
Background
Mammalian target of rapamycin (mTOR) is a key component of phosphatidylinositol 3-kinase (PI3K) pathway that plays an important role of cell growth and proliferation, metabolism, and angiogenesis [1]. Since inhibition of mTOR signaling can abrogate the cellular response to growth factor receptor activation, targeting mTOR activation is an attractive approach for cancer therapy.
Everolimus is a rapamycin analog that is being developed as an inhibitor of mTORC1. Like rapamycin, everolimus binds the cyclophilin FKBP-12, and this complex binds the serine-threonine kinase of mTORC1 and inhibits signaling downstream. Everolimus has been extensively tested for several kinds of tumor types, and showed some significant and durable responses. In renal cell carcinoma, everolimus provided a significant benefit to patients with metastatic renal cell carcinoma after failure of treatment with sorafenib or sunitinib [2]. Adding everolimus to exemestane significantly improved median progression-free survival (PFS) compared with exemestane alone in the treatment of estrogen receptor-positive, HER2-negative advanced breast cancer patients [3]. In addition, everolimus provided a significant reduction in volume and seizure frequency in patients with subependymal giant cell astrocytoma associated with tuberous sclerosis, and prolonged survival in pancreatic neuroendocrine tumor [4, 5].
However, there has not been a validated biomarker for predicting response to everolimus, yet. Previous studies used candidate gene approach for searching mutations and revealed that PI3K/AKT/mTOR pathway had an important role in everolimus sensitivity [6-8]. Recently, whole genome sequencing identified a somatic mutation for everolimus sensitivity in bladder cancer [9]. In this study, mutation of tuberous sclerosis 1 (TSC1) was suggested as a biomarker for everolimus response in bladder cancer. Similarly, one anaplastic thyroid cancer patient who showed sensitivity to everolimus, revealed a nonsense mutation in TSC2, a negative regulator of mTOR [10]. A recent study by Yoon et al. reported that pS6Ser240/4 expression may be a predictive biomarker for everolimus sensitivity in gastric cancer patients [11]. Likewise, we recently discovered one patient who showed exceptional response to everolimus.
We hypothesized that there are driver genetic events, which are clinically actionable and targetable, that occur commonly across different tumor types. In this study, we aim to perform next-generation sequencing (NGS) in patients with different tumor types to explore common genetic aberrations that confer sensitivity to everolimus. We recruited patients who were treated with single-agent everolimus, and sought to find universal biomarkers regardless of tumor types and histologic subtypes.
Results
Everolimus leading to response in a patient with primary ductal adenocarcinoma of the lacrimal gland
Our index patient was a 51-year-old male who diagnosed with primary ductal adenocarcinoma of the lacrimal gland in the left. (Figure 1). The patient received multiple lines of chemotherapy, but patient showed aggravation of primary tumor and progression to bone metastasis. His tumor tissue was sent for targeted next-generation sequencing at Foundation Medicine profiling of 236 cancer-related genes and 47 introns of 19 genes involved in rearrangement. The assay reported TP53 (Q38fs) mutation and NF1 (D1644A) mutation (Supplementary Figure S2A-B). While there were no approved therapies or clinical trials to address for TP53 mutation and NF1 mutation at that time, a therapeutic attempt using everolimus 10mg once per day was initiated in October 2013 on the basis of data published by McGillicuddy et al, supporting the sensitivity of everolimus in loss-of-function mutation in the NF1 tumor suppressor gene [12]. The patient was seen again 1 month after, and we noticed decreased exophthalmus and reduced skin thickening around his left eyelid. In terms of subjective symptoms, he reported improved pain of his bone metastasis lesions in right humerus head and left shoulder. The PET-CT taken after 1 month showed a partial response as per PET response criteria [13], showing more than 25% reduction of SUVmax compared to baseline. He was maintained on everolimus and the CT scan taken after 2 months of therapy showed shrinkage of the measurable tumor lesions (26.5mm to 17.6mm, 33.5% reduction). His tumor showed further decrease in SUVmax in the PET-CT scan taken after 4 months of therapy (Supplementary Figure 3). The partial response was maintained for 8 months.
