
INTRODUCTION
The role of the epidermal growth factor receptor (EGFR) in non-small cell lung cancers (NSCLC) is well-known. Inhibition of the kinase domain of EGFR and the resultant oncogenic cell signaling disruption by small molecule inhibitors have been shown to be particularly beneficial in patients carrying the so-called “sensitizing mutations” such as L858R and the exon-19 deletion [1, 2], which contribute to nearly 90% of lung-cancer-specific EGFR mutations [3, 4]. Unfortunately, approximately 70% of patients respond initially but develop resistance with a median time to progression of 10-16 months [2, 5-8]. In at least 50% of these resistance cases, the emergence of a secondary gate keeper mutation, T790M in exon 20 of EGFR, occurs in combination with an activating mutation [9].
Over the past several decades, various approaches have been developed to target the EGFR signaling pathway, including monoclonal antibodies and small molecule tyrosine kinase inhibitors (TKIs). Small molecule inhibitors that target EGFR are undoubtedly successful, and the first-generation EGFR tyrosine kinase inhibitors (TKIs) (Table 1) such as gefitinib (IRRESA) [10], elotinib (TARCEVA) [11], lapatinib (TYKERB) [12], and icotinib (CONMANA) [13] have efficacy against several types of human cancers. Gefitinib, erlotinib and icotinib were approved for the treatment of NSCLC, while lapatinib, which inhibits EGFR and HER2 kinases, is used in combination therapy for HER2-positive metastatic breast cancer.
Table 1: Transtinib and other TKIs
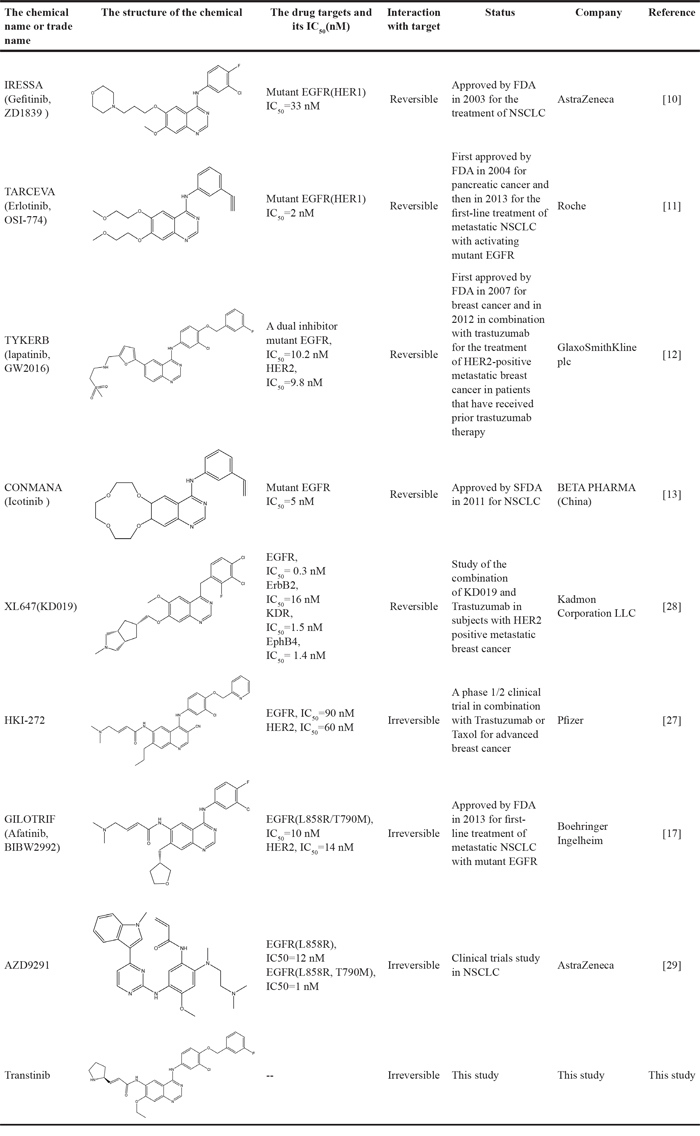
To address the resistance of the first -generation of reversible TKIs, a number of second-generation irreversible inhibitors were developed based on the anilinoquinazoline template of gefitinib (Table 1). These inhibitors contained an acrylamide substituent, a Michael acceptor system that takes advantage of the covalent modification of Cys-797 located at the lip of the ATP binding clef of EGFR, which is thought to overcome the resistance of the first-generation inhibitors. These agents were shown, in pre-clinical models, to be more potent against the second-site mutation than gefitinib or erlotinib [14]. The following are several second generation inhibitors: [(E)-N-(4-(4- ((pyridine – 2 – yl ) methoxy) – 3 -chlorophenylamino) -3-cyano-7- ethoxyquinolin-6-yl) -4-(dimethylamino)but-2-enamide (HKI-272) [15], (E) – N - (7 - ((R) - tetrahydrofuran-3-yloxy) -4-(3-chloro-4- fluorophenylamino) quinazolin-6-yl) – 4 - (dimethylamino)but-2-enamide (BIBW2992) [16, 17], and (E)-N-(4- (3-chloro-4- fluorophenylamino) - 7-methoxyquinazolin-6-yl) -4-(piperidin-1-yl)but-2-enamide (PF-00299804) [18, 19]. However, most of these irreversible EGFR inhibitors failed in clinical-trials because of their toxicities. Thus, clinically acquired drug resistance against EGFR kinase inhibitors is a major challenge in targeted cancer therapies for NSCLC treatment. Herein, we describe some of our work that led to the identification of the clinical candidate Transtinib(13c), a potent inhibitor of both sensitizing and double mutant forms of EGFR.
We focused our development on modifications to the solvent region by attaching cyclic systems to the anilinoquinazoline ring. Therefore, a series of novel EGFR inhibitors were designed and synthesized by incorporating acrylamide-based side chains into the C6 position and alkoxy-based side chains into the C7 position of the 4-anilinoquinazoline scaffold. Different types of anilines from gefitinib, erlotinib, lapatinib and other advanced EGFR inhibitors were substituted into the C-4 position of the quinazoline template. As Edgar R. Wood et.al [20] described, the pyrrolidine is a structural feature that is particularly adept at assisting in protein alkylation in the active site. In our candidate compounds, we predicted that pyrrolidine would promote the formation of covalent binding. Therefore, we introduced a pyrrolidine substituent into the covalent binding structures.
RESULTS AND DISCUSSION
Chemical names and structures
Three anilinoquinazoline compounds (13a-c) were synthesized based on Scheme 1 and 2. To access compound 11, commercial (S)-(-)-1-Boc-2-pyrrolidinemethanol with n=1 was used. To access compounds13a-c, R was setted with n=1. All the structures and names are as follows (Figure 1).
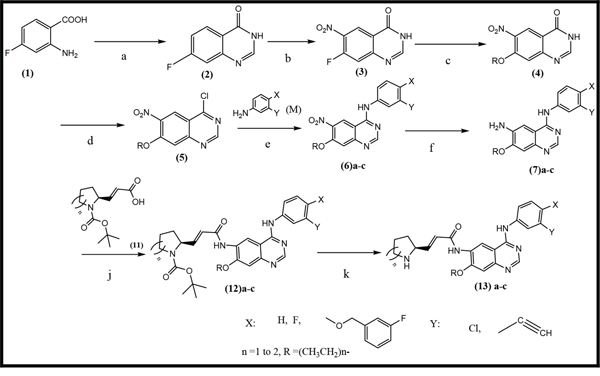
Scheme 1: General synthetic approach to the designed compounds. Reagents, conditions and yields of each procedure are as follows: (a) HCONH2 130-140°C, 20 h, 80%; (b) HNO3 /H2SO4, 0-20-110°C, 91%; (c) Na, CH3CH2OH, reflux, 2.5 h, 73%; (d) SOCl2, POCl3, DMF, 80°C, 3.5 h, 97%; (e) isopropanol, reflux 2 h, 80-90%; (f) Fe, CH3CH2OH, CH3COOH, reflux, 4 h, 75-80%; (j) C2Cl2O2, CHCl3, DMF, NEt3, reflux, 3 h; (k) CF3COOH, 20°C, 1 h. All heating processes were carried out in an oil bath, and the total yield of 13c (Transtinib) was 60%.
