
INTRODUCTION
Hepatocellular carcinoma (HCC) is the second leading cause of cancer-related death and the fifth most common malignancy worldwide [1–3]. Curative interventions such as transplantation, resection, and thermal ablation are applicable for the 30% of patients whose tumors or liver function meet defined criteria [4]. Patients with advanced HCC or liver disease are not suitable for these curative interventions and require systemic chemotherapy [5]. However, these patients frequently develop drug resistance causing their treatments to fail [6, 7].
The mechanisms underlying cancer cell chemoresistance are complex and numerous [8–10], including increased drug efflux, reduction in drug absorption, changes of anticancer drugs’ targets, decreased drug activity, tumor hypoxia, enhancement of DNA repair following damage, abrogation of apoptotic effector pathways by altered gene expression, and changes in signaling pathways. One of the most important causes of resistance is increased expression of the drug transporter family known as the ATP-binding cassette (ABC) transporters. The most common member of this family is the ABCB1 (also known as MDR1 (multidrug resistance gene 1) which encodes for P-glycoprotein. Overexpression of P-glycoprotein is associated with lower accumulation of doxorubicin in HCC cells and with worse prognosis in patients [10]. EMT is also an important process by which HCC acquire 5-FU chemoresistance [11]. Furthermore, the activation of an Oct4-AKT-ABCG2 pathway has been identified to enhance drug resistance in HCC [12, 13]. A comprehensive understanding of mechanisms of chemoresistance in HCC could present opportunities to improve the response to cytotoxic agents and improve clinical outcomes.
The Nogo isoforms-A, -B and -C are members of the reticulon family of proteins. Nogo-A and Nogo-C are highly expressed in the central nervous system (CNS), while Nogo-B is found in most tissues [14, 15]. Nogo-B was previously identified as highly expressed in caveolin-1 enriched microdomains of endothelial cells (EC) [16]. The amino terminus (residues1–200) of Nogo-B (AmNogo-B) serves as a chemoattractant for EC [16]. NgBR was identified as a receptor specific for AmNogo-B by an expression cloning approach [15, 17]. We have previously demonstrated that NgBR is necessary for in vivo angiogenesis in zebrafish via the Akt pathway [18] and NgBR is highly expressed in human breast invasive ductal carcinoma [19]. NgBR breast tumor cell expression is highly correlated with expression of estrogen receptor and survivin [19]. Further study showed that NgBR promotes EMT in breast tumor cells, [20] but any role of NgBR in cancer drug resistance is still unclear. Here, we show that NgBR depletion sensitizes the 5-FU-resistant Bel7402/5FU cells to 5-FU treatment via disruption of the PI3K/Akt/MDM2 signaling pathway and stabilization of p53 protein. Our results suggest that NgBR is a potential novel drug target that can be used to increase the efficacy of conventional chemotherapeutic agents.
RESULTS
NgBR expression is increased in the drug resistant human HCC cells
To confirm the 5-FU chemoresistance phenotype in Bel/5FU cells, the HCC parental cells (Bel7402) and the chemoresistant HCC cells (Bel/5FU) were treated with the indicated concentrations of 5-FU, and cell proliferation and survival were assessed using clonogenic survival assays. As shown in Figure 1A, compared with the Bel7402 control cells, the Bel/5FU cells were resistant to 5-FU. Then, the mRNA and protein levels of NgBR were evaluated in both cell lines. As shown in Figure 1B and 1C, both the NgBR mRNA and protein levels were increased in the Bel/5FU cell lines. These results indicate that higher NgBR expression is associated with chemoresistance in human HCC cell lines.
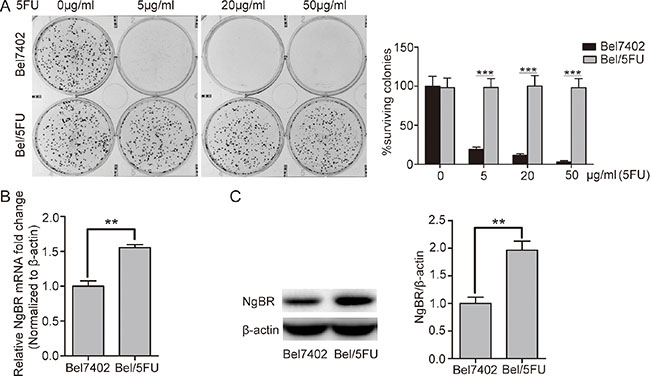
Figure 1: NgBR is highly expressed in the chemoresistant Bel/5FU cells. (A) The 5-FU resistant phenotype was confirmed in Bel/5FU cells. Clonogenic survival assay was used for measuring clonogenicity of Bel7402 and Bel/5FU cells treated with different concentrations of 5-FU (0, 5, 20, and 50 μg/mL) (left panel). The number of untreated cells is set as 100%. The results were analyzed and show the average percentage of surviving colonies (right panel). (B) High mRNA level of NgBR was detected in chemoresistant Bel/FU cells. mRNA level of NgBR was analyzed using real-time RT-PCR and normalized to the β-actin. (C) High NgBR protein level was detected in chemoresistant Bel/5FU cells. Protein level was monitored using western blot (left panel). B and intensities were quantified using Image Lab 5.0 software and were normalized to β-actin (right panel). The data are presented as the mean ± SD of three independent experiments. (**P < 0.01, ***P < 0.001).
NgBR knockdown decreases the chemoresistance of Bel/5FU cells in vitro
To determine whether knockdown of NgBR could decrease the chemoresistance of Bel/5FU cells, NgBR siRNA was used to silence the expression of NgBR. The efficiency of transfection was evaluated by real-time RT-PCR and western blot. The results demonstrate that all three NgBR siRNAs (S1, S2, and S3) effectively reduce the mRNA and protein levels of NgBR(Supplementary Figure S1A and S1B). Clonogenic survival assay showed knockdown of NgBR by all three siRNAs (S1, S2, and S3) reduces colony formation of Bel/5FU cells in response to 5-FU treatment (Supplementary Figure S1C). We used S1 for the following NgBR siRNA knockdown experiment. Clonogenic assay further showed that knockdown of NgBR by siRNA reduces the colony number in response to 5-FU treatment in a dose dependent manner (Figure 2A). In addition, CCK-8 assay results also revealed knockdown of NgBR decreases the cell survival rate of Bel/5FU cells at all 5-FU treatment time points (Figure 2B). These results indicate that NgBR knockdown decreases the chemoresistance of Bel/5FU cells.
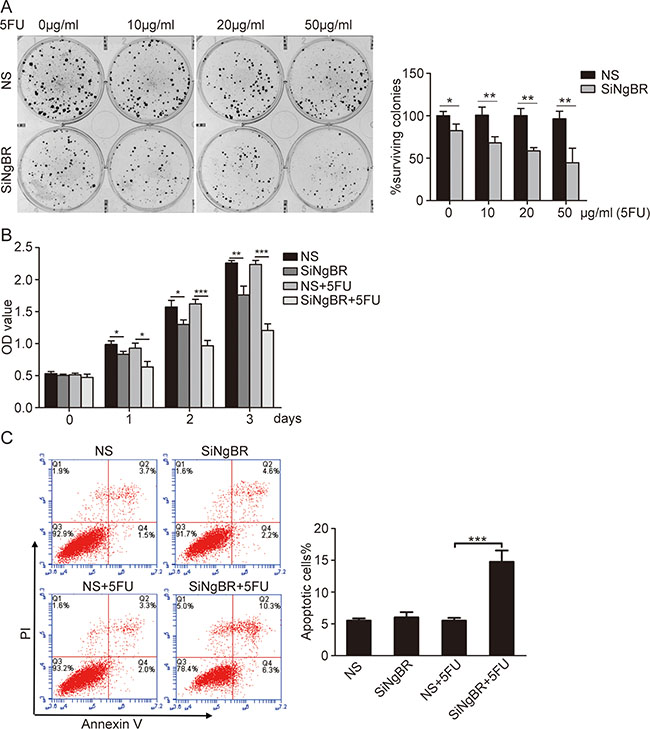
Figure 2: Knockdown of NgBR decreases the chemoresistance of Bel/5FU cells to 5-FU. (A) Knockdown of NgBR decreases the clonogenenicity of Bel/5FU cells. Clonogenic formation assay was used for measuring clonogenicity of Bel/5FU cells treated with different concentrations of 5-FU (0, 10, 20, and 50 μg/mL) (left panel). The number of untreated cells is set as 100% and the results show the average percentage of surviving colonies (right panel). (B) Knockdown of NgBR decreases Bel/5FU cell viability. Cell viability was analyzed by CCK8 assay in Bel/5FU cells treated with 5-FU (0 and 50 μg/mL) for different times 24 h, 48 h, and 72 h. (C) Knockdown of NgBR increases apoptosis of Bel/5FU cells induced by 5-FU. The apoptotic cells were detected by Annexin V-PI dual staining. Representative data from three independent experiments are shown in left panel, the total number of cells in the Q2 and Q4 quadrant was regarded as apoptotic cells. Percentages of apoptotic cells are shown in the bar graph (right panel). The data are means ± SD of three independent assays. (*P < 0.05,**P < 0.01, ***P < 0.001).
