
INTRODUCTION
Radiotherapy has become standard of care for most breast cancer cohorts [1]. Radiation has significantly reduced the risk of local recurrence and also improved overall survival [2]. However, cancer cells can acquire radioresistance with its complementary risk of increased mortality rates [3]. Furthermore, radiotherapy has been shown to increase the risk of cardiovascular diseases [4]. Hence, discovery of targets predicting the response to radiotherapy as well as agents that sensitize cancer cells to ionizing radiation with low side effects, is of great interest.
Various biomarkers have improved the identification of cancer patients who benefit from personalized therapeutic strategies. For example, in breast cancer (BC) progesterone or estrogen receptor positive BC subtypes are generally treated by endocrine ablation therapies in combination with chemotherapy and/or radiotherapy. However, all of those BC subtypes respond equally to radiation and biomarkers helping to define the radiation regime are urgently needed.
In particular, biomarkers for intrinsic or acquired resistance of tumor cells to radiotherapy remain elusive. Mechanisms causing resistance to radiotherapy are diverse and poorly characterized. Recent evidence suggests that aberrant apoptosis may contribute to this phenomenon [6].
p53, a nuclear phosphoprotein, is an important mediator of cell growth control and apoptosis [7]. It is activated by a variety of stress signals, e.g. irradiation, in order to eliminate damaged cells from the host [8]. Increased aggressiveness of cancer is associated with the loss of function of p53 and chemo- and radioresistance have already been correlated with deleted or mutated p53 proteins [9]. Thus, accurate molecular evaluation of the p53 status could be used to stratify patients, who might respond to additional therapies, such as radiotherapy, leading to an improved prognosis. Furthermore, identified mutations in the p53 gene might provide a potential target for clinical intervention strategies. Theoretically, reversion to wild type p53 should restore cell growth control, apoptosis, or radiosensitivity, but has proven to be difficult to achieve [10]. Hence, the identification of downstream effectors of p53 could present novel therapeutic targets to reinforce radiosensitivity. However, the exact p53 affected genes, responsible for radiation induced apoptosis, remain poorly characterized.
Recently, the receptor for hyaluronan-mediated motility (RHAMM) has been identified as a novel effector protein of p53 [11]. RHAMM acts as a cell-surface receptor for hyaluronan (HA) and as intracellular stabilizer of the mitotic spindle [12]. Its functional role is thought to be the response to pathological process and was shown to be increased in various tumors [13]. RHAMM is located on chromosome 5q33.2 and four different isoforms, generated by alternative splicing of its messenger RNA, have been described within the last years. Evidence exists that alternative splicing of RHAMM is involved in promoting formation of metastases of hepatic cancers [14]. As a consequence of its ability to bind HA, an extracellular matrix component known to promote tumorigenesis [14], RHAMM activates signaling pathways which have been implicated in BC progression [15] and cellular survival [16].
Aim of the present study was to investigate the functional role of RHAMM-proteins in BC as well as the relevance of its interaction with p53 with regard to therapeutic interventions supporting radiotherapy-based treatment decisions. In particular, the hypothesis was tested if RHAMM and its binding partner HA are eligible as therapeutic targets to sensitize breast cancer cells to ionizing radiation.
RESULTS
RHAMM is prognostic for overall survival in breast cancer patients and alters cancer cell phenotype in in vitro studies
To characterize the relevance of RHAMM expression in BC progression, mRNA expression data (Affymetrix) from 196 BC tissue samples were analyzed. Patients were stratified into quartiles according to their RHAMM expression for both HMMR probe sets present on the Affymetrix chips. The expression level was correlated to clinical and histological prognostic parameters and patient outcome. Increase in RHAMM expression was significantly correlated with a decrease in overall survival (OS) in both probe sets (Fig. 1A, data of the second probe set not shown) as well as recurrence-free survival (data not shown). Furthermore, a significant relationship between RHAMM and tumor grading was observed (Fig. 1B).
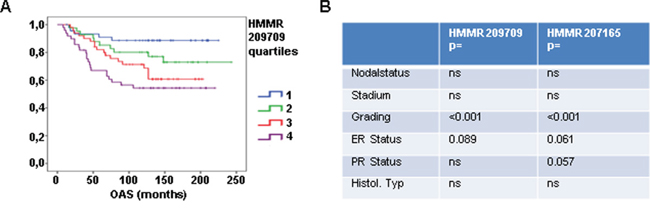
Figure 1: RHAMM is prognostic for patient overall survival. A. Affymetrix analysis of RHAMM expression in 196 tissue samples from breast cancer patients is shown. Patients were stratified into subgroups according their RHAMM expression (low (1), medium (2), high (3), very high (4)) and the subgroups were correlated to overall survival. B. table showing results of statistic tests for clinical parameter in two affymetrix analysis.
Even though in previous studies RHAMM has been proposed as a prognostic marker in BC, its functional role remains largely unknown. Two different BC cell line cells (MCF-7 and MDA-MB-231) were used to test whether RHAMM influences cell proliferation, apoptosis, or migration. It has previously been described that cells of the MCF-7 line harbor high levels of RHAMM whereas cells of the MDA-MB-231 line express low levels of this protein [17, 18]. No effect on cellular proliferation quantified by CFSE and FACS analysis was observed 48h after transient inhibition of all RHAMM splice variants (Fig. 2A-2B). However, sub-G1 analysis revealed that siRHAMM treatment significantly increased the rate of cell death in MCF-7 cells whereas MDA-MB-231 cells were not affected (Fig. 2C). In contrast, knock down of RHAMM led to a decrease of migration in MDA-MB-231 cells whilst no change could be detected in MCF-7 cells (Fig. 2D).
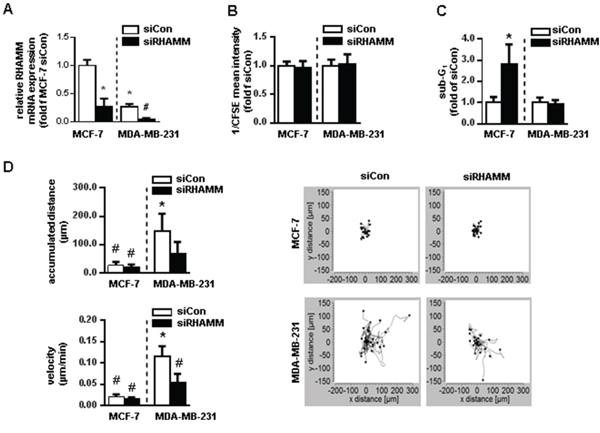
Figure 2: RHAMM has apoptotic and motility characteristics in different cancer cell lines in vitro. A. relative mRNA expression of RHAMM in siRHAMM transfected MCF-7 and MDA-MB-231 cells. B. proliferation rate measured with CFSE staining in MCF-7 and MDA-MB-231 cells 48h after siRNA knockdown of RHAMM. CFSE intensity is assigned reciprocally. C. sub-G1 analysis of MCF-7 and MDA-MB-231 cells 48h after siRNA knockdown of RHAMM. D. accumulated distance and velocity of migration assay with exemplary pictures of one experiment. *, p<0.05 in comparison to MCF-7 siCon; #, p<0.05 in comparison to MDA-MB-231 siCon.
