
INTRODUCTION
BRAF mutations are present in about 50% of melanomas, causing an over-activation of the MAPK/ERK pathway involved in cell proliferation and survival. The most frequent mutation (90% of cases) results in a substitution of a valine in glutamic acid at amino acid 600 (BRAFV600E). In August 2011, the FDA, approved Vemurafenib (Zelboraf; Roche) for treatment of BRAFV600E metastatic melanoma due to improved objective response, progression free survival and overall survival showed in several clinical trial [1]. Inhibition of mutated BRAF and consequently reduction of ERK phosphorylation leads to suppression of cyclin D1, induction of expression of the cell-cycle inhibitor p27, and, eventually, to cell-cycle arrest. Unfortunately, responses to BRAF inhibitors are short-lived, with evidence of disease progression within 6–8 months after the beginning of therapy due to the recovery of MAPK signaling or activation of alternative signaling pathways, such as PI3K/AKT/mTOR by IGF-1R or PDGFRb up-regulation [2]. Mutational activation of NRAS is, instead, a common mechanism of resistance to BRAF inhibitors due to increased formation of RAF dimers, against which the drug cannot work [3]. Furthermore, in cells with mutated NRAS, BRAF inhibitors may induce paradoxical activation of the downstream factor MEK1. Another proposed resistance mechanism to BRAF inhibitors is represented by secondary mutations of MEK1 that may also result in reactivation of the MAPK pathway and cause resistance to BRAF inhibitors [3].
An established strategy to overcome BRAF inhibitor resistance is the combination of BRAF inhibitor with MEK inhibitor that targets another protein in the MAPK pathway as demonstrated in recent clinical trials [4].
Moreover, a latest approach is represented by the combination of immunotherapy and targeted therapy, trying to overcome the great toxicity caused by this combination [5–7].
However, although several studies on genetic alterations have been conducted, the molecular mechanisms underlying this very small range of response time to BRAF inhibitors are to date unknown.
In recent years, Next-Generation Sequencing (NGS) platforms, also known as massive parallel sequencing, have drastically decreased the time and cost associated with a comprehensive cancer genome analysis [8–14]. This sequencing technique allowing whole-genome, whole-exome sequencing but also the screening of specific gene mutations, provides highly relevant advances in a clinical setting since a comprehensive mutational screening of tumors could be useful for the best therapeutic assessment [15, 16]. The sensitivity of NGS is higher than traditional methods such as Sanger sequencing (detection of 2–10% versus 15–25% allele frequency). Furthermore, NGS technologies facilitate the screening of multiple genes with limited starting material derived from blood or FFPE tissues, differently to Sanger’s sequencing method that requires relatively large DNA quantities to assess single gene alterations.
In this study we tested the clinical applicability of the NGS platform Ion Torrent Personal Genome Machine (Life Technologies, Carlsbad, CA), to simultaneously screen metastatic melanoma patients in order to individuate new or already known SNPs and mutations which could be related with different response duration to BRAF inhibitors. We created an Ampliseq Custom Panel (Life Technologies, Carlsbad, CA) including 11 crucial full length genes involved in melanoma carcinogenesis and therapy response pathways.
RESULTS
Alteration frequencies and sensitivity detection of NGS variant calling
All 25 amplified samples showed at least one alteration in at least one of the 11 melanoma cancer-related genes (Figure 1). Querying CLINVAR, 12 patients presented alterations in NRAS and only 7 of these have alterations already evidenced as pathogenic in cancer; 14 patients presented alterations in CTLA4 and 13 of these have alterations already evidenced as a risk factor in pathologies other than cancer; 20 patients showed PIK3CA alterations and 21 patients presented alterations in KIT but none seems to be pathogenic; all patients but one presented alterations in BRAF and 17 have a mutation in codon 600; 12 patients presented alterations in RB1 and 1 of these has alterations already evidenced as pathogenic in retinoblastoma; 16 patients presented alterations in MC1R and 15 of these have alterations already evidenced as associated to pigmentation disorders; furthermore, 16, 9, 14 and 3 patients presented no previously reported alterations in MITF, PTEN, MGMT and CDK4 respectively (see Suppl. Table 1).
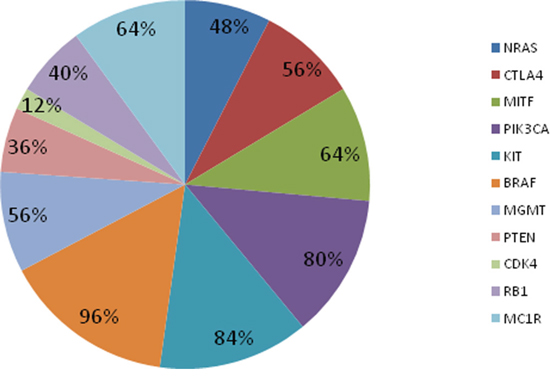
Figure 1: The variant frequency for each gene of the custom panel. MITF, PIK3CA, KIT, CTLA4, BRAF, MGMT and MC1R resulted the more frequently altered genes.
We reported BRAFV600 and NRASQ61 mutations in 17/25 (68%) and 6/25 (24%) samples, respectively. Moreover we more frequently identified the following alterations: PIK3CAI391M (44%) and KITD737N (36%) mutations, CTLA4T17A (52%), MC1RV60L (32%) and MITFS473A (60%) polymorphisms.
Ten nanograms of DNA were processed according to the manufacturers’ protocol. In 25/31 (81%) samples, an adequate library for subsequent sequencing was obtained. No library amplification was observed in 6 FFPE samples probably due to the low quality of the extracted DNA. The sensitivity of our experimental setup was tested analyzing one FFPE sample for the frequent mutation BRAFV600E (c.1799 T>A) and for the polymorphism CTLA4T17A (c.49 A>G). DNA dilution 1:30 corresponding to 3% of tumor DNA content was investigated to verify the ability of Ion Torrent calling. The sample as it was and its dilution showed identical alterations (data not shown).
To verify the variant calling accuracy, BRAF and NRAS mutations were also analyzed by Sanger sequencing and Real-Time PCR ARMS methods respectively. BRAFV600 was detected in 16 samples by both methods, and 1 was missed by Sanger sequencing. NRASQ61 was detected in 4 samples by both methods and 2 were missed by the Real-Time PCR ARMS method. Moreover concordance between Ion Torrent, Sanger and ARMS methods was present in all cases without mutations in BRAF or NRAS. Generally, BRAF and NRAS mutations were mutually exclusive, even if one case of our sample set showed both mutations.
BRAF mutation and risk of concomitant multiple alterations
A generalized linear model analysis considering all alterations detected in all genes included in our Ampliseq custom panel was applied to study the risk of BRAF mutation when other genes resulted altered. Only CTLA4 and KIT resulted significantly associated to BRAF status. In particular, we found that if CTLA4 was altered, the risk of a concomitant BRAFV600 mutation was low, on the contrary this risk was high if KIT was altered [OR, 0.1-95%CI: 0.05%0.74; OR, 12.5- 95%CI: 1.82%125.77 respectively]. These results were confirmed by the Chi-square test evidencing that 59% of patients with BRAFV600 were wild type for CTLA4 (p=0.08), while 88% of patients had KIT altered and BRAFV600 (p=0.03).