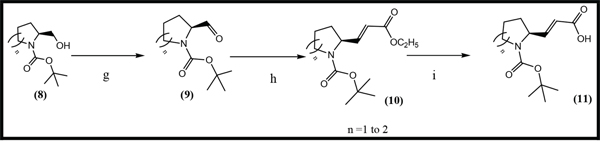
Scheme 2: General synthetic approach to the R3 substituent group. Reagents and conditions are as follows: (g) CH2Cl2, C2Cl2O2, DMSO, NEt3, -78°C, 2 h, [21] (h) THF, NaH, C8H17O5P, N2, 0°C, 2h,[22] (i) CH3CH2OH, LiOH, 15 h[23].
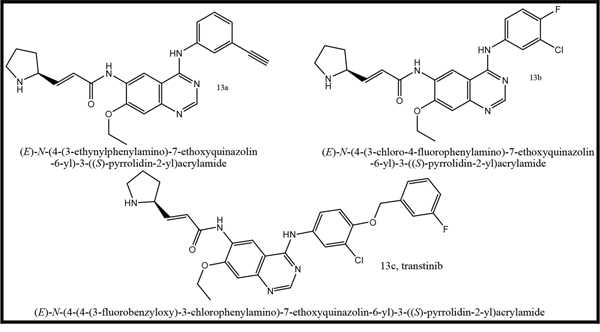
Figure 1: Structures and names of the synthesized compounds (13a-c).
Transtinib potently and selectively targets mutant EGFR cell lines in vitro
Transtinib and other two compounds (13a and, 13b) inhibit cell proliferation in cell lines expressing wild type and mutant EGFR. The in vitro potency of the compounds in comparison to gefitinib was determined in three cancer cell lines. Cell proliferation assays were performed using the MTT assay with increasing concentrations of compounds for 72 h. The results expressed as IC50 are summarized in Table 2. As shown in Table 2, three synthesized compounds demonstrated moderate to excellent anti-proliferative activities against different cancer cell lines, with IC50 values ranging from 34 nM to 95 nM (Table 2). All three compounds were more potent than gefitinib at activating mutation and the gatekeeper mutation (T790M). As expected, none of the compounds, including gefitinib, showed good efficacy against the SW620. In particular, consistent with molecular modeling in the ATP pocket of EGFR, compound 13c (Transtinib) was found to possess higher antitumor activities with an IC50 of 34 nM in the H1975 cell line with both the sensitive mutation L858R and the resistant mutation T790M. Moreover, transtinib exhibited moderate potency against the WT EGFR cell line (A431) with an IC50 of 62 nM.
Table 2: Anti-cancer properties of Transtinib and its derivates in cell models. The IC50 values are the averages of at least three independent experiments
Cell lines |
Mutation |
gefitinib IC50(nM) |
13a IC50(nM) |
13b IC50(nM) |
13c(transtinib) IC50(nM) |
H1975 |
L858R/T790M |
>1000 |
43±12.8 |
50±8.2 |
34±7.6 |
A431 |
Wild type(over-express) |
85±8.8 |
95±13.4 |
75±9.8 |
62±8.3 |
N87 |
HER2(over-express) |
>1000 |
87±8.2 |
80±10.2 |
47±8.8 |
SW620 |
No HER2 and EGFR express |
>1000 |
>1000 |
>1000 |
>1000 |
In addition to assessing the activity against the activating and resistant mutants, we similarly assessed the potency of compounds against the HER2 over-expressing cell line (N87). N87 cells were treated with increasing concentrations of either gefitinib, or 13a, 13b, or transtinib for 72 h. All compounds were effective against WT HER2, except for gefitinib. Transtinib exhibited was more effective at inhibiting the growth of this cell line than 13a or 13b with an IC50 of 47 nM. Thus, transtinib may also be able to target HER2 in tumors. These results indicated that the acrylamide and pyrrolidine substituent groups may increase the binding affinity between compounds and EGFR. Therefore, transtinib was selected for further docking and xenograft model studies.