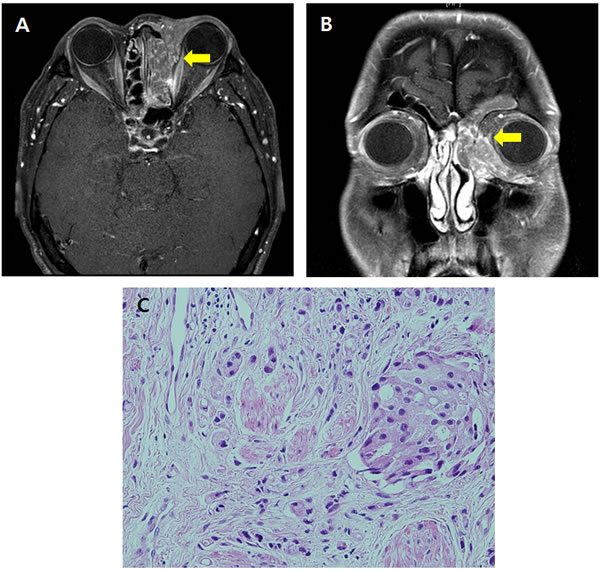
Figure 1: The primary tumor located in the lacrimal gland seen in orbit MRI A. axial view B. coronal view. C. Hematoxylin & eosin staining of the tumor.
Patients’ characteristics
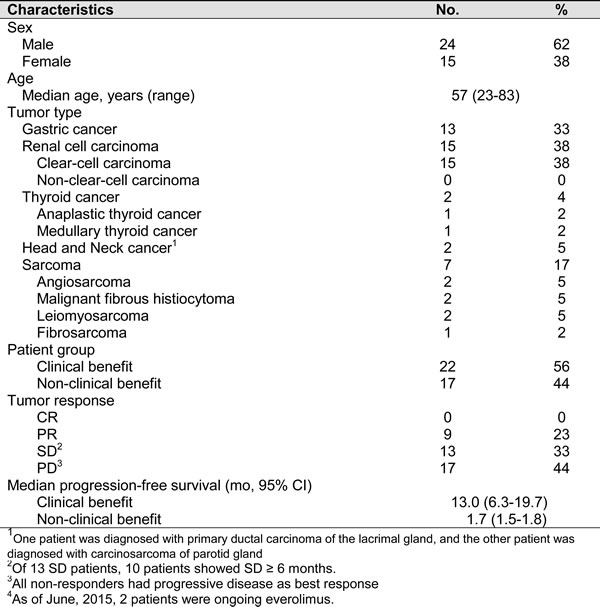
We collected a total of thirty-nine patients with five different tumor types (13 with gastric cancer, 15 with renal cell carcinoma, 2 with thyroid cancer, 2 with head and neck cancer, and 7 with sarcoma) were analyzed by NGS. As shown in Table 1, the median age of all patients was 57, and there were 24 (61.5%) males and 15 (38.5%) females. There were 22 (56.4%) patients with clinical benefit and 17 (43.6%) patients without clinical benefit. Patients with clinical benefit included 9 patients with partial response (PR) and 13 patients with durable stable disease (SD), with 10 patients showing SD ≥ 6 months. All patients without clinical benefit showed progressive disease (PD) as their best response with no tumor shrinkage at all. The median progression-free survival was 13.0 months (95% CI, 6.3-19.7) for patients with clinical benefit and 1.7 months (95% CI, 1.54-1.85) for patients without clinical benefit. As of data cutoff, two patients were still under ongoing everolimus treatment (Supplementary Table 3, 4).
Table 1: Baseline characteristics of all patients (n=39)
Overall genetic alterations
To identify possible genetic mechanisms of sensitivity to everolimus, we performed targeted sequencing of the pre-treatment tumor and paired germline DNA. Tumors were sequenced to a median coverage of 552x. Cancer genomes were characterized by 219 somatic single-nucleotide variants (181 missense, 9 nonsense, 7 splice-site) and 22 frameshift insertions/deletions, with a median of 2.1 mutations per Mb (0 to 12.4 mutations per Mb). Of these alterations, 13 mutations were previously reported in the Catalogue of Somatic Mutations in Cancer. Mutations were further analyzed for functional prediction of amino acid changes using two different prediction algorithms (Provean and SIFT) (Supplementary Table 5). TP53 gene alterations were the most common among all genes and were found in 8 of 38 tumors (21.1%). Recurrently mutated genes such as PBRM1, MTOR, NF1, VHL, PD34DIP, and ARID1A were found in patients with clinical benefit, in the order of frequency. In contrast, recurrently mutated genes such as VHL, TP53, and ARID1A were found in patients with non-clinical benefit. (Supplementary Figure S4). Sanger sequencing for mTOR pathway genes (MTOR/TSC1/TSC2/PIK3CA/NF1/NF2/ PTEN/AKT1) confirmed variants that were found in CCP panel (supplementary Figure S5 & supplementary Table 6).