To confirm that apoptosis contributes to the inhibitory effects of NgBR knockdown on Bel/5FU cell chemoresistance, we used Annexin V-FITC/ propidium iodide (PI) staining-based fluorescence activated cell sorting (FACS) analysis to examine cell apoptosis. Knockdown of NgBR did not increase apoptosis of Bel/5FU cells, and 5-FU treatment alone did not increase apoptosis of the Bel/5FU control cell (non-specific siRNA or NS) either (Figure 2C). However, 5-FU increased apoptosis of Bel/5FU NgBR knockdown cells (siNgBR). AO/EB staining incorporation assay indicates that NgBR knockdown decreased the chemoresistance of the Bel/5FU cell by increasing 5-FU induced HCC cell apoptosis (Supplementary Figure S2).
NgBR knockdown induces the abrogation of S-phase arrest of Bel/5FU cells by increasing p53 protein level
Cell-cycle dysregulation is a hallmark of cancer cells. Cell-cycle checkpoint protein dysfunction can alter the chemoresistance of cancer cells to chemotherapeutics [21]. To explore whether the chemoresistance of Bel/5FU cells is caused by cell cycle change, we examined the percentages of cell cycle distribution by PI staining-based FACS analysis. The results (Figure 3A) show that more Bel/5FU cells undergo S-phase arrest compared with the Bel7402 cells. Interestingly, knockdown of NgBR abrogated the S phase arrest in Bel/5FU cells (Figure 3B). Furthermore, by restoring NgBR expression by transfection of pIRES-NgBR, an NgBR-expressing plasmid, we found that NgBR overexpression rescues the abrogation of S-phase arrest presented in Bel/5FU NgBR knockdown cells (Figure 3C). These results suggest that chemoresistance of Bel/5FU cells may be caused by S phase arrest, and knockdown of NgBR abrogates this S phase arrest thus decreasing the chemoresistance of the Bel/5FU cells.
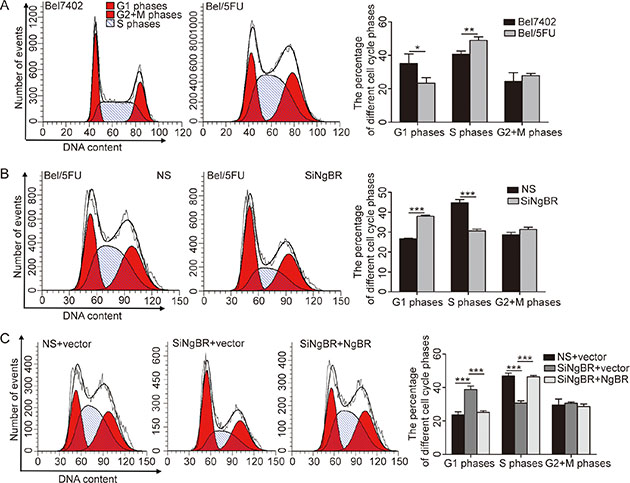
Figure 3: Knockdown of NgBR abrogates the S phase arrest in Bel/5FU cells. (A) Bel/5FU cells undergo S phase arrest. The cell cycle distribution in Bel7402 cells and Bel/5FU cells was analyzed by flow cytometry by using PI staining. Representative figure from three independent experiments is shown in left panel. The percentage of different cell cycle phases was drawn in histogram form to reflect the alteration of cell cycle phases (right panel). The mean and SD obtained from three independent experiments are plotted (*P < 0.05, **P < 0.01). (B) Knockdown of NgBR restores the S phase arrest in Bel/5FU cells. The cell cycle distribution in Bel/5FU cells was analyzed by flow cytometry by using PI staining. Results are shown as the mean ± SD of three independent experiments (***P < 0.001). (C) Overexpression of NgBR rescued the cell cycle distribution impaired by siNgBR in Bel/5FU cells. Bel/5FU cells were co-transfected with siNgBR or NS and NgBR expression vector pIRES-NgBR or pIRES empty vector. Cell cycle was measured using flow cytometry. The mean and SD obtained from three independent experiments are plotted (***P < 0.001).
Given the S phase arrest, p53 protein and downstream p21 protein are the most important G1/S phase checkpoint proteins involved [22]. p21 binds to the CDK6/CyclinD1 complex and blocks the kinase activity of CDKs [23]. The CDK6/CyclinD1 complex increases phosphorylated Rb protein level followed by cell entry into S phase from G1 phase [21]. Therefore, we examined the p53 and p21 protein level in Bel7402 and Bel/5FU cells by western blot. As shown in Figure 4A, the p53 protein and downstream protein p21 protein level were decreased in Bel/5FU cells as compared to the control Bel7402 cells. Consequently, CDK6 and CyclinD1 protein expression as well as phosphorylated Rb protein levels are increased in Bel/5FU cells as compared to the control Bel7402 cells (Figure 4A). On the contrary, knockdown of NgBR in Bel/5FU cells increased p53 and p21 protein levels and consequently decreased CDK6, CyclinD1, and phosphorylated Rb protein levels as compared to the control Bel/5FU NS cells (Figure 4B). Furthermore, these protein level changes in NgBR knockdown Bel/5FU cells can be rescued by co-transfecting pIRES-NgBR plasmid DNA and siNgBR together as shown in Figure 4C. Collectively, these results suggest the altered cell cycle caused by decreased expression of p53 proteins and consequent reduction of p53 downstream signal pathway contributes to the chemoresistance of Bel/5FU cells. Knockdown of NgBR in Bel/5FU cells restores the p53 protein expression as well as normal cell cycle function and thus decreases the chemoresistance of Bel/5FU cells.