Transtinib likely docks into EGFR in a similar manner as HKI272
To give structural insight into the ligand/enzyme interactions and an explanation of the observed activity, we examined the interaction of ligands with the EGFR complex structure by molecular docking of compounds into the ATP binding site of EGFR.
When docking with the WT active state of EGFR, Transtinib (Figure 2B) was more potent than gefitinib (Figure 2A). The quinazoline ring is oriented with the 1-N back of the ATP binding pocket, a hydrogen bond formed between the Cl substituent and the Met793 residue, and the van der Waals forces occur among the side chain of Transtinib and the residues Val726, Leu792, Glu804, and Pro794. While docking with the DFG-out state, bearing the T790M mutation, all of the docking results were poor (data not shown). Thus, we adopted the C-helix out inactive mode, and as illustrated in Figure 2C and 2D, our analysis suggested that Transtinib favorably fitted into EGFR in a similar manner as HKI272. Moreover, a number of key interactions with the mutant EGFR (T790M) protein likely contributed to this compound’s potency. The quinazoline ring is oriented with the 1-N in the back of the ATP-binding pocket, and the covalent Michael acceptor (acrylamide group) is located in close proximity to Cys797, which is feasible for covalent bond formation. Furthermore, the quinazoline core of the inhibitor forms hydrogen bond (1.9 Å) interactions with the backbone amide of the hinge residue Met790 directed toward the gatekeeper. Two other hydrogen bonds form among the side chain of Transtinib and the residues Arg841 (1.8 Å) and Thr854 (2.5 Å) of EGFR. We speculated that the pyrrolidine group may have potential advantages for the interaction with Arg841 and Thr854, which would further strengthen the ligand’s affinity towards the gatekeeper mutant.
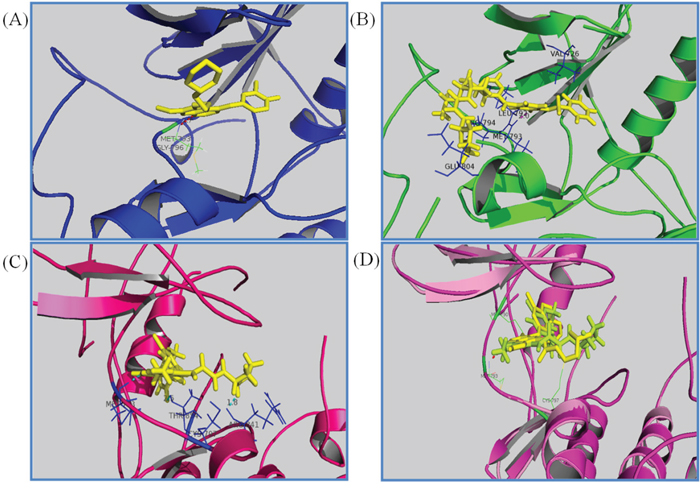
Figure 2: Drug docking studies in the WT and mutant EGFR kinases. The binding mode of Transtinib (B, C, D), gefitinib (A) and HKI272 (D) are compared in the WT (A, B) and T790M mutation kinases (C, D). Key interactions are labeled, the inhibitors are shown in stick form, and hydrogen bonds are indicated with dashed lines. A. gefitinib binding with the active form of EGFR (2ITY [10]), B. Transtinib binding with the active form of EGFR, hydrogen bond formed between the Cl substituent and the Met793 residue C. Transtinib binding with the C-helix out inactive mode with the T790M mutation of EGFR, three hydrogen bonds were formed with Met793, Arg841 and Thr854. D. HKI272 (yellow) superposed on Transtinib (lemon) in 2JIV.
Transtinib demonstrates a significant prolonged response in mutant EGFR xenograft models in vivo
To explore the in vivo activity of Transtinib, we dispensed the drug lead as mono-therapy against mutant EGFR xenografts on behalf of clinical NSCLC scenarios. Once daily dosing of Transtinib induced significant dose-dependent decreases in tumor size in both A431 and H1975 tumor xenograft models, with 90% tumor reduction observed at doses of 5.0 mg/kg/day in both models after 4 weeks (Figure 3A and 3B). Similar tumor reductions were observed after applying of 5 mg/kg/day Gefitinib. These studies showed that Transtinib can induce significant decreases in tumor size at low doses against both EGFR drug-sensitized and T790M/L858R- resistant EGFR mutant disease animal models.
