
INTRODUCTION
Myelodysplastic syndromes (MDS) comprise a diverse group of clonal hematopoietic stem cell malignancies characterized by dysplasia, peripheral cytopenias, ineffective hematopoiesis, and risk of progression to acute myeloid leukemia (AML). It is the common understanding that MDS originates from a founding driver mutation within a cell having the capacity to differentiate into mature myeloid cells [1]. The nature of the founding mutation is likely to be involved in determining clinical phenotype and outcome. For instance, mutations in SF3B1 are strongly associated with refractory anemia with ring sideroblasts (RARS) and a good prognosis [2, 3], whereas SRSF2 and U2AF1 (U2AF35) mutations are mainly observed in refractory cytopenia with multi lineage dysplasia (RCMD) and refractory anemia with excess blasts (RAEB), and associated with high risk of leukemic transformation [4].
The founding mutation will eventually be present in almost all bone marrow cells due to the selective advantage gained by the cell in which the mutation occurred. These clonal cells also carry hundreds of benign somatic passenger mutations captured at the start of clonal proliferation [5]. Most often the driver mutations are found in genes involved in RNA splicing, such as SF3B1, SRSF2, U2AF35, and ZRSR2 or in epigenetic regulation, such as TET2, DNMT3A, IDH1/2, ASXL1, and EZH2 [6–8]. As opposed to mutations in pro-apoptotic genes and growth signaling pathways these mutations do not, by themselves, promote clonal proliferation. However, they may affect the expression of a large number of downstream protein-coding- and microRNA (miRNAs) genes, which lead to expansion of the mutated clone.
In addition to the genetic alterations observed in MDS, several miRNAs have been found to be aberrantly expressed in this disease [9]. Most miRNAs are processed through the canonical pathway involving the nuclease Drosha and its cofactor DGCR8. These canonical miRNAs may be located in intergenic regions or located within introns and exons of protein-coding genes and share their regulatory elements. However, some miRNAs are transcribed from the opposite strand in the reverse direction of the gene, which may lead to an inverse correlation between the expression of the gene and the miRNA [10]. In addition, an emerging number of miRNAs have recently been shown to bypass Drosha by utilizing the spliceosome instead. These are located within introns of protein encoding genes and are known as mirtrons when the 5′ and 3′ ends of the precursor miRNA transcript is defined by splice sites, and tailed mirtrons when either the 5′ or the 3′ end is defined by a splice site [11–13]. Thus, tailed mirtrons are processed by both splicing and exonuclease digestion. Finally, a competitive interaction between the splicing machinery and the Drosha/DGCR8 complex has been discovered for miRNAs with precursors that are splice site overlapping (SO miRNAs) [14, 15].
Here, we studied the effects of spliceosome mutations on miRNA expression in primary bone marrow samples from MDS patients. To avoid potential confounding effects only patients with refractory anemia RA with or without ringsideroblasts were included in the study. High-resolution melting (HRM) assays covering all mutational hotspots of SF3B1, SRSF2, and U2AF35 were developed. All mutations detected by HRM in the patient samples were subsequently confirmed by Sanger sequencing. In total, 76 miRNAs were studied, including intragenic and intergenic canonical miRNAs previously implicated in MDS, as well as mirtrons, tailed mirtrons, and SO miRNAs.
RESULTS
Spliceosome mutations in the patient samples
All patients were screened for mutations within SF3B1, SRSF2, and U2AF35 using HRM analysis. Since we observed a higher frequency of SF3B1 mutations than expected [6], the samples were also tested for mutations in SF3B1 using denaturing gradient gel electrophoresis (DGGE) analyses (data not shown). The results were 100% concordant between the HRM and DGGE analyses, and all mutations were confirmed by Sanger sequencing (Supplementary Table S1, Figure 1). Twenty-two of the patients had a spliceosome mutation and 16 of these were found in SF3B1, five in SRSF2 and one in U2AF35. The mutations were unevenly distributed across RARS, RA, and del 5q subtypes. Among the patients with RARS 14 out of 15 (93%) patients carried a mutation in SF3B1 and one carried a mutation in SRSF2 (7%). Among the patients with RA two out of 18 carried a mutation in SF3B1 (11%), four carried a mutation in SRSF2 (22%), and one carried a mutation in U2AF35 (6%). All mutations were mutually exclusive (Figure 2). Because SF3B1 mutations have a tendency to coexist with DNMT3A mutations [8] the samples were also screened for mutations within this gene (Supplementary Table S1, Supplementary Figure S1). In total, five of the patients carried a mutation in DNMT3A. Three of these patients also carried an SF3B1 mutation and one carried a mutation in SRSF2 (Figure 2).
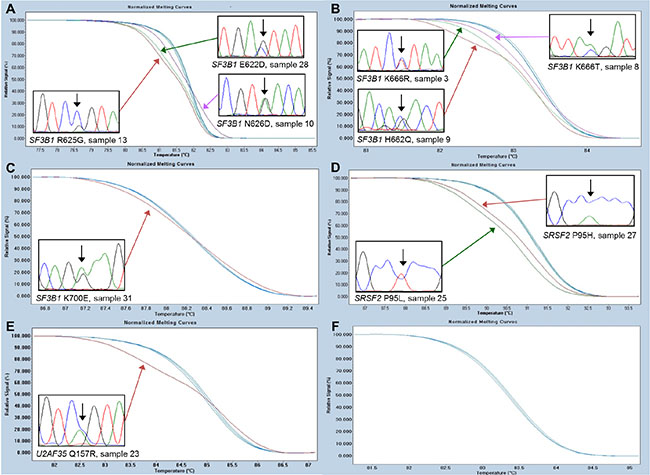
Figure 1: High-resolution melting assays for the detection of SF3B1, SRSF2, and U2AF35 mutations. Representative data for each of the hotspots are shown. (A) SF3B1 hotspot containing the N626D, R625G, and E622D detected in sample 10, 13, and 28, respectively. (B) SF3B1 hotspot containing the K666R, K666T, and H662Q mutations detected in sample 3, 8, and 9, respectively. (C) SF3B1 hotspot containing the K700E mutation detected in sample 31. (D) SRSF2 hotspot containing the P95L and P95H mutations detected in sample 25 and 27, respectively. (E) U2AF35 hotspot containing the Q157R mutation detected in sample 23. (F) U2AF35 hotspot containing the S34 mutations not detected in any of the samples.
