF expression.directly correlates with poor outcome in prostate cancer [7-11]. Moreover, reduced SPDEF protein expression during prostate and breast cancer progression has been attributed to dysregulated miRNAs [6]. To decipher the critical steps involved in prostate cancer metastasis, we investigated the relevance of regulation of SPDEF protein stability in invasion and migration of prostate cancer cells and report now a new regulatory mechanism of SPDEF expression and activity that impact on prostate cancer metastasis (Figure 8C). Our migration and invasion data shown here indicate that silencing CDK11p58 and consequent inhibition of SPDEF degradation reduce prostate cancer cell migration. In contrast, CDK11p58 overexpression induced cancer cell migration and invasion, while re-expression of SPDEF or GADD45α, which we demonstrate here to bind CDK11p58 and inhibit its kinase activity, reduced migration and invasion.
We identified CDK11p58 as an interaction partner of GADD45α by co-immunoprecipitation analysis. We corroborated these data by co-immunoprecipitation in vitro translation assays, showing that all three members of the GADD45 family interact with CDK11p58. GADD45 proteins have been shown to interact with and inhibit the kinase activity of CDK1 [23, 36]. Here we demonstrate that GADD45α overexpression inhibits CDK11p58 kinase activity and impedes SPDEF degradation mediated by CDK11p58, inhibiting prostate cancer cell migration and invasion. These results clearly demonstrate a new GADD45α mechanistic role during prostate cancer cell migration. The GADD45 gene family (GADD45α, β and γ) has been implicated in regulation of several cellular functions including DNA repair, cell cycle control, apoptosis, senescence and genotoxic stress. Emerging functional evidence implies that GADD45 proteins serve as tumor suppressors in response to diverse stimuli [22] and our results support this notion as GADD45α inhibited SPDEF degradation and thereby reduced migration and invasion. We previously demonstrated that NF-κB-mediated repression of GADD45α and γ is essential for prostate cancer survival [18], and GADD45α expression in prostate cancer is repressed due to methylation [17]. Enhanced GADD45α expression sensitized prostate cancer cells to docetaxel [26]. Some of these critical activities of GADD45α in prostate cancer may be mediated through its regulation of SPDEF protein stability. In summary, the regulation of protein degradation of SPDEF by CDK11p58 may be a crucial factor for inducing prostate cancer cell migration and invasion, with major consequences in prostate cancer metastasis and systemic dissemination. Additionally, GADD45’s role in the regulation of CDK11p58 mediating SPDEF degradation introduces a novel activity of GADD45 in cancer cell migration and invasion.
materials and Methods
Cell culture
DU145 and HEK 293T cells were obtained from American Type Culture Collection (Rockville, MD) and maintained in Dulbecco’s Modified Eagle Medium (DMEM) supplemented with 5% fetal bovine serum (FBS) (Gibco, Invitrogen) and 1% penicillin (5000u/ml)/streptomycin (500μg/ml) (Lonza, Walkersville, MD, USA) at 37°C in a humidified atmosphere of 5% CO2. All cell lines were obtained from ATCC and authenticated via short tandem repeat (STR) profiling performed by the company. The experiments were carried out within 6 months of their resuscitation
Vectors
SPDEF-Flag and SPDEF-myc constructs were obtained by cloning the SPDEF coding sequence into pcDNA3-Flag and pcDNA3-myc vectors, respectively. SPDEF-GST plasmid was obtained by cloning the SPDEF into PGEX-3 vector. Mutations in the SPDEF gene were generated by substitution of serine to alanine using the Quick site-directed mutagenesis II kit (Agilent) and SPDEF-Flag vector as DNA template. The specific locations of the mutated serine residues in the SPDEF protein were: 44/46, 61, 81/82, 220, 231/234, 238/242/243, 308. All the mutated SPDEF sites were sequenced and the mutations confirmed (data not shown). The vectors expressing the CDK11p58-HA and the CDK11p46-myc were kindly donated by Dr. Jianxin Gu, Shanghai Medical Center of Fudan University, P.R China.
Transfection
Transfections of 5 x 105 cells were carried out with 1μg plasmid DNA using Lipofectamine Plus (Invitrogen). Cells were incubated for 24 or 48 hours in the presence or absence of 1mM MG132.