Considering the more frequent variations above reported, our linear model analysis revealed that a BRAFV600 patient could have a low risk of presenting also CTLA4T17A, MITFS473A or MC1RV60L alteration and PIK3CAI391M mutation [OR: 0.077- 95%CI: 0.003%0.58; OR: 0.11- 95%CI: 0.005%0.83; OR:0.51- 95%CI: 0.08%3.37; OR: 0.13- 95%CI: 0.015%0.83 respectively]. The Chi-square test evidenced that 65% of BRAFV600 patients lacked CTLA4T17A (p=0.04). On the contrary, the mutation KITD737N resulted associated to BRAFV600 [OR: 2.1-95%CI: 0.35%17.33].
Genetic pattern related to clinical outcome
A subset of 17 BRAF mutated patients, treated with Vemurafenib and Dabrafenib, was followed up until progression. Progression free survival (PFS) was considered with respect to alterations in CTLA4, MITF, PIK3CA, KIT and MC1R by Kaplan–Meier curves and log-rank analysis.
Considering the presence of any alteration in CTLA4 and KIT genes, statistical analysis evidenced that patients without variations had a higher median PFS (4 vs 7 and 7 vs 21, respectively). Interestingly, when we considered the most frequent alteration (PIK3CAI391M, KITD737N, CTLA4T17A, MC1RV60L and MITFS473A), analyses showed that BRAFV600 patients without any of these had a higher median PFS, even if the statistical significance was not reached probably due to the small number of patients analyzed.
The Cox proportional hazards regression model was used to determine the risk that the identified alterations could lead to a progression event (Table 1). The univariate Cox hazard regression model demonstrated a possible role for altered KIT and CTLA4 and for CTLA4T17A, MC1RV60L and MITFS473A as risk factors. Multivariate analysis that considered all CTLA4 and KIT alterations or the most frequent alteration for each gene with respect to gender, age and stage evidenced all single alterations as a risk marker of progression. In particular MC1RV60L results significantly associated with melanoma progression (p=0.004).
Table 1: Univariate and multivariate analyses through the COX regression model
UNIVARIATE |
MULTIVARIATE |
|||
|---|---|---|---|---|
HR |
95% CI |
HR |
95% CI |
|
CTLA4_any alteration |
2.285 |
0.81%6.39 |
2.02 |
0.54%7.51 |
KIT_any alteration |
2.402 |
0.31%18.38 |
1.15 |
0.11%11.80 |
CTLA4T17A |
1.917 |
0.67%5.43 |
1.46 |
0.39%5.45 |
PIK3CAI391M |
0.67 |
0.21%2.13 |
2.77 |
0.52%14.63 |
MITFS473A |
1.91 |
0.70%5.54 |
2.54 |
0.40%15.90 |
KITD737N |
0.94 |
0.33%2.66 |
1.69 |
0.45%6.25 |
MC1RV60L |
2.735 |
0.81%9.18 |
14.43* |
2.35%88.49 |
The analyses considered any CTLA4 or KIT alterations or the single reported more frequent alterations with respect to gender, age and stage. The significant results (p<0.05) are indicated (*)
ROC curves and the AUC (Area Under Curve) parameter were evaluated to exploit the role of the five alterations more frequently found, in predicting duration of response to anti-BRAF therapy. In particular, PFS was taken into account considering alterations both as individually and as concomitantly present in patients. In Figure 2, best curves are represented. In particular, the best accuracy in discriminating short and long PFS was reached by CTLA4T17A, PIK3CAI391M and KITD737N (AUC: 0.7). CTLA4T17A, when simultaneously present with MITFS473A, showed a lower accuracy in predicting disease progression (AUC: 0.63).
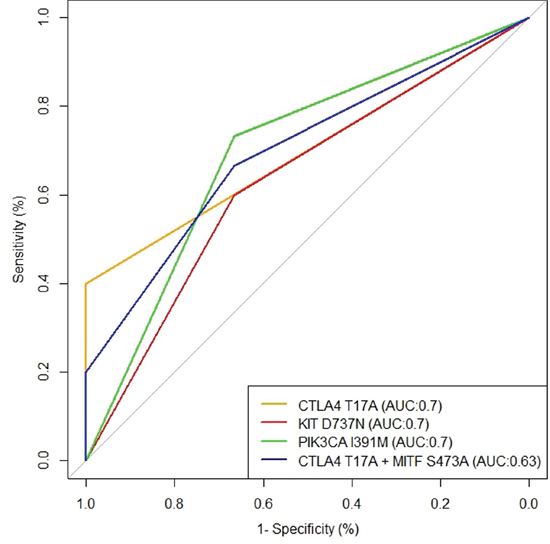
Figure 2: The best ROC curves evaluated to exploit the role of the five alterations more frequently found in relation to anti-BRAF therapy response. The best accuracy in discriminating short and long PFS was reached by CTLA4T17A, PIK3CAI391M and KITD737N (AUC:0.7). CTLA4T17A when simultaneously present with MITFS473A showed a lower accuracy in predicting disease progression (AUC:0.63).
Prediction and structural analysis of the most frequent alterations
To assess the hypothetical pathogenic role of the above alterations (CTLA4T17A, MITFS473A, PIK3CAI391M, MC1RV60L and KITD737N), we used some bioinformatic tools (SIFT, Polyphen-2, CONDEL, SNP&GO and PANTHER). The PIK3CAI391M and KITD737N were predicted to be deleterious with 3/5 tools, while the CTLA4T17A and MITFS473A were considered benign. MC1RV60L was considered damaging only by Polyphen-2.
Moreover to truly clarify the biological characteristic of the 5 alterations, we performed a structural analysis in silico. Codon 17 in the CTLA4 gene is located in the signal peptide, whose secondary structure is important for its functionality. SignalIP indicated positions 37-38 to be the most likely cleavage site. Figure 3a shows that the Thr17 is located in the n-region, which interacts with the translocation machinery and with the negatively charged phospholipids in the lipidic bilayer of the membrane. The T17A signal peptide showed higher hydrophobicity, measured through the Kyte and Doolittle scale, than the wild-type (Figure 3b). Moreover, we observed a higher propensity to form α-helix in the Chou and Fasman scale for the altered signal sequence (Figure 3c).
