
Introduction
First described by James Ewing in 1921 as “diffuse endothelioma of bone,” Ewing sarcoma is the third most common malignant bone neoplasia diagnosed in children and adolescents [1, 2]. While the histogenesis of Ewing sarcoma remains enigmatic, 85% of cases are defined by the t(11;22)(q24;q12) chromosomal translocation, resulting in a pathognomonic chimeric fusion gene, EWSR1-FLI1, which encodes the EWS-FLI protein [3]. Treatment protocols continue to rely upon conventional multidisciplinary approaches coupling intensive chemotherapy with surgery and/or radiotherapy. The implementation of chemotherapy has achieved event-free survival rates nearing 75% for patients with localized disease, however those presenting with overt metastasis (seen in 20-30% of patients at diagnosis) or recurrent disease have poor outcomes, with under 30% disease-free survival [4, 5]. Furthermore, successful chemotherapeutic regimes are associated with significant cumulative and late toxicities [6]. Given the failure of systemic chemotherapy to improve durable remission rates for patients with metastatic disease, translation of novel therapeutic strategies to increase overall survival rates remains imperative.
Several challenges persist in the development of targeted therapies for Ewing sarcoma. Although advances in next generation sequencing have augmented our understanding of disease mechanisms driven by EWS-FLI, the cell-of-origin is unknown. Large-scale genomic sequencing efforts have demonstrated Ewing sarcoma possesses one of the lowest mutation rates amongst all cancers (0.15 mutations/Mb) [7, 8], yielding a paucity of pharmacologically actionable mutations. Indeed, Ewing sarcoma tumor samples showed recurrent, though low frequency, mutations only in the cohesin complex subunit STAG2 (21.5%), the tumor suppressor TP53 (6.2%) and homozygous deletion of the cyclin-dependent kinase inhibitor CDKN2A (13.8%) [7]. It appears possible Ewing sarcoma cells require large-scale epigenetic alteration to maintain malignant programming which disrupts normal developmental processes [9-15]. Notably, EWS-FLI blocks mesenchymal differentiation and promotes neuronal programs, which is in turn, dampened by EWSR1 and REST [13, 16]. Morphological studies suggest Ewing sarcoma cells strike a delicate balance between proliferative growth and metastatic capacity along the mesenchymal differentiation axis [17]. The transcription factor ZEB2 is critical to block expression of genes characteristic of an epithelial lineage [18]. Taken together, the oncogenic interplay of EWS-FLI with varied developmental pathways is marked by complexity. If Ewing sarcoma is to be placed within a Waddington landscape, perhaps it is best categorized as lost in the wilderness.
The importance of epigenomic misregulation in cancer and development of pharmacological tools to probe epigenetic mechanisms have advanced significantly in the past decade. However, the field faces technical hurdles in both collecting data and approaching the complexity in gathered data. Ewing sarcoma and other mutationally quiet pediatric malignancies have emerged as interesting model systems to further probe epigenetic aberrations conspiring in oncogenesis [7, 8, 19-21]. EWS-FLI expression affects the transcriptome, epigenome, and proteome to reprogram cells into a malignant developmental limbo [7, 8, 22-37]. Conversely, several studies suggest cellular context, both epigenetic and otherwise, influences the effects of EWS-FLI, as enforced expression in animal models leads to phenotypically variant tumors [38-40]. Moreover, expression of EWS-FLI in human pediatric mesenchymal stem cells failed to produce tumors in xenograft models, despite recapitulation of disease-specific transcriptomic and epigenomic phenotypes [31]. Rational design and implementation of improved therapeutic regimens requires more comprehensive understanding of disease mechanisms influenced by EWS-FLI and other FET/ETS fusions. Toward this end, recent work has described the epigenomic landscape of EWS-FLI in patient-derived cell lines and primary tumor samples [23, 29, 30, 35]. Additional lines of inquiry have further defined an important role for EWS-FLI in altering transcript splice selection [32, 33]. Notably, disruption of either epigenetic mechanisms or alternative splicing mechanisms delay tumor growth in xenograft models [22, 33].
Methylation is an important and subtle chemical modification which regulates chromatin status and is observed on both DNA and histones. Indeed, the significance of DNA methylation in both cancer initiation and progression has been appreciated for a number of years, resulting in the approval of two agents for the treatment of patients with myelodysplastic syndrome, azacitidine/Vidaza [41] (nucleoside analogue) and decitabine/Dacogen [42], (irreversible inhibitor of DNA methyltransferase enzymes DNMT1 and DNMT3). Histone methylation, a mechanism to modify chromatin structure, dynamically regulates cellular processes including transcription and genomic stability. Until a decade ago, histone methylation was considered an immutable modification, defining programs in concert with DNA methylation and other histone post-translational modification. However, the discovery of the first histone demethylase, lysine-specific demethylase 1 (LSD1) in 2004 [43], challenged this notion and proved lysine methylation is dynamically regulated. LSD1 (also known as KDM1A, AOF2 and BHC110), is a flavin adenine dinucleotide (FAD) dependent amine oxidase with important epigenetic eraser function, specifically catalyzing oxidative demethylation of mono- and dimethyl-lysine at histone H3 lysines 4 and 9 (H3K4me1/2 and H3K9me1/2) [43], generating formaldehyde and hydrogen peroxide. In addition, LSD1 is reported to demethylate modified lysines on a myriad of non-histone proteins such as DNMT1 (residue K1096) [44], E2F1 (K185 residue) [45], MYPT1 (residue K442) [46], p53 (K370 residue) [47] and STAT3 (K140 residue) [48]. Importantly, LSD1 activity is highly context dependent. Several protein interaction partners are reported, including REST corepressor (CoREST) and MTA2, the nucleosome remodeling and deacetylase (NuRD) complex, and nuclear hormone receptors [49, 50]. As such, the epigenetic effects of LSD1 are implicated in diverse biologic processes pertinent to adipogenesis [51], chromosome segregation [52], cell proliferation [53], embryonic development [54], epithelial-mesenchymal transition (EMT) [55], hematopoiesis [56, 57], and regulation of stem cell pluripotency [58].
Owing to its high expression in several solid malignancies including breast [59-61], colorectal [62, 63], lung [62, 64], ovarian cancer [65], undifferentiated neuroblastoma [66], prostate carcinoma [67, 68], urothelial carcinoma [62], and sarcomas (Ewing, chondrosarcoma, osteosarcoma, rhabdomyosarcoma, and synovial) [69, 70], specific small molecule inhibitors of LSD1 have been aggressively pursued as potential therapeutics. Recently, our laboratory demonstrated EWS-FLI mediated transcriptional repression is facilitated through direct binding of a NuRD-LSD1 complex [71]. Furthermore, treatment of Ewing sarcoma cell lines with the potent and reversible LSD1 inhibitor HCI-2509, comprehensively reversed the transcriptional profiles driven by both EWS-FLI and EWS-ERG and significantly delayed tumorigenesis in vivo [22]. As such, this review will cover the rationale for LSD1 inhibition as a therapeutic strategy for Ewing sarcoma and the recent advances made by the scientific and pharmaceutical community to deliver potent LSD1 inhibitors.
Targeting histone demethylation in cancer
A variety of histone modifications, both written and erased by specific enzymes, are recognized by chromatin regulatory complexes to modulate target gene expression [72]. Methylation of histone H3 at lysine 4 (H3K4) and lysine 9 (H3K9) is linked to transcriptional activity at nearby genes [73]. In actively transcribing genes, H3K4me3 is strongly enriched at the transcription start site, with H3K4me2 and H3K4me1 peaks more broadly spread [74]. The methylation state of H3K4 is established through dynamic and coordinated activities of histone lysine methyltransferases, such as Set1/COMPASS family proteins, and histone lysine demethylases, such as FAD-dependent LSD1 and the JmjC domain containing JARID1 family [43, 75-77]. Notably, histone methylation is appreciated to function in precise cell- and tissue-specific manners [78, 79]. This may provide superior opportunities for therapeutic disruption in cancer, due to improved off-target profiles as compared to those observed with the use of histone deacetylase inhibitors [78-82]. LSD1 displays highly context dependent function, making it an attractive target to modulate epigenetic misregulation in cancer.
LSD1 demethylates H3K4 and H3K9 as well as non-histone protein substrates
The core structure of LSD1 comprises three domains, a small alpha-helical Swi3, RSC8, and MOIRA (SWIRM) domain and amine oxidase-like (AOL) domain form a closely packed structure and a protruding tower domain unique to LSD1 [49, 83]. The crystal structure of LSD1 shows similarity to other FAD-dependent oxidases and reveals a large substrate binding cavity to accommodate several residues of the N-terminal histone H3, positioning K4 for demethylation. This binding conformation is critical for the demethylation of mono- and di-methyl modifications, though the catalytic mechanism precludes activity against trimethylated lysine. In contrast to other SWIRM domain-containing proteins such as ADA2α and Swi3, the SWIRM domain of LSD1 lacks conserved DNA-binding residues, thus requiring other interaction partners to bind and demethylate native nucleosomes [83-85]. The tower domain directly protrudes from the catalytic center and binds LSD1-interacting proteins such as CoREST, MTA2, and HDAC1/2.
LSD1 was observed to demethylate the H3K9me1/2 marks in association with the androgen receptor, promoting target gene expression [50]. However, the mechanism by which LSD1 achieves this dual substrate specificity for both H3K4me1/2 and H3K9me1/2 marks was unclear until recently [86]. An alternatively spliced isoform of LSD1, LSD1+8a, can specifically demethylate H3K9me1/2, but not H3K4me1/2. This isoform was previously reported to be predominantly expressed in neuronal cell types and is involved in neuronal maturation [87]. Additional binding of the protein supervilin improved LSD1 catalytic activity toward H3K9me1/2, suggesting some interactions may favor specificity for one substrate over another. The expression of different LSD1 isoforms remains unexplored in Ewing sarcoma, but constitutes an important consideration in both cultured cells and patient-derived samples. Additionally, LSD2/KDM1B is a homolog of LSD1 (31% sequence similarity), lacking the tower domain while possessing additional zinc finger domains. LSD2 specifically acts within the gene bodies of target genes, displaying distinct localization of function as compared to LSD1, which acts primarily on promoter and enhancer regions [88, 89]. Expression of LSD2 in Ewing sarcoma likewise remains unexplored.
