
INTRODUCTION
Colorectal cancer (CRC) is a complex disease in which various types of molecular alterations are implicated [1]. Gene expression is largely deregulated [2], following diverse patterns that allow molecular subtyping into groups with diverse prognosis [3]. Stage I and II CRC patients have a moderate risk of relapse after surgical resection, whereas stage III patients have a higher chance of recurrence. Taking advantage of genome-wide techniques, a large number of studies have performed gene expression profiling to identify molecular prognosis biomarkers useful to discriminate between good and poor prognosis CRC tumors [4, 5], although no marker has been yet adopted in routine clinical practice [6]. In addition to gene expression, diverse aberrations at DNA level have been widely described to contribute to CRC progression such as copy number aberrations, point mutations or altered methylation [7, 8]. Also, increasing data support the main role of tumor microenvironment in CRC progression and prognosis [9, 10]. Tumor microenvironment is composed by a heterogeneous population of stromal cells such as fibroblasts, extracellular matrix components, and also immune cells [11]. Indeed, evasion of immune surveillance and/or suppression of immune system have been described as a hallmark of cancer [12].
The cellular machinery is able to recognize mutant proteins and cleave them via the proteasome. Then, the generated peptides bind to MHC molecules to be presented to T cells [13]. In cancer cells, somatic mutations generate cancer-specific antigens that could be targeted [14]. In fact, an association between HLA expression and prognosis has been described in various types of cancer, including CRC [15]. Immune system activation has been specifically recognized in CRC tumors with DNA mismatch repair deficiencies such as microsatellite instable (MSI) CRC, that accumulate an elevated number of point mutations [16]. The high mutational load in MSI tumors creates many tumor-specific neoantigens, typically 10 to 50 times more than microsatellite stable (MSS) tumors [17]. Indeed, it has been proposed that the higher level of neoantigens and lymphocytic reaction in MSI tumors may contribute to a better patient survival [18]. The MSI subset of colorectal tumors is therefore a good candidate for checkpoint immunotherapy [19], however not as much research has been devoted to study the role of immune system activation in response to mutations in the subset of MSS tumors. Even for MSS tumors, a high number of neoepitopes could be associated a better prognosis.
Exome sequencing has been revealed as a useful technique for mutation discovery in cancer [20, 21]. This technique is also useful to predict, from DNA mutations, putative short peptides (neopeptides) capable to elicit immunoreactivity through their presentation at the cell surface as major histocompatibility complex (MHC) ligands. Recently, our group performed an exome sequencing analysis of a homogenous group of microsatellite stable (MSS) stage II colorectal tumors and described the landscape of somatic mutations [22]. Here we aim to compare the mutational profile of good and poor prognosis stage II MSS CRC tumors, and to explore whether the activation of the immune system in response to somatic mutations is related to prognosis.
RESULTS
No significant association was found between single nucleotide variants (SNVs) and prognosis
A total of 3,597 potentially functional single nucleotide variants (SNVs) were found across the 42 sequenced tumors. No differences on the total number of mutations between good and poor prognosis groups were detected (Figure 1A). When SNVs were classified by functional effect, only “missense near splice site” mutations were marginally significant (P=0.04, Supplementary Figure 1). Good prognosis tumors accumulated 1961 SNVs (median 98, range 45 to 153) whereas relapsing tumors accumulated 1675 SNVs (median 86, range 2 to 146). The median difference was 12 (Mann-Whitney Test p-value=0.23). From these, only 23 were recurrent (found in at least 2 tumors): 16 were shared by good and poor prognosis groups, 4 were good-exclusive (2 located in WASL gene, 1 in ERP27 and 1 in C3orf27) and 3 were poor-exclusive (located in AGTRAP, SYNPO2, and APC genes). No single mutation was associated with prognosis, though the low frequency of recurrent mutations derived in a low power to detect specific differences.
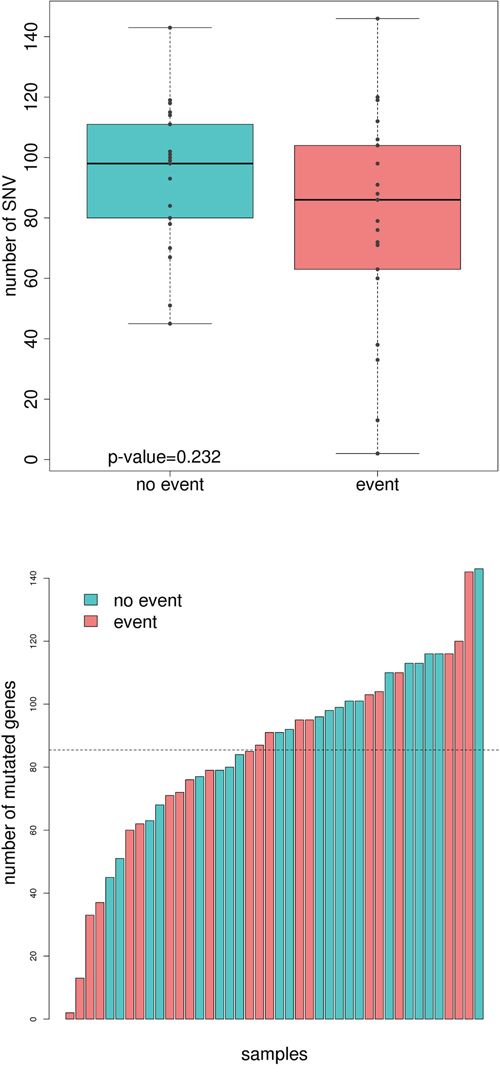
Figure 1: Comparison of total number of mutations and mutated genes between good and poor-prognosis tumors. A. Boxplot showing the number of mutations in the group of tumors with no event (turquoise) and event (light red). B. Number of mutated genes in each sequenced sample. Those tumors from patients with no event are painted in turquoise whereas the ones who relapse are painted in light red.
Prognosis association of SNVs cumulated by genes, pathways, and their position into the human interactome
SNVs were mapped into genes harboring them. The average number of mutated genes per sample was 83 (range: 2-136). In line with SNV results, no differences between the total number of mutated genes between good and poor prognosis groups were detected (Figure 1B). The good prognosis tumors had 1618 mutated genes (median 96, range 45 to 143) whereas the tumors with worse prognosis accumulated 1426 (median 85, range 2 to 142, median difference 11, Mann-Whitney test p-value=0.21). Both groups shared 270 genes (8%), when the expected was less than 1%, so they were probably related to common pathways in carcinogenesis. Regarding recurrently and exclusively mutated genes, 12 genes were found to be mutated in more than 3 poor-prognosis tumors including BRAF and POLE. On the other hand, good-prognosis tumors had 15 exclusively-mutated genes including NOTCH3 (Table 1). Not surprisingly, the well-known APC, TP53, KRAS, along with FAT4, FBXW7, and RYR3 were equally mutated in both groups.