In vitro transcription/translation
GADD45, SPDEF and CDK11p58 proteins were produced in vitro using the TnT® Quick Coupled Transcription/Translation Systems (Promega) according to the manufacterer’s instructions and were used immediately for binding and in vitro kinase studies.
Immuoprecipitation and mass spectrometry analysis to identify GADD45 interacting proteins
Immuprecipitations were performed as previously described [18] using anti-FLAG, anti-HA agarose or anti-myc agarose (Sigma) and eluted using Flag peptide (150μg /ml) or HA peptide (100μg /ml) (Sigma). For mass spectrometry studies, the eluted samples were subjected to SDS-polyacrylamide gel, stained by colloidal Coomassie (Bio-Rad). Prominent bands were cut out, digested with trypsin overnight and analyzed by mass spectrometry as described before [18]. The acquired peptide masses were interrogated by ProFound, a protein identification database. For direct interaction studies proteins were produced by in vitro transcription/translation kit (Promega) following the manufacturer’s protocol and mixed in the presence of the appropriate agarose beads.
In vitro kinase assays
HEK 293T cells were transfected with 1μg of plasmid expressing CDK11p58-HA using Lipofectamine Plus (Invitrogen) in a 6-well plate and incubated for 24 hours. The cells were then washed with PBS and scraped in the presence of lysis buffer (Cell Signaling) and a protease inhibitor cocktail (cOmplete Protease Inhibitor Cocktail, Roche). The lysate was centrifuged with lysis buffer and 200μg of the total protein was immunoprecipitated with anti- HA agarose (Sigma) for 16 hours. The HA-agarose beads were washed 5 times in PBS and once with in vitro kinase buffer (50 mM Tris-HCl (pH 7.5), 15 mM MgCl2, 1 mM DDT). The resultant immunoprecipitate was analyzed for CDK11p58 kinase activity in in vitro kinase buffer, 50 mM ATP, and SPDEF-GST (2-5 mg/ml). SPDEF phosphorylation was analyzed by 12% SDS polyacrylamide gel and Western blotting using a phospho-serine (pSer) antibody (Chemicon, AB1603). In order to test the inhibitory potential of GADD45, we produced the GADD45 and SPDEF proteins using the TnT® Quick Coupled Transcription/Translation Systems (Promega) and mixed in the in vitro kinase reaction.
GST Pull-down assay
GST or GST fusion proteins were expressed in BL21 cells, and equal amounts of bacterial lysates were incubated with 25μl of glutathione-Sepharose beads for 30 min. The beads were then washed three times with GST Lysis buffer (20 mM Tris-Cl (pH 8.0), 200 mM NaCl, 1 mM EDTA (pH 8.0), 0.5% Triton X-100), and incubated with 5μl of in vitro translated (TnT® Quick Coupled Transcription/Translation Systems - Promega) proteins in protein binding buffer for 16 hours at 4°C. The beads were then washed five times with binding buffer and boiled in SDS sample buffer.
Immunofluorescence
Formaldehyde-fixed, permeabilized cells cultured on glass coverslips were incubated with primary antibodies, anti-myc-Cy3 (Sigma), anti-Flag produced in rabbit (Sigma), and anti-HA-FITC (Sigma) for 1 hour at 37 oC, followed by incubation with secondary anti-rabbit-Cy5 (KPL) and DAPI. Cells were analysed on a confocal laser scanning microscope (Karl Zeiss).
Generation of stable cell lines
DU145 CDK11p58 knock-down (DU145CDK11p58-/-) cells were generated by transducing DU145 cells with the MISSION Lentiviral Transduction Particles (Sigma-Aldrich) encoding siRNA against CDK11p58 (clone ID: TRCN0000006992 - target sequence: CGTATAGAAGAGAAGACTCAA) and selected using 10μg /ml puromycin (Invitrogen). DU145 cells overexpressing the CDK11p58 protein (DU145CDK11p58+/+) were generated by transfection with the CDK11p58-HA expression vector and selected with 1 mg/ml G418 (Sigma-Aldrich).