Apart from demethylating histone lysine residues, LSD1 has multiple non-histone substrates with diverse cellular functions, including p53, E2F1 and DNMT1. LSD1-mediated demethylation of p53 at K370me1/2 represses apoptotic activity, revealing a dynamic mode of p53 regulation [47]. Stabilization of E2F1 in p53-deficient tumor cells through demethylation at K185 inhibits DNA-damage induced cell death [45]. In an interesting link between the chromatin and DNA methylation machinery, LSD1 is critical for the maintenance of global DNA methylation patterns through regulation of DNMT1 stability during gastrulation in mouse embryos via demethylation at K1096 [90].
Association of LSD1 with multiprotein complexes regulates functional responses
The activity of LSD1 is predominantly in concert with specific chromatin regulatory complexes found in distinct cell types and several examples illustrate the functional specificity conferred through recruitment by specific factors. LSD1 commonly associates with CoREST and HDAC1/2 containing complexes for demethylation of nucleosomal substrates [91, 92]. In breast cancer cells, LSD1 interacts with MTA2 to forms an essential component of the NuRD complex containing MTA1/2/3 and HDAC1/2 to regulate key signaling mechanisms, including TGFβ signaling, involved in cell proliferation, metastasis and EMT [93]. During embryonic stem cell differentiation, LSD1 is required for the decommissioning and silencing of the pluripotency enhancers for normal differentiation, in cooperation with NuRD components [94]. Additionally, LSD1 is shown to play a crucial role in EMT via formation of a ternary Snail1-CoREST-LSD1 complex, in which LSD1 interacts with the SNAG domain of Snail1 to regulate target gene expression involved in the suppression of cell migration and invasion [95]. Other transcription factors which possess the SNAG domain, homologous to the N-terminus of histone H3, recruit LSD1 as well. During hematopoietic stem cell differentiation, the LSD1-CoREST complex is recruited by the SNAG domain of Gfi-1 to mediate repression of lineage-specific target genes [96]. The wide range of LSD1 interaction partners remains beyond the scope of this review. However, LSD1 is critical for execution of various differentiation programs including adipogenesis, skeletal muscle differentiation and pituitary organogenesis, highlighting the importance of histone demethylation in regulating cell-type specific gene expression patterns [51, 97, 98].
Although LSD1 regulates the expression of several downstream targets, the upstream regulators of LSD1 have not been extensively studied. miR-137 was shown to regulate the protein levels of LSD1 in neural cells through targeting the 3'UTR of LSD1, resulting in negative regulation of neural cell proliferation and increased neural differentiation [99]. miR-137 was also shown to be a tumor suppressor in neuroblastoma by downregulation of LSD1 [100]. Posttranslational control of LSD1 expression is dynamically achieved by ubiquitination. Stabilized levels of LSD1 by deubiquitinase USP28 conferred stem cell like properties to breast cancer cells [101]. Another study showed that an E3 ubiquitin ligase, Jade-2 (jade family PHD finger 2) can negatively regulate LSD1 in developing mouse cortical neurons and zebrafish embryos [102]. It remains to be explored whether these regulatory mechanisms are active in different cancers, including Ewing sarcoma.
High LSD1 expression promotes cell proliferation and metastasis in cancer
LSD1 is overexpressed in both solid and non-solid tumors such as breast, lung, colon, prostate, gastric cancers and acute myeloid leukemia (AML), playing significant roles in cell proliferation, cell migration and metastasis [59, 62, 67, 69, 103-106]. Functional downregulation of LSD1 expression or pharmacological inhibition significantly reduces tumor cell proliferation and metastatic progression in several malignancies in vitro. Although overexpression of LSD1 (mRNA and/or protein) is reported across numerous malignancies, few studies have evaluated whether LSD1 expression correlates with either cancer progression or overall survival. For those studies that have investigated the role of LSD1, limited patient cohort size and lack of clinical follow-up data has generally impeded the ability of studies to achieve statistical significance. In neuroblastoma [66], prostate [68] and ovarian [65] cancer, low LSD1 mRNA levels were predictive of event free survival. In breast cancer, LSD1 expression levels increased considerably during tumor progression from pre-invasive to invasive ductal breast carcinoma [60]. Conversely, in urothelial carcinoma, LSD1 expression levels were significantly high even in early grade (G1) tumors [62], implying that LSD1 is involved in tumor initiation for this malignancy. Wu et al., recently performed a meta-analysis to assess the association between LSD1 expression and overall survival in 1,149 cancer patients (hepatocellular carcinoma, esophageal, colon, breast, melanoma and tongue cancer). Analysis from nine studies, particularly enriched for Asian cohorts suggested that LSD1 overexpression was associated with poor overall survival, particularly for esophageal cancer patients (P = 0.000) [107].
Although expression of LSD1 has been evaluated in a large cohort of sarcomas, there is currently no evidence supporting the role of LSD1 in sarcoma tumor progression or disease-free survival. Immunohistochemical staining of a cohort of 468 sarcomas by Schildhaus et al., reported pronounced LSD1 expression specifically in highly malignant tumor groups including synovial sarcomas, rhabdomyosarcomas, desmoplastic small round cell tumors and malignant peripheral nerve sheath tumors (MPNST) [70]. A subsequent report by Bennani-Baiti confirmed these findings and extended the high expressing LSD1 sarcoma groups to Ewing sarcoma, chondrosarcoma and osteosarcoma [69], subtypes which were not investigated by Schildhaus et al. Although LSD1 expression in Ewing sarcoma was comparable to that of rhabdomyosarcoma, the most significant LSD1-expressing sarcoma subtype, no studies have hitherto to date examined the prognostic value of LSD1 expression in Ewing sarcoma.
Translation of LSD1 inhibitors
The well-defined active site cavity of LSD1 has enabled the development of numerous high-affinity and selective small-molecule inhibitors, reviewed extensively by Zheng et al., [108] and Mould et al., [109]. Unfortunately, numerous candidates failed to satisfy the stringent physicochemical and toxicological requirements for clinical development or were subsequently proven to possess poor LSD1 specificity profiles. Several peptide based LSD1 inhibitors (linear and cyclic) have also been generated [110-112]; however the clinical development pathway for peptide-based therapeutics remains uncertain.
As single-agent therapy increases the likelihood of the emergence of resistant cancer cell clones, the ability of LSD1 inhibitors to synergize with current treatment regimens will be imperative for their implementation into standard treatment protocols. The strongest evidence for potential combinatorial agents is for HDAC inhibitors. Co-treatment of glioblastoma cells with tranylcypromine and vorinostat led to a marked (6-fold) increase in caspase 3 activity [113]. In addition, treatment of primary AML blasts with the pan-HDAC inhibitor panobinostat significantly enhanced HCI-2509 induced apoptosis in vitro and significantly improved the median survival of mice, compared to treatment with HCI-2509 or panobinostat alone [114].
Preclinical evaluation of LSD1 inhibitors in sarcoma
Previously prescribed as an anxiolytic and antidepressant to patients with anxiety or mood disorders [115], tranylcypromine (TCP) was the first monoamine oxidase (MAO) inhibitor identified [116]. TCP exerts its inhibitory activity by covalently binding to FAD, forming an tetracyclic adduct in the amine oxidase-like (AOL) domain binding pocket [116, 117]. In addition to MAOs, TCP is also able to inhibit the demethylase activity of LSD1 and LSD2, with a Ki of 242μM and 180μM, respectively [88, 118]. Treatment of neuroblastoma [66] and breast cancer [59] cell lines in vitro and in vivo with early derivatives of TCP resulted in significant growth inhibition. However, these inhibitory effects were only achieved at supraphysiologic concentrations of TCP, 20-30 fold higher than the enzymatic IC50 for LSD1. In the context of sarcomas, Bennani-Baiti et al., reported TCP inhibits the proliferation of Ewing, osteosarcoma, rhabdomyosarcoma and chrondrosarcoma cell lines, albeit in millimolar ranges which cannot be reasonably achieved in clinical settings [69]. Schildhaus, et al. also demonstrated growth inhibition of synovial sarcoma cell lines following treatment with TCP and clorgyline [70]. In consideration of TCP’s adverse toxicity and poor potency/selectivity (IC50: 20.7µM, 0.2µM and 0.95µM; LSD1, MAO A and MAO B respectively), several analogues of tranylcypromine were synthesized with enhanced potency and target selectivity. This was achieved through the addition of bulky, branched side chains and the modification of the phenyl group using crystal structures of the LSD1 substrate cavity for rational design [119, 120]. Pargyline was initially cited as a suicide inactivator of monoamine oxidases, blocking the demthylation of H3K9 by LSD1 during androgen-induced transcription [50]. However, two subsequent studies demonstrated that pargyline failed to inhibit LSD1 activity toward demethylation of H3K4 [118, 121].
Recently, our laboratory investigated the therapeutic potential of a novel reversible and non-competitive LSD1 inhibitor (HCI-2509, Salarius Pharmaceuticals) for the treatment of Ewing sarcoma. HCI-2509, an N’-(1-phenylethylidene)-benzohydrazide small molecule (LSD1 IC50 13nM; Figure 1), was originally identified through structure-based virtual screening [122]. Treatment of Ewing sarcoma cell lines with HCI-2509 treatment disrupted the global oncogenic activity of EWS-ETS fusions and induced apoptosis at physiologically relevant concentrations near 1µM, as will be discussed in detail below [22, 71]. In addition, cells expressing EWS-FLI were approximately 10-fold more sensitive to HCI-2509 treatment compared to cells with shRNA-mediated depletion of EWS-FLI, underscoring the specificity of LSD1 inhibition for Ewing sarcoma cells. Beyond Ewing sarcoma, HCI-2509 has also demonstrated single agent in vitro and in vivo efficacy in models of breast cancer [1 23], AML [114], poorly differentiated endometrial carcinoma [124] and castration resistant prostate cancer [125]. Clinical formulations of HCI-2509 analogues are currently being assessed and are expected to enter Phase I clinical testing within the near future.