Table 1: Recurrently and exclusively mutated genes
Good prognosis |
|||
|---|---|---|---|
Gene |
# Tumors |
Description |
Reported in literature |
DNHD1 |
4 |
Involved in microtubule motor activity |
--- |
PCDH17 |
4 |
Potential calcium-dependent cell-adhesion protein |
Hu X et al. Protocadherin 17 acts as a tumour suppressor inducing tumour cell apoptosis and autophagy, and is frequently methylated in gastric and colorectal cancers. J Pathol. 2013 Jan;229(1):62-73. |
USH2A |
4 |
Involved in hearing and vision |
Kim N et al. Somatic mutaome profile in human cancer tissues. Genomics Inform. 2013 Dec;11(4):239-44. doi: 10.5808/GI.2013.11.4.239. Epub 2013 Dec 31. |
ADCY2 |
3 |
Membrane-bound, calmodulin-insensitive adenylyl cyclase |
Fang LT et al. Comprehensive genomic analyses of a metastatic colon cancer to the lung by whole exome sequencing and gene expression analysis. Int J Oncol. 2014 Jan;44(1):211-21. |
ATP1B1 |
3 |
ATPase activator activity |
Selvakumar P et al. Epigenetic silencing of Na, K-ATPase β 1 subunit gene ATP1B1 by methylation in clear cell renal cell carcinoma. Epigenetics. 2014 Apr;9(4):579-86. |
CAD |
3 |
This protein is a “fusion” protein encoding four enzymatic activities of the pyrimidine pathway |
--- |
ELFN1 |
3 |
Postsynaptic protein that regulates circuit dynamics in the central nervous system |
--- |
ERP27 |
3 |
Endoplasmic reticulum resident protein 27 |
--- |
ESR1 |
3 |
Nuclear hormone receptor |
Caiazza F, et al. Estrogen receptors and their implications in colorectal carcinogenesis. Front Oncol. 2015 Feb 2;5:19. |
F7 |
3 |
Coagulation factor |
Naderi A. Coagulation factor VII is regulated by androgen receptor in breast cancer. Exp Cell Res. 2015 Feb 1;331(1):239-50. |
ITIH1 |
3 |
May act as a carrier of hyaluronan in serum or as a binding protein between hyaluronan and other matrix protein |
Hamm A et al. Frequent expression loss of Inter-alpha-trypsin inhibitor heavy chain (ITIH) genes in multiple human solid tumors: a systematic expression analysis. BMC Cancer. 2008 Jan 28;8:25. |
LHX5 |
3 |
Plays an essential role in the regulation of neuronal differentiation and migration |
--- |
NFATC1 |
3 |
Plays a role in the inducible expression of cytokine genes in T-cells |
Tripathi MK et al. Nuclear factor of activated T-cell activity is associated with metastatic capacity in colon cancer. Cancer Res. 2014 Dec 1;74(23):6947-57. |
NOTCH3 |
3 |
Functions as a receptor for membrane-bound ligands Jagged1, Jagged2 and Delta1 to regulate cell-fate determination |
Wang XW et al. MicroRNA-206 attenuates tumor proliferation and migration involving the downregulation of NOTCH3 in colorectal cancer. Oncol Rep. 2015 Mar;33(3):1402-10. |
PTPRS |
3 |
Receptor-type tyrosine-protein phosphatase implicated in cell adhesion |
Wang ZC et al. PTPRS Acts as A Metastatic Suppressor in Hepatocellular Carcinoma by Control of EGFR Induced Epithelial-Mesenchymal Transition. Hepatology. 2015 May 22. |
Poor prognosis |
|||
Gene |
# Tumors |
Description |
Reported in literature |
CSMD1 |
5 |
Membrane receptor |
Farrell C et al. Somatic mutations to CSMD1 in colorectal adenocarcinomas. Cancer Biol Ther. 2008 Apr;7(4):609-13. |
BRAF |
3 |
Protein kinase involved in the transduction of mitogenic signals from the cell membrane to the nucleus. |
Clancy C et al. BRAF mutation is associated with distinct clinicopathological characteristics in colorectal cancer: a systematic review and meta-analysis. Colorectal Dis. 2013 Dec;15(12):e711-8. |
CACNA1G |
3 |
Involved in a variety of calcium-dependent processes, including gene expression, cell motility, cell division and cell death |
Berg M et al. Molecular subtypes in stage II-III colon cancer defined by genomic instability:early recurrence-risk associated with a high copy-number variation and loss of RUNX3 and CDKN2A. PLoS One. 2015 Apr 16;10(4):e0122391. |
CCDC168 |
3 |
Coiled-coil domain-containing protein 168 |
--- |
DPYD |
3 |
degradation of the chemotherapeutic drug 5-fluorouracil |
Innocenti F. DPYD variants to predict 5-FU toxicity: the ultimate proof. J Natl Cancer Inst. 2014 Nov 7;106(12). |
DSCAML1 |
3 |
Cell adhesion molecule that plays a role in neuronal self-avoidance. |
--- |
NRG1 |
3 |
Transcriptional repressor that binds NRG1 response elements (NRE) of target promoters |
Lim B, et al. Genome-wide mutation profiles of colorectal tumors and associated liver metastases at the exome and transcriptome levels. Oncotarget. 2015 Jun 1. |
PAPPA2 |
3 |
Metalloproteinase which specifically cleaves IGFBP-5 |
--- |
POLE |
3 |
Participates in DNA repair and in chromosomal DNA replication |
Haraldsdottir S, et al. Colon and endometrial cancers with mismatch repair deficiency can arise from somatic, rather than germline, mutations. Gastroenterology. 2014 Dec;147(6):1308-1316. |
ROBO2 |
3 |
Receptor implicated in cellular migration |
Je EM, et al. Frameshift mutations of axon guidance genes ROBO1 and ROBO2 in gastric and colorectal cancers with microsatellite instability. Pathology. 2013 Dec;45(7):645-50. |
SLITRK5 |
3 |
Suppresses neurite outgrowth |
--- |
SPTA1 |
3 |
The major constituent of the cytoskeletal network underlying the erythrocyte plasma membrane |
Iwakawa R, et al. Expression and clinical significance of genes frequently mutated in small cell lung cancers defined by whole exome/RNA sequencing. Carcinogenesis. 2015 Jun;36(6):616-21. |
Next, a pathway enrichment analysis was performed, stratified by prognosis group. Both groups were enriched in mutated genes that mapped to pathways classically related to cancer, like p53 signaling or Wnt pathway. However, tumors in poor prognosis group had more mutations in pathways related to DNA repair and polymerase activity whereas good prognosis tumors had more mutations in genes related to cell cycle or apoptosis, among others (Table 2 and Supplementary Table 1).