Pathways relevant to everolimus sensitivity
We examined the sequencing data for biologically plausible mechanisms of sensitivity to everolimus, and identified multiple mutations in the mTOR-pathway (Figure 2).
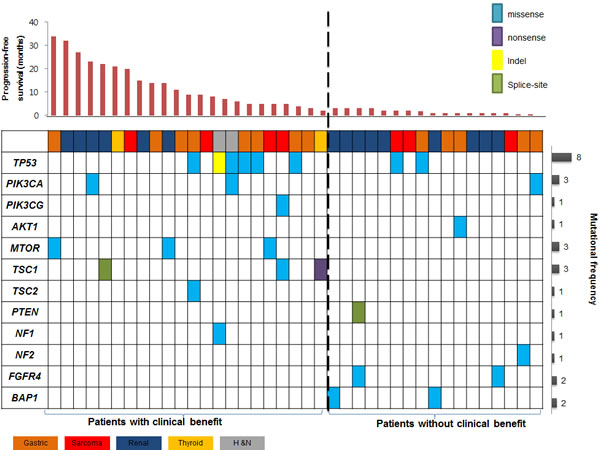
Figure 2: Landscape genomic profile of patients is seen. Genomic alterations which may activate mTOR signaling were identified in 10 of 22 (45%) patients with clinical benefit. Recurrently mutated genes of FGFR4 and BAP1 were noted only in patients without clinical benefit (P = NS).
Two PIK3CA mutations were detected in patients with clinical benefit. A mutation in the helical domain of PIK3CA (p.E542K), which is known to be constitutively activating and selectively sensitive to everolimus [14], was found in a renal cell carcinoma patient with the PFS of 23.9 months. In addition, a kinase domain mutation of PIK3CA (p.H1047R) was found in a head and neck cancer patient with partial response. Alterations in TSC1 gene were noted in three patients: a patient with anaplastic thyroid cancer who harbored a nonsense mutation in TSC1 (p.Trp103*) and a renal cell carcinoma patient with a TSC1 splicing variant (c.1029+1G>A). In addition, a malignant histiosarcoma patient had TSC1 missense mutation (p.A307V) and showed significant tumor reduction (-24.3%). One TSC2 missense mutation (p.E588K) was found in a gastric cancer patient. Three MTOR missense mutations (N1421D, K1771R, I1973F) were found in patients with gastric cancer, angiosarcoma and renal cell carcinoma, respectively. All three mutations were located in FAT (FRAP, ATM, TRAP) domain of MTOR.
Overall, genomic alterations which may activate mTOR signaling were identified in 10 of 22 (45%) patients with clinical benefit (Figure 3A). In particular, TSC1/TSC2/MTOR mutations were key components in determining everolimus sensitivity (Figure 3B). The incidence of these mutations were 31.8% (7/22) in patients with clinical benefit as compared with 0% in those with non-clinical benefit (P=0.012). The prevalence of these recurrently mutated genes and their correlation with clinical benefit strongly suggest that they confer sensitivity to everolimus.
While TSC1/TSC2/MTOR alterations were exclusively found in patients with clinical benefit, we also searched for recurrently mutated genes that were exclusively identified in patients with non-clinical benefit. Mutations in chromatin remodeling gene (BAP1; n=2, 12%) and receptor tyrosine kinase signaling (FGFR4; n=2, 12%) were noted, but not statistically significant.