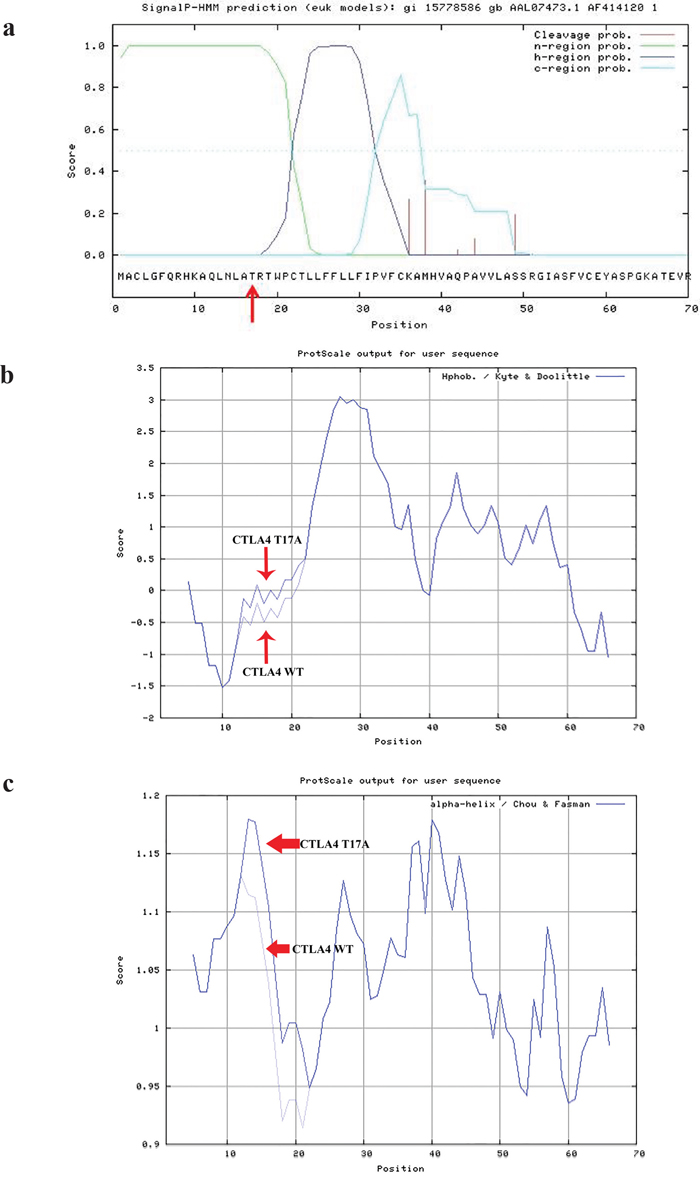
Figure 3: CTLA4 structural analysis. The CTLA4T17A variation prediction analysis has been performed through SignalIP 3.0 (http://www.cbs.dtu.dk/services/SignalP-3.0/) a. This variant is located in the n-region which interacts with the translocation machinery and with the negatively charged phospholipids in the lipidic bilayer of the membrane a. The T17A signal peptide showed higher hydrophobicity, measured through the Kyte and Doolittle scale than the wild-type b. A higher propensity to form an α-helix in the Chou and Fasman scale, due to the variant CTLA4T17A, was shown c.
The MITF gene alteration frequently identified in our study is S473A. The importance of phosphorylation for the functionality of this protein in different conditions is well known. NetPhos 2.0 results indicated that MITF has 25 serines which could be potential phosphorylation sites. Interestingly, Ser473 (Figure 4) is included in the prediction, suggesting a potential influence of S473A alteration in the function of MITF.
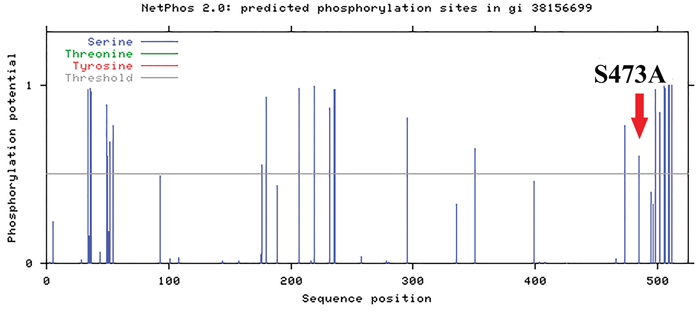
Figure 4: MITF structural analysis. MITFS473 is included in the 25 serines which could be potential phosphorylation sites. This analysis has been conducted through NetPhos 2.0.
We analyzed in detail the structure of PIK3CA and KIT proteins and the potential influence of I391M and D737N alterations, respectively. PIK3CAI391 is located in the C2 domain, responsible for phospholipid membrane binding. In particular, I391 is the only residue that works as a loop between the β-sandwich, which is the core structure of the C2 domain, and an α-helix (Figure 5). The KITD737N mutation is located in the cytoplasmic portion which includes the kinase domain. In Figure 6, D737 residue is shown in green and it could be observed that it is in a loop region near to catalytic tyrosine and serine residues, in particular to S741, whose phosphorylation by PKC/PRKCA is important for the kinase down-regulation. It could be argued that the substitution of Asp737 with an Asn residue could lead to a topological change due to their different polarity.
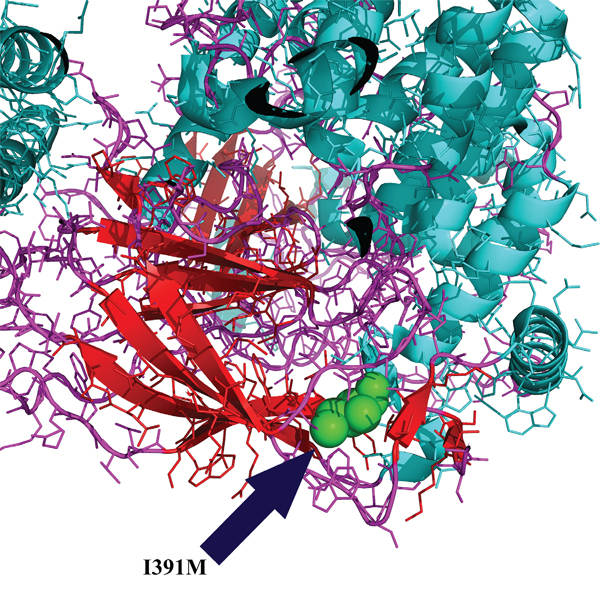
Figure 5: PIK3CA structural analysis. PIK3CAI391 (residue in green) is located in the C2 domain, responsible for phospholipid membrane binding. I391 resulted the only residue to work as a loop between the core structure of the C2 domain and an α-helix. The analysis has been conducted through the PyMOL Molecular Graphics System (Version 1.4.1 Schrödinger, LLC).
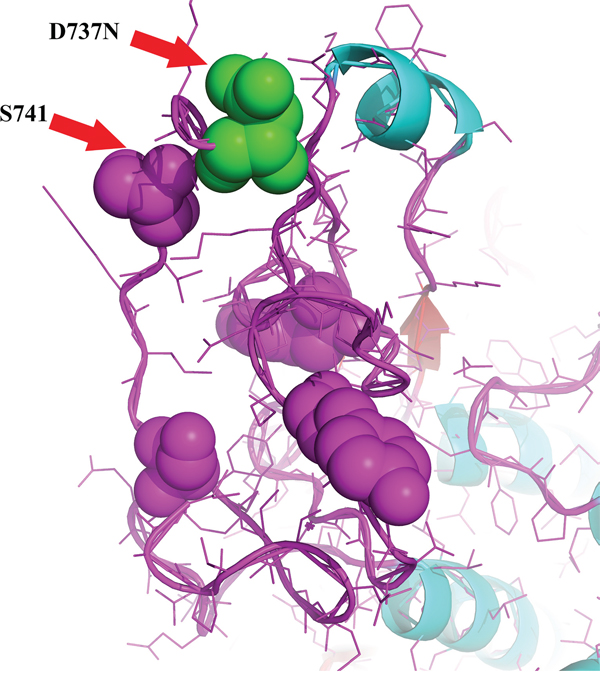
Figure 6: KIT structural analysis. KITD737 (residue in green) is located in a loop region near to catalytic tyrosine and serine residues, in particular to S741, crucial for kinase down-regulation. The analysis has been conducted through the PyMOL Molecular Graphics System (Version 1.4.1 Schrödinger, LLC).