Table 2: Mutated pathways and functions in each group of patients
Good prognosis |
||||
|---|---|---|---|---|
FUNCTION |
score good |
score poor |
dif.score |
p-val |
APOPTOSIS (KEGG) |
0.903 |
0.432 |
-0.472 |
0.001 |
SMALL CELL LUNG CANCER (KEGG) |
0.828 |
0.452 |
-0.376 |
0.012 |
DILATED CARDIOMYOPATHY (KEGG) |
0.837 |
0.540 |
-0.297 |
0.039 |
VEGF SIGNALING PATHWAY (KEGG) |
0.817 |
0.538 |
-0.279 |
0.037 |
CELL CYCLE (KEGG) |
0.621 |
0.365 |
-0.256 |
0.020 |
LIPID KINASE ACTIVITY (GO) |
1.863 |
0.487 |
-1.376 |
0.002 |
INOSITOL OR PHOSPHATIDYLINOSITOL KINASE ACTIVITY (GO) |
1.242 |
0.325 |
-0.917 |
0.002 |
Poor prognosis |
||||
FUNCTION |
score good |
score poor |
dif.score |
p-val |
NUCLEOTIDE EXCISION REPAIR (KEGG) |
0.113 |
0.398 |
0.285 |
0.050 |
TIGHT JUNCTION (KEGG) |
0.389 |
0.654 |
0.265 |
0.019 |
DNA POLYMERASE ACTIVITY (GO) |
0.000 |
0.812 |
0.812 |
0.008 |
DNA DIRECTED DNA POLYMERASE ACTIVITY (GO) |
0.000 |
0.626 |
0.626 |
0.008 |
Finally, genes harboring mutations were mapped into the human interactome and analyzed to test the hypothesis that relevant genes might have central positions in networks (i.e act as hubs). However, no significant differences were found in the centrality measures degree (P=0.65), betwenness (P=0.47), closeness (P=0.56), and eccentricity (P= 0.38), between the two groups of tumors.
Number of neoantigens and expression levels of genes involved in antigen presentation machinery is associated with prognosis, and this association changes within molecular subtypes
Somatic missense mutations can activate T cytotoxic responses. Thus, we used bioinformatics tools to predict putative neoantigens by combining information about HLA genotypes and missense mutations in each patient. Then, we assessed if a relationship existed between the number of hypothetical neoantigens generated by the tumor and the patients’ outcome. No significant differences between good and poor prognosis groups of tumors were found (Figure 2), even if the level of expression of genes harboring missense mutations were taken into account (Supplementary Figure 2). However, a trend was identified when extremes were compared: 4 out of 5 tumors with 0 predicted neoantigens had relapsed whereas 5 out of 6 tumors with more than 8 predicted neoantigens had not relapsed. We were intrigued by the relapse of the tumor with the highest number of predicted neoantigens (n=16). Taking advantage of gene expression data, we realized that this tumor had a marked under-expression of indispensable genes for the neoantigen presentation such as HLA-A, HLA-B, HLA-C, TAP1, and TAP2. Consequently, under the hypothesis that neoantigens only activate immune response if correctly presented by antigen presentation machinery, the levels of expression of these genes were analyzed in the complete sample of 98 tumors of the Colonomics study with expression data that included those 42 analyzed by NGS. Indeed, gene expression levels of genes related to antigen presentation were associated with prognosis. Poor outcome was observed for tumors under-expressing HLA-B (P=0.01), and TAP1 (P=0.02) as shown in Figure 3. Genes of HLA class II (HLA-DPA1, HLA-DPB1, HLA-DQA1, and HLA-DQB1) were also analyzed, although no association with prognosis was found (Supplementary Figure 3). For a more comprehensive analysis of the role of immune response genes, we used all these variables to perform a principal component analysis (PCA). As a result, two extreme group of samples emerged. On one hand, patients with poor prognosis under-expressed HLA and TAP genes (regardless of the number of neoantigens). On the other hand, samples with good prognosis over-expressed HLAs and contained higher levels of predicted neoantigens (Supplementary Figure 4).
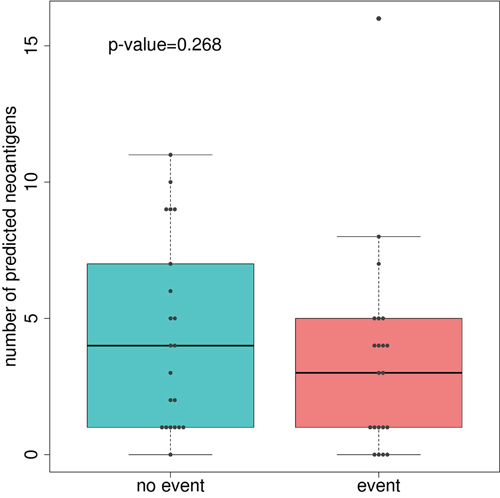
Figure 2: Boxplot comparing the number of predicted neoantigenes between good and poor-prognosis tumors. P-value was calculated using Man-Whitney U test.
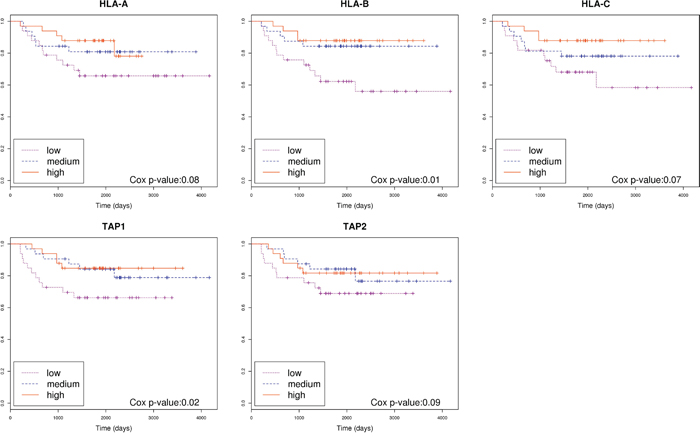
Figure 3: Association of genes related to antigen presentation machinery with prognosis. Kaplan-Meier curves separating patients based on the level of expression of genes HLA-A, HLA-B, HLA-C, TAP1 and TAP2. Cox p-value was calculated for each gene.