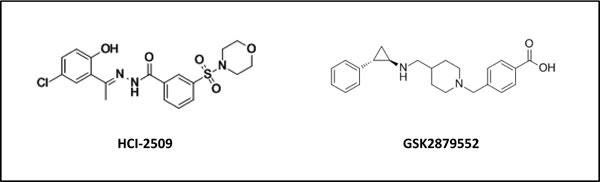
Figure 1: Chemical structure of HCI-2509 and GSK-2879552, reversible and irreversible inhibitors of LSD1 respectively.
Clinical evaluation of LSD1 inhibitors to date
Although the development of specific LSD1 inhibitors is still in its infancy, three agents (tranylcypromine, GSK2879552 and ORY-100) are currently undergoing clinical evaluation, primarily in AML patients (Table 1). To date, the most promising pre-clinical data for LSD1 inhibition has come from mouse models of human AML (MLL-translocated). Foundational studies demonstrate the requirement of LSD1 for clonogenic and leukemia stem cell potential of MLL-AF9 AML cells and LSD1 action at genomic loci bound by MLL-AF9 to sustain expression of AML-associated oncogenic programs which prevent apoptosis and differentiation [126].
Table 1: Current LSD1 inhibitor trials
Compound |
Study Title |
Phase/ Identifier |
Sponsor |
Stage |
|
GSK2879552 |
Dose Escalation Study for GSK2879552 in Subjects With Relapsed/Refractory Acute Myeloid Leukemia |
I NCT02177812 |
GlaxoSmithKline |
Recruiting |
|
Investigation of GSK2879552 in Subjects With Relapsed/Refractory Small Cell Lung Carcinoma |
I NCT02034123 |
GlaxoSmithKline |
Recruiting |
|
Tranylcypromine |
Dose Escalation Study of Tranylcypromine (TCP) in Combination With ATRA (Tretinoin) for Adult Patients With Acute Myelogenous Leukemia and Myelodysplastic Syndromes |
I NCT02273102 |
University Of Miami |
Not yet recruiting |
|
Pilot Trial of ATRA (Tretinoin) and TCP (Tranylcypromine) in Patients With Relapsed or Refractory Acute Myeloid Leukemia when no Intensive Treatment is Possible |
I/II NCT02261779 |
Martin-Luther-Universität Halle-Wittenberg |
Recruiting |
|
ORY-100 |
Study of Human Pharmacokinetics and Safety of ORY-100 in relapsed or refractory acute leukemia |
I/IIa 2013-002447-29 |
Oryzon Genomics |
Ongoing |
ORY-1001 (structure undisclosed), a trans-2-phenylcyclopropylamine-based LSD1 inactivator (IC50 < 20nM) from Oryzon Genomics is reported to be 1,000 times more potent than TCP and highly selective over related enzymes, including LSD2. At sub-nanomolar concentrations (EC50< 1nM) ORY-1001 was shown to reduce leukaemic stem cell potential, colony formation and induce differentiation of AML cell lines [127]. GSK2879552 (GlaxoSmithKline) an N-substituted tranylcypromine derivative (Figure 1), is the first irreversible LSD1 inhibitor to be evaluated clinically in a solid tumor context (Small Cell Lung Carcinoma, SCLC). The recent screening of 165 cancer cell lines of varying histology by Mohammad et al., revealed that the anti-proliferative activity of GSK2879552 was largely restricted to SCLC and AML cell lines (EC50 2-240nM), with genomic analyses revealing elevated MYC expression or amplification was correlated with resistance to GSK2879552, whereas global DNA hypomethylation was correlated with sensitivity [64]. In addition, treatment of AML cell lines promoted the expression of cell surface markers (CD11b and CD86) associated with a differentiated immunophenotype, and induced potent growth inhibition in patient derived bone marrow samples (EC50 205nM) [128]. The results from these trials are eagerly awaited and will help further validate whether targeting the roles of LSD1 in cancer represents a tractable therapeutic option for patients.
LSD1 mutational status in sarcoma
Accelerated cancer genome sequencing and high-throughput functional screen campaigns have significantly expanded our understanding of the abnormal biology and complex genetics of cancer cells. The resulting efforts have governed the discovery and development of targeted small molecules and laid the foundation for personalized medicine. Although molecularly targeted agents aimed specifically at drivers of pathogenesis have had some success, one mechanism of patient non-response can be attributed to pre-existing genetic mutation of the target gene itself. Fortunately, innate mutation of LSD1 is seldom observed. A recent search of cBioPortal revealed that mutation of LSD1 across numerous cancer subtypes is rare, with the highest mutation rates documented in urothelial bladder carcinoma (4/130 patients, 3.1%) [129], and medulloblastoma (1/37 patients, 2.7%) [130]. In congruence with this observation, mutation of LSD1 was not detected in either pediatric or adult Ewing sarcoma patients across five sequencing studies (n = 338 patients) (Table 2).
Drugging the undruggable: Understanding the mechanisms of aberrant EWS-ETS transcription factors
The function of EWS-FLI, and other FET/ETS fusions characterizing Ewing sarcoma, is multifaceted and remains incompletely understood. What is clear is the related fusions sit atop a hierarchy of direct and indirect events which alter the composition of expressed genes, through disruption of both transcriptional and post-transcriptional processes in the cell. The downstream effects culminate in establishment and maintenance of an oncogenic phenotype as shown in Figure 2. Pharmacologically targeting protein-protein interactions and transcription factors remains a major challenge, but in Ewing sarcoma several persistent efforts are beginning to bear fruit. Here we discuss the mechanisms utilized by EWS-FLI that present tractable strategies for pharmacological blockade, specifically inhibition of chromatin modifications with a focus on LSD1.
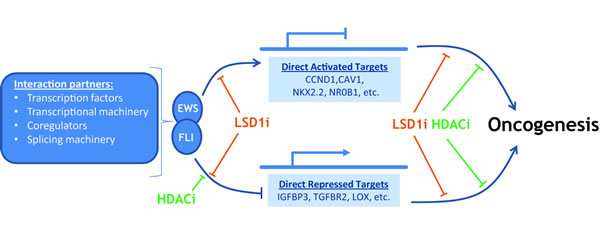
Figure 2: EWS-FLI interacts with multiple partners to cause gene specific activation and repression on the road to oncogenesis. LSD1 inhibition (LSDi) negatively impacts direct transcriptional targets of EWS-FLI, in a manner distinct from HDAC inhibition (HDACi). Moreover, there is data to suggest additional roles for both LSD1 and HDACs in the downstream effects leading to oncogenesis, and these remain an area of active study.
EWS-FLI functions as an aberrant transcriptional activator
Molecular mechanisms animating oncogenesis in Ewing sarcoma arise from the fusion of the N-terminal transcriptional modulation domain (NTD) of EWS with the C-terminal ETS-family DNA binding domain, which binds DNA elements possessing a core 5’-GGA(A/T)-3’ consensus motif [3]. The transcriptional activation induced by enforced EWS-FLI expression in NIH3T3 fibroblasts is sufficient for transformation [131]. Indeed, deregulation of the FLI transcription factor through N-terminal fusion with other strong activation domains, like VP16, shows transforming activity, highlighting an important role for transcriptional activation in Ewing sarcoma tumorigenesis [132]. Unlike full-length EWS, the NTD is shown to interact with the RNA polymerase II (PolII) complex subunit hsRPB7, nucleate the PolII complex, and recruit corroborating transcription factors, such as E2F3 and AP-1, and chromatin modifiers, like CBP/p300, to activate transcription [23, 30, 133-138].
Beyond the transcriptional deregulation resulting from imposition of the NTD in place of the regulatory N-terminal domain of FLI1, EWS-FLI shows emergent properties as an aberrant transcription factor at GGAA-microsatellites throughout the genome [26, 139, 140]. While several ETS-family members can bind GGAA repetitive elements in vitro, only EWS-FLI is able to both bind and activate transcription at nearby genes [139]. Recent deep sequencing studies interrogating the chromatin state at these GGAA microsatellites describe EWS-FLI recruitment of CBP/p300 to acetylate H3K27 and promote formation of enhancer elements at these loci [30, 35]. Several EWS-FLI activated targets indispensable for transformation, including NKX2.2, CAV1, GSTM4, and NR0B1 are regulated through EWS-FLI activity at nearby microsatellites, though none are currently candidates for therapeutic development [26, 30, 140-142].
The transcriptional hierarchy of EWS-FLI includes gene repression
Transcriptional profiling of patient-derived Ewing sarcoma cell lines using RNAi-mediated knockdown of EWS-FLI revealed a transcriptional signature which surprisingly showed more genes repressed by the fusion than activated. Several activated targets of EWS-FLI are, in fact, transcriptional repressors, including BCL11B, NKX2.2, NR0B1, and the long noncoding RNA EWSAT1 [27, 143-146]. Globally, the gene targets affected by these transcriptional repressors comprise only part of the EWS-FLI downregulated signature. Further investigation showed that EWS-FLI directly represses a subset of targets whose downregulation is required for tumorigenesis, including IGFBP3, TGFBR2, and LOX [71, 147, 148]. Moreover, EWS-FLI directly interacts with CHD4, MTA2, HDAC2, and HDAC3 in a NuRD-like complex at genes to enact transcriptional repression [71].