Regarding the potential structural implication of the MC1RV60L variant, there are no crystallographic data on the entire MC1R protein. Therefore, taking into account the approach of Ibarrola-Villava et al [17], we aligned the sequences of β-adrenergic receptor structure and the MC1R consensus sequence in order to identify the position of the MC1R V60 residue in the β-adrenergic receptor sequence (V54). We observed that this residue is located in the Helix I of the seven transmembrane domain structure of the receptor (Figure 7). Moreover, the change to a leucine residue leads to a lower Kyte and Doolittle hydrophobicity score (data not shown). These observations seemed to suggest a possible implication of this variant in the correct folding of Helix I of MC1R.
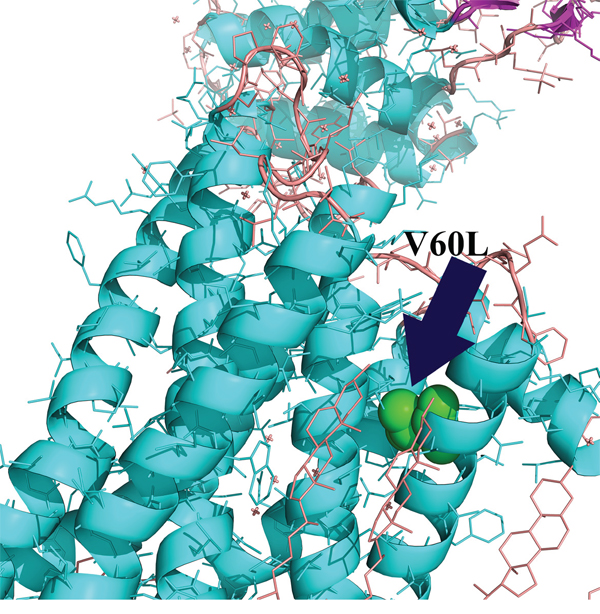
Figure 7: MC1R structural analysis. Since there is no crystallographic data on the entire MC1R protein, we aligned the sequences of β-adrenergic receptor structure and the MC1R consensus sequence in order to identify the position of the variant (residue in green) in the β-adrenergic receptor sequence (V54). The residue MC1RV60 is located in the Helix I of the seven transmembrane domain structure of the receptor. The analysis has been conducted through the PyMOL Molecular Graphics System (Version 1.4.1 Schrödinger, LLC).
DISCUSSION
The identification of biomarkers able to predict therapeutic response is an area of intense research due to the lack of an established algorithm which defines what and when BRAF target therapy has to be placed in the treatment of MM patients. To date in absence of sequential prospective studies, the choice of the correct agent is mostly guided empirically by clinical features such as bulk of disease and its evolutional speed as well as patient characteristics such as performance status, age and presence of comorbidities. All together these issues explain how urgent is to explore new pathways to search for biomarkers which could determine the effective treatment sequence in the single patient. Furthermore, the only BRAF mutation is insufficient to predict the time of response to BRAF. Disease progression often happens within 6–8 months after the beginning of therapy due to the recovery of MAPK signaling or activation of alternative signaling pathways. Therefore, a multigene diagnostic approach, starting from a limited amount of DNA, has become mandatory in routine clinical practice for the better comprehension of the molecular mechanisms underlying this very small range of response time. However, the low amount of material available in most cases does not allow a comprehensive molecular characterization by conventional techniques. In the present study, we showed that targeted NGS using the Ion Torrent technology provides simultaneously information about multiple genes starting from a very limited amount of DNA and in a short time. Furthermore, we implemented a custom gene panel able to evidence possible other prognostic factors associated to BRAF inhibitors response. All our analyzed samples showed at least one mutation/SNP among the 11 melanoma-related genes which constituted our panel. We demonstrated a higher accuracy of NGS methods in the detection of BRAF and NRAS mutations (in 68% and 24% of samples, respectively) than Sanger and ARMS analysis. This was probably due to the low proportion of mutated alleles not detectable by traditional sequencing methods. Apart from the mutations known to be associated with metastatic melanoma, we have more frequently identified two potential pathologic mutations, PIK3CAI391M and KITD737N, and three polymorphisms, MITFS473A, CTLA4T17A and MC1RV60L. The alterations in PIK3CA, KIT, CTLA4 and MC1R were already known. Moreover, for the first time, we have demonstrated in a subset of BRAFV600 patients treated with BRAF inhibitors that those without these alterations had a higher median PFS. Usually these genes resulted low mutated in cutaneous melanomas, since the entire coding regions of these genes have not been well investigated. In fact, in melanomas, hotspot regions or amplification of KIT, PIK3CA, MITF and CTLA4 are often analyzed [18–22]. MC1RV60L is often associated with pigmentary phenotypes and risk of malignant melanoma [23–24]. Thus this study represents the first comprehensive analysis of these genes in metastatic melanoma patients in relation to the treatment with BRAF inhibitors.
The KIT gene encodes for a receptor tyrosine kinase that can activate multiple downstream signalling pathways, including the RAS-RAF-MEK-ERK and PI3K-AKT axis. The role of KIT in the development of malignant melanoma has been largely discussed. Authors have demonstrated in melanomas wild type for NRAS and BRAF, that activating constitutively KIT mutations may be functionally equivalent to constitutive activation of the RAS-RAF-MEK-ERK pathway due to NRAS or BRAF [25]. KIT somatic mutations are present mainly in mucosal melanoma, but they can also be found in cutaneous melanomas [26, 27]. The most frequently investigated KIT mutations in melanoma concern the exons 11 and 13 that resulted partially responsive to KIT inhibitors [28]. In fact, melanoma patients with a KIT mutation affecting a recurrent hot spot, such as the L576P or K642E mutation, have better clinical outcomes than those without a hotspot mutation [29]. Mutations in the gene encoding the catalytic subunit of PI3K, PIK3CA, occur at very low frequencies (<5%) in melanoma [30], although are very frequent in some human cancers leading to constitutive AKT activation [31]. The PI3K-AKT pathway is an important regulator of cell growth, proliferation, differentiation, metabolism, motility, and survival. Moreover, it is highly active in most metastatic melanomas, and its inhibition, together with the RAS-RAF-MEK-ERK pathway block, can lead to suppression of melanoma growth. The most frequent melanoma-associated genetic events in PI3K-AKT signaling pathway are inactivating mutations (often related only to exons 9 and 20 of PIK3CA) or PTEN loss of expression or mutations and activating NRAS mutations [32]. However, it has been demonstrated that PIK3CA mutations may contribute to RAF inhibitor resistance [33]. The MAPK pathway regulation seems to be strongly linked to MITF expression and function [34]. In fact it has been shown that the hyper-activation of RAS-RAF-MEK-ERK signalling causes a significant reduction in MITF levels and therefore melanocytes and melanoma cells proliferation [35]. Amplification of MITF is observed in 10% of primary and in 15–20% of metastatic melanomas and so it may be considered an oncogene in melanoma. In addition, MITF mutations, such as the recurrent E318K, can predispose to melanoma development [36, 37]. Regarding the function of MITF in therapy response, growing evidence has highlighted a role in innate and acquired resistance to MAPK pathway inhibitors. MITF up-regulation, related to increased tolerance and resistance to MAPK pathway inhibitors, is supported by the fact that some patients relapse with a MITF gene amplification [33]. However, this up-regulation correlates with an increased lymphocyte T CD8+ infiltration [38], and it could be advantageous for an immunotherapy approach. In our study, we demonstrated that MITFS473A in association to CTLA4T17A showed a higher sensitivity and specificity (AUC 0.7) in predicting shorter PFS of the considered patients, as reported for the single alterations in CTLA4, PIK3CA and KIT.