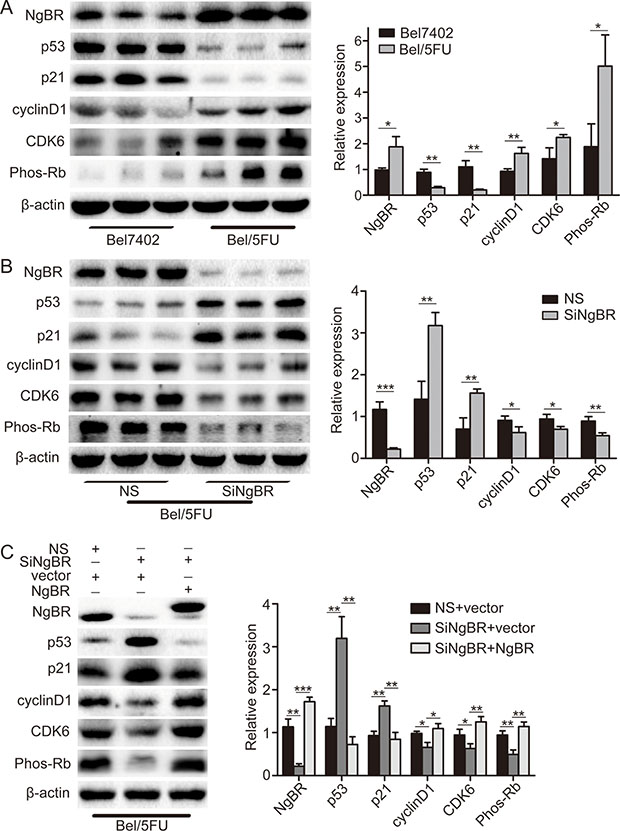
Figure 4: NgBR regulates p53 protein expression in Bel/5FU cells. (A) p53 protein was decreased in Bel/5FU cells. p53, p21, cyclinD1, CDK6, and Phos-Rb protein levels in Bel7402 cells and Bel/5FU cells were determined using western blot analysis (left panel). Band intensities were quantified using Image Lab 5.0 software and were normalized to β-actin (right panel). The data are presented as the mean ± SD of three separate experiments. (*P < 0.05, **P < 0.01). (B)Knockdown of NgBR increases p53 protein level in Bel/5FU cells. The expression of p53, p21, cyclinD1, CDK6, and Phos-Rb in Bel/5FU cells were detected by western blot assay (left panel). The quantitative measurement is shown in right panel. The mean and SD obtained from three independent experiments are plotted (*P < 0.05, **P < 0.01, ***P < 0.001). (C) Overexpression of NgBR rescued the p53 protein level change induced by NgBR siRNA in Bel/5FU cells. The Bel/5FU cells were co-transfected with siNgBR or NS as well as pIRES-NgBR or pIRES empty vector. Expression of p53, p21, cyclinD1, CDK6, and Phos-Rb proteins was then examined using western blot analysis (left panel). The quantitative measurement is shown in right panel. The mean and SD obtained from three independent experiments are plotted (*P < 0.05, **P < 0.01, ***P < 0.001).
NgBR promotes the ubiquitination of p53 via Akt-mediated MDM2 phosphorylation
To further assess the mechanisms by which NgBR reduces p53 protein level, we investigated whether the p53 mRNA levels were changed in Bel/5FU cells first. The results showed that the p53 mRNA levels were not changed in Bel/5FU cells as compared with Bel7402 cells. Nor does knockdown of NgBR in Bel/5FU cells change the p53 mRNA level (Figure 5A). These results indicate that NgBR does not affect p53 transcript levels.
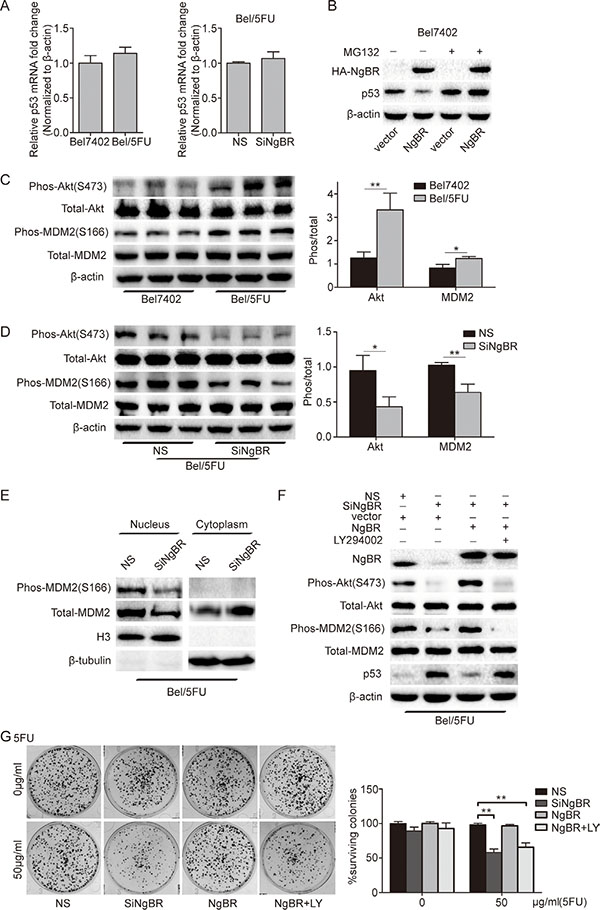
Figure 5: NgBR inhibits p53 expression by activating the PI3K/Akt/MDM2 mediated ubiquitin proteasome pathway. (A) The mRNA level of p53 is not changed in Bel/5FU cells. The mRNA level of p53 in Bel7402 cells and Bel/5FU cells was analyzed using real-time RT-PCR (left panel). Bel/5FU cells were transfected with the indicated siRNA and then the mRNA level of p53 was analyzed by real-time RT-PCR (right panel). The relative amount of p53 mRNA level was normalized to the β-actin. (B) NgBR negatively regulates p53 expression in a proteasome dependent manner. Bel7402 cells were transfected with pIRES-NgBR or pIRES empty vector plasmid DNA for 48 h and then incubated with or without MG132 (20 μM) for an additional 4 h. Whole-cell lysates were analyzed by western blotting. (C) Phos-Akt and phos-MDM2 levels are increased in Bel/5FU cells. The phos-Akt and phos-MDM2 levels were assessed by western blotting. Total Akt and total MDM2 protein levels were used as a loading control (left panel). Band intensities were quantified using Image Lab 5.0 software(right panel). Results are shown as the mean ± SD of three independent experiments (*P < 0.05, **P < 0.01). (D) Knockdown of NgBR decreases the phos-Akt and phos-MDM2 level in Bel/5FU cells. Phos-Akt and phos-MDM2 levels were detected by western blot assay (left panel). The expression of each protein was quantified as the densitometry value analyzed by Image Lab 5.0 software and is normalized to total Akt and total MDM2 (right panel). The data are presented as the mean ± SD of three separate experiments. (*P < 0.05, **P < 0.01). (E) NgBR knockdown decreases the translocation of MDM2 from the cytoplasm to the nucleus. Localization of total MDM2 and phos-MDM2 proteins in nuclear (N) and cytoplasmic (C) cell fractions from Bel/5FU NS and Bel/5FU siNgBR cells was examined by western blot assay. Histone H3 and β-tublin were used as internal markers for the nucleus and cytoplasm, respectively. (F) NgBR regulates phos-MDM2 via Akt pathway. The Bel/5FU cells were co-transfected with siNgBR or NS with pIRES–NgBR plasmid or pIRES empty vector for 24 h and then incubated with or without LY294002 (25 μM) for an additional 48 h. The levels of phos-Akt, phos-MDM2, and p53 were detected by western blot assay. Total Akt, total MDM2, and β-actin protein levels were used as loading control. (G) NgBR regulates chemoresistance of Bel/5FU cells via Akt pathway. Clonogenic formation assay was used for measuring clonogenicity of Bel/5FU cells. The number of untreated cells is set as 100% and the results show the average percentage of surviving colonies. The data are presented as the mean ± SD of three independent experiments. (**P < 0.01, ***P < 0.001).
Previous reports [24, 25] showed that p53 protein undergoes degradation via the ubiquitin proteasome pathway. To determine the role of NgBR in regulating ubiquitination and degradation of p53 proteins in HCC cells, the degradation dynamics assay was used to detect the p53 protein level in Bel7402 and Bel/5FU cells. Cycloheximide(CHX) inhibits new protein translation. Our results show that the p53 protein diminished in control Bel/5FU NS cells 30 minutes after CHX treatment. However, the half-life of the p53 protein was increased in the NgBR knockdown Bel/5FU cells, and p53 protein expression was still detected after 2 hours of CHX treatment (Supplementary Figure S3A). In addition, we found that p53 protein was decreased in Bel7402 cells with pIRES-NgBR plasmid overexpression of NgBR (Figure 5B). However, if we treated the cells with the proteasome inhibitor MG132, NgBR overexpression does not change the p53 protein level as compared to Bel7402 cells transfected with control empty vector (Figure 5B). Furthermore, ubiquitin antibody immunoprecipitation results show that NgBR knockdown reduces p53 ubiquitination in Bel/5FU cells (Supplementary Figure S3B). These results suggest that NgBR regulates p53 protein level by promoting the degradation of p53 via ubiquitin proteasome pathway.