Recently, a group of experts has reported a robust classification of CRC for future clinical stratification that group tumors in four subtypes based on common molecular and clinical characteristics [3]. Under the hypothesis that our results could vary within subtypes, we assessed the prognosis value of genes related with antigen presentation stratifying by consensus molecular subtypes. We found that the expression of genes of immune response were not associated to prognosis for tumors classified as CMS2 “canonical” subtype. However, the association was very strong among the CMS4 “mesenchymal” and in CMS3 “metabolic” and subtypes. Tumors overexpressing genes involved in antigen presentation in these subtypes had better prognosis (Figure 4). Of note, because all our tumors were MSS, only 6 tumors were classified as CMS1 “MSI subtype” and were not analyzed, although 2 of them had relapsed.
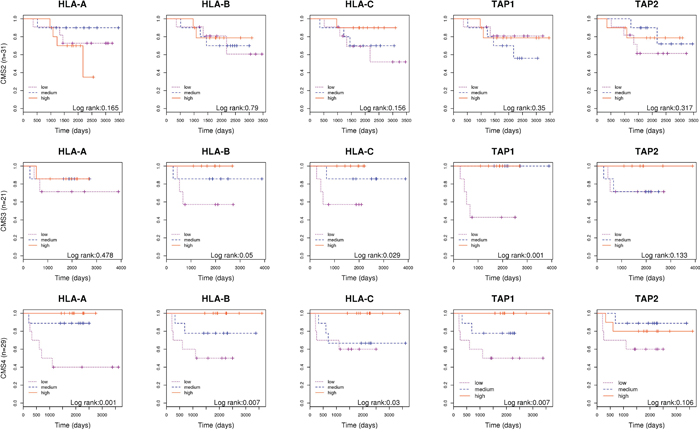
Figure 4: Molecular subtyping. Kaplan-Meier curves separating patients based on the level of expression of genes HLA-A, HLA-B, HLA-C, TAP1 and TAP2; in tumors classified as CMS2, CMS3 and CMS4 separately.
Good-prognosis tumors showed higher levels of lymphocytic infiltration
Apart from neoantigen generation and presentation, a third player is necessary to activate an immune response against the tumor cell: T-cell receptors should bind to MHC-antigen complexes. Under this hypothesis, the level of expression of CD8A was used as a marker of T-cell infiltration and compared between the two groups of tumors [28]. CD8 is a specific marker for T-cells binding MHC Class I but not MHC Class II. As expected, good prognosis tumors showed higher levels of this marker than poor prognosis ones (p-value=0.026) suggesting that not only antigen presentation but also proper recognition by T-cells is necessary for the host immune system to attack the tumor (Figure 5A). Indeed, a Kaplan-Meier curve plotting patients classified on the basis of CD8A expression (Supplementary Figure 5) clearly identified a group of 13 patients over-expressing the gene who did not experience relapse (Figure 5B).
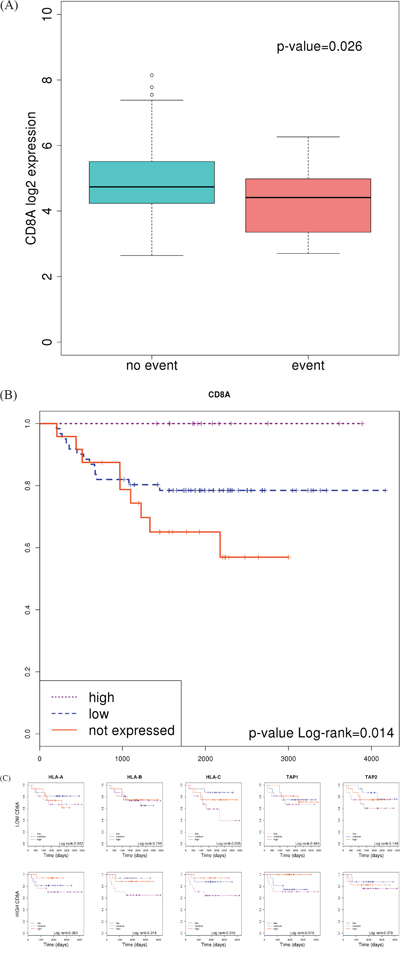
Figure 5: Association of CD8A expression with prognosis. A. Boxplot showing differences between patients who experienced relapse or not. P value was calculated using a t-test B. Kaplan-Meier curve separating a cluster of patients of good prognosis who over-express CD8A. C. Kaplan-Meier curves separating patients based on the level of expression of genes HLA-A, HLA-B and TAP1; in tumors classified as “high CD8A” and “low CD8A”, separately.
Based on this result, we stratified patients into “high” and “low” groups according to CD8A expression. Then, we looked for the prognosis value of HLAs and TAP genes in each group. As expected, survival curves were significantly different in those patients showing T-cell infiltration (high CD8A levels); whereas no differences were found in those patients without or with low T-cell infiltration (Figure 5C). This result suggested that, in order to be recognized and eliminated by the immune system, T-cells needs to infiltrate the tumor (indicated by high CD8A levels).
Based on this, we hypothesized that colon tumors evade immune responses underexpressing genes involved in antigen presentation and/or preventing the infiltration of T-cells into the tumor bulk. Indeed, an analysis comparing the levels of expression of MHC class I and TAPs genes, as well as CD8A gene, showed that, compared with adjacent normal mucosa, tumors underexpressed HLA genes (P=4e-4 for HLA-A, P=3e-7 for HLA-B, and P=4e-5 for HLA-C). CD8A was also underexpressed (P=1e-9), but not TAP1 nor TAP2 genes (Figure 6).