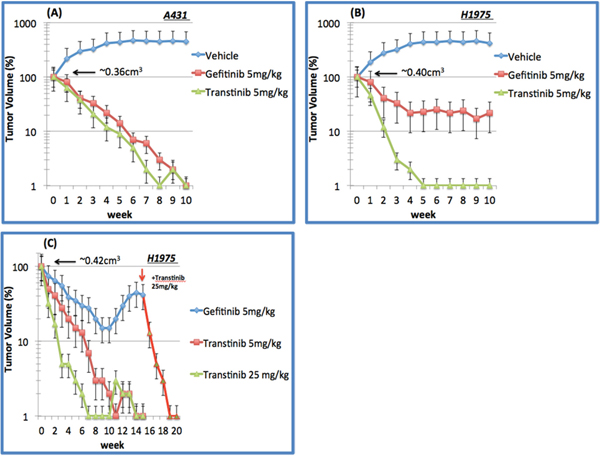
Figure 3: In vivo tumor suppression of Transtinib in xenograft models of EGFR-TKI sensitizing A431 and T790M/L858R resistant H1975 non-small cell lung cancer. A. A431 and B. H1975 xenograft following 10 weeks of daily 5 mg/kg gefitinib (n=6) and Transtinib treatment (n=8 and 10 mice, respectively). C. H1975 following chronic daily oral dosing of 5 and 25 mg/kg Transtinib (n=10 and 8, respectively). Additionally, 25 mg/kg Transtinib was applied to the 5 mg/kg Gefitinib treatment group after 15 weeks to restore the anti-cancer efficacy. Data are plotted as the mean ± standard error.
We then challenged the durability of tumor reduction through 16-20 week long- term daily oral dosing of Transtinib in 8-10 H1975 xenografts (Figure 3C). As a comparison, gefitinib at 5 mg/kg/day induced less tumor reduction and tumors began to re-grow after approximately 15 weeks, but an increased dose of 25 mg/kg/day Transtinib triggered tumor reductions, suggesting that re-growth was still driven by T790M/L858R-resistant EGFR mutants. In H1975 xenografts, 5 mg/kg/day Transtinib resulted in almost complete responses in 9 of 10 tumors at week 11. No visible tumors were observed after 7 weeks of dosing at 25 mg/kg/day Transtinib. The complete responses were maintained for the duration of the study period with no tumor recurrence during the 20 weeks of treatment. Moreover, no growth was observed for an additional 5 weeks after Transtinib treatment was terminated.
In comparison, the efficacy against wild-type and mutant EGFR xenografts was examined. Transtinib did moderately inhibit tumor growth in A431. However, this same 5 mg/kg/day dose induced complete tumor reduction in H1975 mutant EGFR tumor xenografts, suggesting that Transtinib possesses a novel selectivity margin over WT EGFR.
MATERIALS AND METHODS
Chemistry
A general approach to synthesize the designed quinazoline compounds is shown in Scheme 1, starting from commercially available 2-amino-4-fluorobenzoic acid (1). Unless otherwise noted, all reagents and solvents were purchased from Sigma or Aldrich and used without further purification. Dry solvents were purchased as anhydrous reagents from commercial suppliers.
All of the structures of the compounds were evaluated by 1H NMR spectroscopy at 400 MHz or 300 MHz, and by MS (BRUKER Autoflex TOF/TOF). 1H chemical shifts are reported in δ (ppm) as s (singlet), d (doublet), dd (doublet of doublet), t (triplet), q (quartet), m (multiplet), and br s (broad singlet) and are referenced to the residual solvent signal: CDCl3 (7.26) or DMSO-d6 (2.50). The compounds (11) were synthesized according to Scheme 2.