MDM2, an ubiquitin ligase for p53, plays a central role in the regulation of p53 protein stability. Previous studies have demonstrated Akt mediated phosphorylation of MDM2, allowing ubiquitination and degradation of p53 [26–29]. And It has been demonstrated that phosphorylation of Akt increased the translocation of MDM2 from the cytoplasm to the nucleus [30]. Our previous work has reported that NgBR regulates the Akt phosphorylation [18, 20]. Therefore, we hypothesized that NgBR regulates p53 protein stability via the PI3K/Akt/MDM2 signaling pathway in HCC cells. To test this hypothesis, we first used western blot analysis to detect the phosphorylation levels of both Akt and MDM2 in Bel7402 and Bel/5FU cells. The results showed that the phos-Akt and phos-MDM2 levels in Bel/5FU cells were higher than in the control Bel7402 cells, while the total Akt and MDM2 remained unchanged (Figure 5C). Knockdown of NgBR in Bel/5FU cells by siRNA significantly decreased the phosphorylation of both Akt and MDM2 levels while the total Akt and MDM2 remained unchanged (Figure 5D). In addition, NgBR knockdown decreases the translocation of MDM2 from the cytoplasm to the nucleus in Bel/5FU cells (Figure 5E). Furthermore, overexpression of NgBR by co-transfected pIRES-NgBR plasmid together with siNgBR in Bel/5FU cells can rescue impaired phosphorylation of both Akt and MDM2 levels in NgBR knockdown Bel/5FU cells (Figure 5F). While treatment with the PI3K inhibitor, LY294002, inhibits the levels of p-Akt and p-MDM2 in Bel/5FU cells highly expressing NgBR (Figure 5F). Meanwhile, a clonogenic survival assay showed that LY294002 also inhibits the rescue effects of NgBR overexpression in NgBR knockdown Bel/5FU cells (Figure 5G). Collectively, our results demonstrate that NgBR inhibits p53 expression by activating the PI3K/Akt/MDM2 pathway to promote the degradation of p53 via ubiquitin proteasome pathway and thus increases the chemoresistance of Bel/5FU cells.
Knockdown of NgBR decreases tumor chemoresistance to 5-FU by increasing p53 protein leves in hepatocellular carcinoma in vivo
To further examine the in vivo role of NgBR in HCC chemoresistance to 5-FU treatment, we formed tumor xenografts by injecting Bel/5FU cells into nude mice. As shown in Figure 6A–6C, the growth of tumor xenografts was slower in the NgBR siRNA injected group, and 5-FU treatment further decreased the tumor size and weight dramatically in NgBR siRNA injected tumor xenografts. However, 5-FU treatment alone did not decrease tumor size or weight in the NS siRNA injected tumor xenografts, which confirms the chemoresistance of the Bel/5FU tumor xenografts in vivo. Further analysis of the tumor samples by IHC (Figure 6D) confirmed that NgBR reduction by injection of siNgBR significantly stabilized p53 expression in tumor xenografts. Collectively, our data strongly suggest that NgBR knockdown reduces the chemoresistance of human hepatocellular drug-resistant tumors to 5-FU through stabilizing p53 protein levels in vivo.
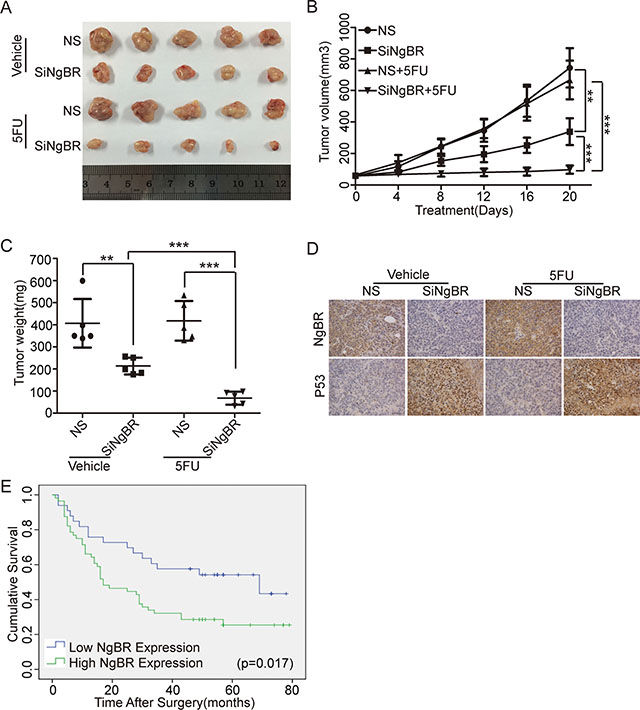
Figure 6: NgBR deficiency reverses the chemoresistance of human hepatocellular drug-resistant tumor to 5-FU through regulating p53 expression in vivo. (A) Knockdown of NgBR decreases the tumor size. The representative images of Bel/5FU xenograft tumors injected intratumorally with non-silencing control siRNA (NS) or NgBR siRNA (siNgBR) and treated with vehicle or 5-FU for 3 weeks. (B) Knockdown of NgBR decreases the tumor volumes. Tumor volumes of different tumor and treatment groups were calculated as described in methods. The data are presented as the mean ± SD of three independent experiments. (**P < 0.01, ***P < 0.001). (C) Knockdown of NgBR decreases the tumor weights. The data are presented as the mean ± SD of three independent experiments. (**P < 0.01, ***P < 0.001). (D) Knockdown of NgBR increases p53 protein level in vivo. Representative immunohistochemical staining images showing the staining of NgBR and p53 in tumors tissue samples obtained from treatment groups at the end point. Scale bars, 50 μm. (E) NgBR expression is negatively associated with overall survival of human HCC patients. Kaplan-Meier analysis of overall survival with high or low NgBR expression of 89 primary human HCC patients (P = 0.017, log-rank test).
NgBR is highly expressed in human HCC patient tissues and associated with a poor prognosis of HCC patients
To explore whether NgBR is an important factor in determining clinical outcomes of HCC patients, we examined the expression of NgBR in 89 primary HCC patient tissues and their corresponding adjacent liver tissues in a tissue microarray by IHC. Positive immunoreactivity for NgBR was observed primarily in the cell membrane and cytoplasm (Supplementary Figure S4A). As shown in Table 1, NgBR was highly expressed in HCC tissues compared with their corresponding adjacent liver tissues (P = 0.002). Segregation of patients into NgBR-positive and NgBR-negative groups did not reveal NgBR expression significant correlations with clinical pathological parameters of sex, gender, hepatitis history, liver cirrhosis, maximal tumor size, tumor number, vascular invasion, or TNM stage (Table 2). Furthermore, the overall survival analysis indicated patients with low NgBR expression owned significantly higher survival rates compared to the patients with high NgBR expression (P = 0.017) (Figure 6E). In addition, multivariate analyses revealed that vascular invasion and TNM stage, which are the established prognostic predictors for HCC [31], are independent prognostic factors for patient survival (Table 3). Our results indicate that NgBR is an independent prognostic factor for overall survival (hazard ratio, 2.255; 95% confidence interval, 1.209–4.206; P = 0.011) (Table 3). These results demonstrate that high NgBR expression is important in tumor progression and serves as an independent molecular marker for poor HCC prognosis.