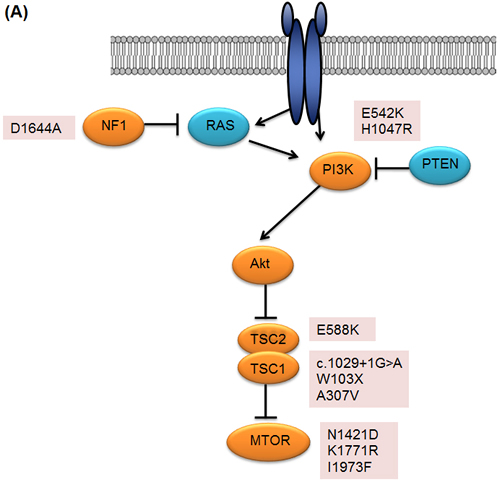
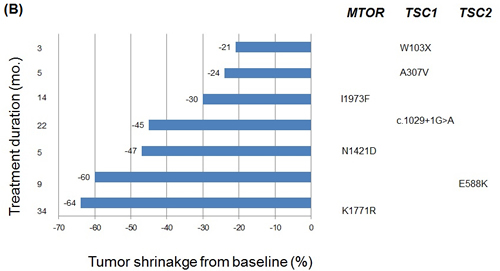
Figure 3: A. Genetic alterations identified in patients with clinical benefit B. Best overall response of patients with clinical benefit, with negative values indicating tumor shrinkage and the vertical axis indicating treatment duration in months. Nonsynonymous mutations for each patient are marked accordingly.
Discussion
Here, we described multiple activating mutations in the mTOR pathway found in patient tumors that showed exquisite sensitivity to everolimus. TSC1/TSC2/MTOR alterations were exclusively found in patients who showed extended clinical benefit, which suggest that they confer sensitivity to everolimus. NF1 mutation together with TP53 mutation presents an aggressive tumor behavior, but tumor growth is effectively inhibited by everolimus.
In theory, dependency on the mTOR pathway may render enhanced sensitivity to mTOR inhibitors. Three different mutations (N1421D, K1771R, I1973F) in the FAT domain of MTOR have not been previously reported in the publicly available genomic database [15]. Previously, it has been proposed that FAT domain consist of TRD1, TRD2 and TRD3 domain and interacts to yield a configuration that exposes the catalytic domain [16, 17]. Especially, N1421D was located in TRD1 domain which is important for mTOR function and both Provean and SIFT predicted that our mutation was deleterious mutation. In addition, K1771R mutation is located in TRD3 which is interact with the C lobe on one side of the kinase domain. Mutation of I1973 amino acid was previously reported to be hyperactivating mutation which involved in restricting active site access and both Provean and SIFT reported that I1973F is deleterious mutation [17].
TSC1 and TSC2 are upstream key negative regulators of mTORC1. They form a heterotrimer, a GTPase-activating protein for Rheb (Ras homologue enriched in brain), a GTP-binding protein that activates mTORC1 by binding to it. As the loss of TSC1-TSC2 function activates mTORC1 pathway, strong scientific rationale exists for the use of mTOR inhibitors in this setting. Sirolimus has shown promising effects in patients with inactivating mutations in TSC1, TSC2 and STK11 in hamartoma syndromes such as tuberous sclerosis complex and Peutz-Jeghers Syndrome [18-20]. Moreover, 2 out of 3 patients with malignant perivascular epithelioid cell tumors who had clinical response to sirolimus showed TSC1/2 loss [21]. There was a recent report of renal cell carcinoma patients with extended benefit from mTOR inhibitor showed that TSC1 and TSC2 offer explanation for treatment response [22]. Similarly, we identified 1 TSC1 splicing variant (renal cell carcinoma), 1 TSC1 nonsense mutation (anaplastic thyroid cancer), 1 TSC1 missense mutation (sarcoma) and 1 TSC2 missense mutation (angiosarcoma). Given that heterozygous loss of TSC1 is common in renal cell carcinoma (>30%) [23], TSC1 and TSC2 may be screened as predictive biomarkers of everolimus in renal cell carcinoma patients who progressed on VEGF-targeted therapy.