Ionizing radiation induces cell death in MCF-7 cells and leaves MDA-MB-231 cells unaffected
Since RHAMM appears to be implicated in tumor progression, the question was raised whether RHAMM has any implications in the cellular response to ionizing radiation. Initially, the susceptibility of both MCF-7 and MDA-MB-231, to 2Gy of ionizing radiation was characterized. As functional readout the rate of proliferation and cell death were chosen. The number of living cells was significantly reduced in MCF-7 cells after irradiation (Fig. 3A), whereas the proliferative capacity was not altered by 2Gy of ionizing radiation of both cell lines (Fig. 3B). MCF-7 cells revealed a significant increase in the apoptotic rate as measured by sub-G1 analysis explaining the decrease of the total cell number (Fig. 3C-3D). In comparison, MDA-MB-231 cells were found to be radioresistant (Fig. 3A-3D). In order to investigate the underlying mechanism of increased cellular death in MCF-7 cells, a protein array was performed analyzing the phosphorylation pattern of proteins involved in apoptosis. p53 and p38 were significantly increased in MCF-7 cells 48h after initial radiation (Fig. 3E-3G). This effect was not seen in MDA-MB-231 cells. Of note, MCF-7 cells harbor wild type p53 whereas MDA-MB-231 cells harbor a mutated form of the protein leading to its accumulation in the nucleus [19]. The involvement of p53 in the induction of apoptosis in MCF-7 cells was further validated by treatment with siRNA against p53 prior to radiation. Short interfering RNA specific for p53 abolished the pro-apoptotic effect of radiation in MCF-7 cells (Fig. 3H). Inhibition of p38 by SB202190 did not abrogate irradiation induced cell death. These results indicate that MCF-7 cell death in response to ionizing radiation is induced by the p53 pathway.
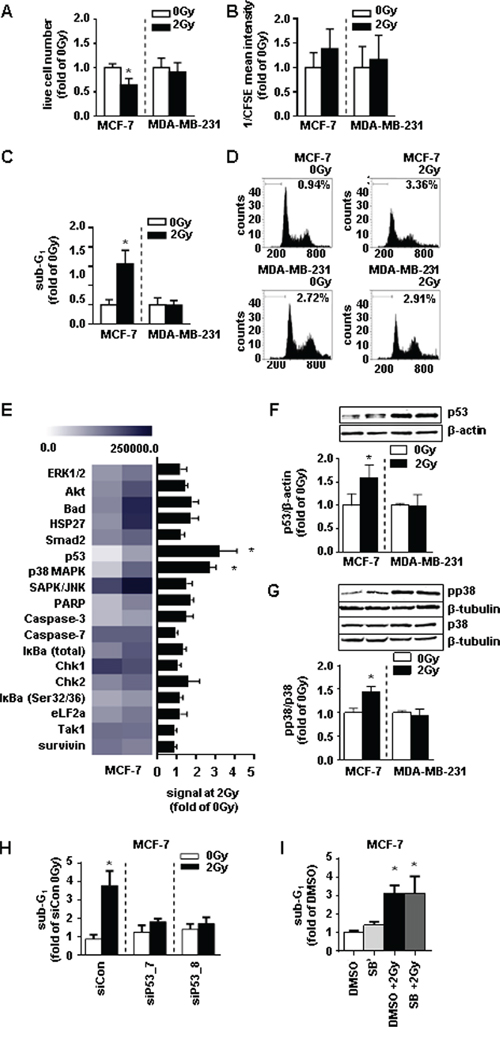
Figure 3: MDA-MB-231 cells are not radiosensitive at a dose of 2Gy whereas MCF-7 cells are radiosensitive via activation of p53 and p38. A. live cell number, B. proliferation rate of via CFSE (assigned reciprocally), and C. sub-G1 analysis of MCF-7 and MDA-MB-231 cells 48h after irradiation with 2Gy measurement. D. representative histograms of sub-G1 analyses. E. absolute expression levels of proteins analyzed via the stress and apoptosis assay depicted in a heatmap (left) and the fold changes of these proteins in MCF-7 cells 48h after irradiation compared to the non-irradiated control (right). F. western blot analyses of MCF-7 cell lysates for p53 and G. pp38 and p38 protein expression. H. sub-G1 analysis in MCF-7 cells transfected with siRNA against p53 48h after irradiation with 2Gy. I. proliferation rate of MCF-7 cells treated with SB (SB202190, p38-inhibitor, 10 μM) 4h before irradiation with 2Gy. *, p<0.05 in comparison with untreated MCF-7 0Gy (A-G, J, K)/siCon (H, I).
Since both analyzed BC cell lines cells exhibit a divergent estrogen receptor (ER) status (MCF7:positive; MDA-MB-231 negative [18]), we next tested whether the ER is involved in the radiosusceptibility. However, treatment of MCF-7 cells with the unspecific ER-antagonist ICI182780 did not abrogate the pro-apoptotic effect of ionizing radiation in this cell type (Suppl. Fig. S1).
RHAMM is regulated by radiation in a p53 correlated manner
To investigate the role of RHAMM in response to radiation, both cell lines were irradiated with 2Gy and RHAMM mRNA level was measured by qRT-PCR (Fig. 4A). RHAMM mRNA was significantly reduced in MCF-7 cells, likewise shown by immunocytochemical staining of RHAMM (Fig. 4B). No change was detected in MDA-MB-231 cells resembling the results on the apoptotic response (Fig. 3C). Immunocytochemistry staining of p53 showed increased signals in irradiated MCF-7 cells compared to sham-irradiated controls. Accumulation of p53 in MDA-MB-231 cells was found independent of ionizing radiation (Fig. 4B). Alternative splicing of RHAMM might be responsible for different cellular functions. The regulation four different protein isoforms regulation of four by ionizing radiation was analyzed by western blot. Of note, only splice variants v1/v2 which both run at 85kDa were decreased in MCF-7 cells while v3 and v4 were not reduced. In contrast, MDA-MB-231 cells displayed a significantly lower expression of all tested isoforms and were not further decreased by radiation (Fig. 4C-4D). Recently, RHAMM has been shown to be transcriptionally repressed by p53 [11]. Treatment of both cell lines with short interfering RNA against p53 confirmed these results (Fig. 4E-4G). The endogenously increased level of p53 in the nucleus of MDA-MB-231 cells could therefore explain the reduced occurrence of RHAMM protein in this cell line. Of note, reduction of RHAMM by siRNA did not change the level of p53 in both cell lines (Suppl. Fig. S2).