Targeting mechanisms of gene regulation
Both indirect repression, for example by NKX2.2, and direct repression depend upon the activity of histone deacetylases [71, 145, 146]. Thus, one potential therapeutic route blocks downstream epigenetic regulators which execute the repressive transcriptional program of EWS-FLI. Treatment with the HDAC inhibitor vorinostat derepresses gene targets of NKX2.2, BCL11B, and EWS-FLI and impairs cell viability and transformation in soft agar assays [71, 145, 146]. We further investigated whether targeting LSD1 might have similar effects in Ewing sarcoma models, as it commonly co-localizes with NuRD in the nucleus. Our initial investigation showed LSD1 inhibition with the small molecule HCI-2509 derepressed EWS-FLI-repressed targets and decreased cell viability in a manner comparable to HDAC inhibition [71]. More thorough transcriptional profiling in A673 and TTC-466 Ewing sarcoma cells intriguingly showed LSD1 inhibition flips both sides of the transcriptional profile for both EWS-FLI and EWS-ERG, respectively, upregulating repressed targets and vice versa [22] (Figure 2). This contrasts the HDAC inhibitor vorinostat, which diminished only EWS-FLI-driven gene repression and hints at roles for LSD1 in Ewing sarcoma biology beyond its documented corepressor activity, though the mechanisms of this activity remain unknown. For both HDAC and LSD1 inhibition, the effects on cell viability and transcription were mitigated in the context of RNAi-mediated EWS-FLI depletion, suggesting a disease-specific function for these enzymes [22, 71,]. Importantly, LSD1 inhibition with HCI-2509 showed single agent efficacy across multiple xenograft models for Ewing sarcoma [22].
Table 2: Frequency of LSD1 mutation in Ewing sarcoma
Study |
Sequencing platform |
Frequency of LSD1 mutation in Ewing sarcoma patient cohort |
||
Tirode, 2014 21 |
WGS |
0/112 (0%) |
||
Crompton, 2014 8 |
WES |
0/92 (0%) |
||
Brohl, 2014 7 |
WGS |
0/65 (0%) |
||
Agelopoulos, 2015 154 |
WES |
0/50 (0%) |
||
Huether, 2014 155 |
WGS |
0/19 (0%) |
||
WES: Whole Exome Sequencing, WGS: Whole Genome Sequencing
While several groups have proposed a model for EWS-FLI activity whereby EWS-FLI binds microsatellite DNA and induces enhancer like features to promote gene activation at critical targets, the data to fully support any particular mechanistic model remain lacking [30, 35]. This complicates interpretation of the downregulation of activated targets by LSD1 inhibition considerably. However, LSD1 plays an important role in enhancer biology in embryonic stem cells (ESCs) and is required to repress the enhancers of pluripotency regulators during differentiation [94, 149, 150]. LSD1 is also reported to localize to enhancers of activated genes in ESCs and whether LSD1 is important for maintained gene activation or fine tuning of transcript levels in these contexts remains unclear [94]. Recent studies in more differentiated cell systems have shown cell-specific factors can toggle LSD1 substrate specificity between H3K4 and H3K9 at regulatory regions of target genes to promote either activating or repressive activity [151, 152]. In our hands, HCI-2509 showed more pronounced effects on global H3K9 methylation status in Ewing sarcoma, though the genomic implications of this result as well as a narrowed focus on H3K4 at regions of interest remain the work of continued studies.
Conclusions and Future Challenges
Exploiting the genetic addictions, vulnerabilities and esoteric dependencies of cancer cells has fueled a paradigm shift in conceiving new therapeutic strategies. Molecularly-targeted treatments informed by patient-specific characteristics are ascendant over non-specific cytotoxic therapies, where possible. Precision medicine aims to better address individuals’ diseases while simultaneously reducing deleterious side effects. The quiet mutational landscape of Ewing sarcoma, coupled with documented overexpression of LSD1 and epigenetic misregulation, highlights the clinical potential of epigenetic inhibitors for the treatment of this aggressive malignancy. Indeed, small molecule blockade of DNA methyltransferase and histone deacetylases has proven epigenetic inhibitors are useful drug candidates. The low overall incidence of Ewing sarcoma 2.93 cases/1,000,000 [153], presents several challenges for the translation of novel agents into the clinic, and underscores the importance of global multi-center trial efforts to investigate the therapeutic potential of LSD1 inhibitors.
Although the use of LSD1 inhibitors for Ewing sarcoma shows promise, there are no reliable molecular biomarkers to predict either clinical activity or resistance to LSD1 therapy. Whether resistance will arise due to genetic mutation of LSD1 itself, activation of adaptive feedback loops or engagement of compensatory survival mechanisms outside the biological pathways targeted by LSD1 inhibitors remains unknown. It is widely accepted that single agent chemotherapy cannot constitute sole therapeutic intervention for Ewing sarcoma patients. As such, future work entails the evaluation of synergetic combinations which potentiate current Ewing sarcoma chemotherapeutic cassettes to provide a clear strategy for further LSD1 inhibitor clinical trials which mitigate drug resistance and achieve maximal effect. Although the initial signs are promising, results from ongoing clinical trials are eagerly anticipated. The rational coupling of mechanistic insight with translational science will ultimately determine whether these new epigenetic therapies comprise a meaningful addition to Ewing sarcoma treatment protocols.
Acknowledgments and Grant Support
Stephen L. Lessnick was supported by the NIH/NCI grants R01 CA140394 and R01 CA183776. Kathleen I. Pishas acknowledges financial support from the University of Adelaide Florey Medical Research Foundation Clinical Cancer Research Fellowship.
Conflicts of Interest
Stephen L. Lessnick is a consultant/advisory board member for Salarius Pharmaceuticals. No potential conflicts of interest were disclosed by the other authors.
References
1. Burningham Z, Hashibe M, Spector L and Schiffman JD. The epidemiology of sarcoma. Clin Sarcoma Res. 2012; 2: 14.
2. Ewing J. Classics in oncology. Diffuse endothelioma of bone. James Ewing. Proceedings of the New York Pathological Society, 1921. CA Cancer J Clin. 1972; 22: 95-8.
3. Delattre O, Zucman J, Plougastel B, Desmaze C, Melot T, Peter M, Kovar H, Joubert I, de Jong P, Rouleau G, et al. Gene fusion with an ETS DNA-binding domain caused by chromosome translocation in human tumours. Nature. 1992; 359: 162-5.
4. Cotterill SJ, Ahrens S, Paulussen M, Jurgens HF, Voute PA, Gadner H and Craft AW. Prognostic factors in Ewing’s tumor of bone: analysis of 975 patients from the European Intergroup Cooperative Ewing’s Sarcoma Study Group. J Clin Oncol. 2000; 18: 3108-14.
5. Rodriguez-Galindo C, Navid F, Liu T, Billups CA, Rao BN and Krasin MJ. Prognostic factors for local and distant control in Ewing sarcoma family of tumors. Ann Oncol. 2008; 19: 814-20.
6. Longhi A, Ferrari S, Tamburini A, Luksch R, Fagioli F, Bacci G and Ferrari C. Late effects of chemotherapy and radiotherapy in osteosarcoma and Ewing sarcoma patients: the Italian Sarcoma Group Experience (1983-2006). Cancer. 2012; 118: 5050-9.
7. Brohl AS, Solomon DA, Chang W, Wang J, Song Y, Sindiri S, Patidar R, Hurd L, Chen L, Shern JF, Liao H, Wen X, Gerard J, et al. The genomic landscape of the Ewing Sarcoma family of tumors reveals recurrent STAG2 mutation. PLoS Genet. 2014; 10: e1004475.
8. Crompton BD, Stewart C, Taylor-Weiner A, Alexe G, Kurek KC, Calicchio ML, Kiezun A, Carter SL, Shukla SA, Mehta SS, Thorner AR, de Torres C, Lavarino C, et al. The genomic landscape of pediatric Ewing sarcoma. Cancer Discov. 2014; 4: 1326-41.
9. Cavazzana AO, Miser JS, Jefferson J and Triche TJ. Experimental evidence for a neural origin of Ewing’s sarcoma of bone. Am J Pathol. 1987; 127: 507-18.
10. Chaturvedi A, Hoffman LM, Welm AL, Lessnick SL and Beckerle MC. The EWS/FLI Oncogene Drives Changes in Cellular Morphology, Adhesion, and Migration in Ewing Sarcoma. Genes Cancer. 2012; 3: 102-16. doi: 10.1177/1947601912457024.
11. Hu-Lieskovan S, Zhang J, Wu L, Shimada H, Schofield DE and Triche TJ. EWS-FLI1 fusion protein up-regulates critical genes in neural crest development and is responsible for the observed phenotype of Ewing’s family of tumors. Cancer Res. 2005; 65: 4633-44.
12. Lipinski M, Hirsch MR, Deagostini-Bazin H, Yamada O, Tursz T and Goridis C. Characterization of neural cell adhesion molecules (NCAM) expressed by Ewing and neuroblastoma cell lines. Int J Cancer. 1987; 40: 81-6.
13. Sankar S, Gomez NC, Bell R, Patel M, Davis IJ, Lessnick SL and Luo W. EWS and RE1-Silencing Transcription Factor Inhibit Neuronal Phenotype Development and Oncogenic Transformation in Ewing Sarcoma. Genes Cancer. 2013; 4: 213-23. doi: 10.1177/1947601913489569.
14. Teitell MA, Thompson AD, Sorensen PH, Shimada H, Triche TJ and Denny CT. EWS/ETS fusion genes induce epithelial and neuroectodermal differentiation in NIH 3T3 fibroblasts. Lab Invest. 1999; 79: 1535-43.
15. Tirode F, Laud-Duval K, Prieur A, Delorme B, Charbord P and Delattre O. Mesenchymal stem cell features of Ewing tumors. Cancer Cell. 2007; 11: 421-9.
16. Zhou Z, Yu L and Kleinerman ES. EWS-FLI-1 regulates the neuronal repressor gene REST, which controls Ewing sarcoma growth and vascular morphology. Cancer. 2014; 120: 579-88.