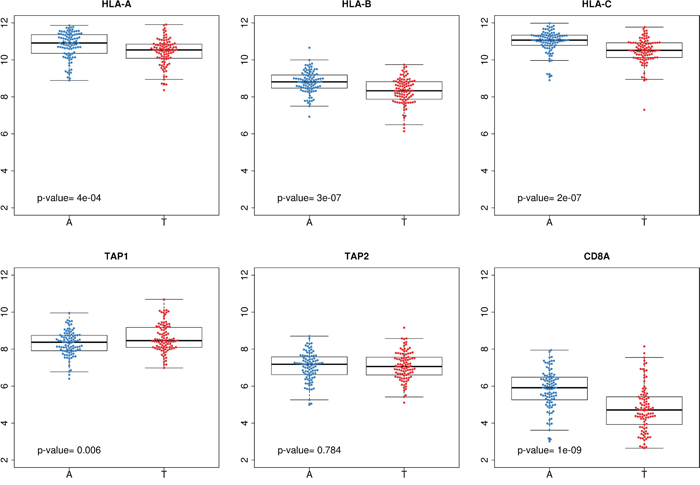
Figure 6: Comparison between tumor and adjacent normal mucosa. Gene expression levels of HLA class I genes, TAP1, TAP2 and CD8A in adjacent mucosa (blue) and paired tumor tissue (red).
DISCUSSION
Our study has shown that there are no major differences between the exome mutational landscape of good and poor-prognosis group of tumors. However, our analyses revealed that tumors able to properly present neoantigens and showing T-cell infiltration have better prognosis. Indeed, increasing data support the idea that the endogenous T cell compartment is able to recognize peptide epitopes that are displayed by major histocompatibility complexes (MHCs) on the surface of the tumor cells [29]. We searched for patient-specific neoantigens that might be generated from both driver and passenger mutations acquired during tumorigenesis. Then, we tested the hypothesis that recognition of such neoantigens by the host immune cells could influence patients’ survival. Certainly, our results pointed to a better outcome in those patients not only with a higher number of neoantigens (although not statistically significant, a trend was observed), but also over-expressing genes involved in antigen presentation machinery. In agreement with our results, an association between the number of predicted neoantigens in a tumor with increased patient survival has been recently described in a meta-analysis including six different types of tumors [30]. Thus, we hypothesize that poor-prognosis MSS stage II CRC tumors are unable to generate an immune response in the host via MHC class I antigen presentation.
It is well known that HLA class I functional abrogation represents a mechanism by which tumors circumvent immune surveillance [31]. In CRC, HLA class I loss has been described as a more frequent event in MSI tumors than in MSS tumors [32]. Regarding non-classical HLA class I molecules, their over-expression is one of the immunosuppressive strategies used by tumors [33]. Zeestraten et al. postulated that patients whose CRC tumors showed loss of HLA-E and HLA-G had better overall survival [15]. However, we have not reproduced this result in our data. We have found that expression of CD8A, considered a marker of lymphocyte infiltration, is higher in patients with better prognosis, probably indicating an activation of immune response against the tumor. This result was concordant with other studies correlating CD8+ lymphocytic infiltration with better survival in colon cancer [34, 35]. In addition, a stratification of our samples into “high CD8A” and “low CD8A” showed that levels of HLA and TAP1 genes are only relevant predicting survival in “high CD8A” tumors. This leads us to the conclusion that proper antigen presentation is only relevant for the inhibition of growth in those tumors showing T-cell infiltration. Indeed, it has been recently reported that loss of tapasin correlates with a reduction in CD8+ t-cell immunity and is associated with tumor progression in CRC [36]. It is important to note that patients in our series had not been treated with chemotherapy. In consequence, we consider that our results mirror the original response of the patients against the residual disease without treatment perturbing the host immune response.
Tumor-specific antigens that are generated by somatic mutation –neoantigens- can influence patient response to immunotherapy treatment and contribute to tumor reduction [37]. It is well known that microsatellite instable (MSI) CRC tumors are frequently characterized by inflammatory lymphocytic infiltration which is associated with a better outcome than microsatellite stable (MSS) CRC, probably reflecting a more effective immune response [38]. Although lymphocyte infiltration is characteristic of MSI tumors, it also has been shown to predict better prognosis in MSS tumors [39-41]. It is important to highlight that in this work all the analyzed CRC tumors are MSS, and a weak immune response should be expected, al least in comparison with MSI tumors. Nevertheless, a recent work assessing immunogenicity of somatic mutations in gastrointestinal cancers demonstrated that a clinically relevant anti-mutation T cell response could be also triggered against tumors with a low mutational load [42]. Thus, we postulate that MSS tumors with high HLA, mutational load and antigen processing pathways acquire a phenotype that helps cancer cells to evade the immune system of the host. Indeed, a comparison between tumor and adjacent normal mucosa revealed that tumors significantly underexpressed HLA class I genes and CD8A, suggesting a poorer ability for antigen presentation and recognition.
We were interested in analyzing our data according to the recent proposal of Consensus Molecular Subtypes that classifies CRC tumors into CMS1 “MSI immune”, CMS2 “Canonical”, CMS3 “Metabolic” and CMS4 “Mesenchymal” [3]. While tumors belonging to CMS1 (not included in our dataset) are more prone to induce a host immune response, our results showed that CMS3 and especially CMS4 tumors might also elicit an immune response. In agreement, recent studies conclude that CMS4 “mesenchymal” phenotype is characterized by a high stromal infiltration [43, 44]. This subgroup usually has worse prognosis than others. So, this stratification inside subgroups might be useful to further selection of patients requiring more aggressive or specific therapies in CRC. It is also important to note that the CMS2 “canonical” subtype comprising almost 40% of CRC tumors seems not to be able to elicit an immune response.
Although no single SNV was found to be associated with prognosis, several genes have been found to be exclusively and recurrently mutated in the poor-prognosis group of tumors such as classically CRC related genes POLE and BRAF. Mutations in POLE have been related to shorter patient survival [45]. Also in agreement with our results, BRAF mutations have been associated with a decrease in survival rate in CRC patients [46-48] and specifically in MSS CRC tumors [49]. We have found CACNA1G missense mutations in three tumors, all of them exhibiting poor-prognosis. Interestingly, a recent paper by Berg et al. reported that losses of CACNA1G by copy number changes were associated with early recurrence [50]. Thus, we hypothesized that loss of this gene could be a poor prognosis biomarker. Regarding functions in which mutated genes are implicated, several pathways have been found exclusively mutated in good and poor group of tumors. This result suggests that diverse molecular alterations could converge in similar phenotypes necessary for cancer progression and invasion.