Loss-of-function mutations in the NF1 tumor suppressor gene affects mTOR pathway, leading to constitutive activation of mTOR. This activation depends on Ras and PI3 kinase, and is mediated by the inactivation of the TSC2-encoded protein tuberin by AKT [24]. In this way, NF1-deficient tumors alone may show exquisite sensitivity to mTOR inhibitors. In conjunction with our index patient with TP53 and NF1 mutation, it has been reported that NF1 heterozygosity cooperates with TP53 mutation to promote tumorigenesis. Homozygous null NF1 mutations may occur, which mediate sensitivity to mTOR inhibitors [12]. Although rare in tumors such as gastric cancer, sarcoma, and renal cell carcinoma, recent finding that NF1 mutations are enriched (11%) in oncogene-negative subset of lung adenocarcinomas, and our finding suggests novel therapeutic opportunities for the subset of patients with NF1 mutations.
Conceptually, activating mutations within the phosphoinositide 3-kinase, catalytic subunit α (PI3K-p110 α) encoded by the PIK3CA gene, lead to mTOR pathway activation [6, 25]. In this study, activating PIK3CA mutations in helical domain (p.E542K), and kinase domain (p.H1047R) were found in a renal cell carcinoma and a parotid gland choriocarcinoma patient, respectively. However, one patient with a PIK3CA mutation (p.H1047R) showed non-clinical benefit to everolimus, showing rapid increase of ascites within 1 month of everolimus treatment. That activating PIK3CA mutation does not always lead to response was previously reported by Janku et al. where they described patients with H1047R PIK3CA mutation experienced a response rate of 44% [26].
Our study has a few limitations. Although prespecified, the selected patients with clinical benefit in this study may only explain a portion of mechanisms of sensitivity. Other genetic or epigenetic alterations that are not covered in the CCP may be missed out even if they regulate sensitivity or resistance to everolimus. Because study was retrospective in nature, and the number of patients analyzed was relatively small, these data must be interpreted cautiously
However, to the best of our knowledge, this is the first report that identified histology-independent mechanisms of sensitivity to mTOR inhibitor in multiple tumor types. Regardless of histology, mTOR pathway-activating mutations confer exceptional response to everolimus, and this provides the rationale for the development of so-called basket trials. The current study highlights the fact that mutational analyses of somatic variants could allow sub-classification of patients for optimal treatment, and provides a basis for a basket trial in the future. These efforts will eventually increase the likelihood of success of drug trials especially in patients with rare cancer types.
In conclusion, the genomic information derived from patients with exceptional clinical benefit may provide a basis for everolimus sensitivity. Overall, it seems that screening for TSC1/TSC2/MTOR mutations warrants further investigation in application of mTOR inhibitors in the clinic.
Materials and Methods
Patients
We recruited tissue samples and matched blood samples among patients who participated in the clinical trials using everolimus and renal cell carcinoma patients treated with everolimus as a standard second line of therapy (Supplementary Table 1). Patients with extended clinical benefit and non-benefit were selected for this study. The criteria for extended clinical benefit were: 1) complete response or 2) partial response for more than 6 months or 3) durable stable disease with PFS on everolimus ≥ 1.5 x PFS of prior treatment. The criteria for non-benefit were 1) no shrinkage in tumor diameter and 2) progressive disease as best response.
Clinical information including age, sex, treatment duration, best response to treatment, percent change in tumor size, previous treatment history and survival data were collected. The study protocol was approved by each center’s independent ethics committee or institutional review board and was conducted in accordance with the Declaration of Helsinki and Good Clinical Practice. All patients provided written informed consent for genomic testing used for this study. Specimens were evaluated by pathologists (H.K.K) to identify tumor-bearing areas for DNA extraction.
Targeted sequencing
Tumor genomic DNA was extracted from formalin-fixed, paraffin-embedded (FFPE) tumor tissue blocks, unstained 10 um thick tissue sections. All samples were micro-dissected to ensure ≥70% tumor contents. Normal genomic DNA was extracted from either peripheral blood mononuclear cells or histologically confirmed normal tissue. Library preparation for each sample was performed using Ion AmpliSeqTM Comprehensive Cancer Panel (CCP, Life Technology) following the manufacturer’s instructions. The pooled capture library was quantified by Qubit (Invitrogen) and Tape station (Agilent) and sequenced by Ion ProtonTM System. Two patients (anaplastic thyroid cancer and renal cell carcinoma) were sequenced by Sanger sequencing using primers for all coding region of TSC1.