The CTLA4, a member of the CD28 superfamily, is not constitutively expressed on T cells but it is induced after CD28 binding and activation. It is responsible for the attenuation of immune response by binding to ligands (B7-1 and B7-2) expressed on the surface of antigen presenting cells. CTLA-4 gene polymorphisms have been associated with numerous autoimmune conditions, including diabetes and inflammatory bowel disease [39]. Among the numerous CTLA4 polymorphisms, the recurrent T17A substitution (rs231775) can be considered one cause of an altered endoplasmic reticulum trafficking and/or processing of CTLA4 leading to its differential expression on the cell surface [40]. We showed a higher hydrophobicity and propensity to form α-helix for CTLA4T17A peptide than the wild-type one. Moreover, this non-synonymous polymorphism seems to be associated with the anti-CTLA4 therapy response [41]. There is evidence that the BRAF inhibition exerts an influence on immunological landscape of cutaneous melanoma [42–44]. In fact, BRAF inhibitors treatment is associated with decreased production of the immunosuppressive factor IL10 and enhanced expression of tumor-specific antigens [45] and so BRAF inhibitors might induce T-cell infiltration before treatment with immunotherapy. In our study, we found that patients without the CTLA4T17A variant had a higher median PFS after BRAF inhibitors treatment. In future studies, it would be interesting to see whether this variant is also associated with lower immune response due to BRAF inhibitors treatment.
MC1R is a central control point in skin and hair pigmentation. Moreover, inherited variation in the MC1R gene is considered a genetic marker for moderately increased risk of melanoma [23]. In a recent study, MC1R variants were correlated with tumor characteristics suggesting that inherited variation in MC1R may play a role in determining the anatomic site of melanomas and may differ with respect to skin pigmentation phenotype [46]. The red hair colour phenotype is due to the production of more pheomelanin than eumelanin, which is a result of an altered function of MC1R. High-penetrance R alleles are the variants D84E, R151C, R160W and D294H, strongly associated with red hair and fair skin phenotypes; while the variants V60L, V92M, and R163Q are low penetrance r alleles. These three variants are often predicted neutral and tolerant by most tools [47] as demonstrated also by our group for V60L. However, for the first time, we reported a significant association of MC1RV60L to melanoma progression.
In conclusion, the application of an Ampliseq Custom Panel on the Ion Torrent Personal Genome Machine for a routine clinical practice needs validation in a larger series of cases. However, these preliminary results evidenced a higher sensitivity and specificity in detecting a wide range of genetic alterations than traditional sequencing methods. Therefore a NGS approach with a custom panel may provide crucial and new information to evaluate tumor-specific therapeutic susceptibility and individual prognosis to improve the care of metastatic melanoma patients.
MATERIALS AND METHODS
Ethics statement
The study was approved by the local Ethics Committee of the Istituto Tumori “Giovanni Paolo II” of Bari (prot. no. 515/EC of May 12, 2015) and was conducted in accordance with the international standards of good clinical practice. All patients signed an informed consent. Moreover, all samples and medical data used in this study have been anonymized.
Patient information
A series of 25 cutaneous melanoma patients (12 female and 13 male) with histologically confirmed stage IV melanoma who started a first line treatment at the Oncology Department of the IRCCS Istituto Tumori ‘Giovanni Paolo II’ in Bari (Italy) were retrospectively evaluated. Clinical features of patients are listened in Table 2. The period of enrollment was from July 2011 to February 2013.
Table 2: Baseline clinical features of all 25 cutaneous metastatic melanoma patients
Overall |
BRAF inhibitors treated patients |
Patient not treated with target therapy |
|
|---|---|---|---|
Patients |
25 |
17 |
8 |
Median age |
58 (30-82) |
58 (36-82) |
58 (30-71) |
Female |
11 |
8 |
3 |
Male |
14 |
9 |
5 |
Site of primary melanoma |
|||
skin |
25 |
14 |
8 |
unknown |
0 |
3 |
0 |
Stage IV* |
|||
M1a |
6 |
3 |
3 |
M1b |
4 |
1 |
3 |
M1c |
15 |
13 |
2 |
Brain involvement |
3 |
2 |
1 |
ECOG status |
|||
0 |
18 |
12 |
6 |
1 |
5 |
5 |
0 |
2 |
2 |
0 |
2 |
* According to the AJCC melanoma staging system
Seventeen from this population of MM patients were treated with BRAF inhibitors according to the presence of BRAF V600 mutation (V600E in 14 patients and V600K in 3 patients). Target therapy consisted of Vemurafenib (15 patients) and Dabrafenib (2 patients) at the standard dose of 960 mg and 150 mg respectively twice daily until progression. This subset of patients was included in our analysis. Patients were selected if they had measurable lesions; adequate renal, hepatic and bone marrow functions; an Eastern Cooperative Oncology Group (ECOG) performance status ≤ 2; a life expectancy of more than 12 weeks and it did not need dose reduction or withholdings of doses of BRAF inhibitors for related toxicities.
Patients were underwent to clinical and laboratory exams every 4 weeks and radiological evaluation with tumor assessments at baseline and then approximately every 12 weeks in order to evaluate therapeutic effectiveness. Response Evaluation Criteria In Solid Tumors (RECIST) version 1.1 was used for efficacy assessment [48]. We assessed the best response during BRAF inhibitors as complete response (CR), partial response (PR), stable disease lasting for at least 12 weeks (SD) and progressive disease (PD). We also measured PFS, defined as the length of time (in months) from the start of the treatment until disease progression.
As best response we assessed 2 CR, 10 PR and 5 PD. Median follow-up was 8 months and at the time of the final observation (January 2015) the median value of PFS was 7 months (range 1-36+ months). At the final observation, all patients but two had progressed after BRAF inhibitor treatment.
DNA preparation
DNA was isolated from FFPE samples, after deparaffinization in xylene of 10 mm thick paraffin sections (two sections/each sample), using the QIAamp DNA FFPE Tissue Kit (Qiagen) according to the manufacturer’s instructions.
Ion Torrent PGM library preparation and sequencing
The custom panel, developed with Ion Ampliseq Designer tool (www.ampliseq.com), was used to analyze the entire coding region of 11 genes (BRAF, NRAS, PTEN, MITF, CDK4, MGMT, CTLA4, PIK3CA, MC1R, KIT, RB1) with a coverage of 93.85%. We verified that the gene regions lost in the panel were not the exon regions of our interest. These genes are crucial because they are involved in melanoma carcinogenesis and treatment response. The panel size was 39.08Kb, contained 303 amplicons, and for the analysis an input of 20ng of FFPE DNA (10 nanograms/each primer pool) was required. Ion Torrent adapter-ligated libraries were made following the manufacturer’s protocol for the Ion AmpliSeq Library Kit 2.0 (Life Technologies). AMPure beads (Beckman Coulter) were used to purify the resulting libraries. To determine the concentration and size of the libraries, we used two methods: the Agilent 2100 BioAnalyzer (Agilent Technologies) with the Agilent BioAnalyzer DNA High-Sensitivity kit (Agilent Technologies) and the Ion Library Quantitation kit (Life technologies).