Molecular docking study
The wild type (WT) and various mutant forms of the EGFR kinase domain have been structurally characterized. Analysis of previously published structures of TKI binding to EGFR revealed two binding modes. The first mode is the DFG-out state, which is characterized by the core structure of inhibitors forming strong interactions with the hinge region in EGFR and the other moiety of inhibitors extending to (or close to) the solvent exposure area, such as erlotinib (Figure 4A), gefitinib, and BIBW2992. The second mode is the C-helix out inactive mode. In this second mode, the core structure of inhibitors, such as HKI272 (2JIV) [15] (Figure 4B), forms a single H-bond and hydrophobic interactions with the hinge region, including the mutant gatekeeper residue Met790, while the lipophilic moiety of the inhibitors expands to the back pocket of ATP binding and disrupts the salt bridge between the glutamate residue on helix αC and the lysine residue on the N-lobe. In addition to these noncovalent interactions, the covalent bond is formed between Cys797 and the crotonamide Michael-acceptor group on the inhibitor.
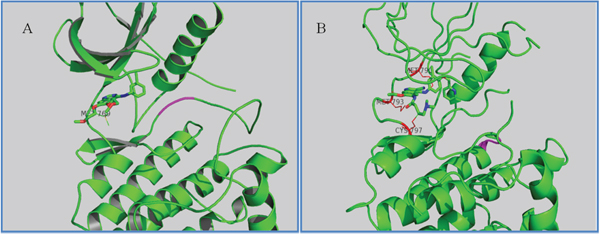
Figure 4: Two inactive states of EGFR. A. Crystallographic structure of EGFR with erlotinib (1M17, DFG-out state). The interaction residue Met769 is labeled and the DFG motif is labeled in magenta. B. Crystallographic structure of EGFR with HKI272 (2JIV, C-helix out inactive mode). The interaction residues Met790/793 and Cys797 are labeled.
To predict the possible binding mode in the ErbB family enzyme active site, Transtinib underwent in silico screening, and the autoDock4.2 package [24] was used for molecular docking. In addition, the structures of HKI-272 and gefitinib were considered as controls. The docking was performed using the receptor from the 2JIV (HKI272) and 2ITZ (gefitinib) structures from the RCSC Protein Data Bank. The co-crystal inhibitors were removed from the initial X-ray structures, water molecules were then removed, and polar hydrogens and Gasteiger charges were added using the Autodock Tool. Ligands were optimized for energy and geometry using MM2 force fields. The rotational bonds of all proteins were regarded as being rigid, while the rotational bonds of the ligands were treated as flexible. All figures were produced using the open source PyMOL 1.5 package.
Anti-proliferative activities of compounds 13a-13c
To assess their potential antitumor activity, compounds 13a, 13b, and 13c (Transtinib) were tested at a range of concentrations and evaluated in four cell lines harboring either wild-type or mutant EGFR. The first generation TKI (gefitinib) was employed as the positive control. For EGFR (T790M/L858R), the human NSCLC NCI-H1975 was obtained and cultured in RPMI-1640 with 10% fetal bovine serum (FBS) and 1% penicillin-streptomycin. For HER2 expression and no expression of EGFR and HER2, the human gastric carcinoma cell line NCI-N87 (HER2 over-expressing) and the human colorectal adenocarcinoma cell line SW620 (no expression of EGFR and HER2) were obtained from the American Type Culture Collection and cultured in RPMI-1640 with 10% FBS and 1% penicillin-streptomycin. For EGFR (wild-type), the human epidermoid A431 carcinoma cell line (EGFR overexpression) was obtained and cultured in high-glucose DMEM with 10% FBS and 1% penicillin-streptomycin. The ability of the compounds to inhibit proliferation was measured by the MTT assay. Cell lines were plated in 96-well plates at between 1,000 and 10,000 cells per well. The cells were allowed to attach overnight at 37°C under 5% CO2. Twenty-four hours after the cells were plated, they were exposed to compounds at titrations concentration and the treated cells were incubated for a further 72 h at 37°C under 5% CO2. Fresh MTT was added to each well at a terminal concentration of 5 mg/mL and incubated with the cells at 37°C for 4 h. The formazan crystals were dissolved by adding 100μl of DMSO to each well, and the absorbance at 492 nm (for the absorbance of MTT formazan) and 630 nm (for the reference wavelength) were measured with the ELISA reader. All of the compounds were tested three times in each of the cell lines. The results were expressed as IC50 (inhibitory concentration 50%) and were the averages of three determinations.