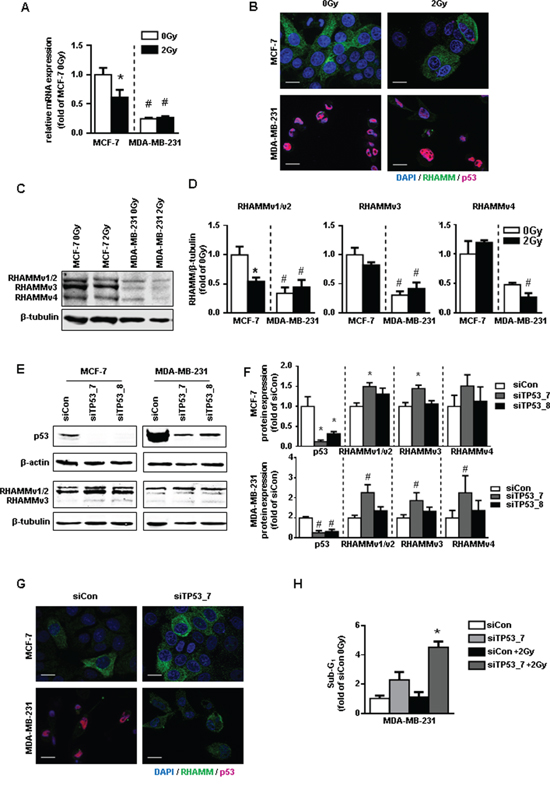
Figure 4: p53 and RHAMM variant status of MCF-7 and MDA-MB-231 in response to 2Gy irradiation. A. relative mRNA expression of RHAMMpan. B. RHAMM (green) and p53 (pink) immunofluorescence staining 48h after irradiation with 2Gy. Scale bars: 20μm. C. western blot analysis of RHAMM protein expression and its quantification and D. quantification. E. exemplary blots of protein expression of p53 and RHAMM of p53 depleted MCF-7 and MDA-MB-231 cells and F. quantification. G. RHAMM (green) and p53 (pink) immunofluorescence staining 48h after siRNA knockdown of p53. Scale bars: 20μm. H. MDA-MB-231 were transfected with sip53. 48h after transfection cells were irradiated. Sub-G1 was analyzed further 48h later.*, p<0.05 in comparison to MCF-7 siCon, #, p<0.05 MDA-MB-231 in comparison to MCF-7 siCon.
Next it was investigated whether the increase of RHAMM v1/v2 in MDA-MB-231 cells after p53 knock down would restore radiosensitivity. Knock down of p53 and subsequent upregulation of RHAMM v1/v2 increased the rate of cellular death in MDA-MB-231 cells. Of note, the apoptotic effect was even further enhanced by subsequent ionizing irradiation (Fig. 4H).
RHAMM splice variants increase the radiosensitivity of breast cancer cell lines
To establish the radiosensitizing ability of RHAMM - observed in terms of apoptosis - and to investigate the involvement of the different RHAMM splice variants, cells were treated with siRNA against the respective mRNAs (Suppl. Fig. S3) and subsequently irradiated. In MCF-7 cells siRHAMMpan as well as siRNA against all individual RHAMM splice variants increased the rate of apoptosis (white bars Fig. 5A).
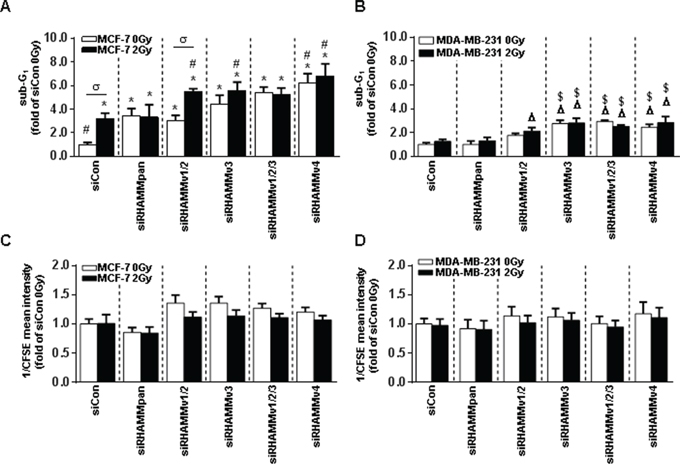
Figure 5: RHAMMpan and RHAMM variant knock-down in MCF-7 and MDA-MB-231 cells leads to increased radiosensitivity after irradiation with 2Gy. A. sub-G1 cell cycle analysis of siRHAMM variants transfected MCF-7 and B. MDA-MB-231 cells 48h after irradiation with 2Gy. C. analysis of proliferation rate via CFSE measurement (assigned reciprocally) in siRHAMM variant transfected MCF-7 and D. MDA-MB-231 cells 48h after irradiation with 2Gy. *, p<0.05 in comparison to MCF-7 siCon 0Gy, #, p<0.05 in comparison to MCF-7 siCon 2Gy, σ, p<0.05 MCF-7 siRHAMMv1/2 2Gy in comparison to MCF-7 siRHAMMv1/2 0Gy, Δ, p<0.05 in comparison to MDA-MB-231 siCon 0Gy, $, p<0.05 in comparison to MDA-MB-231 siCon 2Gy.
Again, irradiation induced the percentage of apoptotic cells. An additional apoptotic effect was observed in cells which were treated with siRHAMM v1/v2 and irradiation. Knock down of RHAMM v3 and v4 did not alter this effect (Fig. 5A).
In MDA-MB-231 cells neither siRHAMMpan nor siRHAMMv1/v2 revealed a significant induction of cell death (Fig. 5B). However, siRNA against RHAMMv3 and v4 induced cell death in MDA-MB-231 cells. Irradiation did not increase MDA-MB-231 cell death in any treatment group.
As expected from the previous experiments, the proliferation rate was not altered by either ionizing radiation or siRNA treatment (Fig. 5C-5D).