Analysis of molecular aberrations
Ion AmpliSeq™ Comprehensive Cancer Panel (CCP, Life Technologies) was used for cancer specific target sequencing, targeting 409 genes with ~16,000 amplicons. Sequencing was processed by Ion PGMTM system and sequencing data was analyzed by PGMTM built in TSS (Torrent Suite Software) 4.0.2 version (Supplementary Fig S1). Variants acquired from the CCP panel were filtered by germline variants acquired from our patients and 1000 genome data. Mutations with less than 50x depth and less than 10% variant frequency were filtered out [27]. Quality score, a parameter of variant call format using phred scale, was used to filter out the variants and Q30 was used for cut-off value [27]. Then, variants were annotated using ANNOVAR [28] and non-coding region and synonymous variants were filtered out. Remaining variants were assessed using the Integrative Genomics Viewer (IGV) [29] and loci were further analyzed for functional prediction of amino acid changes using two different prediction algorithms (Provean and SIFT) [30, 31]. Mutations in MTOR, TSC1, TSC2, PIK3CA, NF1, NF2, PTEN, AKT1 were validated by Sanger sequencing or pyrosequencing. Sanger sequencing and pyrosequencing primers listed in the supplementary table 2.
Statistical analyses
Tumor response was evaluated using the RECIST v1.1 [32] and progression-free survival (PFS) was defined as the time from the start of everolimus to disease progression or death from any cause. Statistical significance of preferential association of somatic variations with specific clusters was assessed with chi-square test or Fisher’s exact test. All statistical analyses were performed using SPSS 20.0 (SPSS, Chicago, IL, USA).
Abbreviations
PFS: progression-free survival; NGS: next-generation sequencing; FFPE: formalin-fixed, paraffin-embedded; CCP: Comprehensive Cancer Panel; IGV: Integrative Genomics Viewer; RECIST: Response evaluation criteria in solid tumor; PR: partial response; SD: stable disease; PD: progressive disease
Conflicts of Interest
SMA and JRG are employees of and have equity interest in Foundation Medicine, Inc.
Grant Support
This study was supported by a grant of the Korean Health Technology R&D Project, Ministry of Health & Welfare, Republic of Korea. (HI12C1440).
References
1. Cantley LC. The phosphoinositide 3-kinase pathway. Science (New York, NY). 2002; 296:1655-1657.
2. Motzer RJ, Escudier B, Oudard S, Hutson TE, Porta C, Bracarda S, Grunwald V, Thompson JA, Figlin RA, Hollaender N, Urbanowitz G, Berg WJ, Kay A, Lebwohl D and Ravaud A. Efficacy of everolimus in advanced renal cell carcinoma: a double-blind, randomised, placebo-controlled phase III trial. Lancet. 2008; 372:449-456.
3. Baselga J, Campone M, Piccart M, Burris HA, 3rd, Rugo HS, Sahmoud T, Noguchi S, Gnant M, Pritchard KI, Lebrun F, Beck JT, Ito Y, Yardley D, Deleu I, Perez A, Bachelot T, et al. Everolimus in postmenopausal hormone-receptor-positive advanced breast cancer. The New England journal of medicine. 2012; 366:520-529.
4. Krueger DA, Care MM, Holland K, Agricola K, Tudor C, Mangeshkar P, Wilson KA, Byars A, Sahmoud T and Franz DN. Everolimus for subependymal giant-cell astrocytomas in tuberous sclerosis. The New England journal of medicine. 2010; 363:1801-1811.
5. Yao JC, Shah MH, Ito T, Bohas CL, Wolin EM, Van Cutsem E, Hobday TJ, Okusaka T, Capdevila J, de Vries EG, Tomassetti P, Pavel ME, Hoosen S, Haas T, Lincy J, Lebwohl D, et al. Everolimus for advanced pancreatic neuroendocrine tumors. The New England journal of medicine. 2011; 364:514-523.
6. Janku F, Tsimberidou AM, Garrido-Laguna I, Wang X, Luthra R, Hong DS, Naing A, Falchook GS, Moroney JW, Piha-Paul SA, Wheler JJ, Moulder SL, Fu S and Kurzrock R. PIK3CA mutations in patients with advanced cancers treated with PI3K/AKT/mTOR axis inhibitors. Molecular cancer therapeutics. 2011; 10:558-565.