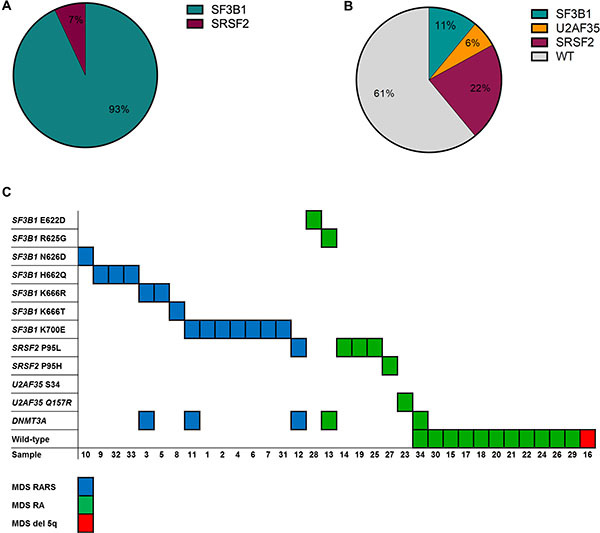
Figure 2: Overview of the spliceosome and DNMT3A mutations detected in the MDS samples. (A) Distribution of the mutations detected in the RARS patients. (B) Distribution of the mutations detected in the RA and del 5q patient samples. (C) Number and nature of the mutations detected in individual patients of different subtype.
Identification of accurate reference miRNA genes for the RT-qPCR analysis
For normalization of the miRNA expression data the identification of stably expressed reference genes are of paramount importance [16]. For this purpose we used the GeNorm and NormFinder algorithms [17, 18]. Data for 29 miRNAs and three commonly used reference genes (SNORD44, SNORD47, and RNU6B), which were expressed in all samples, were included in the analyses. Overall, there was a striking resemblance between the data obtained by the two algorithms (Supplementary Figure S2). The potential reference genes RNU6B, SNORD44, and SNORD47 were ranked ninth, 13th, and 22nd, respectively, by NormFinder, and 11th, 15th, and 21st, respectively, by GeNorm. Both algorithms identified miR-10b-5p and miR-486-5p as the least stable genes, while miR-106b-3p was the most stable gene in both analyses. NormFinder identified miR-29a-3p and miR-130b-3p as the best combination of any two reference genes. These two miRNAs were also among the most stably expressed genes in the GeNorm analysis (Supplementary Figure S2). As NormFinder takes into account that different biological subgroups exists within the cohort, we decided to use the geometric mean of the Ct-values for miR-29a-3p and miR-130b-3p when calculating relative expression quantities for each miRNA of interest.
Overall influences of spliceosome mutations on miRNA expression levels
When performing hierarchical clustering for all samples and miRNAs we observed that 14 out of 17 samples clustering together contain a spliceosome mutation within SF3B1 or SRSF2 (Figure 3). This was unlikely to have happened by chance (P = 0.018). In addition, we observed that most mirtrons clustered together (P < 0.001). Sample 15 clustered separately from all other samples as it had most miRNAs overexpressed, with the exception of miR-145-5p, miR-146a-5p (located on chromosome 5q), miR-125b-5p, miR-10a-5p, miR-10b-5p, and miR-27b-3p. This patient did not carry a spliceosome mutation or a mutation in DNMT3A, and was the youngest of all the patients in this cohort (Supplementary Table S1).
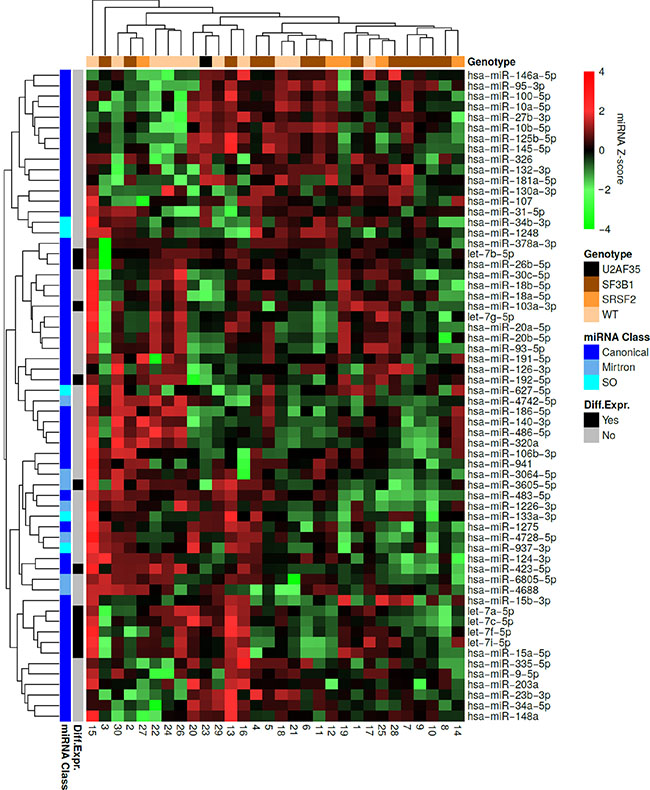
Figure 3: Hierarchical clustering of the MDS patient samples and miRNAs. Each row represents a miRNA and each column represents a patient sample. Green indicates low expression relative to the mean expression level of the samples and red indicates high expression. The mutational status of each sample is indicated at the top. The miRNA class of each miRNA as well as differentially expressed miRNAs between wild-type MDS samples and SF3B1 mutated samples are indicated to the left. It is observed that 14 out of 17 samples containing a spliceosome mutation within SF3B1 or SRSF2 clustered together. Most of the miRNAs, which were differentially expressed between wild-type MDS samples and samples containing an SF3B1 mutation clustered together. Finally, all mirtrons except miR-4742-5p were found within the same cluster.
We also performed hierarchical clustering for each class of miRNA separately. Among the canonical miRNAs, we observed that 8 out of 12 samples clustering together contain an SF3B1 mutation in (Supplementary Figure S3). This was unlikely to have happened by chance (P = 0.04). When analyzing the mirtrons we observed that 11 out of 12 samples clustering together contain a spliceosome mutation (Supplementary Figure S4A). This was unlikely to have happened by chance (P = 0.01). No obvious clustering of spliceosome mutated samples could be observed when analyzing SO miRNAs (Supplementary Figure S4B).
Overall, canonical miRNAs were downregulated in spliceosome mutated samples versus wild-type MDS samples (P = 0.002), and there was a tendency for mirtrons to be downregulated as well (P = 0.11), while this was not the case for SO miRNAs (P = 0.63) (Figure 4A). Because each spliceosome mutation may have a different impact on the expression of miRNAs, SF3B1 mutated samples and SRSF2 mutated samples were analyzed separately. In SF3B1 mutated samples canonical miRNAs were downregulated (P = 0.004), and there was a tendency for mirtrons to be downregulated (P = 0.16), while SO miRNAs were not (P = 0.44) (Figure 4B). Finally, it was observed that canonical miRNAs were downregulated in SRSF2 mutated samples (P = 0.002), while mirtrons (P = 0.22) and SO miRNAs (P = 0.31) were not (Figure 4C).