Sample emulsion PCR and enrichment were performed using the Ion PGM Template OT2 200 Kit, according to the manufacturer’s instructions. The Ion 314 and 316 Chip Kit v2 (Life Technologies) were used for sequencing on the Ion Torrent PGM barcoded samples. The Ion PGM 200 Sequencing Kit was used for sequencing reactions (Life Technologies).
Variant calling and experimental validation
Data from the PGM runs were processed initially using the Ion Torrent platform-specific pipeline software Torrent Suite to generate sequence reads, trim adapter sequences, filter, and remove poor signal-profile reads. Initial variant calling from the Ion AmpliSeq sequencing data was generated using Torrent Suite Software v4.2 with a plug-in “variant caller v4.2” program. In order to eliminate errors in base calling, Somatic-High Stringency parameters setting was used to generate the final variant calling. Filtered variants were annotated using the Ion Reporter software v4.2 (Life Technologies). Mutations were visually examined using Integrative Genomics Viewer (IGV) software (http://www.broadinstitute.org/igv).
BRAFV600 and NRASQ61 missense mutations were confirmed by Sanger’s sequencing and the Real-Time PCR ARMS method (Qiagen) respectively.
Prediction tools analysis
Six computational tools were used to predict the effect of aminoacid substitutions on CTLA4, MITF, PIK3CA, KIT and MC1R. In detail, SIFT [49], Polyphen-2 [50], PROVEAN [51], CONDEL [52], SNPS&GO [53] and Panther [54] were used to perform predictions. They are based on phylogenetic and structural information, which allow to obtain a score indicating how aminoacid substitutions could change the protein structure.
Protein structural analysis
To analyze CTLA4T17A variation, we started performing prediction through SignalIP 3.0 (http://www.cbs.dtu.dk/services/SignalP-3.0/) [55]. To clarify its influence in secondary structure, we analyzed both hydropathicity in the Kyte and Doolittle scale [56] and α-helix propensity in the Chou and Fasman scale [57] (http://web.expasy.org/protscale/) [58]. The NetPhos 2.0 (http://www.cbs.dtu.dk/services/NetPhos/) was the bioinformatic tool used to predict phosphorylation sites of MITF [59].
The PyMOL Molecular Graphics System (Version 1.4.1 Schrödinger, LLC) was used to tridimensionally evaluate PIK3CA, KIT and MC1R structure. In detail, PDB IDs for PIK3CA was 3HIZ [60]. KIT structure was downloaded by Protein Model Portal (http://www.proteinmodelportal.org/) [61] and provided by SwissModel (http://swissmodel.expasy.org/) [62]. KIT ID was 4hvsA. To analyze the structure of MC1R, the recent β-adrenergic receptor structure (PDB ID: 2RH1) has been taken into account [63]. The structural evaluation of the V60L variants was performed firstly aligning through BLASTP [64] β-adrenergic G protein-coupled receptor to the consensus sequence of MC1R.
Statistical analysis
Statistical analyses were carried out through R v3.2.0. The Chi-square test was performed through the chisq.test command. The univariate and multivariate analyses were carried out fitting generalized linear model through the glm() function of R package “MASS”. The “survival” R-package was used to fulfill Cox regression analysis taking into account PFS. PFS probability was computed by comparing the Kaplan-Meier curves through the log rank test by GraphPad Prism 5.0.1 (GraphPad Prism 5.01, San Diego, CA, USA). Results from all statistical analyses were considered to be significant at a level of p-values less than 0.05.
Receiver operating characteristic (ROC) curves and the area under the ROC curve (AUC) were used to assess the sensitivity and specificity of the most frequent alterations and their combination with respect to clinical response to BRAF inhibitors treatment (partial response/stable disease or progression established through RECIST criteria). The ROC analysis was performed through R-package “pROC” (available at http://expasy.org/tools/pROC/ under the GNU General Public License).
ACKNOWLEDGMENTS
The authors would like to thank Giorgio Pea for technical support and Caroline Oakley for manuscript revision. This work was supported by PON 01.01297 “VIRTUALAB”. No additional external funding received for this study.
CONFLICTS OF INTEREST
The authors declare no conflicts of interest
REFERENCES
1. Chapman PB, Hauschild A, Robert C, Haanen JB, Ascierto P, Larkin J, Dummer R, Garbe C, Testori A, Maio M, Hogg D, Lorigan P, Lebbe C, et al. Improved survival with vemurafenib in melanoma with BRAF V600E mutation. The New England journal of medicine. 2011; 364:2507–2516.
2. Tentori L, Lacal PM, Graziani G. Challenging resistance mechanisms to therapies for metastatic melanoma. Trends in pharmacological sciences. 2013; 34:656–666.
3. Trunzer K, Pavlick AC, Schuchter L, Gonzalez R, McArthur GA, Hutson TE, Moschos SJ, Flaherty KT, Kim KB, Weber JS, Hersey P, Long GV, Lawrence D, et al. Pharmacodynamic effects and mechanisms of resistance to vemurafenib in patients with metastatic melanoma. J Clin Oncol. 2013; 31:1767–1774.
4. Long GV, Stroyakovskiy D, Gogas H, Levchenko E, de Braud F, Larkin J, Garbe C, Jouary T, Hauschild A, Grob JJ, Chiarion-Sileni V, Lebbe C, Mandala M, et al. Dabrafenib and trametinib versus dabrafenib and placebo for Val600 BRAF-mutant melanoma: a multicentre, double-blind, phase 3 randomised controlled trial. Lancet. 2015; 386:444–451.
5. Azijli K, Stelloo E, Peters GJ, AJ VDE. New developments in the treatment of metastatic melanoma: immune checkpoint inhibitors and targeted therapies. Anticancer research. 2014; 34:1493–1505.
6. Ascierto PA, Marincola FM, Atkins MB. What’s new in melanoma? Combination! Journal of translational medicine. 2015; 13:213.
7. Ribas A, Hodi FS, Callahan M, Konto C, Wolchok J. Hepatotoxicity with combination of vemurafenib and ipilimumab. The New England journal of medicine. 2013; 368:1365–1366.
8. Bai X, Zhang E, Ye H, Nandakumar V, Wang Z, Chen L, Tang C, Li J, Li H, Zhang W, Han W, Lou F, Zhang D, et al. PIK3CA and TP53 gene mutations in human breast cancer tumors frequently detected by Ion Torrent DNA sequencing. PloS one. 2014; 9:e99306.
9. Cai X, Sheng J, Tang C, Nandakumar V, Ye H, Ji H, Tang H, Qin Y, Guan H, Lou F, Zhang D, Sun H, Dong H, et al. Frequent mutations in EGFR, KRAS and TP53 genes in human lung cancer tumors detected by Ion Torrent DNA sequencing. PloS one. 2014; 9:e95228.
10. van Engen-van Grunsven AC, Kusters-Vandevelde H, Groenen PJ, Blokx WA. Update on Molecular Pathology of Cutaneous Melanocytic Lesions: What is New in Diagnosis and Molecular Testing for Treatment? Frontiers in medicine. 2014; 1:39.