Real-time PCR
Total RNA was harvested using QIAshredder (Qiagen, Valencia, CA) and the RNeasy mini kit (Qiagen). Real time PCR analysis of Cytokeratin 18 and E-cadherin was performed as described [5].
Invasion and migration assays
Cell migration and invasion assays were performed as described [5] using a modified transwell chamber migration assay and invasion assay matrigel-coated membrane (BD Biosciences Bedford, MA).
Wound healing assay
Wound healing assays were carried out in DU145 and DU145-CDK11p58-/- cells. A scratch was created in near-confluent cell cultures using the tip of a P-1000 pipetman. The plates were photographed at 0, 8, 16 and 20h using microscope digital camera (Olympus sc30) and Analysis GetIt software at three different points down the length of the scratch. The images were then analysed using WCIF Image J software.
Western blot analysis
Western blot analysis was performed as described [5, 18]. The primary antibodies used for Western blot analysis were anti-phospho-serine (Chemicon), β-actin (Santa Cruz), anti-Flag-HRP (Sigma), anti-HA-HRP (Sigma), anti-myc-HRP (Sigma), anti-SPDEF (Santa Cruz), and anti-CDK11p58 (Santa Cruz).
Statistical analysis
Statistical analysis was performed using Student’s t-test was used to evaluate the statistical difference among the groups compared to the control.
CoNFLICTS OF interests
The authors declare that they have no competing interests.
Financial Support
International Centre for Genetic Engineering and Biotechnology (LZ); National Institute of Health Grants CA006516 (TAL) and CA85467 (TAL); Department of Defense grant PC051217 (TAL); Prostate Cancer Foundation Award (TAL). R.E.T. and JDP were recipients of the ICGEB post-doctoral fellowship.
Editorial note
This paper has been accepted based in part on peer-review conducted by another journal and the authors’ response and revisions as well as expedited peer-review in Oncotarget.
References
1. Wu Y, Rosenberg JE, Taplin ME. Novel agents and new therapeutics in castration-resistant prostate cancer. Curr Opin Oncol. 2011; 23: 290-296.
2. Yu J, Yu J, Mani RS, Cao Q, Brenner CJ, Cao X, Wang X, Wu L, Li J, Hu M, Gong Y, Cheng H, Laxman B, et al. An integrated network of androgen receptor, polycomb, and tmprss2-erg gene fusions in prostate cancer progression. Cancer Cell. 2010; 17: 443-454.
3. Kunderfranco P, Mello-Grand M, Cangemi R, Pellini S, Mensah A, Albertini V, Malek A, Chiorino G, Catapano CV, Carbone GM. Ets transcription factors control transcription of ezh2 and epigenetic silencing of the tumor suppressor gene nkx3.1 in prostate cancer. PLoS One. 2010; 5: e10547.
4. Tomlins SA, Laxman B, Varambally S, Cao X, Yu J, Helgeson BE, Cao Q, Prensner JR, Rubin MA, Shah RB, Mehra R, Chinnaiyan AM. Role of the tmprss2-erg gene fusion in prostate cancer. Neoplasia. 2008; 10: 177-188.
5. Gu X, Zerbini LF, Otu HH, Bhasin M, Yang Q, Joseph MG, Grall F, Onatunde T, Correa RG, Libermann TA. Reduced pdef expression increases invasion and expression of mesenchymal genes in prostate cancer cells. Cancer Res. 2007; 67: 4219-4226.
6. Turner DP, Findlay VJ, Moussa O, Semenchenko VI, Watson PM, LaRue AC, Desouki MM, Fraig M, Watson DK. Mechanisms and functional consequences of pdef protein expression loss during prostate cancer progression. Prostate. 2011; 71: 1723-1735.
7. Ghadersohi A, Sharma S, Zhang S, Azrak RG, Wilding GE, Manjili MH, Li F. Prostate-derived ets transcription factor (pdef) is a potential prognostic marker in patients with prostate cancer. Prostate. 2011; 71: 1178-1188.
8.Haller AC, Tan W, Payne-Ondracek R, Underwood W, Tian L, Morrison C, Li F. High spdef may identify patients who will have a prolonged response to androgen deprivation therapy. Prostate. 2014; 74: 509-519.