Transtinib anti-tumor activity in in vivo xenograft models
To explore the in vivo activity of transtinib, we administered the drug as monotherapy against various mutant EGFR xenografts representing clinical NSCLC scenarios.
Four-six-week-old SCID female nude mice were purchased from the Animal facility, Institute of Molecular Biology, Academic Sinica, and housed in ventilated cages. All standard methods for animal model and assays were approved by the School of Pharmaceutical Sciences, Xiamen University. Xenografts were bred by injecting 5~7x106 cells (H1975 and A431) with 50% Matrigel in total volume of 0.5 ml per mice. Before treatment, mice were randomized to receive Transtinib, gefitinib and the vehicles. Reagents were diluted with 0.5% lactic acid and were given by oral gavages. Tumor size was measured twice a week from an approximate mean starting volume of 0.32-0.42 cm3. The average tumor size was calculated by the formula π / 6 × (large diameter, mm) × (small diameter, mm)2. Statistical significance was evaluated using a one-tailed Student’s t test.
Experimental methods
General methods for the synthesis of the compounds
7-fluoroquinazolin-4(3H)-one (2)[25] A solution of 20 g of chemical (1) in 60 ml methanamide was heated to 130-140°C for 20 h, and then cooled to room temperature. The solid was collected and washed with ethanol and petroleum benzin then air-dried. MS (m/z) 164.5; 1H NMR (400 MHz, DMSO-d6) δ 12.37 (s, 1H), 8.16 (m, 2H), 7.37-7.73 (m, 2H) ppm.
7-fluoro-6-nitroquinazolin-4(3H)-one (3) A solution of 17 g of (2) in100ml H2SO4 was cooled to 0°C in an ice bath for over 30 min by intensely stirring and then a mixture solution of 20 ml of fuming nitric acid (fresh) and 20 ml of concentrated sulfuric acid was added dropwise. Then, the solution was reacted at room temperature with stirring for 1.5-2 h. The mixture was heated to 110°C with stirring for 2 h and poured onto ice with stirring. The solid was collected and washed with ice water and absolute ethyl alcohol, and air-dried. MS (m/z) 209.6; 1H NMR (400 MHz, DMSO-d6) δ 12.08-13.28 (b, 1H), 8.70-8.74 (d, 1H), 8.30 (s, 1H), 7.75-7.78 (d, 1H) ppm.
7-ethoxy-6-nitroquinazolin-4(3H)-one (4) 5.3 g of Na in a flask and 200 ml of anhydrous ethanol were added dropwise with reflux and stirring. After the Na was completely dissolved, stirring was continued for over 1 h, and 15.5 g of (3) was added. Heating at reflux was continued for 2.5 h. The reaction was cooled to room temperature, filtered and the solid was washed with ethanol and air-dried. MS (m/z) 233.6.
4-chloro-7-ethoxy-6-nitroquinazoline(5) [25] To 11.3 g of (4) in a flask, 88 ml of SOCl2 and 17 ml of POCl3 were added dropwise, respectively, and then the reaction was heated to over 80°C and intensely refluxed for 3.5 h. Then, the solution was cooled to room temperature, and the excess SOCl2 and POCl3 were removed by rotary evaporation. The solid residue was washed in toluene three times. The toluene was removed by rotary evaporation, and the solid was air-dried. MS (m/z) 254.8; 1HNMR (400 MHz, CDCl3-d1) δ 9.05 (s, 1H), 8.6 (s, 1H), 4.30-4.40 (d, 2H), 1.50-1.6 (m, 3H) ppm.
N-(3-Y-4-X-phenyl)-7-ethoxy-6-nitroquinazolin-4-amine (6)a-c A suspension of 13 g of (5) in 200 ml of isopropanol was stirred, and an equimolar of M was added. Next, the reaction was heated to over 85°C and refluxed for 4 h. After cooling to room temperature, the reaction was filtered and the solid was washed with isopropanol and air-dried. 6a MS (m/z) 334.8, 6b MS (m/z) 362.8, 6c MS (m/z) 468.9.