17. Chaturvedi A, Hoffman LM, Jensen CC, Lin YC, Grossmann AH, Randall RL, Lessnick SL, Welm AL and Beckerle MC. Molecular dissection of the mechanism by which EWS/FLI expression compromises actin cytoskeletal integrity and cell adhesion in Ewing sarcoma. Mol Biol Cell. 2014; 25: 2695-709.
18. Wiles ET, Bell R, Thomas D, Beckerle M and Lessnick SL. ZEB2 Represses the Epithelial Phenotype and Facilitates Metastasis in Ewing Sarcoma. Genes Cancer. 2013; 4: 486-500. doi: 10.1177/1947601913506115.
19. Jiang Y, Subbiah V, Janku F, Ludwig JA, Naing A, Benjamin RS, Brown RE, Anderson P and Kurzrock R. Novel secondary somatic mutations in Ewing’s sarcoma and desmoplastic small round cell tumors. PLoS One. 2014; 9: e93676.
20. Lawrence MS, Stojanov P, Polak P, Kryukov GV, Cibulskis K, Sivachenko A, Carter SL, Stewart C, Mermel CH, Roberts SA, Kiezun A, Hammerman PS, McKenna A, et al. Mutational heterogeneity in cancer and the search for new cancer-associated genes. Nature. 2013; 499: 214-8.
21. Tirode F, Surdez D, Ma X, Parker M, Le Deley MC, Bahrami A, Zhang Z, Lapouble E, Grossetete-Lalami S, Rusch M, Reynaud S, Rio-Frio T, Hedlund E, et al. Genomic landscape of Ewing sarcoma defines an aggressive subtype with co-association of STAG2 and TP53 mutations. Cancer Discov. 2014; 4: 1342-53.
22. Sankar S, Theisen ER, Bearss J, Mulvihill T, Hoffman LM, Sorna V, Beckerle MC, Sharma S and Lessnick SL. Reversible LSD1 inhibition interferes with global EWS/ETS transcriptional activity and impedes Ewing sarcoma tumor growth. Clin Cancer Res. 2014; 20: 4584-97.
23. Bilke S, Schwentner R, Yang F, Kauer M, Jug G, Walker RL, Davis S, Zhu YJ, Pineda M, Meltzer PS and Kovar H. Oncogenic ETS fusions deregulate E2F3 target genes in Ewing sarcoma and prostate cancer. Genome Res. 2013; 23: 1797-809.
24. Embree LJ, Azuma M and Hickstein DD. Ewing sarcoma fusion protein EWSR1/FLI1 interacts with EWSR1 leading to mitotic defects in zebrafish embryos and human cell lines. Cancer Res. 2009; 69: 4363-71.
25. Erkizan HV, Schneider JA, Sajwan K, Graham GT, Griffin B, Chasovskikh S, Youbi SE, Kallarakal A, Chruszcz M, Padmanabhan R, Casey JL, Uren A and Toretsky JA. RNA helicase A activity is inhibited by oncogenic transcription factor EWS-FLI1. Nucleic Acids Res. 2015; 43: 1069-80.
26. Guillon N, Tirode F, Boeva V, Zynovyev A, Barillot E and Delattre O. The oncogenic EWS-FLI1 protein binds in vivo GGAA microsatellite sequences with potential transcriptional activation function. PLoS One. 2009; 4: e4932.
27. Marques Howarth M, Simpson D, Ngok SP, Nieves B, Chen R, Siprashvili Z, Vaka D, Breese MR, Crompton BD, Alexe G, Hawkins DS, Jacobson D, Brunner AL, et al. Long noncoding RNA EWSAT1-mediated gene repression facilitates Ewing sarcoma oncogenesis. J Clin Invest. 2014; 124: 5275-90.
28. Nakatani F, Ferracin M, Manara MC, Ventura S, Del Monaco V, Ferrari S, Alberghini M, Grilli A, Knuutila S, Schaefer KL, Mattia G, Negrini M, Picci P, et al. miR-34a predicts survival of Ewing’s sarcoma patients and directly influences cell chemo-sensitivity and malignancy. J Pathol. 2012; 226: 796-805.
29. Patel M, Simon JM, Iglesia MD, Wu SB, McFadden AW, Lieb JD and Davis IJ. Tumor-specific retargeting of an oncogenic transcription factor chimera results in dysregulation of chromatin and transcription. Genome Res. 2012; 22: 259-70.
30. Riggi N, Knoechel B, Gillespie SM, Rheinbay E, Boulay G, Suva ML, Rossetti NE, Boonseng WE, Oksuz O, Cook EB, Formey A, Patel A, Gymrek M, et al. EWS-FLI1 utilizes divergent chromatin remodeling mechanisms to directly activate or repress enhancer elements in Ewing sarcoma. Cancer Cell. 2014; 26: 668-81.
31. Riggi N, Suva ML, De Vito C, Provero P, Stehle JC, Baumer K, Cironi L, Janiszewska M, Petricevic T, Suva D, Tercier S, Joseph JM, Guillou L, et al. EWS-FLI-1 modulates miRNA145 and SOX2 expression to initiate mesenchymal stem cell reprogramming toward Ewing sarcoma cancer stem cells. Genes Dev. 2010; 24: 916-32.
32. Sanchez G, Bittencourt D, Laud K, Barbier J, Delattre O, Auboeuf D and Dutertre M. Alteration of cyclin D1 transcript elongation by a mutated transcription factor up-regulates the oncogenic D1b splice isoform in cancer. Proc Natl Acad Sci U S A. 2008; 105: 6004-9.
33. Selvanathan SP, Graham GT, Erkizan HV, Dirksen U, Natarajan TG, Dakic A, Yu S, Liu X, Paulsen MT, Ljungman ME, Wu CH, Lawlor ER, Uren A, et al. Oncogenic fusion protein EWS-FLI1 is a network hub that regulates alternative splicing. Proc Natl Acad Sci U S A. 2015; 112: E1307-16.
34. Spahn L, Siligan C, Bachmaier R, Schmid JA, Aryee DN and Kovar H. Homotypic and heterotypic interactions of EWS, FLI1 and their oncogenic fusion protein. Oncogene. 2003; 22: 6819-29.
35. Tomazou EM, Sheffield NC, Schmidl C, Schuster M, Schonegger A, Datlinger P, Kubicek S, Bock C and Kovar H. Epigenome mapping reveals distinct modes of gene regulation and widespread enhancer reprogramming by the oncogenic fusion protein EWS-FLI1. Cell Rep. 2015; 10: 1082-95.
36. Wei GH, Badis G, Berger MF, Kivioja T, Palin K, Enge M, Bonke M, Jolma A, Varjosalo M, Gehrke AR, Yan J, Talukder S, Turunen M, et al. Genome-wide analysis of ETS-family DNA-binding in vitro and in vivo. EMBO J. 2010; 29: 2147-60.
37. Yang L, Chansky HA and Hickstein DD. EWS.Fli-1 fusion protein interacts with hyperphosphorylated RNA polymerase II and interferes with serine-arginine protein-mediated RNA splicing. J Biol Chem. 2000; 275: 37612-8.
38. Leacock SW, Basse AN, Chandler GL, Kirk AM, Rakheja D and Amatruda JF. A zebrafish transgenic model of Ewing’s sarcoma reveals conserved mediators of EWS-FLI1 tumorigenesis. Dis Model Mech. 2012; 5: 95-106.
39. Lin PP, Pandey MK, Jin F, Xiong S, Deavers M, Parant JM and Lozano G. EWS-FLI1 induces developmental abnormalities and accelerates sarcoma formation in a transgenic mouse model. Cancer Res. 2008; 68: 8968-75.
40. Torchia EC, Boyd K, Rehg JE, Qu C and Baker SJ. EWS/FLI-1 induces rapid onset of myeloid/erythroid leukemia in mice. Mol Cell Biol. 2007; 27: 7918-34.
41. Raj K and Mufti GJ. Azacytidine (Vidaza(R)) in the treatment of myelodysplastic syndromes. Ther Clin Risk Manag. 2006; 2: 377-88.
42. Saba HI. Decitabine in the treatment of myelodysplastic syndromes. Ther Clin Risk Manag. 2007; 3: 807-17.
43. Shi Y, Lan F, Matson C, Mulligan P, Whetstine JR, Cole PA and Casero RA. Histone demethylation mediated by the nuclear amine oxidase homolog LSD1. Cell. 2004; 119: 941-53.
44. Wang J, Hevi S, Kurash JK, Lei H, Gay F, Bajko J, Su H, Sun W, Chang H, Xu G, Gaudet F, Li E and Chen T. The lysine demethylase LSD1 (KDM1) is required for maintenance of global DNA methylation. Nat Genet. 2009; 41: 125-9.
45. Kontaki H and Talianidis I. Lysine methylation regulates E2F1-induced cell death. Mol Cell. 2010; 39: 152-60.
46. Cho HS, Suzuki T, Dohmae N, Hayami S, Unoki M, Yoshimatsu M, Toyokawa G, Takawa M, Chen T, Kurash JK, Field HI, Ponder BA, Nakamura Y, et al. Demethylation of RB regulator MYPT1 by histone demethylase LSD1 promotes cell cycle progression in cancer cells. Cancer Res. 2011; 71: 655-60.
47. Huang J, Sengupta R, Espejo AB, Lee MG, Dorsey JA, Richter M, Opravil S, Shiekhattar R, Bedford MT, Jenuwein T and Berger SL. p53 is regulated by the lysine demethylase LSD1. Nature. 2007; 449: 105-8.
48. Yang J, Huang J, Dasgupta M, Sears N, Miyagi M, Wang B, Chance MR, Chen X, Du Y, Wang Y, An L, Wang Q, Lu T, et al. Reversible methylation of promoter-bound STAT3 by histone-modifying enzymes. Proc Natl Acad Sci U S A. 2010; 107: 21499-504.