11. Siroy AE, Boland GM, Milton DR, Roszik J, Frankian S, Malke J, Haydu L, Prieto VG, Tetzlaff M, Ivan D, Wang WL, Torres-Cabala C, Curry J, et al. Beyond BRAF(V600): clinical mutation panel testing by next-generation sequencing in advanced melanoma. The Journal of investigative dermatology. 2015; 135:508–515.
12. Scarpa A, Sikora K, Fassan M, Rachiglio AM, Cappellesso R, Antonello D, Amato E, Mafficini A, Lambiase M, Esposito C, Bria E, Simonato F, Scardoni M, et al. Molecular typing of lung adenocarcinoma on cytological samples using a multigene next generation sequencing panel. PloS one. 2013; 8:e80478.
13. Simbolo M, Mian C, Barollo S, Fassan M, Mafficini A, Neves D, Scardoni M, Pennelli G, Rugge M, Pelizzo MR, Cavedon E, Fugazzola L, Scarpa A. High-throughput mutation profiling improves diagnostic stratification of sporadic medullary thyroid carcinomas. Virchows Arch. 2014; 465:73–78.
14. Grunewald I, Vollbrecht C, Meinrath J, Meyer MF, Heukamp LC, Drebber U, Quaas A, Beutner D, Huttenbrink KB, Wardelmann E, Hartmann W, Buttner R, Odenthal M, Stenner M. Targeted next generation sequencing of parotid gland cancer uncovers genetic heterogeneity. Oncotarget. 2015; 6:18224–18237. doi: 10.18632/oncotarget.4015.
15. Gagan J, Van Allen EM. Next-generation sequencing to guide cancer therapy. Genome medicine. 2015; 7:80.
16. Jones S, Anagnostou V, Lytle K, Parpart-Li S, Nesselbush M, Riley DR, Shukla M, Chesnick B, Kadan M, Papp E, Galens KG, Murphy D, Zhang T, et al. Personalized genomic analyses for cancer mutation discovery and interpretation. Science translational medicine. 2015; 7:283ra253.
17. Ibarrola-Villava M, Pena-Chilet M, Llorca-Cardenosa MJ, Oltra S, Cadenas CM, Bravo J, Ribas G. Modeling MC1R rare variants: a structural evaluation of variants detected in a Mediterranean case-control study. The Journal of investigative dermatology. 2014; 134:1146–1149.
18. Hartman ML, Czyz M. MITF in melanoma: mechanisms behind its expression and activity. Cell Mol Life Sci. 2015; 72:1249–1260.
19. Glitza IC, Davies MA. Genotyping of cutaneous melanoma. Chinese clinical oncology. 2014; 3:27.
20. Breunis WB, Tarazona-Santos E, Chen R, Kiley M, Rosenberg SA, Chanock SJ. Influence of cytotoxic T lymphocyte-associated antigen 4 (CTLA4) common polymorphisms on outcome in treatment of melanoma patients with CTLA-4 blockade. Journal of immunotherapy. 2008; 31:586–590.
21. Queirolo P, Morabito A, Laurent S, Lastraioli S, Piccioli P, Ascierto PA, Gentilcore G, Serra M, Marasco A, Tornari E, Dozin B, Pistillo MP. Association of CTLA-4 polymorphisms with improved overall survival in melanoma patients treated with CTLA-4 blockade: a pilot study. Cancer investigation. 2013; 31:336–345.
22. Manca A, Lissia A, Capone M, Ascierto PA, Botti G, Caraco C, Stanganelli I, Colombino M, Sini M, Cossu A, Palmieri G. Activating PIK3CA mutations coexist with BRAF or NRAS mutations in a limited fraction of melanomas. Journal of translational medicine. 2015; 13:37.
23. Guida S, Bartolomeo N, Zanna PT, Grieco C, Maida I, De Summa S, Tommasi S, Guida M, Azzariti A, Foti C, Filotico R, Guida G. Sporadic melanoma in South-Eastern Italy: the impact of melanocortin 1 receptor (MC1R) polymorphism analysis in low-risk people and report of three novel variants. Archives of dermatological research. 2015; 307:495–503.
24. Haddadeen C, Lai C, Cho SY, Healy E. Variants of the melanocortin-1 receptor: do they matter clinically? Experimental dermatology. 2015; 24:5–9.
25. Lennartsson J, Jelacic T, Linnekin D, Shivakrupa R. Normal and oncogenic forms of the receptor tyrosine kinase kit. Stem cells. 2005; 23:16–43.
26. Kim KB, Alrwas A. Treatment of KIT-mutated metastatic mucosal melanoma. Chinese clinical oncology. 2014; 3:35.
27. Glitza IC, Davies MA. Genotyping of cutaneous melanoma. Chinese clinical oncology. 2014; 3:27.
28. Hodi FS, Corless CL, Giobbie-Hurder A, Fletcher JA, Zhu M, Marino-Enriquez A, Friedlander P, Gonzalez R, Weber JS, Gajewski TF, O’Day SJ, Kim KB, Lawrence D, et al. Imatinib for melanomas harboring mutationally activated or amplified KIT arising on mucosal, acral, and chronically sun-damaged skin. J Clin Oncol. 2013; 31:3182–3190.
29. Kim KB, Alrwas A. Treatment of KIT-mutated metastatic mucosal melanoma. Chinese clinical oncology. 2014; 3:35.
30. Omholt K, Krockel D, Ringborg U, Hansson J. Mutations of PIK3CA are rare in cutaneous melanoma. Melanoma research. 2006; 16:197–200.
31. Lai K, Killingsworth MC, Lee CS. Gene of the month: PIK3CA. Journal of clinical pathology. 2015; 68:253–257.
32. Shull AY, Latham-Schwark A, Ramasamy P, Leskoske K, Oroian D, Birtwistle MR, Buckhaults PJ. Novel somatic mutations to PI3K pathway genes in metastatic melanoma. PloS one. 2012; 7:e43369.
33. Van Allen EM, Wagle N, Sucker A, Treacy DJ, Johannessen CM, Goetz EM, Place CS, Taylor-Weiner A, Whittaker S, Kryukov GV, Hodis E, Rosenberg M, McKenna A, et al. The genetic landscape of clinical resistance to RAF inhibition in metastatic melanoma. Cancer discovery. 2014; 4:94–109.
34. Wellbrock C, Arozarena I. Microphthalmia-associated transcription factor in melanoma development and MAP-kinase pathway targeted therapy. Pigment cell & melanoma research. 2015; 28:390–406.
35. Wellbrock C, Marais R. Elevated expression of MITF counteracts B-RAF-stimulated melanocyte and melanoma cell proliferation. The Journal of cell biology. 2005; 170:703–708.
36. Bertolotto C, Lesueur F, Giuliano S, Strub T, de Lichy M, Bille K, Dessen P, d’Hayer B, Mohamdi H, Remenieras A, Maubec E, de la Fouchardiere A, Molinie V, et al. A SUMOylation-defective MITF germline mutation predisposes to melanoma and renal carcinoma. Nature. 2011; 480:94–98.