9. Johnson TR, Koul S, Kumar B, Khandrika L, Venezia S, Maroni PD, Meacham RB, Koul HK. Loss of pdef, a prostate-derived ets factor is associated with aggressive phenotype of prostate cancer: Regulation of mmp 9 by pdef. Mol Cancer. 2010; 9:148.
10. Pal M, Koul S, Koul HK. The transcription factor sterile alpha motif (sam) pointed domain-containing ets transcription factor (spdef) is required for e-cadherin expression in prostate cancer cells. J Biol Chem. 2013; 288: 12222-12231.
11. Steffan JJ, Koul S, Meacham RB, Koul HK. The transcription factor spdef suppresses prostate tumor metastasis. J Biol Chem. 2012; 287: 29968-29978.
12. Findlay VJ, Turner DP, Yordy JS, McCarragher B, Shriver MR, Szalai G, Watson PM, Larue AC, Moussa O, Watson DK. Prostate-derived ets factor regulates epithelial-to- mesenchymal transition through both slug-dependent and independent mechanisms. Genes Cancer. 2011; 2: 120-129. doi: 10.1177/1947601911410424.
13. Ghadersohi A, Pan D, Fayazi Z, Hicks DG, Winston JS, Li F. Prostate-derived ets transcription factor (pdef) downregulates survivin expression and inhibits breast cancer cell growth in vitro and xenograft tumor formation in vivo. Breast Cancer Res Treat. 2007; 102: 19-30.
14. Horst D, Gu X, Bhasin M, Yang Q, Verzi M, Lin D, Joseph M, Zhang X, Chen W, Li YP, Shivdasani RA, Libermann TA. Requirement of the epithelium-specific ets transcription factor spdef for mucous gland cell function in the gastric antrum. J Biol Chem. 2010; 285: 35047-35055.
15. Moussa O, Turner DP, Feldman RJ, Sementchenko VI, McCarragher BD, Desouki MM, Fraig M, Watson DK. Pdef is a negative regulator of colon cancer cell growth and migration. J Cell Biochem. 2009; 108: 1389-1398.
16. Oettgen P, Finger E, Sun Z, Akbarali Y, Thamrongsak U, Boltax J, Grall F, Dube A, Weiss A, Brown L, Quinn G, Kas K, Endress G et al. Pdef, a novel prostate epithelium- specific ets transcription factor, interacts with the androgen receptor and activates prostate-specific antigen gene expression. J Biol Chem. 2000; 275: 1216-1225.
17. Zerbini LF, Libermann TA. Gadd45 deregulation in cancer: Frequently methylated tumor suppressors and potential therapeutic targets. Clin Cancer Res. 2005; 11: 6409-6413.
18. Zerbini LF, Wang Y, Czibere A, Correa RG, Cho JY, Ijiri K, Wei W, Joseph M, Gu X, Grall F, Goldring MB, Zhou JR, Libermann TA. Nf-kappa b-mediated repression of growth arrest- and dna-damage-inducible proteins 45alpha and gamma is essential for cancer cell survival. Proc Natl Acad Sci U S A. 2004; 101: 13618-13623.
19. Chi Y, Hong Y, Zong H, Wang Y, Zou W, Yang J, Kong X, Yun X, Gu J. Cdk11p58 represses vitamin d receptor-mediated transcriptional activation through promoting its ubiquitin-proteasome degradation. Biochem Biophys Res Commun. 2009; 386: 493-498.
20. Kong X, Gan H, Hao Y, Cheng C, Jiang J, Hong Y, Yang J, Zhu H, Chi Y, Yun X, Gu J. Cdk11p58 phosphorylation of pak1 ser174 promotes dlc2 binding and roles on cell cycle progression. J Biochem. 2009; 146: 417-427.
21. Zong H, Chi Y, Wang Y, Yang Y, Zhang L, Chen H, Jiang J, Li Z, Hong Y, Wang H, Yun X, Gu J. Cyclin d3/ cdk11p58 complex is involved in the repression of androgen receptor. Mol Cell Biol. 2007; 27: 7125-7142.