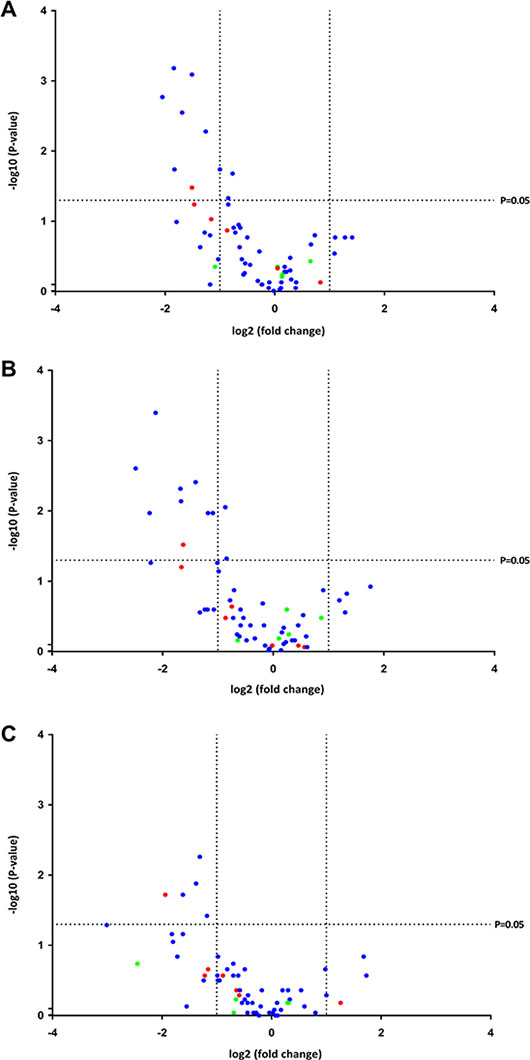
Figure 4: Volcano plots of the P-values and fold changes in expression of the miRNAs. Each miRNA is represented by a colored dot indicating its class. Blue represents canonical miRNA, red represents mirtron, and green represents SO miRNA. (A) Spliceosome mutated samples versus wild-type samples. It can be observed that all differentially expressed miRNAs were downregulated in the spliceosome mutated samples. (B) SF3B1 mutated samples versus wild-type samples. It can be observed that all differentially expressed miRNAs were downregulated in the samples containing an SF3B1 mutation. (C) SRSF2 mutated samples versus wild-type samples. It can be observed that all differentially expressed miRNAs were downregulated in the samples containing an SRSF2 mutation.
Identification of differentially expressed miRNAs in spliceosome mutated samples
SF3B1 was the most frequently mutated gene and we decided to mainly focus on these mutations in the subsequent analyses. Several of the studied miRNAs were differentially expressed between wild-type samples and samples with SF3B1 mutations, and all were downregulated in the SF3B1 mutated samples. Most of these were canonical miRNAs, with the exception of one mirtron (miR-3605-5p). Nine of the 10 canonical miRNAs were located in host genes. Furthermore, most of these miRNAs have previously been shown to have tumor suppressor properties (Table 1).
Table 1: Differentially expressed miRNAs between SF3B1 mutated patients and wild-type patients
MiRNA |
miRNA-class |
Chr. |
Host Gene |
Transcribed direction |
LFC |
Tumor Suppressor |
P-value |
|---|---|---|---|---|---|---|---|
let-7c-5p |
Canonical |
21 |
MIR99AHG |
Same |
−2.13 |
Yes |
0.001 |
let-7b-5p |
Canonical |
22 |
MIRLET7BHG |
Same |
−2.49 |
Yes |
0.002 |
let-7f-5p let-7f-1 let-7f-2 |
|
|
|
|
−1.4 |
Yes |
0.004 |
Canonical |
9 |
JB153432 |
Same |
|
|
|
|
Canonical |
x |
HUWE1 |
Same |
|
|
|
|
let-7a-5p let-7a-1 let-7a-2 let-7a-3 |
|
|
|
|
−1.67 |
Yes |
0.005 |
Canonical |
9 |
JB153432 |
Same |
|
|
|
|
Canonical |
11 |
MIRLET100 |
Same |
|
|
|
|
Canonical |
22 |
MIRLET7BHG |
Same |
|
|
|
|
hsa-miR-423-5p |
Canonical |
17 |
NSRP1 |
Same |
−1.66 |
Yes |
0.007 |
hsa-miR-103a-3p hsa-miR-103a-1 hsa-miR-103a-2 |
|
|
|
|
−0.87 |
Yes |
0.009 |
Canonical |
5 |
PANK3 |
Same |
|
|
|
|
Canonical |
20 |
PANK2 |
Same |
|
|
|
|
hsa-miR-26b-5p |
Canonical |
2 |
CTDSP1 |
Same |
−2.24 |
Yes |
0.011 |
let-7i-5p |
Canonical |
12 |
No host gene |
N/A |
−1.09 |
Yes |
0.011 |
hsa-miR-15a-5p |
Canonical |
13 |
DLEU2 |
Same |
−1.18 |
Unknown |
0.011 |
hsa-miR-3605-5p |
Mirtron |
1 |
PHC2 |
Same |
−1.23 |
Unknown |
0.030 |
hsa-miR-192-5p |
Canonical |
11 |
AB429224 |
Same |
−0.84 |
Yes |
0.047 |
Abbreviations, Chr: Chromosome, LFC: log2 fold change.
Hierarchical clustering was performed for the differentially expressed miRNAs, and the majority of the miRNAs were downregulated in the majority of the SF3B1 mutated samples (Figure 5). Sample 1 and 13 were exceptions to this trend. Of note, sample 13 carried a DNMT3A mutation, and diverted the most from the other SF3B1 mutated samples. Interestingly, this sample clustered together with wild-type samples, and was the only sample carrying an R625G mutation.
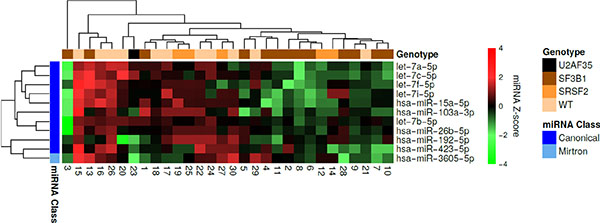
Figure 5: Hierarchical clustering of the MDS patient samples and differentially expressed miRNAs between wild-type samples and samples containing an SF3B1 mutation. Each row represents a miRNA and each column represents a patient sample. Green means low expression relative to the mean expression level of the samples and red means high expression. The mutational status of each sample is indicated at the top. It can be observed that the majority of the miRNAs were downregulated in the majority of the SF3B1 mutated samples.