7. Meric-Bernstam F, Akcakanat A, Chen H, Do KA, Sangai T, Adkins F, Gonzalez-Angulo AM, Rashid A, Crosby K, Dong M, Phan AT, Wolff RA, Gupta S, Mills GB and Yao J. PIK3CA/PTEN mutations and Akt activation as markers of sensitivity to allosteric mTOR inhibitors. Clinical cancer research. 2012; 18:1777-1789.
8. Nishi T, Iwasaki K, Ohashi N, Tanaka C, Kobayashi D, Nakayama G, Koike M, Fujiwara M, Kobayashi T and Kodera Y. Phosphorylation of 4E-BP1 predicts sensitivity to everolimus in gastric cancer cells. Cancer letters. 2013; 331:220-229.
9. Iyer G, Hanrahan AJ, Milowsky MI, Al-Ahmadie H, Scott SN, Janakiraman M, Pirun M, Sander C, Socci ND, Ostrovnaya I, Viale A, Heguy A, Peng L, Chan TA, Bochner B, Bajorin DF, et al. Genome sequencing identifies a basis for everolimus sensitivity. Science (New York, NY). 2012; 338:221.
10. Wagle N, Grabiner BC, Van Allen EM, Amin-Mansour A, Taylor-Weiner A, Rosenberg M, Gray N, Barletta JA, Guo Y, Swanson SJ, Ruan DT, Hanna GJ, Haddad RI, Getz G, Kwiatkowski DJ, Carter SL, et al. Response and acquired resistance to everolimus in anaplastic thyroid cancer. The New England journal of medicine. 2014; 371:1426-1433.
11. Yoon DH RM, Park YS, Lee HJ, Lee C, Ryoo BY, Lee JL, Chang HM, Kim TW, Kang YK. Phase II study of everolimus with biomarker exploration in patients with advanced gastric cancer refractory to chemotherapy including fluoropyrimidine and platinum. British Journal of Cancer. 2012; 106:1039-1044.
12. McGillicuddy LT, Fromm JA, Hollstein PE, Kubek S, Beroukhim R, De Raedt T, Johnson BW, Williams SM, Nghiemphu P, Liau LM, Cloughesy TF, Mischel PS, Parret A, Seiler J, Moldenhauer G, Scheffzek K, et al. Proteasomal and genetic inactivation of the NF1 tumor suppressor in gliomagenesis. Cancer cell. 2009; 16:44-54.
13. Wahl RL, Jacene H, Kasamon Y and Lodge MA. From RECIST to PERCIST: Evolving Considerations for PET response criteria in solid tumors. Journal of nuclear medicine. 2009; 50 Suppl 1:122S-150S.
14. Di Nicolantonio F, Arena S, Tabernero J, Grosso S, Molinari F, Macarulla T, Russo M, Cancelliere C, Zecchin D, Mazzucchelli L, Sasazuki T, Shirasawa S, Geuna M, Frattini M, Baselga J, Gallicchio M, et al. Deregulation of the PI3K and KRAS signaling pathways in human cancer cells determines their response to everolimus. The Journal of clinical investigation. 2010; 120:2858-2866.
15. Grabiner BC, Nardi V, Birsoy K, Possemato R, Shen K, Sinha S, Jordan A, Beck AH and Sabatini DM. A diverse array of cancer-associated MTOR mutations are hyperactivating and can predict rapamycin sensitivity. Cancer discovery. 2014; 4:554-563.
16. Hay N and Sonenberg N. Upstream and downstream of mTOR. Genes & development. 2004; 18(16):1926-1945.
17. Haijuan Yang DGR, Joseph D. Koos. mTOR kinase structure, mechanism and regulation. Nature. 2012; 497:217-224.
18. Davies DM, de Vries PJ, Johnson SR, McCartney DL, Cox JA, Serra AL, Watson PC, Howe CJ, Doyle T, Pointon K, Cross JJ, Tattersfield AE, Kingswood JC and Sampson JR. Sirolimus therapy for angiomyolipoma in tuberous sclerosis and sporadic lymphangioleiomyomatosis: a phase 2 trial. Clinical cancer research. 2011; 17:4071-4081.