Table 1: NgBR expression in tumor and adjacent tissues of liver cancer
|
|
Adjacent tissues |
n |
P |
|
|---|---|---|---|---|---|
Low |
High |
||||
Tumor tissues |
Low |
21 |
12 |
33 |
0.002* |
High |
16 |
40 |
56 |
|
|
n |
37 |
52 |
89 |
|
|
Table 2: Correlation between NgBR expression determined by immunohistochemical staining and clinicopathological parameters in patients with liver cancer
Clinicopathological parameters |
n |
NgBR expression |
P |
||
|---|---|---|---|---|---|
Low |
High |
||||
Age (y) |
|
|
|
|
|
|
≤ 54 |
47 |
22 |
25 |
0.051 |
|
> 54 |
42 |
11 |
31 |
|
Gender |
|
|
|
|
|
|
Male |
80 |
29 |
51 |
0.906 |
|
Female |
9 |
4 |
5 |
|
Hepatitis history |
|
|
|
|
|
|
No |
27 |
8 |
19 |
0.474 |
|
Yes |
62 |
25 |
37 |
|
Cirrhosis |
|
|
|
|
|
|
No |
27 |
8 |
19 |
0.474 |
|
Yes |
62 |
25 |
37 |
|
Tumor size |
|
|
|
|
|
|
≤ 5 cm |
38 |
13 |
25 |
0.306 |
|
> 5 cm |
51 |
20 |
31 |
|
Tumor number |
|
|
|
|
|
|
Single |
46 |
15 |
31 |
0.389 |
|
Multiple |
43 |
18 |
25 |
|
Vascular invation |
|
|
|
|
|
|
No |
78 |
30 |
48 |
0.700 |
|
Yes |
11 |
3 |
8 |
|
TNM stage |
|
|
|
|
|
|
I + II |
46 |
19 |
27 |
0.511 |
|
III + IV |
43 |
14 |
29 |
|
Table 3: Multiple analysis of factors associated with overall survival
Variable |
Hazard Ratio |
95% CI |
P |
|---|---|---|---|
NgBR expression |
2.255 |
1.209 to 4.206 |
0.011* |
Age |
0.993 |
0.967 to 1.019 |
0.580 |
Gender |
1.184 |
0.455 to 3.079 |
0.730 |
Hepatitis history |
2.025 |
0.944 to 4.345 |
0.070 |
Cirrhosis |
2.025 |
0.944 to 4.345 |
0.070 |
Tumor size |
1.010 |
0.944 to 1.080 |
0.775 |
Tumor number |
0.719 |
0.359 to 1.437 |
0.350 |
Vascular invation |
3.117 |
1.385 to 7.018 |
0.006* |
TNM stage |
2.145 |
1.323 to 3.479 |
0.002* |
DISCUSSION
The development of drug resistance is the major obstacle in successful and effective chemotherapeutic treatment of HCC [32]. HCC frequently and easily acquires chemoresistance. Therefore, conventional chemotherapy treatments achieve poor efficacy in patients with advanced HCC and often do not show any benefit to survival [33]. The mechanism by which HCC acquires chemoresistance is still not well understood. In this study, we demonstrate that NgBR expression is upregulated in HCC cells with acquired chemoresistance and knockdown of NgBR in the chemoresistant Bel/5FU cells reduces the chemoresistance in HCC cells. These results indicate that NgBR could play an important role in the development of chemoresistance in HCC cells.
5-Fluorouracil (5-FU) is widely used in the treatment of a variety of tumors. It interferes with nucleoside metabolism and results in DNA synthesis disorders and RNA dysfunction, leading to cytotoxicity [34]. However, chemotherapeutic drugs that function by inducing DNA damage frequently fail due to tumors acquiring the ability to repair the DNA damage and therefore escaping cell death. Cell cycle dysregulation has been demonstrated to be involved in the growth inhibition and DNA repair by DNA-damaging drugs [35]. Alterations in expression of proteins that control progression through the cell cycle have been demonstrated to affect chemosensitivity [36]. Mutated forms of p53 and/or Rb have been shown to be associated with increased resistance of tumor cells to various anticancer drugs and irradiation, mainly because of cell cycle dysregulation [37]. Furthermore, alterations in other cell cycle regulators such as the cyclins, cyclin-dependent kinases (CDKs), and their inhibitors, p21Cip1/WAF1 and p27Kip1, may also play an important role in the regulation of drug sensitivity [38]. In this study, we show that chemoresistant Bel/5FU cells undergo an S-phase arrest not seen in Bel7402 parental cells. Surprisingly, we found that knockdown of NgBR abrogates the S-phase arrest in Bel/5FU cells. In addition, we also clearly demonstrate that altered expression of these cell cycle regulators is triggered by NgBR, which is consistent with alterations of the cell cycle distribution. Therefore, we conclude that targeting NgBR could partially reverse the drug resistance of Bel/5FU cells by regulating cell cycle regulators, which results in alterations of the cell cycle progression.
p53 suppresses cancer progression through the induction of cell cycle arrest, apoptosis, or senescence in response to a variety of cellular stimuli. As we know, p53 acts as the most important checkpoint protein to suppress cell growth by inhibiting G1 progression to S phase while the cell attempts to repair the damage or to promote apoptosis in cells that fail to repair [39, 40]. Cancer cells harboring wild-type p53 are generally sensitive to antitumor agents [41]. In contrast, loss of p53 function in cells, either through mutation or post-translational modification, might therefore be expected to lead to unchecked proliferation, tumor growth, and therapeutic resistance [42, 43]. In addition, a report showed that p53 gene transfection not only induced suppression of cell growth, but also increased the sensitivity of Bel/5FU cells to 5-FU, vincristine, and doxorubicin [32]. In this study, we found a robust loss of p53 in chemoresistant HCC cells (Bel/5FU) compared with their corresponding parental cells (Bel7402). Interestingly, we found that p53 protein levels increased upon knock down of NgBR in Bel/5FU cells.
Previous studies have demonstrated that Akt mediates phosphorylation of MDM2 and induces its nuclear translocation, allowing ubiquitination and degradation of p53 [26–29]. Our previous work has reported that NgBR regulates the Akt phosphorylation [18, 20]. In this study, we found that NgBR increases the level of phos-Akt; phos-Akt then phosphorylates MDM2, and phos-MDM2 facilitates the ubiquitination and degradation of p53 proteins. In conclusion, we show that NgBR facilitates ubiquitination and degradation of p53 proteins via the PI3K/Akt/MDM2 mediated ubiquitin proteasome pathway and thus increases the drug resistance of Bel/5FU cells. The proposed working model showing the roles of NgBR in regulating the chemoresistance of HCC cells is shown in Figure 7.
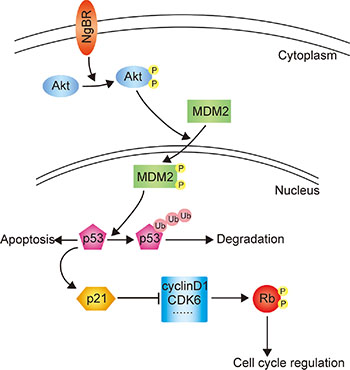
Figure 7: The proposed working model elucidating the roles of NgBR in regulating the chemoresistance of HCC cells.
In summary, our results demonstrate that higher NgBR expression in HCC cells contributes to 5-FU chemoresistance both in vitro and in vivo. This study provides compelling evidence in support of targeting NgBR in combination with 5-FU as a viable option for improved treatment of drug resistant HCC.
MATERIALS AND METHODS
Chemicals and reagents
5-Fluorouracil (5-FU) was purchased from Sigma Aldrich (St. Louis, MO, USA). The Cell Counting Kit-8 and Annexin V-FITC/PI Apoptosis Detection Kit were purchased from Dojindo (Kumamoto, Japan). Co-immunoprecipitation Kit was purchased from Thermo (Rockford, IL, USA). LY294002 and MG132 were purchased from Selleck (Houston, TX, USA). All the other chemicals and reagents were purchased from Sigma (St. Louis, MO, USA) unless otherwise specified. All the water used in experiments was purified using a Milli-Q system (Millipore, Bedford, MA, USA).