This study also has several limitations. Besides the limited sample size, we have not a direct evidence that the mutated proteins generate neoantigens, but have used instead bioinformatics prediction tools. Moreover, it has been reported that HLA inference program HLAMiner achieves a good performance with RNA-seq or whole genome data rather than with exome sequencing data [26]. Finally, it is well known that each gene could codify several protein isoforms. However, in this work, to predict neoantigens only those isoforms reported in UniProt repository as the most common ones have been taken into account. Also, tumor infiltrating lymphocytes have not been measured directly, but CD8A expression has been used as a surrogate variable.
In conclusion, this analysis of prognosis in relation to somatic mutations in CRC has shown that although no single mutation was significantly associated with prognosis, some genes were found to be exclusively mutated in poor-prognosis tumors. Moreover, neither the total number of mutations nor the number of predicted neoantigens were different between good and poor-prognosis groups of tumors. However, those patients with higher levels of CD8A (as a marker of lymphocytic infiltration) and HLA-class I genes expression showed better prognosis. These results pave the way for future studies assessing the putative role of genes implicated in tumor-specific antigen presentation and recognition as prognostic biomarkers in pre-metastatic tumors, specifically in the subset of tumors classified as “metabolic” or “mesenchymal”. In addition, they suggest that personalized immunotherapy could stand for a treatment opportunity in patients suffering MSS CRC tumors.
MATERIALS AND METHODS
Patients and samples
This study included a subset of 42 paired adjacent normal and tumor tissues (84 samples) from a previously described set of 100 patients with colon cancer diagnosed at stage II and microsatellite stable tumors (colonomics project –CLX-: www.colonomics.org; NCBI BioProject PRJNA188510). The series of 42 patients was selected to include the 21 patients who experienced relapse and 21 without metastatic progression after a minimal follow-up of 3 years (Supplementary Table 2). None of them received chemotherapy. All patients were recruited at the Bellvitge University Hospital (Barcelona, Spain). Written informed consent was obtained from all patients and the Institution’s Ethics Committee approved the protocol. DNA was extracted using a standard phenol-chloroform protocol. Moreover, gene expression profiles of these tumors have been performed [23] and is available in GEO repository (GSE44076).
Exome sequencing and somatic single nucleotide (SNV) variants selection
Exome sequencing pipeline was extensively described in a previous work [22]. Briefly, Genomic DNA from the set of 42 adjacent-tumor paired samples was sequenced in the National Center of Genomic Analysis, Barcelona, Spain (CNAG) using the Illumina HiSeq-2000 platform. Exome capture was performed with the commercial kit Sure Select XT Human All Exon 50MB (Agilent). After data alignment and processing, high quality single nucleotide variants (SNVs) were identified using GATK software and germline variants were filtered. Finally, somatic SNVs were annotated using the SeattleSeq Variant Annotation web tool. Only potentially functional single nucleotide variants (SNVs) were taken into account in this work. Those include variants annotated as: coding-synonymous-near-splice, missense, missense-near-splice, splice-3, splice-5, stop-gained, stop-gained-near-splice, stop-lost, utr-3 and utr-5.
Functional analysis
Gene sets containing function and pathway information from “KEGG”, “Biocarta”, “Reactome”, and “GO” were downloaded from the Molecular Signatures Database [24]. For each gene set, an enrichment score was calculated for each tumor by dividing the number of mutations mapping into genes constituting the gene set by the number of genes in such gene set. The score was then compared for good and poor group of tumors. To assess if differences in scores were significant, a p-value was calculated by randomly permuting the tumor group label. The number of permutations was calculated in each case to ensure that the minimum p-value was at least as small as required by the Bonferroni correction at nominal 0.05 significance level.
Interactome analysis
Human pairs of protein-protein interactions were downloaded from Hippie database [25] (last updated: 09/05/14). Only highly reliable interactions were taken into account (score > 0.72) to construct a human protein-protein interaction network containing 9,963 nodes and 49,847 edges. This network was used as a framework in which mutated genes were mapped. To each mutated gene in each tumor, centrality parameters degree, betweenness, closeness and eccentricity were calculated using igraph library in R.
HLA genotyping and neoantigens prediction
Exome sequencing data from normal samples was used to infer MHC class I alleles of HLA-A and HLA-B (genes that predominantly mediates anti-tumor response) using HLAminer software [26].
FASTA sequences extracted from UniProt were used to translate information about DNA missense mutations into an aminoacid change level (only isoforms 1 were taken into account). For each missense mutation, a sequence of 21-aminoacids centered on the mutation and the corresponding wild type peptide were analyzed for potential neoantigens. NetMHCpan 2.8 Server [27] was used to infer putative immunogenic peptides (8 to 11 aminoacids) combining information about HLAs genotypes and peptides harboring missense mutations. An immunogenic epitope was defined as a mutated peptide with high affinity for one HLA allele of the patient (IC50<50nM) and low affinity for the wild-type counterpart (IC50>500nM). Information about the level of expression of each gene generating an immunopeptide was also computed.
Survival analyses
Kaplan–Meier curves were plotted to represent the results, dividing the range of gene expression into quartiles and log-rank test were computed. Also, Cox models were fitted to consider multiple variables simultaneously and control for potential confounding.
Molecular subtyping
The CMSclassifier R package [3] was used to classify our samples into the four CRC consensus molecular subtypes CMS1, CMS2, CMS3 and CMS4, using a Random Forest approach. Also, a principal components analysis (PCA) was performed to explore dispersion in our data based on the number of predicted neoantigens and expression level of genes of interest.
ACKNOWLEDGMENTS
We would like to thank Carmen Atencia, Pilar Medina, and Isabel Padrol for their help with the clinical annotation of the samples.
CONFLICTS OF INTEREST
The authors declare no conflicts of interests
GRANT SUPPORT
This study was supported by the Instituto de Salud Carlos III, co-funded by FEDER funds –a way to build Europe (grants PI09-01037, PI11-01439 and PIE13-00022), CIBERESP CB07/02/2005, the Spanish Association Against Cancer (AECC) Scientific Foundation, the Catalan Government DURSI grant 2014SGR647, and National Institutes of Health grant U19CA148107 (CORECT). Sample collection was supported by the Xarxa de Bancs de Tumors de Catalunya sponsored by Pla Director d’Oncología de Catalunya (XBTC) and ICOBIOBANC, sponsored by the Catalan Institute of Oncology.
REFERENCES
1. Hornberg JJ, Bruggeman FJ, Westerhoff HV, Lankelma J. Cancer: a Systems Biology disease. Biosystems. 2006; 83:81-90.
2. Cordero D, Sole X, Crous-Bou M, Sanz-Pamplona R, Pare-Brunet L, Guino E, Olivares D, Berenguer A, Santos C, Salazar R, et al. Large differences in global transcriptional regulatory programs of normal and tumor colon cells. BMC Cancer. 2014; 14:708.