Pharmacologic inhibition of hyaluronan synthesis increases the response to ionizing radiation
Finding pharmacological approaches which increase the susceptibility of cancer cells to radiation and thereby reduce the rate of recurrence is of great interest. HA is the main intra- and extracellular ligand of RHAMM thereby regulating both functional aspects [20]. Its pharmacological inhibition can be realized by treatment of cells with the HA-synthase inhibitor 4-methylumbelliferone (4-MU). 4-MU treatment suppressed HA synthesis in irradiated and non-irradiated breast cancer cells (Fig. 6A). Importantly, incubation of MCF-7 cells with 4-MU increased the radiosensitivity of the cells with respect to apoptosis fourfold. Whereas MDA-MB-231 cells did not respond to 4-MU treatment alone, the susceptibility of the cells after additional radiation was increased (Fig. 6B-6C).
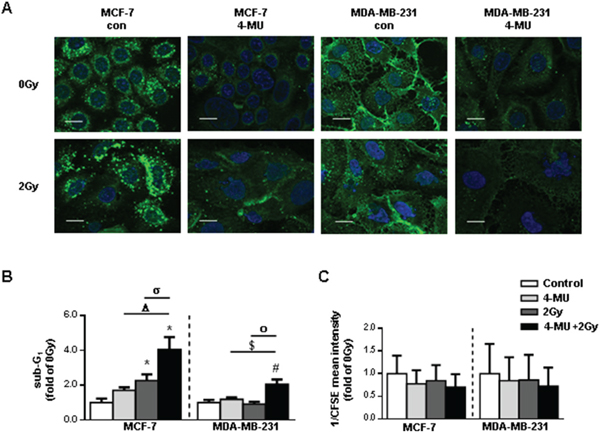
Figure 6: Pharmacological inhibition of HA system via 4-MU in MCF-7 and MDA-MB-231 leads to increased radiosensitivity. A. affinitycytochemistry of HA 48h after irradiation with 2Gy. Scale bars: 20μm. B. sub-G1 cell cycle analysis of 4-MU treated MCF-7 and MDA-MB-231 cells 48h after irradiation with 2Gy. C. analysis of proliferation rate via CFSE staining of 4-MU treated MCF-7 and MDA-MB-231 cells 48h after irradiation with 2Gy. CFSE intensity is assigned reciprocally. *, p<0.05 in comparison to MCF-7 0Gy, Δ, p<0.05 in comparison to MCF-7 4-MU, σ, p<0.05 in comparison to MCF-7 2Gy, #, p<0.05 in comparison to MCF-7 0Gy, $, p<0.05 in comparison to MCF-7 4-MU, ο, p<0.05 in comparison to MCF-7 2Gy.
In conclusion, we provide evidence, that RHAMM is involved in the malignant phenotype of BC cells. Detection of RHAMM isoform expression in correlation with the p53 mutation status might allow to predict the responsiveness to radiation. Importantly, pharmacological inhibition of HA, the main binding partner of RHAMM, could help to increase the radiosensitivity of both p53 wild type and mutated cancer types (Fig. 7).

Figure 7: Schematic model of mamma-ca radiosensitization. Breast cancer cells with wildtype p53 status are radiosensitive and can be forced into apoptosis upon RHAMM downregulation. Mutant p53 breast cancer cells are only radiosensitive if treated with 4-MU which affects RHAMM ligand inhibition.
DISCUSSION
The receptor for hyaluronan-mediated motility (RHAMM) exhibits at least two distinct functions. As intracellular protein it is involved in maintaining the stability of the mitotic spindle [12]. In addition, by attachment to a GPI-anchor, it is associated with the cellular membrane and acts as cellular HA receptor promoting cell motility and invasion [15, 21]. Various studies report overexpression of RHAMM during tumor development and suggest a prognostic significance of RHAMM expression in e.g. leukemia, bladder cancer, and BC [13, 22, 23]. These findings have fostered the idea to use RHAMM for therapy in acute myeloid leukemia and multiple myeloma, now being evaluated as vaccination against RHAMM in small clinical trials [24]. However, its value as prognostic marker and therapeutic target remains indeterminate.
In agreement with other reports, our data show that the level of RHAMM in tumor biopsies derived from BC patients is correlated with recurrence-free and OS. Interestingly in vitro its functional role also appears to depend on the expression level. Whereas in RHAMMhigh MCF-7 cells the cellular survival depends on the RHAMM expression, MDA-MB-231 cells (RHAMMlow) survive independently of RHAMM. Of note, this circumstance is reversed with regard to the migratory capacity of both cell types. One possible explanation is that RHAMM is transported to the membrane in invasive cancer cell lines [21] and thereby increases migratory behavior. This suggests that (i) the amount of RHAMM mRNA is important for the prognosis of the disease and (ii) that the subcellular localization might be informative for the functional role. Future studies will have to be conducted to clarify this open issue.
Several factors may be associated with altered cellular radiosensitivity and the probability of cells to be killed by apoptotic mechanisms including p53, FAS-mediated pathways, and the BCL-2 gene family [25]. The relationship between the level of endogenous apoptosis of tumors and their radiosensitivity has been investigated leading to conflicting results [26, 27]. However, it has been shown that cells from radiosensitive tumors are more susceptible to ionizing radiation compared to cells from unresponsive tumors [28]. Hence, the apoptotic response may serve as predictive assay for intrinsic radiosensitivity [29]. In the present study, a threefold increase in the apoptotic rate of RHAMMhigh MCF-7 was detected. In comparison, RHAMMlow cells (MDA-MB-231) were found to be radioresistant. Apoptosis in caspase-3 deficient cells (MCF-7) has been reported to depend on alternative pathways like caspase-7 activation of DFF40 like DNAse [30]. In this study analysis of proteins involved in radiation induced cell death of MCF-7 cells revealed an involvement of p53 in this process. Of interest MDA-MB-231 cells harbor mutations in the p53 gene [31], whereas MCF-7 express wild-type p53 [32]. A number of tumors acquire p53 mutations with a high frequency [33-36]. In general, patients with these mutations respond poorly to treatment with ionizing radiation.
In genome wide mRNA screens RHAMM was observed to be decreased when p53 is active [7, 37, 38]. Furthermore, it was recently described that p53 can repress RHAMM expression via its promotor including the first exon and intron [11].