Antibodies
NgBR rabbit monoclonal antibody (CloneID: EPR8668) was generated by Epitomics (Burlingame, CA, USA) as a collaboration project. NgBR rabbit polyclonal antibody was generated as described previously [19] and was used for immunohistochemistry analysis. Rabbit polyclonal antibodies for phospho-AKT (Ser473), total Akt, Phospho-MDM2 (Ser166), cyclinD1, p21, phos-Rb (Ser807/811), HA-Tag rabbit monoclonal antibody, Histone H3, and β-actin and all the secondary antibodies were purchased from Cell Signaling (Danvers, MA, USA). Rabbit polyclonal antibodies for p53, ubiquitin, CDK6, and β-tublin were purchased from ProteinTech (Chicago, IL, USA). Rabbit polyclonal antibody for total MDM2 was purchased from Anbo Biotechnology (San Francisco, CA, USA).
Cell lines and cell culture
Human HCC cell lines Bel7402 and 5-FU-selected drug-resistant Bel7402/5FU (Bel/5FU) were obtained from KeyGen Biotech Co., Ltd (Nanjing, China). The cell lines were cultured in RPMI Medium 1640 (Gibco, USA) supplemented with antibiotics(1 × penicillin/streptomycin 100 U/ml, Gibco, USA) and 10% fetal bovine serum (Gibco, USA). To maintain the drug-resistant phenotype, the medium for the Bel/5FU cells was supplemented with 20 μg/ml 5-FU. Cells were incubated at 37°C in a humidified atmosphere containing 5% CO2.
Transfection of siRNA and plasmid DNA
NgBR siRNA1 (S1forward: GGAAAUACAUAGA CCUACA, S1reverse: UGUAGGUCUAUGUAUUUCC), NgBR siRNA2 (S2 forward: GUAUGGAAAUAAACU UAUA, S2 reverse: UAUAAGUUUAUUUCCAUAC) and NgBR siRNA3 (S3 forward: GCUGAUUCUUAGAU AGAAA, S3 reverse: UUUCUAUCUAAGAAUCAGC) oligonucleotides with 3′dTdT overhangs were synthesized by Shanghai GenePharma Co. (Shanghai, China). Control siRNA in experiments refers to an All-Star non-silencing siRNA (forward sequence:GGGUAUCGACGAUUACA AAUU, reverse sequence: UUUGUAAUCGUCGAUA CCCUG) synthesized by Shanghai GenePharma Co. (Shanghai, China). Lipofectamine RNAiMAX reagent (Invitrogen, Carlsbad, CA, USA) was used for the transfection of siRNA according to the manufacturer’s instructions. Lipofectamine 2000 (Invitrogen, Carlsbad, CA, USA) was used for transfection of NgBR expression plasmid pIRES-NgBR. The specificity of NgBR siRNA and pIRES-NgBR has been validated in our previous publications [17, 18].
Quantitative real-time polymerase chain reaction
Total RNA was extracted from cells by using TRIzol reagent according to the manual (TaKaRa Bio, Dalian, China) and cDNA was reverse-transcribed using the PrimeScript RT Reagent Kit (TaKaRa Bio, Dalian, China) according to the manufacturer’s instructions. Real-time PCR was performed using QuantiTect SYBR Green PCR kit (TaKaRa Bio, Dalian, China) and was run on Stratagene MX3000P (Agilent, CA, USA). The relative mRNA expression of each gene was normalized to β-actin RNA levels. The primers were synthesized by Invitrogen (Carlsbad, CA, USA). The forward and reverse primers for NgBR are 5′-tgccagttagtagcccagaagcaa-3′ and 5′-tgatgtgccagggaagaaagccta-3′, respectively. The forward and reverse primers for p53 are 5′-gcgtgtggagtatttggatgac-3′ and 5′-agtgtgatgatggtgaggatgg-3′, respectively. The forward and reverse primers for β-actin are 5′-ttctacaat gagctgcgtgtggct-3′ and 5′-tagcacagcctggatagcaacgta-3′ respectively.
Western blot
Cells were harvested and lysed in immune precipitation assay buffer (KeyGen Biotech Co., Ltd, Nanjing, China) supplemented with 1 mM phenylmethylsulfonyl floride (KeyGen Biotech Co., Ltd, Nanjing, China) and 1 mM phosphatase inhibitor cocktail (KeyGen Biotech Co., Ltd) for extracting whole cell protein samples. For detecting the cellular localization of total MDM2 and phos-MDM2, nuclear and cytoplasmic fractions were isolated using the Nuclear and Cytoplasmic Protein Extraction kit (Thermo Fisher Scientific) according to the instructions of the manufacturer. Protein concentration was determined using a BCA protein assay kit (KeyGen Biotech Co., Ltd). Same amount protein samples were separated on 10% SDS-PAGE gels and then transferred to a polyvinylidene difluoride membrane (Pall Corporation, Port Washington, NY, USA). The protein band intensities were evaluated using ECL western blotting kit (Advansta, Menlo Park, CA, USA) and were normalized to those of β-actin. All experiments were performed at least three times.
Clonogenic survival assay
Cells were seeded in triplicate into a 6-well culture dish (1000 cells/well). Cells were transfected with NgBR siRNA and/or NgBR expressing vector pIRES-NgBR. At 24 hours after transfection, cells were treated with 5-FU and/or Akt inhibitor LY294002 at the indicated doses for 72 hours. Then, the cells were maintained for 2 weeks. The cell colonies were washed three times with phosphate buffered saline buffer (PBS), fixed in methanol for 15 minutes, and stained with Crystal Violet (Sigma-Aldrich, St. Louis, MO, USA) for 15 minutes at room temperature.
Apoptosis assay by AO/EB staining
The cells cultured in 6-well plates were treated with different concentrations of 5-FU for 48 hours. After indicated treatment times, the cells were stained with acridine orange (AO, 200 μg/mL) and ethidium bromide (EB, 200 μg/mL) for 2 min and then washed with PBS to remove background staining. After that, cells were observed under a fluorescence microscope (Nikon Ti-S, Nikon Inc, Japan). The normal cells and early apoptotic cells can be stained by AO to display bright green fluorescence, while the late apoptotic cells can be stained by EB to display orange fluorescence.
Apoptosis measured by annexin V-FITC/propidium iodide (PI) staining
An Annexin V-FITC/PI apoptosis kit was used to quantify the percentage of cells undergoing apoptosis. Cells cultured in 6-well plates were treated with 5-FU (50 μg/mL) for 48 hours. Cells were then harvested and stained with 5 μL Annexin V-FITC and 5 μL PI in 500 uL of apoptosis reaction solution at room temperature in the dark for 30 min. Accuri C6 flow cytometer (Accuri Cytometers, Inc., Ann Arbor, MI, USA) was used to detect apoptotic cells. Cell population in different quadrants was calculated statistically.
Cell cycle analysis
The cells were treated with 5-FU at the indicated concentrations, harvested by trypsinization, and fixed overnight with 70% cold ethanol at −20°C. Fixed cells were stained with the 1 ml 50 μg/ml PI solution containing 0.1% Triton X-100 and 0.1 mg/ml RNase in the dark at 37°C for 30 min. The cell cycle profiles were obtained by Accuri C6 flow cytometer. ModFit LT software (Verify Software, Topsham, MN, USA) was used to calculate the percentages of cells in each cycle phase.
Cell viability assay
Cell viability was determined using the CCK-8 assay according to the manufacturer’s instructions. Cells were seeded in 96-well plates at 5000 cells/well. At 24 hours after seeding, cells were transfected with All Star NS siRNA or NgBR siRNA. Then, after the cells were treated with 5-FU (50 μg/ml) for 24, 48, or 72 hours, the medium was exchanged for 100 μl of RPMI-1640 and 10 μl of CCK-8 reagent was added. The cells were incubated for 3 hours at 37°C. The optical density was measured using an EnSpire™ 2300 Multilabel Reader (Perkin Elmer, Waltham, MA, USA) at 450 nm. Three replicates were prepared for each condition.