3. Guinney J, Dienstmann R, Wang X, de Reynies A, Schlicker A, Soneson C, Marisa L, Roepman P, Nyamundanda G, Angelino P, et al. The consensus molecular subtypes of colorectal cancer. Nat Med. 2015; 21:1350-1356.
4. Salazar R, Roepman P, Capella G, Moreno V, Simon I, Dreezen C, Lopez-Doriga A, Santos C, Marijnen C, Westerga J, et al. Gene expression signature to improve prognosis prediction of stage II and III colorectal cancer. J Clin Oncol. 2011; 29:17-24.
5. Webber EM, Lin JS, Evelyn PW: Oncotype DX tumor gene expression profiling in stage II colon cancer. Application: prognostic, risk prediction. PLoS Curr. 2010; 2. pii: RRN1177.
6. Sanz-Pamplona R, Berenguer A, Cordero D, Riccadonna S, Sole X, Crous-Bou M, Guino E, Sanjuan X, Biondo S, Soriano A, et al. Clinical value of prognosis gene expression signatures in colorectal cancer: a systematic review. PLoS One. 2012; 7:e48877.
7. Jasmine F, Rahaman R, Dodsworth C, Roy S, Paul R, Raza M, Paul-Brutus R, Kamal M, Ahsan H, Kibriya MG. A genome-wide study of cytogenetic changes in colorectal cancer using SNP microarrays: opportunities for future personalized treatment. PLoS One. 2012; 7:e31968.
8. Loo LW, Tiirikainen M, Cheng I, Lum-Jones A, Seifried A, Church JM, Gryfe R, Weisenberger DJ, Lindor NM, Gallinger S, et al. Integrated analysis of genome-wide copy number alterations and gene expression in microsatellite stable, CpG island methylator phenotype-negative colon cancer. Genes Chromosomes Cancer. 2013; 52:450-466.
9. Berdiel-Acer M, Sanz-Pamplona R, Calon A, Cuadras D, Berenguer A, Sanjuan X, Paules MJ, Salazar R, Moreno V, Batlle E, et al. Differences between CAFs and their paired NCF from adjacent colonic mucosa reveal functional heterogeneity of CAFs, providing prognostic information. Mol Oncol. 2014; 8:1290-1305.
10. Calon A, Espinet E, Palomo-Ponce S, Tauriello DV, Iglesias M, Cespedes MV, Sevillano M, Nadal C, Jung P, Zhang XH, et al. Dependency of colorectal cancer on a TGF-beta-driven program in stromal cells for metastasis initiation. Cancer Cell. 2012; 22:571-584.
11. Quante M, Varga J, Wang TC, Greten FR. The gastrointestinal tumor microenvironment. Gastroenterology. 2013; 145:63-78.
12. Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011; 144:646-674.
13. Margalit H, Altuvia Y. Insights from MHC-bound peptides. Novartis Found Symp. 2003; 254:77-90; discussion 91-101, 216-122, 250-102.
14. Brusic V, Petrovsky N. Immunoinformatics--the new kid in town. Novartis Found Symp. 2003; 254:3-13; discussion 13-22, 98-101, 250-102.
15. Zeestraten EC, Reimers MS, Saadatmand S, Goossens-Beumer IJ, Dekker JW, Liefers GJ, van den Elsen PJ, van de Velde CJ, Kuppen PJ. Combined analysis of HLA class I, HLA-E and HLA-G predicts prognosis in colon cancer patients. Br J Cancer. 2014; 110:459-468.
16. Vilar E, Gruber SB. Microsatellite instability in colorectal cancer-the stable evidence. Nat Rev Clin Onco.l 2010; 7:153-162.
17. Llosa NJ, Cruise M, Tam A, Wicks EC, Hechenbleikner EM, Taube JM, Blosser RL, Fan H, Wang H, Luber BS, et al. The vigorous immune microenvironment of microsatellite instable colon cancer is balanced by multiple counter-inhibitory checkpoints. Cancer Discov. 2015; 5:43-51.
18. Gubin MM, Zhang X, Schuster H, Caron E, Ward JP, Noguchi T, Ivanova Y, Hundal J, Arthur CD, Krebber WJ, et al. Checkpoint blockade cancer immunotherapy targets tumour-specific mutant antigens. Nature. 2014; 515:577-581.
19. Xiao Y, Freeman GJ. The microsatellite instable subset of colorectal cancer is a particularly good candidate for checkpoint blockade immunotherapy. Cancer Discov. 2015; 5:16-18.
20. TCGA Consortium. Comprehensive molecular characterization of human colon and rectal cancer. Nature. 2012; 487:330-337.
21. Wang L, Wheeler DA. Genomic sequencing for cancer diagnosis and therapy. Annu Rev Med. 2014; 65:33-48.
22. Sanz-Pamplona R, Lopez-Doriga A, Pare-Brunet L, Lazaro K, Bellido F, Alonso MH, Ausso S, Guino E, Beltran S, Castro-Giner F, et al. Exome sequencing reveals AMER1 as a frequently mutated gene in colorectal cancer. Clin Cancer Res. 2015; 21:4709-18.
23. Sanz-Pamplona R, Berenguer A, Cordero D, Mollevi DG, Crous-Bou M, Sole X, Pare-Brunet L, Guino E, Salazar R, Santos C, et al. Aberrant gene expression in mucosa adjacent to tumor reveals a molecular crosstalk in colon cancer. Mol Cancer. 2014; 13:46.
24. Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub TR, Lander ES, Mesirov JP. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci U S A. 2005; 102:15545-15550.
25. Schaefer MH, Fontaine JF, Vinayagam A, Porras P, Wanker EE, Andrade-Navarro MA. HIPPIE: Integrating protein interaction networks with experiment based quality scores. PLoS One. 2012; 7:e31826.
26. Warren RL, Choe G, Freeman DJ, Castellarin M, Munro S, Moore R, Holt RA. Derivation of HLA types from shotgun sequence datasets. Genome Med. 2012; 4:95.
27. Hoof I, Peters B, Sidney J, Pedersen LE, Sette A, Lund O, Buus S, Nielsen M. NetMHCpan, a method for MHC class I binding prediction beyond humans. Immunogenetics. 2009; 61:1-13.