22. Tamura RE, de Vasconcellos JF, Sarkar D, Libermann TA, Fisher PB, Zerbini LF. Gadd45 proteins: Central players in tumorigenesis. Curr Mol Med. 2012; 12: 634-651.
23. Vairapandi M, Balliet AG, Hoffman B, Liebermann DA. Gadd45b and gadd45g are cdc2/cyclinb1 kinase inhibitors with a role in s and g2/m cell cycle checkpoints induced by genotoxic stress. J Cell Physiol. 2002; 192: 327-338.
24. Chi Y, Wang L, Xiao X, Wei P, Wang Y, Zhou X. Abnormal expression of cdk11p58 in prostate cancer. Cancer Cell Int. 2014; 14:2.
25. Turner DP, Findlay VJ, Kirven AD, Moussa O, Watson DK. Global gene expression analysis identifies pdef transcriptional networks regulating cell migration during cancer progression. Mol Biol Cell. 2008; 19: 3745-3757.
26. Ramachandran K, Gopisetty G, Gordian E, Navarro L, Hader C, Reis IM, Schulz WA, Singal R. Methylation- mediated repression of gadd45alpha in prostate cancer and its role as a potential therapeutic target. Cancer Res. 2009; 69: 1527-1535.
27. Beyaert R, Kidd VJ, Cornelis S, Van de Craen M, Denecker G, Lahti JM, Gururajan R, Vandenabeele P, Fiers W. Cleavage of pitslre kinases by ice/casp-1 and cpp32/casp-3 during apoptosis induced by tumor necrosis factor. J Biol Chem. 1997; 272: 11694-11697.
28. Qi CJ, Qian KQ, Ning YL, Ma HB, Wang SZ, Zhang XG. Ligation or cross-linking of cd40 has different effects on ags gastric cancer cells. Cell Immunol. 2009; 259: 135-140.
29. Chen HH, Wang YC, Fann MJ. Identification and characterization of the cdk12/cyclin l1 complex involved in alternative splicing regulation. Mol Cell Biol. 2006; 26: 2736- 2745.
30. Hu D, Valentine M, Kidd VJ, Lahti JM. Cdk11(p58) is required for the maintenance of sister chromatid cohesion. J Cell Sci. 2007; 120: 2424-2434.
31. Duan Z, Zhang J, Choy E, Harmon D, Liu X, Nielsen P, Mankin H, Gray NS, Hornicek FJ. Systematic kinome shrna screening identifies cdk11 (pitslre) kinase expression is critical for osteosarcoma cell growth and proliferation. Clin Cancer Res. 2012; 18: 4580-4588.
32. Tiedemann RE, Zhu YX, Schmidt J, Yin H, Shi CX, Que Q, Basu G, Azorsa D, Perkins LM, Braggio E, Fonseca R, Bergsagel PL, Mousses S, et al. Kinome-wide rnai studies in human multiple myeloma identify vulnerable kinasetargets, including a lymphoid-restricted kinase, grk6. Blood. 2010 ;115: 1594-1604.
33. Giannelli G, Bergamini C, Fransvea E, Sgarra C, Antonaci S. Laminin-5 with transforming growth factor-beta1 induces epithelial to mesenchymal transition in hepatocellular carcinoma. Gastroenterology. 2005; 129: 1375-1383.
34. Sood AK, Saxena R, Groth J, Desouki MM, Cheewakriangkrai C, Rodabaugh KJ, Kasyapa CS, Geradts J. Expression characteristics of prostate-derived ets factor support a role in breast and prostate cancer progression. Hum Pathol. 2007; 38: 1628-1638.
35. Cho JY, Lee M, Ahn JM, Park ES, Cho JH, Lee SJ, Kim BG, Heo SH, Park HJ, Zerbini LF, Hwang D, Libermann TA. Proteomic analysis of a pdef ets transcription factor- interacting protein complex. J Proteome Res. 2009; 8: 1327-1337.
36. Zhan Q, Antinore MJ, Wang XW, Carrier F, Smith ML, Harris CC, Fornace AJ, Jr. Association with cdc2 and inhibition of cdc2/cyclin b1 kinase activity by the p53- regulated protein gadd45. Oncogene. 1999; 18: 2892-2900.