Only five miRNAs (let-7a-5p, let-7b-5p, let-7c-5p, miR-335-5p, and miR-4728-5p) were differentially expressed in SRSF2 mutated compared to wild-type MDS samples. Except for one mirtron (miR-4728-5p) all of these miRNAs were intragenic canonical miRNAs transcribed in the same direction as their host genes and downregulated in the mutated samples. Three of these miRNAs (let-7a-5p, let-7b-5p, and let-7c-5p) were also significantly downregulated in SF3B1 mutated samples relative to wild-type samples (Table 1). When considering all mutated samples together, eight miRNAs (let-7c-5p, let-7a-5p, let-7b-5p, miR-423-5p, let-7f-5p, miR-26b-5p, miR-15a-5p, and miR-3605-5p) were differentially expressed compared to wild-type samples. Except for one mirtron (miR-3605-5p), all of these miRNAs were intragenic canonical miRNAs downregulated in the mutated samples. All of these were also significantly downregulated in SF3B1 mutated samples relative to wild-type samples, which is not surprising as most of the spliceosome mutated samples had an SF3B1 mutation.
Target and pathway analyses of the differentially expressed miRNAs
miRNA target predictions were performed for the differentially expressed miRNAs between wild-type samples and SF3B1 mutated samples. The miRNA target predictions of the differentially expressed miRNAs were compared to target predictions of the remaining miRNAs. These analyses revealed many putative oncogenes among the significantly enriched targets (See Supplementary Table S2), underscoring the notion that the differentially expressed miRNAs are tumor suppressors.
Next, it was investigated which cellular processes and human diseases the significantly enriched predicted targets are involved in using the Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway database. Several KEGG pathways related to MDS were found among the top significant hits (P < 0.001) including apoptosis, hematopoietic cell lineage, acute myeloid leukemia, JAK-STAT signaling pathway, spliceosome, MAPK signaling pathway, p53 signaling pathway, chemokine signaling pathway, and several cancers. In addition, several metabolic pathways were found among the top hits including glutathione metabolism, beta-alanine metabolism, alanine, aspartate and glutamate metabolism, glycolysis/gluconeogenesis, amino sugar and nucleotide sugar metabolism, and citrate cycle (TCA cycle) (See Supplementary Table S3).
Host gene analyses
To investigate whether the observed downregulation of miRNAs could be a result of their respective host genes being downregulated each canonical miRNA was classified as intergenic or intragenic (Supplementary Figure S5A). Only one of the significantly downregulated miRNAs was intergenic and, overall, there was no difference in expression between intergenic and intragenic miRNAs (P = 0.86) (Supplementary Figure S5B). Next, the expression of four host genes MIRLET7BHG, CTDSP1, PANK2, and PANK3 were measured. MIRLET7BHG is host gene for let-7b and CTDSP1 for miR-26b, while PANK2 and PANK3 host miR-103a-2 and miR-103a-1, respectively. A weak but non-significant positive correlation between let-7b and MIRLET7BHG was observed (P = 0.08), while no correlations were observed between the expression of CTDSP1 and miR-26b and between PANK2 and PANK3 and miR-103a (Supplementary Figure S6).
Validation of the custom plate miRNA data using single TaqMan assays
Individual cDNA syntheses were performed twice for all samples and analyzed by single TaqMan assays for the following miRNAs: let-7b, miR-10b-5p, miR-26b-5p, miR-29a-5p, miR-103-3p, miR-130b-3p, miR-145-5p, and miR-146a-5p. A high reproducibility of the raw Ct-values obtained after RT-qPCR performed on individual cDNA syntheses for each miRNA was observed (Supplementary Figure S7A). Furthermore, relatively good correlations were observed between the normalized data of the single TaqMan assays and the normalized data from the 96-well custom plate (Supplementary Figure S7B). Finally, all of these miRNAs were downregulated in patients with SF3B1 mutations relative to wild-type patients as expected, with the exception of miR-10b-5p, which was also upregulated in the previous analysis (Supplementary Figure S8).
DISCUSSION
A high frequency of spliceosome mutations are observed in MDS [8]. Therefore, it is likely that abnormal RNA splicing plays an important role in MDS pathogenesis. Spliceosome mutations in MDS are generally gain-of-function in nature, but little is known about their potential effects on the transcriptome [19]. It has, however, been shown that the vast majority of differentially expressed genes in SF3B1 mutated samples compared to wild-type samples are downregulated [20, 21]. This is likely due to the use of cryptic 3′ splice sites leading to retention of an intronic sequence containing in frame stop codons, which, in turn, may lead to nonsense-mediated mRNA decay (NMD) [20]. Alternatively, it may be due to key regulatory genes (e.g. encoding transcription factors or epigenetic modifiers) being aberrantly spliced. Splicing abnormalities of the epigenetic modifiers ASXL1 and EZH1, the transcription factor RUNX3, as well as the RAS pathway gene CBL have been observed in cases with SF3B1 mutations [21, 22]. Likewise, a splicing abnormality of RUNX1, a recurrently mutated transcription factor in MDS, has been observed in bone marrow cells from two cases with SRSF2 mutations, and aberrant splicing of the epigenetic modifier TET2 has been associated with mutations in U2AF35 [4]. In addition, SF3B1 mutations often coexist with mutations in DNMT3A [8], and for this reason it can be difficult to decipher the in vivo effects of each individual mutation.
Our study is the first to analyze the impact of spliceosome mutations on the expression of miRNAs. We decided to focus on MDS as spliceosome mutations are common in this disease and on miRNAs since they play important roles in both solid and hematologic cancers [9, 23]. We studied a large panel of miRNAs, including intragenic and intergenic canonical miRNAs previously implicated in MDS, as well as mirtrons, tailed mirtrons, and SO miRNAs. To accurately quantify the expression of the miRNAs we used the GeNorm and NormFinder algorithms to identify stably expressed miRNA genes to normalize the RT-qPCR data. These two algorithms are inherently different, but can be expected to give similar results [24]. Indeed, we observed a striking similarity between the data obtained by each algorithm. Thus, the miRNA genes we identified to be stably expressed in MDS may be useful for normalization of RT-qPCR data in future miRNA studies. On the other hand, the commonly used references, SNORD44 and SNORD47, were not among the most stable genes in either of the analyses.
Overall, our results show that miRNA expression is affected by spliceosome mutations in MDS. In line with the observations by Visconte and colleagues [21], we observed a strong association between spliceosome mutations and miRNA downregulation. In fact, we found none of the miRNAs to be significantly upregulated in SF3B1 or SRSF2 mutated samples compared to wild-type samples. Because mutations within SF3B1 coexist with mutations in DNMT3A the observed miRNA downregulation could, in theory, be caused by altered methylation patterns of regulatory regions associated with the miRNAs and/or their host-genes. Thus, we tested all samples for mutations within DNMT3A. However, only three of the SF3B1 mutated samples carried a DNMT3A mutation, making it highly unlikely that DNMT3A mutations are responsible for the observed downregulation of miRNAs in the spliceosome mutated samples. Furthermore, two of the samples (patient 3 and 13), which carried both an SF3B1 mutation and a DNMT3A mutation, clustered together with wild-type samples. This cluster analysis also indicates that spliceosome mutations have an overall effect on the transcription of miRNAs, as most spliceosome mutated samples clustered together. Finally, it should be noted that the spliceosome is a very complex macromolecule consisting of numerous proteins and snRNAs [25], and it cannot be ruled out that wild-type samples carry a mutation in other spliceosome genes apart from the most commonly mutated studied here.