49. Chen Y, Yang Y, Wang F, Wan K, Yamane K, Zhang Y and Lei M. Crystal structure of human histone lysine-specific demethylase 1 (LSD1). Proc Natl Acad Sci U S A. 2006; 103: 13956-61.
50. Metzger E, Wissmann M, Yin N, Muller JM, Schneider R, Peters AH, Gunther T, Buettner R and Schule R. LSD1 demethylates repressive histone marks to promote androgen-receptor-dependent transcription. Nature. 2005; 437: 436-9.
51. Musri MM, Carmona MC, Hanzu FA, Kaliman P, Gomis R and Parrizas M. Histone demethylase LSD1 regulates adipogenesis. J Biol Chem. 2010; 285: 30034-41.
52. Lv S, Bu W, Jiao H, Liu B, Zhu L, Zhao H, Liao J, Li J and Xu X. LSD1 is required for chromosome segregation during mitosis. Eur J Cell Biol. 2010; 89: 557-63.
53. Scoumanne A and Chen X. The lysine-specific demethylase 1 is required for cell proliferation in both p53-dependent and -independent manners. J Biol Chem. 2007; 282: 15471-5.
54. Foster CT, Dovey OM, Lezina L, Luo JL, Gant TW, Barlev N, Bradley A and Cowley SM. Lysine-specific demethylase 1 regulates the embryonic transcriptome and CoREST stability. Mol Cell Biol. 2010; 30: 4851-63.
55. McDonald OG, Wu H, Timp W, Doi A and Feinberg AP. Genome-scale epigenetic reprogramming during epithelial-to-mesenchymal transition. Nat Struct Mol Biol. 2011; 18: 867-74.
56. Li Y, Deng C, Hu X, Patel B, Fu X, Qiu Y, Brand M, Zhao K and Huang S. Dynamic interaction between TAL1 oncoprotein and LSD1 regulates TAL1 function in hematopoiesis and leukemogenesis. Oncogene. 2012; 31: 5007-18.
57. Sprussel A, Schulte JH, Weber S, Necke M, Handschke K, Thor T, Pajtler KW, Schramm A, Konig K, Diehl L, Mestdagh P, Vandesompele J, Speleman F, et al. Lysine-specific demethylase 1 restricts hematopoietic progenitor proliferation and is essential for terminal differentiation. Leukemia. 2012; 26: 2039-51.
58. Zhou H, Li W, Zhu S, Joo JY, Do JT, Xiong W, Kim JB, Zhang K, Scholer HR and Ding S. Conversion of mouse epiblast stem cells to an earlier pluripotency state by small molecules. J Biol Chem. 2010; 285: 29676-80.
59. Lim S, Janzer A, Becker A, Zimmer A, Schule R, Buettner R and Kirfel J. Lysine-specific demethylase 1 (LSD1) is highly expressed in ER-negative breast cancers and a biomarker predicting aggressive biology. Carcinogenesis. 2010; 31: 512-20.
60. Serce N, Gnatzy A, Steiner S, Lorenzen H, Kirfel J and Buettner R. Elevated expression of LSD1 (Lysine-specific demethylase 1) during tumour progression from pre-invasive to invasive ductal carcinoma of the breast. BMC Clin Pathol. 2012; 12: 13.
61. Derr RS, van Hoesel AQ, Benard A, Goossens-Beumer IJ, Sajet A, Dekker-Ensink NG, de Kruijf EM, Bastiaannet E, Smit VT, van de Velde CJ and Kuppen PJ. High nuclear expression levels of histone-modifying enzymes LSD1, HDAC2 and SIRT1 in tumor cells correlate with decreased survival and increased relapse in breast cancer patients. BMC Cancer. 2014; 14: 604.
62. Hayami S, Kelly JD, Cho HS, Yoshimatsu M, Unoki M, Tsunoda T, Field HI, Neal DE, Yamaue H, Ponder BA, Nakamura Y and Hamamoto R. Overexpression of LSD1 contributes to human carcinogenesis through chromatin regulation in various cancers. Int J Cancer. 2011; 128: 574-86.
63. Huang Z, Li S, Song W, Li X, Li Q, Zhang Z, Han Y, Zhang X, Miao S, Du R and Wang L. Lysine-specific demethylase 1 (LSD1/KDM1A) contributes to colorectal tumorigenesis via activation of the Wnt/beta-catenin pathway by down-regulating Dickkopf-1 (DKK1) [corrected]. PLoS One. 2013; 8: e70077.
64. Mohammad HP, Smitheman KN, Kamat CD, Soong D, Federowicz KE, Van Aller GS, Schneck JL, Carson JD, Liu Y, Butticello M, Bonnette WG, Gorman SA, Degenhardt Y, et al. A DNA Hypomethylation Signature Predicts Antitumor Activity of LSD1 Inhibitors in SCLC. Cancer Cell. 2015; 28: 57-69.
65. Konovalov S and Garcia-Bassets I. Analysis of the levels of lysine-specific demethylase 1 (LSD1) mRNA in human ovarian tumors and the effects of chemical LSD1 inhibitors in ovarian cancer cell lines. J Ovarian Res. 2013; 6: 75.
66. Schulte JH, Lim S, Schramm A, Friedrichs N, Koster J, Versteeg R, Ora I, Pajtler K, Klein-Hitpass L, Kuhfittig-Kulle S, Metzger E, Schule R, Eggert A, et al. Lysine-specific demethylase 1 is strongly expressed in poorly differentiated neuroblastoma: implications for therapy. Cancer Res. 2009; 69: 2065-71.
67. Kahl P, Gullotti L, Heukamp LC, Wolf S, Friedrichs N, Vorreuther R, Solleder G, Bastian PJ, Ellinger J, Metzger E, Schule R and Buettner R. Androgen receptor coactivators lysine-specific histone demethylase 1 and four and a half LIM domain protein 2 predict risk of prostate cancer recurrence. Cancer Res. 2006; 66: 11341-7.
68. Kashyap V, Ahmad S, Nilsson EM, Helczynski L, Kenna S, Persson JL, Gudas LJ and Mongan NP. The lysine specific demethylase-1 (LSD1/KDM1A) regulates VEGF-A expression in prostate cancer. Mol Oncol. 2013; 7: 555-66.
69. Bennani-Baiti IM, Machado I, Llombart-Bosch A and Kovar H. Lysine-specific demethylase 1 (LSD1/KDM1A/AOF2/BHC110) is expressed and is an epigenetic drug target in chondrosarcoma, Ewing’s sarcoma, osteosarcoma, and rhabdomyosarcoma. Hum Pathol. 2012; 43: 1300-7.
70. Schildhaus HU, Riegel R, Hartmann W, Steiner S, Wardelmann E, Merkelbach-Bruse S, Tanaka S, Sonobe H, Schule R, Buettner R and Kirfel J. Lysine-specific demethylase 1 is highly expressed in solitary fibrous tumors, synovial sarcomas, rhabdomyosarcomas, desmoplastic small round cell tumors, and malignant peripheral nerve sheath tumors. Hum Pathol. 2011; 42: 1667-75.
71. Sankar S, Bell R, Stephens B, Zhuo R, Sharma S, Bearss DJ and Lessnick SL. Mechanism and relevance of EWS/FLI-mediated transcriptional repression in Ewing sarcoma. Oncogene. 2013; 32: 5089-100.
72. Kouzarides T. Chromatin modifications and their function. Cell. 2007; 128: 693-705.
73. Greer EL and Shi Y. Histone methylation: a dynamic mark in health, disease and inheritance. Nat Rev Genet. 2012; 13: 343-57.
74. Barski A, Cuddapah S, Cui K, Roh TY, Schones DE, Wang Z, Wei G, Chepelev I and Zhao K. High-resolution profiling of histone methylations in the human genome. Cell. 2007; 129: 823-37.
75. Shilatifard A. The COMPASS family of histone H3K4 methylases: mechanisms of regulation in development and disease pathogenesis. Annu Rev Biochem. 2012; 81: 65-95.
76. Eissenberg JC and Shilatifard A. Histone H3 lysine 4 (H3K4) methylation in development and differentiation. Dev Biol. 2010; 339: 240-9.
77. Blair LP, Cao J, Zou MR, Sayegh J and Yan Q. Epigenetic Regulation by Lysine Demethylase 5 (KDM5) Enzymes in Cancer. Cancers (Basel). 2011; 3: 1383-404.
78. Islam AB, Richter WF, Lopez-Bigas N and Benevolenskaya EV. Selective targeting of histone methylation. Cell Cycle. 2011; 10: 413-24.
79. Popovic R and Licht JD. Emerging epigenetic targets and therapies in cancer medicine. Cancer Discov. 2012; 2: 405-13.
80. Choudhary C, Kumar C, Gnad F, Nielsen ML, Rehman M, Walther TC, Olsen JV and Mann M. Lysine acetylation targets protein complexes and co-regulates major cellular functions. Science. 2009; 325: 834-40.
81. Kubicek S, Gilbert JC, Fomina-Yadlin D, Gitlin AD, Yuan Y, Wagner FF, Holson EB, Luo T, Lewis TA, Taylor B, Gupta S, Shamji AF, Wagner BK, et al. Chromatin-targeting small molecules cause class-specific transcriptional changes in pancreatic endocrine cells. Proc Natl Acad Sci U S A. 2012; 109: 5364-9.
82. Tian X, Zhang S, Liu HM, Zhang YB, Blair CA, Mercola D, Sassone-Corsi P and Zi X. Histone lysine-specific methyltransferases and demethylases in carcinogenesis: new targets for cancer therapy and prevention. Curr Cancer Drug Targets. 2013; 13: 558-79.
83. Stavropoulos P, Blobel G and Hoelz A. Crystal structure and mechanism of human lysine-specific demethylase-1. Nat Struct Mol Biol. 2006; 13: 626-32.
84. Qian C, Zhang Q, Li S, Zeng L, Walsh MJ and Zhou MM. Structure and chromosomal DNA binding of the SWIRM domain. Nat Struct Mol Biol. 2005; 12: 1078-85.