19. Bissler JJ, McCormack FX, Young LR, Elwing JM, Chuck G, Leonard JM, Schmithorst VJ, Laor T, Brody AS, Bean J, Salisbury S and Franz DN. Sirolimus for angiomyolipoma in tuberous sclerosis complex or lymphangioleiomyomatosis. The New England journal of medicine. 2008; 358:140-151.
20. Klumpen HJ, Queiroz KC, Spek CA, van Noesel CJ, Brink HC, de Leng WW, de Wilde RF, Mathus-Vliegen EM, Offerhaus GJ, Alleman MA, Westermann AM and Richel DJ. mTOR inhibitor treatment of pancreatic cancer in a patient With Peutz-Jeghers syndrome. Journal of clinical oncology. 2011; 29:e150-153.
21. Wagner AJ, Malinowska-Kolodziej I, Morgan JA, Qin W, Fletcher CD, Vena N, Ligon AH, Antonescu CR, Ramaiya NH, Demetri GD, Kwiatkowski DJ and Maki RG. Clinical activity of mTOR inhibition with sirolimus in malignant perivascular epithelioid cell tumors: targeting the pathogenic activation of mTORC1 in tumors. Journal of clinical oncology. 2010; 28:835-840.
22. Voss MH, Hakimi AA, Pham CG, Brannon AR, Chen YB, Cunha LF, Akin O, Liu H, Takeda S, Scott SN, Socci ND, Viale A, Schultz N, Sander C, Reuter VE, Russo P, et al. Tumor genetic analyses of patients with metastatic renal cell carcinoma and extended benefit from mTOR inhibitor therapy. Clinical cancer research. 2014; 20:1955-1964.
23. Cerami E, Gao J, Dogrusoz U, Gross BE, Sumer SO, Aksoy BA, Jacobsen A, Byrne CJ, Heuer ML, Larsson E, Antipin Y, Reva B, Goldberg AP, Sander C and Schultz N. The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer discovery. 2012; 2:401-404.
24. Johannessen CM, Reczek EE, James MF, Brems H, Legius E and Cichowski K. The NF1 tumor suppressor critically regulates TSC2 and mTOR. Proceedings of the National Academy of Sciences of the United States of America. 2005; 102:8573-8578.
25. Zhao JJ, Liu Z, Wang L, Shin E, Loda MF and Roberts TM. The oncogenic properties of mutant p110alpha and p110beta phosphatidylinositol 3-kinases in human mammary epithelial cells. Proceedings of the National Academy of Sciences of the United States of America. 2005; 102:18443-18448.
26. Janku F, Wheler JJ, Westin SN, Moulder SL, Naing A, Tsimberidoue AM, Fu S. PI3K/AKT/mTOR inhibitors in patients with breast and gynecologic malignancies harboring PIK3CA mutations. Journal of Clinical Oncology. 2012; 30:777-782.
27. Wang L, Yamaguchi S, Burstein MD, Terashima K, Chang K, Ng HK, Nakamura H, He Z. Novel somatic and germline mutations in intracranial germ cell tumors. Nature. 2014; 511:241-245.
28. Wang K, Li M and Hakonarson H. ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 2010; 38(16):e164.
29. Robinson JT, Thorvaldsdottir H, Winckler W, Guttman M, Lander ES, Getz G and Mesirov JP. Integrative genomics viewer. Nat Biotechnol. 2011; 29:24-26.
30. Choi Y and Chan AP. PROVEAN web server: a tool to predict the functional effect of amino acid substitutions and indels. Bioinformatics. 2015.
31. Kumar P, Henikoff S and Ng PC. Predicting the effects of coding non-synonymous variants on protein function using the SIFT algorithm. Nat Protoc. 2009; 4:1073-1081.
32. Eisenhauer EA, Therasse P, Bogaerts J, Schwartz LH, Sargent D, Ford R, Dancey J, Arbuck S, Gwyther S, Mooney M, Rubinstein L, Shankar L, Dodd L, Kaplan R, Lacombe D and Verweij J. New response evaluation criteria in solid tumours: revised RECIST guideline (version 1.1). Eur J Cancer. 2009; 45:228-247.