In the present study, we demonstrate that RHAMM expression in MDA-MB-231 was severely reduced in correlation with an accumulation of p53 within the nucleus. Even though it is generally postulated, that mutant p53 cells have lost the ability to bind consensus p53 DNA binding regions, it has been demonstrated that mutated p53 is still able to regulate gene expression directly or through mitochondrial or cytoplasmatic activities [39]. In MDA-MB-231 cells it was previously reported that the p53 mutant is required for cellular survival through yet unknown mechanism [40]. In the present study siRNA against p53 raised RHAMM protein levels indicating that mutated p53 in MDA-MB-231 cells is still able to repress RHAMM expression.
Of interest, not all RHAMM isoforms appear to be decreased by p53 as only RHAMMv1/v2 was reduced in MDA-MB-213 cells and elevated in response to siRNA against p53. Different functions of the RHAMM splice variants have been proposed previously. In multiple myeloma patients the ratio of RHAMMv3:RHAMMv1/v2 has been correlated with poor prognosis [16]. Furthermore RHAMMv3 appears to promote tumor growth and metastasis to lymph nodes and the liver in a mouse model [14]. As RHAMMv1/v2 encodes for longer proteins than RHAMMv3 and v4 and the transport mechanism of RHAMM to the membrane is still unknown, alternative splicing of RHAMM might present one reason for its presence in different subcellular compartments and thereby different cellular functions.
Regarding radiation responses the expression of RHAMM v1/v2 appears to be vital. Whereas RHAMMv1/v2high MCF-7 cells were sensitive to ionizing radiation, RHAMMv1/v2low MDA-MB-231 cells were radioresistant. It appears plausible that RHAMMv1/2 is permanently reduced in p53 mutated cancer cells and other compensatory pathways exist, thereby conferring radioresistance.
CD44 is a widely expressed cell surface membrane receptor which participates in cell-cell and cell-matrix interactions. CD44 facilitates mitogenic/invasive as well as proliferative cellular phenotypes [41]. Conflicting functions of CD44 have been proposed in experimental models of tumorigenesis and –progression in comparison to in vivo data. This may be due to the presence and absence of RHAMM and vice versa [42]. CD44 is known to co-operate with RHAMM and has been reported to compensate for loss of RHAMM. RHAMM and CD44 unify at least two distinct characteristics: i) both have been shown to be transcriptionally repressed by p53 [11, 43] and ii) they share the same binding partner, HA. HA is a glycosaminoglycan and an important component of the extracellular matrix which has been associated with BC progression [44]. In vitro HA induces BC cell motility [45] and survival [46]. HA has been predicted to bind all RHAMM isoforms near the carboxyl-terminus [47]. In the present study treatment with the HA-inhibitor 4-MU augmented the radiation effect in both cell lines. However, 4-MU did only alter MDA-MB-231 cellular survival in combination with irradiation. This hints towards a HA dependent resistance mechanism possibly mediated by CD44 or RHAMM v3/v4. Future studies will have to address the role of CD44 and RHAMM for radiosensitivity of cancer cells, possibly also including further binding partners of both proteins, such as MET [48]. Apart from 4-MU the application of other pharmacological compounds interfering with HA-signaling pathways, as RHAMM/CD44 blocking antibodies or HA-binding peptides, should be considered.
Prognostic markers may help to improve the accuracy of risk stratification of cancer patients and therefore possibly provide important data to optimize therapeutic decisions. In the present study we report that the detection of RHAMM splice variants in correlation with the p53 mutation status might help to pre-evaluate the susceptibility of breast cancer cells towards radiotherapy. Additionally, the data raise the possibility that the response to radiotherapy in selected tumors may be improved by targeting RHAMM and its ligand HA.
MATERIALS AND METHODS
Analysis of mRNA expression data in tumor tissue samples
In order to investigate the mRNA expression of RHAMM, microarray data (Affymetrix HG-U133A) from a cohort of 196 mammary carcinoma enrolled in the Department of Gynecology, Hamburg University medical Center, were analyzed. The clinical and histological characteristics of this cohort as well as technical details have been described elsewhere [49]. All microarray data have been submitted to Gene Expression Omnibus (GEO) under the following accession numbers: GSE26971 (samples GSM663775-GSM663852), GSE31519 (samples GSM782523-GSM782529), GSE31519 (samples GSM782554-GSM782568), GSE46184 (samples GSM1125783-GSM112856) [49]. Informed consent for the scientific use of tissue materials, which was approved by the local ethics committee (Ethik-Kommission der Ärztekammer Hamburg, #OB/V/03), was obtained from all patients. The study was performed in accordance to the principles of the declaration of Helsinki and REMARK criteria [50].
Expression data of two RHAMM probe sets present on the Affymetrix chips (209709_s_at and 207165_at) were retrieved from the files. For statistical analysis, the cohort was divided into quartiles of similar size according to their expression values. χ2-tests were used to examine correlations between RHAMM expression and clinicopathological factors comparing the following groups: histological grading G1 vs. G2 vs. G3; stage pT1 vs. pT2 vs. pT3-4; lymph node involvement N0 vs. N1; estrogen and progesterone receptor status (ER, PR), positive vs. negative; histological type ductal vs. others. Overall survival was analyzed by Kaplan-Meier analysis and Log-Rank-Tests. Probability values less than 0.05 were regarded as statistically significant.
Treatment and transfection of cells
MCF-7 (CLS, Eppelheim) and MDA-MB-231 (CLS, Eppelheim) were cultured in DMEM-high glucose (Gibco, Carlsbad) with 2% FBS (Gibco) and 1% Penicillin/Streptomycin (Gibco). Ionizing radiation was applied with Gulmay RS225 (Xstrahl, Camberley) at 150kV and 15mA with 0.2mm copper filter 24h after treatment or seeding of 5000 cells per cm2. 10nM siRNA (AllStars Negative Control siRNA, siHMMR_9 (siRHAMMpan), siTP53_7 and siTP53_8 from Qiagen, Hilden and custom siRNA for RHAMM variants were purchased from Sigma) was used for transfection with 1:500 RNAiMax (Life Technologies, Carlsbad) in DMEM according to the manufacturer’s manual. siRNA against sequences for knockdown of RHAMM variants are AAAGAGATTCGTGTTCTTCTACA (siRHAMMv1/2), AAGATTCGTGTTCTTCTACAGGA (siRHAMMv3), AAAGTTAAGTCTTCGGAATCAAA (siRHAMMv1/2/3), and AATGACCCTTCTGATTCGTGT (siRHAMMv4). Cells were irradiated 24h after reversed transfection with siRNA against RHAMM or 4-MU (Sigma-Aldrich Chemie, München) treatment. Cells receiving p53 knockdown were irradiated 48h after reversed transfection with siRNA against p53.