37. Yokoyama S, Woods SL, Boyle GM, Aoude LG, MacGregor S, Zismann V, Gartside M, Cust AE, Haq R, Harland M, Taylor JC, Duffy DL, Holohan K, et al. A novel recurrent mutation in MITF predisposes to familial and sporadic melanoma. Nature. 2011; 480:99–103.
38. Frederick DT, Piris A, Cogdill AP, Cooper ZA, Lezcano C, Ferrone CR, Mitra D, Boni A, Newton LP, Liu C, Peng W, Sullivan RJ, Lawrence DP, et al. BRAF inhibition is associated with enhanced melanoma antigen expression and a more favorable tumor microenvironment in patients with metastatic melanoma. Clin Cancer Res. 2013; 19:1225–1231.
39. Gough SC, Walker LS, Sansom DM. CTLA4 gene polymorphism and autoimmunity. Immunological reviews. 2005; 204:102–115.
40. Anjos S, Nguyen A, Ounissi-Benkalha H, Tessier MC, Polychronakos C. A common autoimmunity predisposing signal peptide variant of the cytotoxic T-lymphocyte antigen 4 results in inefficient glycosylation of the susceptibility allele. The Journal of biological chemistry. 2002; 277:46478–46486.
41. Breunis WB, Tarazona-Santos E, Chen R, Kiley M, Rosenberg SA, Chanock SJ. Influence of cytotoxic T lymphocyte-associated antigen 4 (CTLA4) common polymorphisms on outcome in treatment of melanoma patients with CTLA-4 blockade. Journal of immunotherapy. 2008; 31:586–590.
42. Tomei S, Bedognetti D, De Giorgi V, Sommariva M, Civini S, Reinboth J, Al Hashmi M, Ascierto ML, Liu Q, Ayotte BD, Worschech A, Uccellini L, Ascierto PA, et al. The immune-related role of BRAF in melanoma. Molecular oncology. 2015; 9:93–104.
43. Kawakami Y, Yaguchi T, Sumimoto H, Kudo-Saito C, Tsukamoto N, Iwata-Kajihara T, Nakamura S, Nishio H, Satomi R, Kobayashi A, Tanaka M, Park JH, Kamijuku H, Tsujikawa T, Kawamura N. Cancer-induced immunosuppressive cascades and their reversal by molecular-targeted therapy. Annals of the New York Academy of Sciences. 2013; 1284:80–86.
44. Cooper ZA, Frederick DT, Juneja VR, Sullivan RJ, Lawrence DP, Piris A, Sharpe AH, Fisher DE, Flaherty KT, Wargo JA. BRAF inhibition is associated with increased clonality in tumor-infiltrating lymphocytes. Oncoimmunology. 2013; 2:e26615.
45. Sumimoto H, Imabayashi F, Iwata T, Kawakami Y. The BRAF-MAPK signaling pathway is essential for cancer-immune evasion in human melanoma cells. The Journal of experimental medicine. 2006; 203:1651–1656.
46. Taylor NJ, Busam KJ, From L, Groben PA, Anton-Culver H, Cust AE, Begg CB, Dwyer T, Gallagher RP, Gruber SB, Orlow I, Rosso S, Thomas NE, et al. Inherited variation at MC1R and histological characteristics of primary melanoma. PloS one. 2015; 10:e0119920.
47. Hepp D, Goncalves GL, de Freitas TR. Prediction of the damage-associated non-synonymous single nucleotide polymorphisms in the human MC1R gene. PloS one. 2015; 10:e0121812.
48. Eisenhauer EA, Therasse P, Bogaerts J, Schwartz LH, Sargent D, Ford R, Dancey J, Arbuck S, Gwyther S, Mooney M, Rubinstein L, Shankar L, Dodd L, Kaplan R, Lacombe D, Verweij J. New response evaluation criteria in solid tumours: revised RECIST guideline (version 1.1). European journal of cancer. 2009; 45:228–247.
49. Kumar P, Henikoff S, Ng PC. Predicting the effects of coding non-synonymous variants on protein function using the SIFT algorithm. Nature protocols. 2009; 4:1073–1081.
50. Adzhubei IA, Schmidt S, Peshkin L, Ramensky VE, Gerasimova A, Bork P, Kondrashov AS, Sunyaev SR. A method and server for predicting damaging missense mutations. Nature methods. 2010; 7:248–249.
51. Choi Y, Sims GE, Murphy S, Miller JR, Chan AP. Predicting the functional effect of amino acid substitutions and indels. PloS one. 2012; 7:e46688.
52. Gonzalez-Perez A, Lopez-Bigas N. Improving the assessment of the outcome of nonsynonymous SNVs with a consensus deleteriousness score, Condel. American journal of human genetics. 2011; 88:440–449.
53. Calabrese R, Capriotti E, Fariselli P, Martelli PL, Casadio R. Functional annotations improve the predictive score of human disease-related mutations in proteins. Human mutation. 2009; 30:1237–1244.
54. Thomas PD, Campbell MJ, Kejariwal A, Mi H, Karlak B, Daverman R, Diemer K, Muruganujan A, Narechania A. PANTHER: a library of protein families and subfamilies indexed by function. Genome research. 2003; 13:2129–2141.
55. Bendtsen JD, Nielsen H, von Heijne G, Brunak S. Improved prediction of signal peptides: SignalP 3.0. Journal of molecular biology. 2004; 340:783–795.
56. Kyte J, Doolittle RF. A simple method for displaying the hydropathic character of a protein. Journal of molecular biology. 1982; 157:105–132.
57. Chou PY, Fasman GD. Prediction of the secondary structure of proteins from their amino acid sequence. Advances in enzymology and related areas of molecular biology. 1978; 47:45–148.
58. Wilkins MR, Gasteiger E, Bairoch A, Sanchez JC, Williams KL, Appel RD, Hochstrasser DF. Protein identification and analysis tools in the ExPASy server. Methods in molecular biology. 1999; 112:531–552.
59. Blom N, Gammeltoft S, Brunak S. Sequence and structure-based prediction of eukaryotic protein phosphorylation sites. Journal of molecular biology. 1999; 294:1351–1362.
60. Mandelker D, Gabelli SB, Schmidt-Kittler O, Zhu J, Cheong I, Huang CH, Kinzler KW, Vogelstein B, Amzel LM. A frequent kinase domain mutation that changes the interaction between PI3Kalpha and the membrane. Proceedings of the National Academy of Sciences of the United States of America. 2009; 106:16996–17001.
61. Haas J, Roth S, Arnold K, Kiefer F, Schmidt T, Bordoli L, Schwede T. The Protein Model Portal—a comprehensive resource for protein structure and model information. Database (Oxford). 2013; 2013:bat031.
62. Biasini M, Bienert S, Waterhouse A, Arnold K, Studer G, Schmidt T, Kiefer F, Cassarino TG, Bertoni M, Bordoli L, Schwede T. SWISS-MODEL: modelling protein tertiary and quaternary structure using evolutionary information. Nucleic acids research. 2014; 42:W252–258.
63. Cherezov V, Rosenbaum DM, Hanson MA, Rasmussen SG, Thian FS, Kobilka TS, Choi HJ, Kuhn P, Weis WI, Kobilka BK, Stevens RC. High-resolution crystal structure of an engineered human beta2-adrenergic G protein-coupled receptor. Science. 2007; 318:1258–1265.
64. Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. Basic local alignment search tool. Journal of molecular biology. 1990; 215:403–410.