N-4--(3-Y-4-X-phenyl)-7-ethoxyquinazoline-4,6-diamine (7)a-c A mixture of 13 g of (6),200 ml of anhydrous ethanol, 35 ml of glacial acetic acid and 13 g of Fe power was stirred at 60°C for 1 h, and then the reaction was heated to reflux for 4 h. After cooling to room temperature, the reaction mixture was diluted with water and ethyl acetate with stirring for 15 min, and the resulting mixture was extracted with ethyl acetate. The combined organic layer was washed with saturated salt water. The excess glacial acetic acid was removed with saturated NaHCO3 3 times, and the resulting organic was dried over anhydrous sodium sulfate and evaporated in vacuo. 7a MS (m/z) 304.8, 7b MS (m/z) 332.8, 7c MS (m/z) 439.0.
(S)-tert-butyl 2-formylpyrrolidine-1-carboxylate (9) This compound was prepared from (8), which is a commercially available chemical. A 1 L, 3-necked, round-bottomed flask equipped with an overhead stirred and a nitrogen inlet was charged with oxalyl chloride (14 ml) in dichloromethane (DCM, 200 ml). The solution was cooled to -78°C in a dry ice/ ethanol bath. A solution of DMSO (25 ml) in DCM was added dropwise. After 30 min, a solution of alcohol (8) (14 g) in DCM (100 ml) was added dropwise, and the reaction was continued for 1 h. Next, triethylamine (50 ml) was added dropwise and the mixture reacted for 30min. Then, the mixture was transferred to an ice/water bath and stirring was continued for 30 min. Then, the reaction was diluted with DCM and washed successively with water, 1 M HCl, saturated NaHCO3, and saturated NaCl. The DCM layer was dried over MgSO4, filtered and concentrated to afford (9) as an oil.
(S)-tert-butyl 2-((E)-2-(ethoxycarbonyl)vinyl)pyrrolidine-1-carboxylate (10) A 1 L, 3-necked, round-bottomed flask equipped with an overhead stirred and a nitrogen inlet was charged with NaH (10 g) in anhydrous tetrahydrofuran (THF, 100 ml). The solution was cooled in an ice/water bath for 20min, and then a solution of Triethyl phosphonoacetate (21.5 g) in THF (100 ml) was slowly added dropwise. A solution of crude (9) (16.4 g) in THF (100 ml) was added dropwise 1h later, and the reaction was continued for 1 h. After that, 100 ml of saturated NaCl was added dropwise to stop the reaction. The resulting solution was diluted with ethyl acetate and the layers were separated. The ether layer was washed with 1 M HCl and saturated NaCl, dried over anhydrous MgSO4, filtered and concentrated to afford (10) as a yellow oil that was used without further purification.1HNMR (CDCl3, 300 MHz) δ 6.92-6.76 (m, 1H),5.82 (d,1H), 4.56-4.32 (m, 1H), 4.25-4.12 (m, 2H), 3.48-3.27 (m, 2H), 2.20-1.98 (m, 1H), 1.91-1.72 (m, 2H), 1.43 (s, 9H), 1.25 (t,3H) ppm.
(E)-3-((S)-1-(tert-butoxycarbonyl)pyrrolidin-2-yl)acrylic acid (11) To a solution of (10) (26.4 g) in anhydrous ethanol (200 ml), LiOH (10 g) in water (125 ml) was added, and the solution was stirred at room temperature for 15 h. Ethanol was distilled off, and the residue dissolved in water (100 ml). The solution was cooled with an ice/water bath and carefully acidified with dilute HCl. The aqueous layer was extracted with DCM. The organic layer was dried over anhydrous MgSO4, filtered and concentrated to afford (11). 1HNMR (CDCl3, 300 MHz) δ 6.71-6.91 (m, 1H), 5.81 (d, 1H), 4.26-4.38 (m, 1H), 4.16 (q, 2H), 3.40 (brm, 2H), 1.95-2.10 (m, 1H), 1.70-1.90 (m, 3H), 1.4 (s, 9H) and 1.25 (t, 3H) ppm.
°δ
&
–
–
ä
–
–
’
––
&
&