Migration assay
Migration of the cells was investigated via time lapse microscopy starting 24h after ionizing radiation with 2Gy. Three videos per sample were recorded with Axio Observer.Z1 under standard culturing conditions and eight cells per video were tracked. Tracking was performed using ImageJ 1.47t (Wayne Rasband, National Institutes of Health, USA) manual tracking plugin (Fabrice Cordeli, Institut Curie, Orsay, France) and exemplary pictures are processed with Chemotaxis and Migration Tool (Ibidi, Gerhard Trapp, Martinsried, Germany).
Western blotting
Western blots were performed using standard procedures and the following reagents: RHAMM (GTX62573, GeneTex, Irvine), p53 (OP43, Calbiochem, Billerica), p38 (#9212, Cell Signaling Technologies, Danvers), pp38 (#9211, Cell Signaling Technologies, Danvers), β-tubulin (T7816, Sigma-Aldrich Chemie, München) and β-actin (A5316, Sigma-Aldrich Chemie, München). Binding protein for affinitycytochemistry of hyaluronan was biotinylated HABP (Calbiochem, Billerica) and as secondary antibody streptavidin-FITC (Dako, Glostrup) was used. Secondary antibodies for western blotting were IRDye® 800CW goat anti-rabbit IgG, IRDye® 800CW goat anti-mouse IgM, IRDye® 680RD goat anti-rabbit IgG and IRDye® 680LT goat anti-mouse IgM (LI-COR Biotechnology, Bad Homburg). Immunofluorescence secondary antibodies were AF488 goat anti-rabbit IgG (Life Technologies, Carsbad) and AF568 (Fab’)2 fragment of goat anti-mouse IgG (H+L) (Life Technologies, Carsbad).
Immunofluorescence staining and affinitycytochemistry of hyaluronic acid
For RHAMM and p53 immunofluorescent stainings cells were fixed with 4% paraformaldehyde (PFA). For hyaluronic acid affinitycytochemical staining cells were fixed with 70% EtOH/4% PFA/0.5% acidic acid. Stainings were performed as described previously [51, 52].
RNA isolation, cDNA transcription, and qRT-PCR
RNA was isolation was performed according to the PeqLab peqGOLD TriFast (Erlangen) protocol. cDNA transcription was carried out with QuantiTect Reverse Transcriptase Kit (Qiagen, Hilden) according to the instruction manual. qRT-PCR was performed in duplicate on StepOnePlus RealTime PCR System using Platinum SYBR Green qPCR SuperMix-UDG (Life Technologies, Carlsbad) with ROX reference dye according to the manufacturer’s protocol. Primer sequences (5’→3’) for GAPDH f are GTGAAGGTCGGAGTCAACG, GAPDH r TGAGGTCAATGAAGGGGTC, RHAMMpan f GAATTTGAGAATTCTAAGCTTG and RHAMMpan r CCATCATACCCCTCATCTTTGTT were used. Data were analyzed by ΔΔCT-method using GAPDH as reference gene.
Intracellular signaling array
Cells were lysed in Path Scan® Sandwich ELISA Lysis Buffer (Cell Signaling Technology, Danvers) and analyzed using PathScan® Stress and Apoptosis Signaling Antibody Array Kit (Cell Signaling Technology, Danvers) according to the vendor’s protocol. Chemiluminescent signals were detected via Odyssey Infrared Imaging System (application software version 3.0) from Li-Cor biosciences (Bad Homburg).
Sub-G1 cell cycle analysis
Cells were detached via incubation withTrypsin/EDTA (Gibco), washed once with PBS and resuspended in 75μL Lysis buffer (0.1% Sodium citrate, 0.1% Triton X-100). Directly before FACS analysis 25μL GUAVA cell cycle reagent (Millipore, Billerica) were added and doublet-discrimination was performed via signal height/area dot plot. 10000 cells per sample were analyzed employing a guava easyCyte 5 flow cytometer (Millipore).
Proliferation and determination of proliferation rate
Cells were detached and diluted 1:2 with TrypanBlue (Life Technologies, Carlsbad) for live dead discrimination and determination of live cell number in Countess (Invitrogen, Carlsbad).
To evaluate proliferation rate cells were stained with CFSE (Life Technologies, Carlsbad) as shown previously [53]. Cells were harvested 48h after irradiation. Analysis was performed employing the easyCyte5 flow cytometer (Millipore, Billerica). 10000 cells were analyzed per sample. Mean of fluorescence signal was assigned reciprocal.
Statistical analysis
Real-time data were analyzed by logarithmic data transformation. Data were analyzed with GraphPad Prism 6 (GraphPad Software, La Jolla, CA, USA) and are represented as mean ± SEM. Comparison of two groups was analyzed via two-tailed t-test. Comparison of more than two independent variables was analyzed via one-way ANOVA and Sidak’s multiple comparison post hoc test. Statistical significance was considered when p<0.05.
ACKNOWLEDGMENTS
We thank Prof. Reiner Jänicke, Laboratory for molecular Radiooncology, University Hospital Düsseldorf, to provide the Gulmay’s RS225 irradiator, and Petra Rompel for her excellent technical assistance. This work was supported by grant from the DFG (GRK1739/10).
CONFLICTS OF INTEREST
The authors declare no conflicts of interest.
REFERENCES
1. Favourable and unfavourable effects on long-term survival of radiotherapy for early breast cancer: an overview of the randomised trials. Lancet 2000; 355:1757–1770.
2. Mokbel K. Current management of ductal carcinoma in situ of the breast. Int. J. Clin. Oncol. 2003; 8:18–22.
3. Boyages J, Delaney G, Taylor R. Predictors of local recurrence after treatment of ductal carcinoma in situ: a meta-analysis. Cancer 1999; 85:616–28.
4. Hooning MJ, Botma A, Aleman BM, Baaijens MH, Bartelink H, Klijn JG, Taylor CW, van Leeuwen FE. Long-term risk of cardiovascular disease in 10-year survivors of breast cancer. J. Natl. Cancer Inst. 2007; 99:365–75.
5. Wilder RB, Curcio LD, Khanijou RK, Eisner ME, Kakkis JL, Chittenden L, Agustin J, Lizarde J, Mesa AV, Macedo JC, Ravera J, Tokita KM. Results with accelerated partial breast irradiation in terms of estrogen receptor, progesterone receptor, and human growth factor receptor 2 status. Int. J. Radiat. Oncol. Biol. Phys. 2010; 78:799–803.
6. Chang L, Graham PH, Hao J, Ni J, Bucci J, Cozzi PJ, Kearsley JH, Li Y. PI3K/Akt/mTOR pathway inhibitors enhance radiosensitivity in radioresistant prostate cancer cells through inducing apoptosis, reducing autophagy, suppressing NHEJ and HR repair pathways. Cell Death Dis. 2014; 5:e1437.