When analyzing each miRNA class separately it was observed that none of the SO miRNAs were significantly downregulated. This was expected since the Drosha/DGCR8 complex competes with the spliceosome machinery for processing of the precursor RNA molecule. Thus, it is likely that a mutated spliceosome is less competitive, leading to increased production of the miRNA and less production of the mRNA. However, as few SO miRNAs were expressed in the samples we were unable to make firm conclusions regarding the influence of spliceosome mutations on this group of miRNAs. On the other hand, it was expected that less efficient splicing would lead to decreased expression of mirtrons, which are directly dependent on the spliceosome for processing of the precursors. Indeed, it could be observed that most mirtrons were downregulated in spliceosome mutated samples compared to wild-type samples. However, we were surprised to observe that the spliceosome mutations had most profound effect on canonical miRNAs.
Only two of the differentially expressed miRNAs in our study are directly processed by the spliceosome (miR-3605-5p and miR-4728-5p), and we found an intergenic miRNA (let-7i-5p) to be significantly downregulated in the SF3B1 mutated samples. These observations point towards indirect mechanisms mainly being responsible for the aberrant expression of miRNAs in spliceosome mutated MDS. This could potentially be due to aberrant splicing of genes encoding transcription factors or epigenetic modifiers, which, in turn, may have an effect on the expression of miRNA host genes as well as miRNA genes having their own promotors. A weak but non-significant positive correlation between let7b and MIRLET7BHG (P = 0.08) was found, while no correlations were observed between the expression of CTDSP1 and miR-26b and between PANK2 and PANK3 and miR-103a. It was, however, expected that correlations between PANK2 and PANK3 and miR-103a would be weaker as miR-103a has two genes, miR-103a-1 and miR-103a-2, located in PANK3 and PANK2, respectively. Furthermore, both genes have antisense transcripts, which may also influence the expression of the mature miRNA. Nevertheless, strong correlations between miRNAs and their host genes would not be expected as their half-lives are likely to be dissimilar and most host genes have several isoforms which may be differentially expressed.
Interestingly, we found several tumor suppressor miRNAs to be downregulated in patient samples with SF3B1 and SRSF2 mutations. Among these were several miRNAs belonging to the let-7 family, which are well known for their tumor suppressor activities [26, 27], and their essential role in stem cell differentiation and hematopoiesis [28]. In MDS let-7a has been shown to downregulate oncogenic KRAS and to be under-expressed in patients with intermediate or high-risk karyotype [29]. The miR-26 family is also involved in hematopoiesis [9], and miR-423-5p and miR-103a have previously been shown to be downregulated in MDS and to have tumor suppressor properties [30, 31]. Interestingly, a recent study highlighted that several genes involved in early hematopoiesis are aberrantly spliced in a model of mutant SRSF2 [32], including MEIS1 [33]. Thus our data suggest that the ineffective hematopoiesis observed in MDS not only results from aberrant splicing of protein encoding genes, but also from downregulation of miRNAs.
When comparing the predicted targets of the differentially expressed miRNAs with the remaining miRNAs many putative oncogenes were among the significantly enriched targets. These included BACH1, a transcription factor, which is upregulated in several cancers and known to be regulated by the let-7 family [34], HMGA1, which is involved in cell differentiation and apoptosis and upregulated in several cancers [35], SLC4A4, which is upregulated in chronic myeloid leukemia stem cells [36], ZBTB5, which function as an oncogene by repressing the cell cycle arrest gene p21 [37], and ACSL6, which has been shown to be part of a gene fusion in MDS and polycythemia vera [38, 39]. Overall, the significantly enriched predicted targets were found to be involved in apoptosis, hematopoiesis, acute myeloid leukemia, the JAK-STAT-, MAPK-, p53-, and chemokine signaling pathways, the spliceosome, and several cancers. In addition, several metabolic pathways were among the most significant pathways, which corresponds well with the recent finding that let-7a plays a prominent role in regulating energy metabolism in cancer cells [40].
In conclusion, our study is the first to provide evidence that spliceosome mutations play an important role in MDS pathophysiology by affecting the expression of tumor suppressor miRNA genes involved in the development and progression of MDS.
MATERIALS AND METHODS
Patient samples
Ten mL of bone marrow (BM) were aspirated from the posterior iliac crest of 34 MDS patients at the time of diagnosis and three subjects with suspected MDS due to peripheral cytopenia, but without BM dysplasia or chromosomal abnormalities. The diagnoses of MDS were based on morphology supplemented by cytogenetics, and divided into subgroups according to WHO 2008 criteria.
Clinical and cytogenetic data of MDS patients can be found in Supplementary Table S1.
The study was approved by the ethical committee for the Capital Region of Denmark (H-D-2009-003) and the Danish Data Protection Agency (30–1419) and conducted in accordance with the tenants of Helsinki.
Extraction of DNA and miRNA
DNA was purified from the mononuclear fraction of BM samples using the Gentra Puregene Cell Kit (Qiagen) according to the manufacturer’s instructions. miRNA was purified from Formalin-Fixed Paraffin Embedded (FFPE) whole BM biopsies using the Recover All Total Nucleic Acid Isolation Kit for FFPE (Ambion) according to the manufacturer’s instructions.
Mutation detection
HRM assays, covering mutations within SF3B1 (3 hotspots), SRSF2 (1 hotspot), U2AF35 (2 hotspots) and DNMT3A (3 hotspots), were designed and optimized. DGGE assays, covering mutations within SF3B1 (3 hotspots) were designed and optimized. All PCR primer sequences are shown in Supplementary Table S4. The nature of the mutations detected in the samples was confirmed using traditional Sanger sequencing of the PCR products. Additional information can be found in the Supplementary Files.
TaqMan miRNA 96-well custom plate design
The miRNA TaqMan assays used in this study were pre-spotted on a 96-well plate. In total, 67 canonical miRNAs, 11 mirtrons, and 11 SO miRNAs were included, together with four potential noncoding RNA reference genes. A background signal was detected for 17 assays, which were excluded from further analyses, yielding a total of 56 canonical miRNAs, nine mirtrons, 11 SO miRNAs, and three potential noncoding RNA reference genes. The miRNAs were selected following a literature search to ensure that the majority has been implicated in MDS previously. Details regarding each miRNA assay can be found in Supplementary Table S5.