85. Da G, Lenkart J, Zhao K, Shiekhattar R, Cairns BR and Marmorstein R. Structure and function of the SWIRM domain, a conserved protein module found in chromatin regulatory complexes. Proc Natl Acad Sci U S A. 2006; 103: 2057-62.
86. Laurent B, Ruitu L, Murn J, Hempel K, Ferrao R, Xiang Y, Liu S, Garcia BA, Wu H, Wu F, Steen H and Shi Y. A specific LSD1/KDM1A isoform regulates neuronal differentiation through H3K9 demethylation. Mol Cell. 2015; 57: 957-70.
87. Zibetti C, Adamo A, Binda C, Forneris F, Toffolo E, Verpelli C, Ginelli E, Mattevi A, Sala C and Battaglioli E. Alternative splicing of the histone demethylase LSD1/KDM1 contributes to the modulation of neurite morphogenesis in the mammalian nervous system. J Neurosci. 2010; 30: 2521-32.
88. Karytinos A, Forneris F, Profumo A, Ciossani G, Battaglioli E, Binda C and Mattevi A. A novel mammalian flavin-dependent histone demethylase. J Biol Chem. 2009; 284: 17775-82.
89. Ciccone DN, Su H, Hevi S, Gay F, Lei H, Bajko J, Xu G, Li E and Chen T. KDM1B is a histone H3K4 demethylase required to establish maternal genomic imprints. Nature. 2009; 461: 415-8.
90. Yokoyama A, Igarashi K, Sato T, Takagi K, Otsuka IM, Shishido Y, Baba T, Ito R, Kanno J, Ohkawa Y, Morohashi K and Sugawara A. Identification of myelin transcription factor 1 (MyT1) as a subunit of the neural cell type-specific lysine-specific demethylase 1 (LSD1) complex. J Biol Chem. 2014; 289: 18152-62.
91. Shi YJ, Matson C, Lan F, Iwase S, Baba T and Shi Y. Regulation of LSD1 histone demethylase activity by its associated factors. Mol Cell. 2005; 19: 857-64.
92. Lee MG, Wynder C, Cooch N and Shiekhattar R. An essential role for CoREST in nucleosomal histone 3 lysine 4 demethylation. Nature. 2005; 437: 432-5.
93. Wang Y, Zhang H, Chen Y, Sun Y, Yang F, Yu W, Liang J, Sun L, Yang X, Shi L, Li R, Li Y, Zhang Y, et al. LSD1 is a subunit of the NuRD complex and targets the metastasis programs in breast cancer. Cell. 2009; 138: 660-72.
94. Whyte WA, Bilodeau S, Orlando DA, Hoke HA, Frampton GM, Foster CT, Cowley SM and Young RA. Enhancer decommissioning by LSD1 during embryonic stem cell differentiation. Nature. 2012; 482: 221-5.
95. Lin Y, Wu Y, Li J, Dong C, Ye X, Chi YI, Evers BM and Zhou BP. The SNAG domain of Snail1 functions as a molecular hook for recruiting lysine-specific demethylase 1. EMBO J. 2010; 29: 1803-16.
96. Saleque S, Kim J, Rooke HM and Orkin SH. Epigenetic regulation of hematopoietic differentiation by Gfi-1 and Gfi-1b is mediated by the cofactors CoREST and LSD1. Mol Cell. 2007; 27: 562-72.
97. Choi J, Jang H, Kim H, Kim ST, Cho EJ and Youn HD. Histone demethylase LSD1 is required to induce skeletal muscle differentiation by regulating myogenic factors. Biochem Biophys Res Commun. 2010; 401: 327-32.
98. Wang J, Scully K, Zhu X, Cai L, Zhang J, Prefontaine GG, Krones A, Ohgi KA, Zhu P, Garcia-Bassets I, Liu F, Taylor H, Lozach J, et al. Opposing LSD1 complexes function in developmental gene activation and repression programmes. Nature. 2007; 446: 882-7.
99. Sun G, Ye P, Murai K, Lang MF, Li S, Zhang H, Li W, Fu C, Yin J, Wang A, Ma X and Shi Y. miR-137 forms a regulatory loop with nuclear receptor TLX and LSD1 in neural stem cells. Nat Commun. 2011; 2: 529.
100. Althoff K, Beckers A, Odersky A, Mestdagh P, Koster J, Bray IM, Bryan K, Vandesompele J, Speleman F, Stallings RL, Schramm A, Eggert A, Sprussel A, et al. MiR-137 functions as a tumor suppressor in neuroblastoma by downregulating KDM1A. Int J Cancer. 2013; 133: 1064-73.
101. Wu Y, Wang Y, Yang XH, Kang T, Zhao Y, Wang C, Evers BM and Zhou BP. The deubiquitinase USP28 stabilizes LSD1 and confers stem-cell-like traits to breast cancer cells. Cell Rep. 2013; 5: 224-36.
102. Han X, Gui B, Xiong C, Zhao L, Liang J, Sun L, Yang X, Yu W, Si W, Yan R, Yi X, Zhang D, Li W, et al. Destabilizing LSD1 by Jade-2 promotes neurogenesis: an antibraking system in neural development. Mol Cell. 2014; 55: 482-94.
103. Ding J, Zhang ZM, Xia Y, Liao GQ, Pan Y, Liu S, Zhang Y and Yan ZS. LSD1-mediated epigenetic modification contributes to proliferation and metastasis of colon cancer. Br J Cancer. 2013; 109: 994-1003.
104. Kauffman EC, Robinson BD, Downes MJ, Powell LG, Lee MM, Scherr DS, Gudas LJ and Mongan NP. Role of androgen receptor and associated lysine-demethylase coregulators, LSD1 and JMJD2A, in localized and advanced human bladder cancer. Mol Carcinog. 2011; 50: 931-44.
105. Rao M, Chinnasamy N, Hong JA, Zhang Y, Zhang M, Xi S, Liu F, Marquez VE, Morgan RA and Schrump DS. Inhibition of histone lysine methylation enhances cancer-testis antigen expression in lung cancer cells: implications for adoptive immunotherapy of cancer. Cancer Res. 2011; 71: 4192-204.
106. Schenk T, Chen WC, Gollner S, Howell L, Jin L, Hebestreit K, Klein HU, Popescu AC, Burnett A, Mills K, Casero RA, Jr., Marton L, Woster P, et al. Inhibition of the LSD1 (KDM1A) demethylase reactivates the all-trans-retinoic acid differentiation pathway in acute myeloid leukemia. Nat Med. 2012; 18: 605-11.
107. Wu J, Hu L, Du Y, Kong F and Pan Y. Prognostic role of LSD1 in various cancers: evidence from a meta-analysis. Onco Targets Ther. 2015; 8: 2565-70.
108. Zheng YC, Ma J, Wang Z, Li J, Jiang B, Zhou W, Shi X, Wang X, Zhao W and Liu HM. A Systematic Review of Histone Lysine-Specific Demethylase 1 and Its Inhibitors. Med Res Rev. 2015.
109. Mould DP, McGonagle AE, Wiseman DH, Williams EL and Jordan AM. Reversible inhibitors of LSD1 as therapeutic agents in acute myeloid leukemia: clinical significance and progress to date. Med Res Rev. 2015; 35: 586-618.
110. Kakizawa T, Ota Y, Itoh Y, Tsumoto H and Suzuki T. Histone H3 peptide based LSD1-selective inhibitors. Bioorg Med Chem Lett. 2015; 25: 1925-8.
111. Kumarasinghe IR and Woster PM. Synthesis and evaluation of novel cyclic Peptide inhibitors of lysine-specific demethylase 1. ACS Med Chem Lett. 2014; 5: 29-33.
112. Culhane JC, Szewczuk LM, Liu X, Da G, Marmorstein R and Cole PA. A mechanism-based inactivator for histone demethylase LSD1. J Am Chem Soc. 2006; 128: 4536-7.
113. Singh MM, Manton CA, Bhat KP, Tsai WW, Aldape K, Barton MC and Chandra J. Inhibition of LSD1 sensitizes glioblastoma cells to histone deacetylase inhibitors. Neuro Oncol. 2011; 13: 894-903.
114. Fiskus W, Sharma S, Shah B, Portier BP, Devaraj SG, Liu K, Iyer SP, Bearss D and Bhalla KN. Highly effective combination of LSD1 (KDM1A) antagonist and pan-histone deacetylase inhibitor against human AML cells. Leukemia. 2014; 28: 2155-64.
115. Frieling H and Bleich S. Tranylcypromine: new perspectives on an “old” drug. Eur Arch Psychiatry Clin Neurosci. 2006; 256: 268-73.
116. Schmidt DM and McCafferty DG. trans-2-Phenylcyclopropylamine is a mechanism-based inactivator of the histone demethylase LSD1. Biochemistry. 2007; 46: 4408-16.
117. Yang M, Culhane JC, Szewczuk LM, Jalili P, Ball HL, Machius M, Cole PA and Yu H. Structural basis for the inhibition of the LSD1 histone demethylase by the antidepressant trans-2-phenylcyclopropylamine. Biochemistry. 2007; 46: 8058-65.
118. Lee MG, Wynder C, Schmidt DM, McCafferty DG and Shiekhattar R. Histone H3 lysine 4 demethylation is a target of nonselective antidepressive medications. Chem Biol. 2006; 13: 563-7.
119. Mimasu S, Umezawa N, Sato S, Higuchi T, Umehara T and Yokoyama S. Structurally designed trans-2-phenylcyclopropylamine derivatives potently inhibit histone demethylase LSD1/KDM1. Biochemistry. 2010; 49: 6494-503.
120. Ueda R, Suzuki T, Mino K, Tsumoto H, Nakagawa H, Hasegawa M, Sasaki R, Mizukami T and Miyata N. Identification of cell-active lysine specific demethylase 1-selective inhibitors. J Am Chem Soc. 2009; 131: 17536-7.