7. Kho PS, Wang Z, Zhuang L, Li Y, Chew JL, Ng HH, Liu ET, Yu Q. P53-Regulated Transcriptional Program Associated With Genotoxic Stress-Induced Apoptosis. J. Biol. Chem. 2004; 279:21183–21192.
8. Yoon KW, Byun S, Kwon E, Hwang SY, Chu K, Hiraki M, Jo SH, Weins A, Hakroush S, Cebulla A, Sykes DB, Greka A, Mundel P, et al. Control of signaling-mediated clearance of apoptotic cells by the tumor suppressor p53. Science (80-. ). 2015; 349:1261669–1261669.
9. Mcilwrath AJ, Vasey P a, Ross GM, Mcllwrath AJ, Brown R. Cell Cycle Arrests and Radiosensitivity of Human Tumor Cell Lines : Dependence on Wild-Type p53 for Radiosensitivity Advances in Brief Cell Cycle Arrests and Radiosensitivity of Human Tumor Cell Lines : Dependence on Wild-Type p53 for Radiosensitivity’. 1994; 3718–3722.
10. Meyn MS, Strasfeld L, Allen C. Testing the Role of p53 in the Expression of Genetic Instability and Apoptosis in Ataxia-telangiectasia. Int. J. Radiat. Biol. 1994; 66:S141–S149.
11. Sohr S, Engeland K. RHAMM is differentially expressed in the cell cycle and downregulated by the tumor suppressor p53. Cell Cycle 2008; 7:3448–3460.
12. Chen H, Mohan P, Jiang J, Nemirovsky O, He D, Fleisch MC, Niederacher D, Pilarski LM, Lim CJ, Maxwell CA. Spatial regulation of Aurora A activity during mitotic spindle assembly requires RHAMM to correctly localize TPX2. Cell Cycle 2014; 13:2248–2261.
13. Niedworok C, Kretschmer I, Röck K, Vom Dorp F, Szarvas T, Heß J, Freudenberger T, Melchior-Becker A, Rübben H, Fischer JW. The impact of the receptor of hyaluronan-mediated motility (RHAMM) on human urothelial transitional cell cancer of the bladder. PLoS One 2013; 8:e75681.
14. Du Y-CN, Chou C-K, Klimstra DS, Varmus H. Receptor for hyaluronan-mediated motility isoform B promotes liver metastasis in a mouse model of multistep tumorigenesis and a tail vein assay for metastasis. Proc. Natl. Acad. Sci. 2011; 108:16753–16758.
15. Hamilton SR, Fard SF, Paiwand FF, Tolg C, Veiseh M, Wang C, McCarthy JB, Bissell MJ, Koropatnick J, Turley EA. The hyaluronan receptors CD44 and Rhamm (CD168) form complexes with ERK1,2 that sustain high basal motility in breast cancer cells. J. Biol. Chem. 2007; 282:16667–80.
16. Maxwell CA, Rasmussen E, Zhan F, Keats JJ, Adamia S, Strachan E, Crainie M, Walker R, Belch AR, Pilarski LM, Barlogie B, Shaughnessy J Jr, Reiman T. RHAMM expression and isoform balance predict aggressive disease and poor survival in multiple myeloma. Blood 2004; 104:1151–8.
17. Assmann V, Marshall JF, Fieber C, Hofmann M, Hart IR. The human hyaluronan receptor RHAMM is expressed as an intracellular protein in breast cancer cells. J. Cell Sci. 1998; 111:1685–1694.
18. Veiseh M, Kwon DH, Borowsky AD, Tolg C, Leong HS, Lewis JD, Turley EA, Bissell MJ. Cellular heterogeneity profiling by hyaluronan probes reveals an invasive but slow-growing breast tumor subset. Proc. Natl. Acad. Sci. 2014; 111: E1731–E1739.
19. Brosh R, Rotter V. When mutants gain new powers: news from the mutant p53 field. Nat. Rev. Cancer 2009; 9:701–13.
20. Ziebell MR, Zhao ZG, Luo B, Luo Y, Turley E a., Prestwich GD. Peptides that mimic glycosaminoglycans: High-affinity ligands for a hyaluronan binding domain. Chem. Biol. 2001; 8:1081–1094.
21. Hall CL, Yang B, Yang X, Zhang S, Turley M, Samuel S, Lange LA, Wang C, Curpen GD, Savani RC, Greenberg AH, Turley EA. Overexpression of the hyaluronan receptor RHAMM is transforming and is also required for H-ras transformation. Cell 1995; 82:19–26.
22. Crainie M, Belch a R, Mant MJ, Pilarski LM. Overexpression of the receptor for hyaluronan-mediated motility (RHAMM) characterizes the malignant clone in multiple myeloma: identification of three distinct RHAMM variants. Blood 1999; 93:1684–1696.
23. Rhodes DR, Yu J, Shanker K, Deshpande N, Varambally R, Ghosh D, Barrette T, Pandey A, Chinnaiyan AM. ONCOMINE: A Cancer Microarray Database and Integrated Data-Mining Platform1. Neoplasia 2004; 6:1–6.
24. Greiner J, Schmitt A, Giannopoulos K, Rojewski MT, Götz M, Funk I, Ringhoffer M, Bunjes D, Hofmann S, Ritter G, Döhner H, Schmitt M. High-dose RHAMM-R3 peptide vaccination for patients with acute myeloid leukemia, myelodysplastic syndrome and multiple myeloma. Haematologica 2010; 95:1191–1197.
25. Zhivotovsky B, Joseph B, Orrenius S. Tumor radiosensitivity and apoptosis. Exp. Cell Res. 1999; 248:10–7.
26. Sirzén F, Zhivotovsky B, Nilsson A, Bergh J, Lewensohn R. Spontaneous and radiation-induced apoptosis in lung carcinoma cells with different intrinsic radiosensitivities. Anticancer Res. 1997; 18:695–9.
27. Belcheva I, Stoychev T, Karadjov K, Danchev D. Neuropharmacological activity of newly-synthethized derivatives of 3,3-diethyl-2,4-pyridinedione. III. N-Acyl derivatives of 3,3-diethyl-2,4-pyridinedione. Acta Physiol. Pharmacol. Bulg. 1979; 5:75–81.