RT-qPCR
One-hundred ng of RNA from each patient sample were reverse transcribed with the TaqMan® MicroRNA Reverse Transcription Kit (Life Technologies) according to manufacturer’s instructions with a customized primer pool for all miRNA targets. The reactions were performed on a Veriti® 96-well Thermal Cycler (Applied Biosystems) with the following conditions: one cycle of 16°C for 30 min, 42°C for 30 min, and 85°C for 5 min. Prior to qPCR the cDNA samples were pre-amplified using TaqMan® PreAmp Master Mix Kit (Applied Biosystems) according to the manufacturer’s instructions. The pre-amplification was performed on a Veriti® 96-well Thermal Cycler (Applied Biosystems) with the following conditions: one cycle of 95°C for 10 min, 55°C for 2 min, 72°C for 2 min, 12 cycles of 95°C for 15 sec and 60°C for 4 min, and one cycle of 99.9°C for 10 min. Subsequently, the samples were diluted 1:10 in DNAse/RNAse free water. cDNA synthesis and pre-amplification were also performed on a no enzyme control (NEC) sample and a no template control (NTC). The qPCRs were performed using the TaqMan® Universal Master Mix II, no UNG Kit (Life Technologies) in a 96-well format, as described above, according to the manufacturer’s instructions on a StepOnePlus® Real-Time PCR System (Life Technologies). The following negative controls were included; the NEC and NTC from the cDNA synthesis, NTC from the pre-amplification and NTC at the qPCR level.
Host gene analyses
Single TaqMan assays were used for the host gene analysis. In total, four host genes and two reference genes were included in the study. In the RT-qPCR step ten ng of RNA from each patient sample were reverse transcribed with the TaqMan® MicroRNA Reverse Transcription Kit (Life Technologies) according to the manufacturer’s instructions with a primer pool for all target genes. The reactions were performed on a Veriti® 96-well Thermal Cycler (Applied Biosystems) as described above. Prior to qPCR the cDNA samples were pre-amplified using TaqMan® PreAmp Master Mix Kit (Applied Biosystems) according to the manufacturer’s instructions. The pre-amplification was performed as described above using 10 instead of 12 cycles of 95°C for 15 sec and 60°C for 4 min. Subsequently, the samples were diluted 1:5 in DNAse/RNAse free water. The qPCRs were performed using the TaqMan® Universal Master Mix II, no UNG Kit (Life Technologies), according to the manufacturer’s instructions on a StepOnePlus® Real-Time PCR System (Life Technologies). The same negative controls as described above were included in these experiments.
Validation of the custom plate miRNA data using single TaqMan assays
RT-qPCR was performed using single TaqMan assays (Life Technologies) for the following miRNAs; let-7b, miR-10b-5p, miR-26b-5p, miR-29a-5p, miR-103-3p, miR-130b-3p, miR-145-5p, and miR-146a-5p. cDNA synthesis, pre-amplification, and qPCR were performed in duplicates for each patient sample. Ten ng of RNA were reverse transcribed using the TaqMan® MicroRNA Reverse Transcription Kit according to manufacturer’s instructions (Life Technologies). For pre-amplification all eight reverse transcription primers were pooled according to manufacturer’s instructions. The RT reaction was as described above. The pre-amplification was performed as described above using 10 instead of 12 cycles of 95°C for 15 sec and 60°C for 4 min. Subsequently, the samples were diluted 1:5 in DNAse/RNAse free water. The same negative controls as described above were included in these experiments.
Data and statistical analyses
Raw cycle threshold (Ct) values for miRNAs were extracted from the StepOnePlus® Real-Time PCR system (Applied Biosystems). Samples with an overall poor quality (geometric mean (Ctref genes) > 35) were excluded from further analyses. Therefore, it was not due to analysis failure when a sample did not amplify for a miRNA of interest, but rather it meant that the miRNA was not expressed in that sample. Accordingly, samples not amplifying for a given miRNA were given a Ct value of 40. Stably expressed reference genes were determined by importing Ct values transformed into a linear scale expression quantities, and raw Ct values into the NormFinder [17] and GeNorm algorithms [18], respectively. Data for 29 miRNAs and three commonly used reference genes (SNORD44, SNORD47, and RNU6B), which were expressed in all samples, were included in these analyses. For the NormFinder analysis three biological groups (wild-type controls, wild-type MDS, and spliceosome mutated MDS) were specified. miRNA expression data were analyzed with the qBase+ software, version 3.0 (Biogazelle) using the 2-ΔΔCt method [41], where ΔΔCt = (Cttarget-Ctgeometric mean,ref genes)MDS sample-(Cttarget-Ctgeometric mean,ref genes)mean of control samples. One-sided Fisher’s exact tests were used to evaluate if specific clusters were enriched for a given type of sample of miRNA. Differences in miRNA expression between spliceosome mutated samples and wild-type samples were evaluated with a Mann-Whitney U test. Overall differences in miRNA expression between spliceosome mutated samples and wild-type samples were evaluated with a Wilcoxon signed-rank test. For the cluster analyses miRNAs were excluded from the analyses when more than 1/3 of the samples did not amplify. In total, less than five percent of the data points in the unsupervised cluster analysis were derived from samples which did not amplify. The Z-transformed miRNA expression levels were subjected to complete-linkage hierarchical clustering using the Pearson distance metric. miRNA target predictions were retrieved from TargetScan [42], PicTar [43], miRanda [44], and StarMirDB [45] and integrated to improve the precision of the predictions using the same strategy as in STRING [46]. The top 0.1% of the integrated predictions (~100 targets per miRNA) was used to infer enriched targets and associated KEGG pathway database terms (http://www.genome.jp/kegg/pathway.html) of the differentially expressed miRNA between samples with SF3B1 mutations compared to wild-type samples. Enrichments were evaluated using the Fisher’s exact test and the Benjamini and Hochberg correction for multiple testing.
For the host gene analysis, the RT-qPCR data were analyzed as described above, using the mean Ct value of two reference genes (UBC and JUNB). Goodness-of-fit linear regression was used to evaluate possible relations between expression of the host genes and the miRNAs using Prism 6 (GraphPad software, San Diego, CA, USA), which employs an F test to evaluate if the slope is significantly different from zero.
For the validation analysis, the RT-qPCR data were analyzed as described above, using the mean Ct value of the two replicates for each miRNA. However, samples with a standard deviation above two for any of the reference miRNA genes were excluded from the analysis of differentially expressed miRNAs (this applied to sample 18, 23, 29, and 31). Goodness-of-fit linear regression was used to evaluate possible relations between expression of the host genes and the miRNAs using Prism 6 (GraphPad).
Any differences were considered to be statistically significant when the P-value was < 0.05.
ACKNOWLEDGMENTS AND FUNDING
The authors would like to thank Laurine Bente Schram Harsløf for critical reading of the manuscript and The Novo Nordisk Foundation, Rigshospitalets Research Foundation, The Danish Cancer Society, and The Danish Strategic Research Foundation for supporting grants. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Authors’ contributions
DA, MKN, and LSK carried out the molecular genetic studies. DA, CG, KD, and LSK analyzed the data. JWH and ASH collected the clinical data. LDS, MTS, and MBT collected the clinical material. LSK and KG conceived the study. LSK and DA wrote the manuscript. MTS, KG, and LSK contributed reagents and materials. All authors read and approved the final manuscript.