Ubiquitination assays
Cells were transfected with indicated siRNA and cultured for 48 h, and then incubated with 20 μM MG132 for 4 h. Whole-cell lysates were harvested and used for immunoprecipitation (IP) to detect protein ubiquitination. The IP experiments were performed with the Pierce Co-IP Kit (Thermo Scientific) according to the manufacturer’s protocol. 10 μg of the ubiquitin antibody were covalently coupled with the delivered resin. The antibody-coupled resin was incubated with 400 μg of the whole-cell protein lysates overnight at 4°C. The precipitated proteins were denatured and resolved by 2 × SDS-PAGE loading buffer. Ubiquitinated proteins were then detected by anti-p53 antibody.
Tissue microarray and immunohistochemistry analysis
The HCC tissue microarray containing 89 primary HCC samples and their corresponding adjacent liver tissues was purchased from Shanghai Outdo Biotech (Shanghai, China). The overall survival (OS) for the corresponding patients was calculated from the day of surgery to the day of death or to the last follow-up. Immunohistochemistry was performed using a standard methodology to analyze NgBR protein expression. Briefly, the tissue microarray (TMA) slides were heated in citrate buffer (pH 6.0) in a pressure cooker for 6 min and incubated with the rabbit polyclonal anti-NgBR antibody (1:200) overnight at 4°C. The TMA were then incubated with goat anti-rabbit Envision System plus-HRP (Dako Cytomation) for 30 min at room temperature and counter stained with Mayer’s hematoxylin. The degrees of immunostaining were reviewed and scored by two independent pathologists who were blinded to patient outcome. Cytoplasmic or membranous staining for NgBR was considered positive [19]. The proportion of the stained cells and the extent of the staining were used as criteria of evaluation. For each case, at least 1,000 tumor cells were analyzed and the percentage of the tumor cells with positively staining was recorded [44]. The percentage of positive cells was assigned a score from 0 (< 5%), 1 (5–25%), 2 (> 25–50%), 3 (> 50–75%), or 4 (> 75%) and the staining intensities within the respective subcellular locations were noted as 0 = negative, 1 = weak, 2 = moderate, or 3 = strong. A final score was then calculated by multiplying the above two scores. If the final score was equal or bigger than six, the protein expression in the tumor was considered high; otherwise, the protein expression in the tumor was considered low [19].
Animal studies
The male nu/nu mice (4–6 weeks old) were used and all animal experiments were maintained in SPF Laboratory Animal Center at Dalian Medical University. Cholesterol-conjugated All Star non-silencing siRNA (NS) and NgBR siRNA (siNgBR) for in vivo delivery were obtained from Shanghai GenePharmaCo. (Shanghai, China). Bel/5FU cells (5 × 106 in 100 μL PBS) were inoculated subcutaneously into the flank of the nude mice. Two weeks later, when the tumor diameters reached 4 mm to 5 mm, mice were randomly divided into 4 different groups n = 5/group): non-targeting siRNA injected group (NS), NgBR siRNA injected group (siNgBR), non-targeting siRNA injected plus 5-FU treatment group (NS + 5-FU) and NgBR siRNA injected plus 5-FU treatment group (siNgBR + 5-FU). 10 nmol indicated siRNA in 0.1 ml saline buffer was injected intratumorally twice a week for 3 weeks [44, 45]. At the same time, 30 mg/kg 5-FU in saline buffer was injected intraperitoneally three times per week for 3 weeks in the 5-FU treatment groups. Tumors were measured with a caliper every 4 days, and the tumor volume was calculated using the formula V = 1/2 (width2 × length). At 5 weeks after tumor cell inoculation, all mice were terminated with ether anesthesia and the total weight of the tumors in each mouse was measured. Tumor specimens were harvested for NgBR and p53 protein immunohistochemistry staining.
All animal maintenance and procedures were carried out in strict accordance with the recommendations established by the Animal Care and Ethics Committee of Dalian Medical University as well as the guidelines of the U.S. National Institutes of Health Guide for the Care and Use of Laboratory Animals. The protocol was approved by the Animal Care and Ethics Committee of Dalian Medical University.
Statistical analysis
All experiments were repeated three times. The chi squared test, Analysis of Variance, and two-tailed Student t test were performed as appropriate. The cumulative survival probability was evaluated using the Kaplan-Meier method, and the differences were assessed using the log-rank test. Cox multivariate regression analysis was used to determine independent prognostic factors. A p-value < 0.05 defined statistical significance. SPSS 17.0 software was used for all statistical analysis.
ACKNOWLEDGMENTS AND FUNDING
This work was supported by the funds from the National Natural Science Foundation of China (81272368, 81471755, 81470878, 21405153, 31571027) and the clinical capability construction project for Liaoning provincial hospitals (LNCCC-B03-2014). B.Z. thanks the support from the “Hundred Talents Program” of CAS. This work was supported in part by start-up funds from the Medical College of Wisconsin (MCW), NIH R01HL108938, and We Care Fund to QRM.
CONFLICTS OF INTEREST
The authors declare no conflicts of interest and no financial disclosures.
REFERENCES
1. Jemal A, Bray F, Center MM, Ferlay J, Ward E, Forman D. Global cancer statistics. CA Cancer J Clin. 2011; 61:69–90.
2. Siegel R, Naishadham D, Jemal A. Cancer statistics, 2012. CA Cancer J Clin. 2012; 62:10–29.
3. Maluccio M, Covey A. Recent progress in understanding, diagnosing, and treating hepatocellular carcinoma. CA Cancer J Clin. 2012; 62:394–399.
4. Llovet JM, Burroughs A, Bruix J. Hepatocellular carcinoma. Lancet. 2003; 362:1907–1917.
5. Llovet JM, Di Bisceglie AM, Bruix J, Kramer BS, Lencioni R, Zhu AX, Sherman M, Schwartz M, Lotze M, Talwalkar J, Gores GJ. Design and endpoints of clinical trials in hepatocellular carcinoma. J Natl Cancer Inst. 2008; 100:698–711.
6. Tong SW, Yang YX, Hu HD, An X, Ye F, Hu P, Ren H, Li SL, Zhang DZ. Proteomic investigation of 5-fluorouracil resistance in a human hepatocellular carcinoma cell line. J Cell Biochem. 2012; 113:1671–1680.
7. Cheng L, Luo S, Jin C, Ma H, Zhou H, Jia L. FUT family mediates the multidrug resistance of human hepatocellular carcinoma via the PI3K/Akt signaling pathway. Cell Death Dis. 2013; 4:e923.
8. Szakacs G, Paterson JK, Ludwig JA, Booth-Genthe C, Gottesman MM. Targeting multidrug resistance in cancer. Nat Rev Drug Discov. 2006; 5:219–234.
9. Teodori E, Dei S, Martelli C, Scapecchi S, Gualtieri F. The functions and structure of ABC transporters: implications for the design of new inhibitors of Pgp and MRP1 to control multidrug resistance (MDR). Curr Drug Targets. 2006; 7:893–909.
10. Asghar U, Meyer T. Are there opportunities for chemotherapy in the treatment of hepatocellular cancer? J Hepatol. 2012; 56:686–695.
11. Uchibori K, Kasamatsu A, Sunaga M, Yokota S, Sakurada T, Kobayashi E, Yoshikawa M, Uzawa K, Ueda S, Tanzawa H, Sato N. Establishment and characterization of two 5-fluorouracil-resistant hepatocellular carcinoma cell lines. Int J Oncol. 2012; 40:1005–1010.
12. Wang XQ, Ongkeko WM, Chen L, Yang ZF, Lu P, Chen KK, Lopez JP, Poon RT, Fan ST. Octamer 4 (Oct4) mediates chemotherapeutic drug resistance in liver cancer cells through a potential Oct4-AKT-ATP-binding cassette G2 pathway. Hepatology. 2010; 52:528–539.
13. Ma S, Lee TK, Zheng BJ, Chan KW, Guan XY. CD133+ HCC cancer stem cells confer chemoresistance by preferential expression of the Akt/PKB survival pathway. Oncogene. 2008; 27:1749–1758.
14. Huber AB, Weinmann O, Brosamle C, Oertle T, Schwab ME. Patterns of Nogo mRNA and protein expression in the developing and adult rat and after CNS lesions. J Neurosci. 2002; 22:3553–3567.