28. Olive PL, Durand RE. Apoptosis: an indicator of radiosensitivity in vitro? Int. J. Radiat. Biol. 1997; 71:695–707.
29. Decaudin D, Delic J, Dumont J, Tertian G, Blot E, Dubray B, Grandpeix C, Peffault de Latour R, Cosset JM. Clinical efficacy of irradiation in CLL patients: predictive value of in vitro radio-induced apoptosis. Leuk. Lymphoma 2002; 43:827–9.
30. Devarajan E, Sahin AA, Chen JS, Krishnamurthy RR, Aggarwal N, Brun AM, Sapino A, Zhang F, Sharma D, Yang XH, Tora AD, Mehta K. Down-regulation of caspase 3 in breast cancer: a possible mechanism for chemoresistance. Oncogene 2002; 21:8843–8851.
31. Olivier M, Eeles R, Hollstein M, Khan MA, Harris CC, Hainaut P. The IARC TP53 database: new online mutation analysis and recommendations to users. Hum. Mutat. 2002; 19:607–14.
32. Lu X, Errington J, Curtin NJ, Lunec J, Newell DR. The impact of p53 status on cellular sensitivity to antifolate drugs. Clin. Cancer Res. 2001; 7:2114–2123.
33. Stretch JR, Gatter KC, Ralfkiaer E, Lane DP, Harris a L. Expression of mutant p53 in melanoma. Cancer Res. 1991; 51:5976–5979.
34. Chiba I, Takahashi T, Nau MM, D’Amico D, Curiel DT, Mitsudomi T, Buchhagen DL, Carbone D, Piantadosi S, Koga H, et al. Mutations in the p53 gene are frequent in primary, resected non-small cell lung cancer. Lung Cancer Study Group. Oncogene 1990; 5:1603–10.
35. Hinds PW, Finlay CA, Quartin RS, Baker SJ, Fearon ER, Vogelstein B, Levine AJ. Mutant p53 DNA clones from human colon carcinomas cooperate with ras in transforming primary rat cells: a comparison of the “hot spot” mutant phenotypes. Cell Growth Differ. 1990; 1:571–580.
36. Yamaguchi T, Mukai H, Yamashita S, Fujii S, Ushijima T. Comprehensive DNA Methylation and Extensive Mutation Analyses of HER2-Positive Breast Cancer. Oncology 2015; 88:377–84.
37. Spurgers KB, Gold DL, Coombes KR, Bohnenstiehl NL, Mullins B, Meyn RE, Logothetis CJ, McDonnell TJ. Identification of cell cycle regulatory genes as principal targets of p53-mediated transcriptional repression. J. Biol. Chem. 2006; 281:25134–25142.
38. Rother K, Johne C, Spiesbach K, Haugwitz U, Tschöp K, Wasner M, Klein-Hitpass L, Möröy T, Mössner J, Engeland K. Identification of Tcf-4 as a transcriptional target of p53 signalling. Oncogene 2004; 23:3376–3384.
39. Kim E, Deppert W. Transcriptional activities of mutant p53: When mutations are more than a loss. J. Cell. Biochem. 2004; 93:878–886.
40. Hui L, Zheng Y, Yan Y, Bargonetti J, Foster D a. Mutant p53 in MDA-MB-231 breast cancer cells is stabilized by elevated phospholipase D activity and contributes to survival signals generated by phospholipase D. Oncogene 2006; 25:7305–7310.
41. Hiraga T, Ito S, Nakamura H. Cancer stem-like cell marker CD44 promotes bone metastases by enhancing tumorigenicity, cell motility, and hyaluronan production. Cancer Res. 2013; 73:4112–4122.
42. Maxwell CA, McCarthy J, Turley E. Cell-surface and mitotic-spindle RHAMM: moonlighting or dual oncogenic functions? J. Cell Sci. 2008; 121:925–932.
43. Godar S, Ince TA, Bell GW, Feldser D, Donaher JL, Bergh J, Liu A, Miu K, Watnick RS, Reinhardt F, McAllister SS, Jacks T, Weinberg RA. Growth-inhibitory and tumor-suppressive functions of p53 depend on its repression of CD44 expression. Cell 2008; 134:62–73.
44. Edward M, Gillan C, Micha D, Tammi RH. Tumour regulation of fibroblast hyaluronan expression: A mechanism to facilitate tumour growth and invasion. Carcinogenesis 2005; 26:1215–1223.
45. Tzircotis G, Thorne RF, Isacke CM. Chemotaxis towards hyaluronan is dependent on CD44 expression and modulated by cell type variation in CD44-hyaluronan binding. J. Cell Sci. 2005; 118:5119–5128.
46. Auvinen P, Tammi R, Parkkinen J, Tammi M, Agren U, Johansson R, Hirvikoski P, Eskelinen M, Kosma VM. Hyaluronan in peritumoral stroma and malignant cells associates with breast cancer spreading and predicts survival. Am. J. Pathol. 2000; 156:529–536.
47. Yang B, Yang BL, Savani RC, Turley EA. Identification of a common hyaluronan binding motif in the hyaluronan binding proteins RHAMM, CD44 and link protein. EMBO J. 1994; 13:286–96.
48. Ponta H, Sherman L, Herrlich PA. CD44: from adhesion molecules to signalling regulators. Nat. Rev. Mol. Cell Biol. 2003; 4:33–45.
49. Milde-Langosch K, Karn T, Schmidt M, zu Eulenburg C, Oliveira-Ferrer L, Wirtz RM, Schumacher U, Witzel I, Schütze D, Müller V. Prognostic relevance of glycosylation-associated genes in breast cancer. Breast Cancer Res. Treat. 2014; 145:295–305.
50. Mcshane LM, Altman DG, Sauerbrei W, Taube SE, Gion M, Clark GM. REporting recommendations for tumor MARKer prognostic studies (REMARK). 2005; 2:416–422.
51. Röck K, Tigges J, Sass S, Schütze A, Florea AM, Fender AC, Theis FJ, Krutmann J, Boege F, Fritsche E, Reifenberger G, Fischer JW. miR-23a-3p Causes Cellular Senescence by Targeting Hyaluronan Synthase 2: Possible Implication for Skin Aging. J. Invest. Dermatol. 2014; 135:369–377.
52. Twarock S, Tammi MI, Savani RC, Fischer JW. Hyaluronan Stabilizes Focal Adhesions, Filopodia, and the Proliferative Phenotype in Esophageal Squamous Carcinoma Cells. J. Biol. Chem. 2010; 285:23276–23284.
53. Quah BJC, Warren HS, Parish CR. Monitoring lymphocyte proliferation in vitro and in vivo with the intracellular fluorescent dye carboxyfluorescein diacetate succinimidyl ester. Nat. Protoc. 2007; 2:2049–56.