CONFLICTS OF INTEREST
The authors declare that they have no competing interests.
REFERENCES
1. Cazzola M, Della Porta MG, Malcovati L. The genetic basis of myelodysplasia and its clinical relevance. Blood. 2013; 122:4021–4034.
2. Chesnais V, Kosmider O, Damm F, Itzykson R, Bernard OA, Solary E, Fontenay M. Spliceosome mutations in myelodysplastic syndromes and chronic myelomonocytic leukemia. Oncotarget. 2012; 3:1284–1293. doi: 10.18632/oncotarget.749.
3. Malcovati L, Karimi M, Papaemmanuil E, Ambaglio I, Jadersten M, Jansson M, Elena C, Galli A, Walldin G, Della Porta MG, Raaschou-Jensen K, Travaglino E, Kallenbach K, et al. SF3B1 mutation identifies a distinct subset of myelodysplastic syndrome with ring sideroblasts. Blood. 2015; 126:233–41.
4. Makishima H, Visconte V, Sakaguchi H, Jankowska AM, Abu Kar S, Jerez A, Przychodzen B, Bupathi M, Guinta K, Afable MG, Sekeres MA, Padgett RA, Tiu RV, et al. Mutations in the spliceosome machinery, a novel and ubiquitous pathway in leukemogenesis. Blood. 2012; 119:3203–3210.
5. Walter MJ, Shen D, Ding L, Shao J, Koboldt DC, Chen K, Larson DE, McLellan MD, Dooling D, Abbott R, Fulton R, Magrini V, Schmidt H, et al. Clonal architecture of secondary acute myeloid leukemia. The New England journal of medicine. 2012; 366:1090–1098.
6. Yoshida K, Sanada M, Shiraishi Y, Nowak D, Nagata Y, Yamamoto R, Sato Y, Sato-Otsubo A, Kon A, Nagasaki M, Chalkidis G, Suzuki Y, Shiosaka M, et al. Frequent pathway mutations of splicing machinery in myelodysplasia. Nature. 2011; 478:64–69.
7. Papaemmanuil E, Cazzola M, Boultwood J, Malcovati L, Vyas P, Bowen D, Pellagatti A, Wainscoat JS, Hellstrom-Lindberg E, Gambacorti-Passerini C, Godfrey AL, Rapado I, Cvejic A, et al. Somatic SF3B1 mutation in myelodysplasia with ring sideroblasts. The New England journal of medicine. 2011; 365:1384–1395.
8. Haferlach T, Nagata Y, Grossmann V, Okuno Y, Bacher U, Nagae G, Schnittger S, Sanada M, Kon A, Alpermann T, Yoshida K, Roller A, Nadarajah N, et al. Landscape of genetic lesions in 944 patients with myelodysplastic syndromes. Leukemia. 2014; 28:241–247.
9. Rhyasen GW, Starczynowski DT. Deregulation of microRNAs in myelodysplastic syndrome. Leukemia. 2012; 26:13–22.
10. Soes S, Daugaard IL, Sorensen BS, Carus A, Mattheisen M, Alsner J, Overgaard J, Hager H, Hansen LL, Kristensen LS. Hypomethylation and increased expression of the putative oncogene ELMO3 are associated with lung cancer development and metastases formation. Oncoscience. 2014; 1:367–374. doi: 10.18632/oncoscience.42.
11. Curtis HJ, Sibley CR, Wood MJ. Mirtrons, an emerging class of atypical miRNA. Wiley interdisciplinary reviews RNA. 2012; 3:617–632.
12. Sibley CR, Seow Y, Saayman S, Dijkstra KK, El Andaloussi S, Weinberg MS, Wood MJ. The biogenesis and characterization of mammalian microRNAs of mirtron origin. Nucleic acids research. 2012; 40:438–448.
13. Ladewig E, Okamura K, Flynt AS, Westholm JO, Lai EC. Discovery of hundreds of mirtrons in mouse and human small RNA data. Genome research. 2012; 22:1634–1645.
14. Melamed Z, Levy A, Ashwal-Fluss R, Lev-Maor G, Mekahel K, Atias N, Gilad S, Sharan R, Levy C, Kadener S, Ast G. Alternative splicing regulates biogenesis of miRNAs located across exon-intron junctions. Molecular cell. 2013; 50:869–881.
15. Mattioli C, Pianigiani G, Pagani F. Cross talk between spliceosome and microprocessor defines the fate of pre-mRNA. Wiley interdisciplinary reviews RNA. 2014; 5:647–658.
16. Bustin SA, Benes V, Garson JA, Hellemans J, Huggett J, Kubista M, Mueller R, Nolan T, Pfaffl MW, Shipley GL, Vandesompele J, Wittwer CT. The MIQE guidelines: minimum information for publication of quantitative real-time PCR experiments. Clinical chemistry. 2009; 55:611–622.
17. Andersen CL, Jensen JL, Orntoft TF. Normalization of real-time quantitative reverse transcription-PCR data: a model-based variance estimation approach to identify genes suited for normalization, applied to bladder and colon cancer data sets. Cancer research. 2004; 64:5245–5250.
18. Vandesompele J, De Preter K, Pattyn F, Poppe B, Van Roy N, De Paepe A, Speleman F. Accurate normalization of real-time quantitative RT-PCR data by geometric averaging of multiple internal control genes. Genome biology. 2002; 3:RESEARCH0034.
19. Visconte V, Makishima H, Maciejewski JP, Tiu RV. Emerging roles of the spliceosomal machinery in myelodysplastic syndromes and other hematological disorders. Leukemia. 2012; 26:2447–2454.
20. DeBoever C, Ghia EM, Shepard PJ, Rassenti L, Barrett CL, Jepsen K, Jamieson CH, Carson D, Kipps TJ, Frazer KA. Transcriptome sequencing reveals potential mechanism of cryptic 3′ splice site selection in SF3B1-mutated cancers. PLoS computational biology. 2015; 11:e1004105.
21. Visconte V, Rogers HJ, Singh J, Barnard J, Bupathi M, Traina F, McMahon J, Makishima H, Szpurka H, Jankowska A, Jerez A, Sekeres MA, Saunthararajah Y, et al. SF3B1 haploinsufficiency leads to formation of ring sideroblasts in myelodysplastic syndromes. Blood. 2012; 120:3173–3186.
22. Dolatshad H, Pellagatti A, Fernandez-Mercado M, Yip BH, Malcovati L, Attwood M, Przychodzen B, Sahgal N, Kanapin AA, Lockstone H, Scifo L, Vandenberghe P, Papaemmanuil E, et al. Disruption of SF3B1 results in deregulated expression and splicing of key genes and pathways in myelodysplastic syndrome hematopoietic stem and progenitor cells. Leukemia. 2015; 29:1092–1103.