121. Forneris F, Binda C, Vanoni MA, Battaglioli E and Mattevi A. Human histone demethylase LSD1 reads the histone code. J Biol Chem. 2005; 280: 41360-5.
122. Sorna V, Theisen ER, Stephens B, Warner SL, Bearss DJ, Vankayalapati H and Sharma S. High-throughput virtual screening identifies novel N’-(1-phenylethylidene)-benzohydrazides as potent, specific, and reversible LSD1 inhibitors. J Med Chem. 2013; 56: 9496-508.
123. Stephens BJ TE, Warner SL, Sharma S, Bearss DJ. Abstract 1045: Activity of the LSD1 inhibitor HCI-2509 in ER-negative breast cancer cells. Proceedings of the American Association for Cancer Research 103rd Annual Meeting 2012: 31 March −4 April 2012. IL: Chicago; . 2012.
124. Theisen ER, Gajiwala S, Bearss J, Sorna V, Sharma S and Janat-Amsbury M. Reversible inhibition of lysine specific demethylase 1 is a novel anti-tumor strategy for poorly differentiated endometrial carcinoma. BMC Cancer. 2014; 14: 752.
125. Theisen ER BJ, Sorna V, Bearss DJ, Sharma S. Abstract 3: Targeted inhibition of LSD1 in castration-resistant prostate cancer. Proceedings of the American Association for Cancer Research 104th Annual Meeting 2013. DC: Washington;. 2013.
126. Harris WJ, Huang X, Lynch JT, Spencer GJ, Hitchin JR, Li Y, Ciceri F, Blaser JG, Greystoke BF, Jordan AM, Miller CJ, Ogilvie DJ and Somervaille TC. The histone demethylase KDM1A sustains the oncogenic potential of MLL-AF9 leukemia stem cells. Cancer Cell. 2012; 21: 473-87.
127. Maes T, Tirapu I, Mascaro C, Ortega A, Estiarte A, Valls N, Castro-Palomino J, Arjol CB and Kurz G. Preclinical characterization of a potent and selective inhibitor of the histone demethylase KDM1A for MLL leukemia. Journal of Clinical Oncology. 2013; 31.
128. Kruger RG, Mohammad H, Smitheman K, Cusan M, Liu Y, Pappalardi M, Federowicz K, Aller GV, Kasparec J, Tian X, Suarez D, Rouse M, Schneck J, et al. Inhibition Of LSD1 As a Therapeutic Strategy For The Treatment Of Acute Myeloid Leukemia. Blood. 2013; 122: 3964.
129. Network TCGAR. Comprehensive molecular characterization of urothelial bladder carcinoma. Nature. 2014; 507: 315-22.
130. Robinson G, Parker M, Kranenburg TA, Lu C, Chen X, Ding L, Phoenix TN, Hedlund E, Wei L, Zhu X, Chalhoub N, Baker SJ, Huether R, et al. Novel mutations target distinct subgroups of medulloblastoma. Nature. 2012; 488: 43-8.
131. May WA, Lessnick SL, Braun BS, Klemsz M, Lewis BC, Lunsford LB, Hromas R and Denny CT. The Ewing’s sarcoma EWS/FLI-1 fusion gene encodes a more potent transcriptional activator and is a more powerful transforming gene than FLI-1. Mol Cell Biol. 1993; 13: 7393-8.
132. Lessnick SL, Braun BS, Denny CT and May WA. Multiple domains mediate transformation by the Ewing’s sarcoma EWS/FLI-1 fusion gene. Oncogene. 1995; 10: 423-31.
133. Bertolotti A, Melot T, Acker J, Vigneron M, Delattre O and Tora L. EWS, but not EWS-FLI-1, is associated with both TFIID and RNA polymerase II: interactions between two members of the TET family, EWS and hTAFII68, and subunits of TFIID and RNA polymerase II complexes. Mol Cell Biol. 1998; 18: 1489-97.
134. Kim S, Denny CT and Wisdom R. Cooperative DNA binding with AP-1 proteins is required for transformation by EWS-Ets fusion proteins. Mol Cell Biol. 2006; 26: 2467-78.
135. Petermann R, Mossier BM, Aryee DN, Khazak V, Golemis EA and Kovar H. Oncogenic EWS-Fli1 interacts with hsRPB7, a subunit of human RNA polymerase II. Oncogene. 1998; 17: 603-10.
136. Ramakrishnan R, Fujimura Y, Zou JP, Liu F, Lee L, Rao VN and Reddy ES. Role of protein-protein interactions in the antiapoptotic function of EWS-Fli-1. Oncogene. 2004; 23: 7087-94.
137. Schwentner R, Papamarkou T, Kauer MO, Stathopoulos V, Yang F, Bilke S, Meltzer PS, Girolami M and Kovar H. EWS-FLI1 employs an E2F switch to drive target gene expression. Nucleic Acids Res. 2015; 43: 2780-9.
138. Zhou H and Lee KA. An hsRPB4/7-dependent yeast assay for trans-activation by the EWS oncogene. Oncogene. 2001; 20: 1519-24.
139. Gangwal K, Close D, Enriquez CA, Hill CP and Lessnick SL. Emergent Properties of EWS/FLI Regulation via GGAA Microsatellites in Ewing’s Sarcoma. Genes Cancer. 2010; 1: 177-187. doi: 10.1177/1947601910361495
140. Gangwal K, Sankar S, Hollenhorst PC, Kinsey M, Haroldsen SC, Shah AA, Boucher KM, Watkins WS, Jorde LB, Graves BJ and Lessnick SL. Microsatellites as EWS/FLI response elements in Ewing’s sarcoma. Proc Natl Acad Sci U S A. 2008; 105: 10149-54.
141. Luo W, Gangwal K, Sankar S, Boucher KM, Thomas D and Lessnick SL. GSTM4 is a microsatellite-containing EWS/FLI target involved in Ewing’s sarcoma oncogenesis and therapeutic resistance. Oncogene. 2009; 28: 4126-32.
142. Tirado OM, Mateo-Lozano S, Villar J, Dettin LE, Llort A, Gallego S, Ban J, Kovar H and Notario V. Caveolin-1 (CAV1) is a target of EWS/FLI-1 and a key determinant of the oncogenic phenotype and tumorigenicity of Ewing’s sarcoma cells. Cancer Res. 2006; 66: 9937-47.
143. Fadul J, Bell R, Hoffman LM, Beckerle MC, Engel ME and Lessnick SL. EWS/FLI utilizes NKX2-2 to repress mesenchymal features of Ewing sarcoma. Genes Cancer. 2015; 6: 129-43. doi: 10.18632/genesandcancer.57
144. Kinsey M, Smith R, Iyer AK, McCabe ER and Lessnick SL. EWS/FLI and its downstream target NR0B1 interact directly to modulate transcription and oncogenesis in Ewing’s sarcoma. Cancer Res. 2009; 69: 9047-55.
145. Owen LA, Kowalewski AA and Lessnick SL. EWS/FLI mediates transcriptional repression via NKX2.2 during oncogenic transformation in Ewing’s sarcoma. PLoS One. 2008; 3: e1965.
146. Wiles ET, Lui-Sargent B, Bell R and Lessnick SL. BCL11B is up-regulated by EWS/FLI and contributes to the transformed phenotype in Ewing sarcoma. PLoS One. 2013; 8: e59369.
147. Hahm KB. Repression of the gene encoding the TGF-beta type II receptor is a major target of the EWS-FLI1 oncoprotein. Nat Genet. 1999; 23: 481.
148. Prieur A, Tirode F, Cohen P and Delattre O. EWS/FLI-1 silencing and gene profiling of Ewing cells reveal downstream oncogenic pathways and a crucial role for repression of insulin-like growth factor binding protein 3. Mol Cell Biol. 2004; 24: 7275-83.
149. Kearns NA, Pham H, Tabak B, Genga RM, Silverstein NJ, Garber M and Maehr R. Functional annotation of native enhancers with a Cas9-histone demethylase fusion. Nat Methods. 2015; 12: 401-3.
150. Mendenhall EM, Williamson KE, Reyon D, Zou JY, Ram O, Joung JK and Bernstein BE. Locus-specific editing of histone modifications at endogenous enhancers. Nat Biotechnol. 2013; 31: 1133-6.
151. Ambrosio R, Damiano V, Sibilio A, De Stefano MA, Avvedimento VE, Salvatore D and Dentice M. Epigenetic control of type 2 and 3 deiodinases in myogenesis: role of Lysine-specific Demethylase enzyme and FoxO3. Nucleic Acids Res. 2013; 41: 3551-62.
152. Cai C, He HH, Gao S, Chen S, Yu Z, Gao Y, Chen MW, Zhang J, Ahmed M, Wang Y, Metzger E, Schule R, Liu XS, et al. Lysine-specific demethylase 1 has dual functions as a major regulator of androgen receptor transcriptional activity. Cell Rep. 2014; 9: 1618-27.
153. Esiashvili N, Goodman M and Marcus RB, Jr. Changes in incidence and survival of Ewing sarcoma patients over the past 3 decades: Surveillance Epidemiology and End Results data. J Pediatr Hematol Oncol. 2008; 30: 425-30.
154. Agelopoulos K, Richter GH, Schmidt E, Dirksen U, von Heyking K, Moser B, Klein HU, Kontny U, Dugas M, Poos K, Korsching E, Buch T, Weckesser M, et al. Deep sequencing in conjunction with expression and functional analyses reveals activation of FGFR1 in Ewing sarcoma. Clin Cancer Res. 2015; 21:4935-46. doi: 10.1158/1078-0432.CCR-14-2744.
155. Huether R, Dong L, Chen X, Wu G, Parker M, Wei L, Ma J, Edmonson MN, Hedlund EK, Rusch MC, Shurtleff SA, Mulder HL, Boggs K, et al. The landscape of somatic mutations in epigenetic regulators across 1,000 paediatric cancer genomes. Nat Commun. 2014; 5: 3630.