28. Lugli A, Karamitopoulou E, Panayiotides I, Karakitsos P, Rallis G, Peros G, Iezzi G, Spagnoli G, Bihl M, Terracciano L, Zlobec I. CD8+ lymphocytes/ tumour-budding index: an independent prognostic factor representing a ‘pro-/anti-tumour’ approach to tumour host interaction in colorectal cancer. Br J Cancer. 2009; 101:1382-1392.
29. Joyce JA, Fearon DT. T cell exclusion, immune privilege, and the tumor microenvironment. Science. 2015; 348:74-80.
30. Brown SD, Warren RL, Gibb EA, Martin SD, Spinelli JJ, Nelson BH, Holt RA. Neo-antigens predicted by tumor genome meta-analysis correlate with increased patient survival. Genome Res. 2014; 24:743-750.
31. Chang CC, Ferrone S. Immune selective pressure and HLA class I antigen defects in malignant lesions. Cancer Immunol Immunother. 2007; 56:227-236.
32. Dierssen JW, de Miranda NF, Ferrone S, van Puijenbroek M, Cornelisse CJ, Fleuren GJ, van Wezel T, Morreau H. HNPCC versus sporadic microsatellite-unstable colon cancers follow different routes toward loss of HLA class I expression. BMC Cancer. 2007; 7:33.
33. Gomes AQ, Correia DV, Silva-Santos B. Non-classical major histocompatibility complex proteins as determinants of tumour immunosurveillance. EMBO Rep. 2007; 8:1024-1030.
34. Koelzer VH, Lugli A, Dawson H, Hadrich M, Berger MD, Borner M, Mallaev M, Galvan JA, Amsler J, Schnuriger B, et al. CD8/CD45RO T-cell infiltration in endoscopic biopsies of colorectal cancer predicts nodal metastasis and survival. J Transl Med. 2014; 12:81.
35. Zlobec I, Terracciano LM, Lugli A. Local recurrence in mismatch repair-proficient colon cancer predicted by an infiltrative tumor border and lack of CD8+ tumor-infiltrating lymphocytes. Clin Cancer Res. 2008; 14:3792-3797.
36. Sokol L, Koelzer VH, Rau TT, Karamitopoulou E, Zlobec I, Lugli A. Loss of tapasin correlates with diminished CD8(+) T-cell immunity and prognosis in colorectal cancer. J Transl Med. 2015; 13:279.
37. Desrichard A, Snyder A, Chan TA. Cancer Neoantigens and Applications for Immunotherapy. Clin Cancer Res. 2015; PMID:26515495.
38. Boissiere-Michot F, Lazennec G, Frugier H, Jarlier M, Roca L, Duffour J, Du Paty E, Laune D, Blanchard F, Le Pessot F, et al. Characterization of an adaptive immune response in microsatellite-instable colorectal cancer. Oncoimmunology. 2014; 3:e29256.
39. Huh JW, Lee JH, Kim HR. Prognostic significance of tumor-infiltrating lymphocytes for patients with colorectal cancer. Arch Surg. 2012; 147:366-372.
40. Mei Z, Liu Y, Liu C, Cui A, Liang Z, Wang G, Peng H, Cui L, Li C. Tumour-infiltrating inflammation and prognosis in colorectal cancer: systematic review and meta-analysis. Br J Cancer. 2014; 110:1595-1605.
41. Ogino S, Nosho K, Irahara N, Meyerhardt JA, Baba Y, Shima K, Glickman JN, Ferrone CR, Mino-Kenudson M, Tanaka N, et al. Lymphocytic reaction to colorectal cancer is associated with longer survival, independent of lymph node count, microsatellite instability, and CpG island methylator phenotype. Clin Cancer Res. 2009; 15:6412-6420.
42. Tran E, Ahmadzadeh M, Lu YC, Gros A, Turcotte S, Robbins PF, Gartner JJ, Zheng Z, Li YF, Ray S, et al. Immunogenicity of somatic mutations in human gastrointestinal cancers. Science. 2015; 350:1387-1390.
43. Calon A, Espinet E, Palomo-Ponce S, Tauriello DV, Iglesias M, Cespedes MV, Sevillano M, Nadal C, Jung P, Zhang XH, et al. Dependency of colorectal cancer on a TGF-beta-driven program in stromal cells for metastasis initiation. Cancer Cell. 2015; 22:571-584.
44. Isella C, Terrasi A, Bellomo SE, Petti C, Galatola G, Muratore A, Mellano A, Senetta R, Cassenti A, Sonetto C, et al. Stromal contribution to the colorectal cancer transcriptome. Nat Genet. 2015; 47:312-319.
45. Stenzinger A, Pfarr N, Endris V, Penzel R, Jansen L, Wolf T, Herpel E, Warth A, Klauschen F, Kloor M, et al. Mutations in POLE and survival of colorectal cancer patients--link to disease stage and treatment. Cancer Med. 2014; 3:1527-1538.
46. Ahn TS, Jeong D, Son MW, Jung H, Park S, Kim H, Bae SB, Kim HJ, Jeon YW, Lee MS, Baek MJ. The BRAF mutation is associated with the prognosis in colorectal cancer. J Cancer Res Clin Oncol. 2014; 140:1863-1871.
47. Farina-Sarasqueta A, van Lijnschoten G, Moerland E, Creemers GJ, Lemmens VE, Rutten HJ, van den Brule AJ. The BRAF V600E mutation is an independent prognostic factor for survival in stage II and stage III colon cancer patients. Ann Oncol. 2010; 21:2396-2402.
48. Ogino S, Shima K, Meyerhardt JA, McCleary NJ, Ng K, Hollis D, Saltz LB, Mayer RJ, Schaefer P, Whittom R, et al. Predictive and prognostic roles of BRAF mutation in stage III colon cancer: results from intergroup trial CALGB 89803. Clin Cancer Res. 2012; 18:890-900.
49. Landau MS, Kuan SF, Chiosea S, Pai RK. BRAF-mutated microsatellite stable colorectal carcinoma: an aggressive adenocarcinoma with reduced CDX2 and increased cytokeratin 7 immunohistochemical expression. Hum Pathol. 2014; 45:1704-1712.
50. Berg M, Nordgaard O, Korner H, Oltedal S, Smaaland R, Soreide JA, Soreide K. Molecular subtypes in stage II-III colon cancer defined by genomic instability: early recurrence-risk associated with a high copy-number variation and loss of RUNX3 and CDKN2A. PLoS One. 2015; 10:e0122391.