23. Calin GA, Croce CM. MicroRNA signatures in human cancers. Nature reviews Cancer. 2006; 6:857–866.
24. Soes S, Sorensen BS, Alsner J, Overgaard J, Hager H, Hansen LL, Kristensen LS. Identification of accurate reference genes for RT-qPCR analysis of formalin-fixed paraffin-embedded tissue from primary non-small cell lung cancers and brain and lymph node metastases. Lung Cancer. 2013; 81:180–186.
25. Wahl MC, Will CL, Luhrmann R. The spliceosome: design principles of a dynamic RNP machine. Cell. 2009; 136:701–718.
26. Johnson SM, Grosshans H, Shingara J, Byrom M, Jarvis R, Cheng A, Labourier E, Reinert KL, Brown D, Slack FJ. RAS is regulated by the let-7 microRNA family. Cell. 2005; 120:635–647.
27. Esquela-Kerscher A, Trang P, Wiggins JF, Patrawala L, Cheng A, Ford L, Weidhaas JB, Brown D, Bader AG, Slack FJ. The let-7 microRNA reduces tumor growth in mouse models of lung cancer. Cell Cycle. 2008; 7:759–764.
28. Copley MR, Eaves CJ. Developmental changes in hematopoietic stem cell properties. Experimental & molecular medicine. 2013; 45:e55.
29. Vasilatou D, Papageorgiou SG, Kontsioti F, Kontos CK, Tsiotra P, Mpakou V, Pavlou MA, Economopoulou C, Dimitriadis G, Dervenoulas J, Pappa V. Expression analysis of mir-17-5p, mir-20a and let-7a microRNAs and their target proteins in CD34+ bone marrow cells of patients with myelodysplastic syndromes. Leukemia research. 2013; 37:251–258.
30. Erdogan B, Facey C, Qualtieri J, Tedesco J, Rinker E, Isett RB, Tobias J, Baldwin DA, Thompson JE, Carroll M, Kim AS. Diagnostic microRNAs in myelodysplastic syndrome. Exp Hematol. 2011; 39:915–926 e912.
31. Dostalova Merkerova M, Krejcik Z, Votavova H, Belickova M, Vasikova A, Cermak J. Distinctive microRNA expression profiles in CD34+ bone marrow cells from patients with myelodysplastic syndrome. European journal of human genetics. 2011; 19:313–319.
32. Komeno Y, Huang YJ, Qiu J, Lin L, Xu Y, Zhou Y, Chen L, Monterroza DD, Li H, DeKelver RC, Yan M, Fu XD, Zhang DE. SRSF2 is essential for hematopoiesis and its myelodysplastic syndromes-related mutations dysregulate alternative pre-mRNA splicing. Molecular and cellular biology. 2015; 35:3071–82.
33. Ariki R, Morikawa S, Mabuchi Y, Suzuki S, Nakatake M, Yoshioka K, Hidano S, Nakauchi H, Matsuzaki Y, Nakamura T, Goitsuka R. Homeodomain transcription factor Meis1 is a critical regulator of adult bone marrow hematopoiesis. PloS one. 2014; 9:e87646.
34. Dossing KB, Binderup T, Kaczkowski B, Jacobsen A, Rossing M, Winther O, Federspiel B, Knigge U, Kjaer A, Friis-Hansen L. Down-Regulation of miR-129-5p and the let-7 Family in Neuroendocrine Tumors and Metastases Leads to Up-Regulation of Their Targets Egr1, G3bp1, Hmga2 and Bach1. Genes. 2014; 6:1–21.
35. Fusco A, Fedele M. Roles of HMGA proteins in cancer. Nature reviews Cancer. 2007; 7:899–910.
36. Gerber JM, Gucwa JL, Esopi D, Gurel M, Haffner MC, Vala M, Nelson WG, Jones RJ, Yegnasubramanian S. Genome-wide comparison of the transcriptomes of highly enriched normal and chronic myeloid leukemia stem and progenitor cell populations. Oncotarget. 2013; 4:715–728. doi: 10.18632/oncotarget.990.
37. Koh DI, Choi WI, Jeon BN, Lee CE, Yun CO, Hur MW. A novel POK family transcription factor, ZBTB5, represses transcription of p21CIP1 gene. The Journal of biological chemistry. 2009; 284:19856–19866.
38. Yagasaki F, Jinnai I, Yoshida S, Yokoyama Y, Matsuda A, Kusumoto S, Kobayashi H, Terasaki H, Ohyashiki K, Asou N, Murohashi I, Bessho M, Hirashima K. Fusion of TEL/ETV6 to a novel ACS2 in myelodysplastic syndrome and acute myelogenous leukemia with t(5;12)(q31;p13). Genes, chromosomes & cancer. 1999; 26:192–202.
39. Murati A, Adelaide J, Gelsi-Boyer V, Etienne A, Remy V, Fezoui H, Sainty D, Xerri L, Vey N, Olschwang S, Birnbaum D, Chaffanet M, Mozziconacci MJ. t(5;12)(q23-31;p13) with ETV6-ACSL6 gene fusion in polycythemia vera. Leukemia. 2006; 20:1175–1178.
40. Serguienko A, Grad I, Wennerstrom AB, Meza-Zepeda LA, Thiede B, Stratford EW, Myklebost O, Munthe E. Metabolic reprogramming of metastatic breast cancer and melanoma by let-7a microRNA. Oncotarget. 2015; 6:2451–2465. doi: 10.18632/oncotarget.3235.
41. Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods. 2001; 25:402–408.
42. Garcia DM, Baek D, Shin C, Bell GW, Grimson A, Bartel DP. Weak seed-pairing stability and high target-site abundance decrease the proficiency of lsy-6 and other microRNAs. Nature structural & molecular biology. 2011; 18:1139–1146.
43. Krek A, Grun D, Poy MN, Wolf R, Rosenberg L, Epstein EJ, MacMenamin P, da Piedade I, Gunsalus KC, Stoffel M, Rajewsky N. Combinatorial microRNA target predictions. Nature genetics. 2005; 37:495–500.
44. John B, Enright AJ, Aravin A, Tuschl T, Sander C, Marks DS. Human MicroRNA targets. PLoS biology. 2004; 2:e363.
45. Rennie W, Liu C, Carmack CS, Wolenc A, Kanoria S, Lu J, Long D, Ding Y. STarMir: a web server for prediction of microRNA binding sites. Nucleic acids research. 2014; 42:W114–118.
46. Szklarczyk D, Franceschini A, Wyder S, Forslund K, Heller D, Huerta-Cepas J, Simonovic M, Roth A, Santos A, Tsafou KP, Kuhn M, Bork P, Jensen LJ, et al. STRING v10: protein-protein interaction networks, integrated over the tree of life. Nucleic acids research. 2015; 43:D447–452.