15. Josephson A, Trifunovski A, Widmer HR, Widenfalk J, Olson L, Spenger C. Nogo-receptor gene activity: cellular localization and developmental regulation of mRNA in mice and humans. J Comp Neurol. 2002; 453:292–304.
16. Acevedo L, Yu J, Erdjument-Bromage H, Miao RQ, Kim JE, Fulton D, Tempst P, Strittmatter SM, Sessa WC. A new role for Nogo as a regulator of vascular remodeling. Nat Med. 2004; 10:382–388.
17. Miao RQ, Gao Y, Harrison KD, Prendergast J, Acevedo LM, Yu J, Hu F, Strittmatter SM, Sessa WC. Identification of a receptor necessary for Nogo-B stimulated chemotaxis and morphogenesis of endothelial cells. Proc Natl Acad Sci U S A. 2006; 103:10997–11002.
18. Zhao B, Chun C, Liu Z, Horswill MA, Pramanik K, Wilkinson GA, Ramchandran R, Miao RQ. Nogo-B receptor is essential for angiogenesis in zebrafish via Akt pathway. Blood. 2010; 116:5423–5433.
19. Wang B, Zhao B, North P, Kong A, Huang J, Miao QR. Expression of NgBR is highly associated with estrogen receptor alpha and survivin in breast cancer. PLoS One. 2013; 8:e78083.
20. Zhao B, Xu B, Hu W, Song C, Wang F, Liu Z, Ye M, Zou H, Miao QR. Comprehensive proteome quantification reveals NgBR as a new regulator for epithelial-mesenchymal transition of breast tumor cells. J Proteomics. 2015; 112:38–52.
21. Stewart ZA, Westfall MD, Pietenpol JA. Cell-cycle dysregulation and anticancer therapy. Trends Pharmacol Sci. 2003; 24:139–145.
22. Levine AJ. p53, the cellular gatekeeper for growth and division. Cell. 1997; 88:323–331.
23. Wang Y, Fisher JC, Mathew R, Ou L, Otieno S, Sublet J, Xiao L, Chen J, Roussel MF, Kriwacki RW. Intrinsic disorder mediates the diverse regulatory functions of the Cdk inhibitor p21. Nat Chem Biol. 2011; 7:214–221.
24. Hirano Y, Ronai Z. A new function for p53 ubiquitination. Cell. 2006; 127:675–677.
25. Brooks CL, Gu W. p53 ubiquitination: Mdm2 and beyond. Mol Cell. 2006; 21:307–315.
26. Abraham AG, O’Neill E. PI3K/Akt-mediated regulation of p53 in cancer. Biochem Soc Trans. 2014; 42:798–803.
27. Ogawara Y, Kishishita S, Obata T, Isazawa Y, Suzuki T, Tanaka K, Masuyama N, Gotoh Y. Akt enhances Mdm2-mediated ubiquitination and degradation of p53. J Biol Chem. 2002; 277:21843–21850.
28. Zhou BP, Liao Y, Xia W, Zou Y, Spohn B, Hung MC. HER-2/neu induces p53 ubiquitination via Akt-mediated MDM2 phosphorylation. Nat Cell Biol. 2001; 3:973–982.
29. Huang Q, Li J, Xing J, Li W, Li H, Ke X, Zhang J, Ren T, Shang Y, Yang H, Jiang J, Chen Z. CD147 promotes reprogramming of glucose metabolism and cell proliferation in HCC cells by inhibiting the p53-dependent signaling pathway. J Hepatol. 2014; 61:859–866.
30. Zhou M, Gu L, Findley HW, Jiang R, Woods WG. PTEN reverses MDM2-mediated chemotherapy resistance by interacting with p53 in acute lymphoblastic leukemia cells. Cancer Res. 2003; 63:6357–6362.
31. Gao Q, Qiu SJ, Fan J, Zhou J, Wang XY, Xiao YS, Xu Y, Li YW, Tang ZY. Intratumoral balance of regulatory and cytotoxic T cells is associated with prognosis of hepatocellular carcinoma after resection. J Clin Oncol. 2007; 25:2586–2593.
32. Li YX, Lin ZB, Tan HR. Wild type p53 increased chemosensitivity of drug-resistant human hepatocellular carcinoma Bel7402/5-FU cells. Acta Pharmacol Sin. 2004; 25:76–82.
33. Patt YZ, Hassan MM, Aguayo A, Nooka AK, Lozano RD, Curley SA, Vauthey JN, Ellis LM, Schnirer, II, Wolff RA, Charnsangavej C, Brown TD. Oral capecitabine for the treatment of hepatocellular carcinoma, cholangiocarcinoma, and gallbladder carcinoma. Cancer. 2004; 101:578–586.
34. Ma X, Cheng Z, Jin Y, Liang X, Yang X, Dai Z, Tian J. SM5-1-conjugated PLA nanoparticles loaded with 5-fluorouracil for targeted hepatocellular carcinoma imaging and therapy. Biomaterials. 2014; 35:2878–2889.
35. Huhn D, Bolck HA, Sartori AA. Targeting DNA double-strand break signalling and repair: recent advances in cancer therapy. Swiss Med Wkly. 2013; 143:w13837.
36. Hodkinson PS, Elliott T, Wong WS, Rintoul RC, Mackinnon AC, Haslett C, Sethi T. ECM overrides DNA damage-induced cell cycle arrest and apoptosis in small-cell lung cancer cells through beta1 integrin-dependent activation of PI3-kinase. Cell Death Differ. 2006; 13:1776–1788.
37. Gregorc V, Ludovini V, Pistola L, Darwish S, Floriani I, Bellezza G, Sidoni A, Cavaliere A, Scheibel M, De Angelis V, Bucciarelli E, Tonato M. Relevance of p53, bcl-2 and Rb expression on resistance to cisplatin-based chemotherapy in advanced non-small cell lung cancer. Lung Cancer. 2003; 39:41–48.
38. Hui R, Finney GL, Carroll JS, Lee CS, Musgrove EA, Sutherland RL. Constitutive overexpression of cyclin D1 but not cyclin E confers acute resistance to antiestrogens in T-47D breast cancer cells. Cancer Res. 2002; 62:6916–6923.
39. Blagosklonny MV. P53: an ubiquitous target of anticancer drugs. Int J Cancer. 2002; 98:161–166.
40. Shen Y, White E. p53-dependent apoptosis pathways. Adv Cancer Res. 2001; 82:55–84.
41. O’Connor PM, Jackman J, Bae I, Myers TG, Fan S, Mutoh M, Scudiero DA, Monks A, Sausville EA, Weinstein JN, Friend S, Fornace AJ, Jr., et al. Characterization of the p53 tumor suppressor pathway in cell lines of the National Cancer Institute anticancer drug screen and correlations with the growth-inhibitory potency of 123 anticancer agents. Cancer Res. 1997; 57:4285–4300.
42. Levine AJ, Oren M. The first 30 years of p53: growing ever more complex. Nat Rev Cancer. 2009; 9:749–758.
43. Brown CJ, Lain S, Verma CS, Fersht AR, Lane DP. Awakening guardian angels: drugging the p53 pathway. Nat Rev Cancer. 2009; 9:862–873.
44. Xiao Y, Wang J, Qin Y, Xuan Y, Jia Y, Hu W, Yu W, Dai M, Li Z, Yi C, Zhao S, Li M, Du S, et al. Ku80 cooperates with CBP to promote COX-2 expression and tumor growth. Oncotarget. 2015. doi: 10.18632/oncotarget.3508.
45. Hou J, Lin L, Zhou W, Wang Z, Ding G, Dong Q, Qin L, Wu X, Zheng Y, Yang Y, Tian W, Zhang Q, Wang C, et al. Identification of miRNomes in human liver and hepatocellular carcinoma reveals miR-199a/b-3p as therapeutic target for hepatocellular carcinoma. Cancer Cell. 2011; 19:232–243.