
INTRODUCTION
The DNA repair system represents a central mechanism of cellular homeostasis facilitating the detection and consequent repair of exogenous and endogenous DNA damage, thus preventing the perpetuation of potentially detrimental mutations and cancer formation. It consists of multiple DNA repair mechanisms which, depending on the recognized DNA damage pattern and cell cycle phase, activate different DNA repair pathways [1, 2]. Upon recognition of DNA damage, a complex system of events is triggered to restore genetic integrity. However, when DNA damage accumulates beyond the capability of the DNA repair system, apoptosis is triggered [3].
The Fanconi Anemia (FA) pathway represents the central DNA repair pathway for homologous recombination and is activated in response to stalled replication forks during the S-phase of the cell cycle [4]. Inactivation of this pathway occurs in patients suffering from the rare recessive disorder FA and is caused by bi-allelic germline mutations in one of at least 15 FA genes [5]. In addition, FA pathway-inactivation occurs at low frequency in various cancer entities among the general (non-FA) population, indicating a selective advantage during carcinogenesis, either through an early malignant transformation-promoting step due to an increased mutation rate or through an increased cellular tolerance towards abnormal DNA replication [6]. For example, proximal FA pathway inactivation due to rare mutations in FANCC or FANCG has been reported in pancreatic cancer [7, 8] and epigenetic inactivation of FANCF was found in a large variety of different tumors, including bladder cancer [9], breast cancer [10], cervical cancer [11], head and neck cancer [12], lung cancer [12] and ovarian cancer [13]. Even more prominently, inactivation of the distal FA pathway through mutations in the BRCA2 (FANCD1) gene has been reported in breast cancer [14] (familial cases [15–17]), pancreatic cancer [18, 19] and ovarian cancer [20], among others. Due to the well-established hypersensitivity of FA pathway-deficient tumor cells towards DNA-damaging ICL-agents, FA gene defects define patient subpopulations for individualized genotype-based therapies [21–23]. However, due to the side effects of these agents, there is a need to identify additional agents eliciting FA hypersensitivity, which could then be applied either alone or in combination with ICL-agents [23]. This concept was recently substantiated by reports of strong clinically responses of FA pathway-deficient cancers towards ICL-agents and PARP-inhibitors [24–28].
The two functional receptors for TRAIL, TRAIL-receptor-1 (TRAIL-R1) and TRAIL-receptor-2 (TRAIL-R2), are expressed in most human tissues and tumors and possess the particular ability to trigger apoptosis in cancer cells but not in non-malignant cells [29]. This tumor-selective pro-apoptotic effect of TRAIL-R stimulation is thought to reflect the physiological role played by the TRAIL-system during tumor-surveillance, which is regulated by the immune-mediated clearance of malignant and metastatic cells during the development of tumors. This function is supported by studies showing a correlation between loss of TRAIL-R-expression, poor prognosis and tumor recurrence [30–33] and by in vivo studies showing that TRAIL knockout (KO) mice exhibit enhanced primary tumor and metastasis formation [34]. Thus, TRAIL represents a promising novel anti-cancer therapy. Many types of recombinant TRAIL or agonistic antibodies targeting TRAIL-R have been made available for clinical use [29] and are currently tested in clinical trials. However, none of the previously conducted trials with TRAIL-R-targeting compounds reached their endpoint of improving patients’ outcomes (reviewed in [35]). One possible explanation for the failure of such agents to reproduce the effects achieved in preclinical experiments could be represented by the heterogeneity of the distribution of cell surface-bound cell receptors, as we previously suggested [31–33]. This idea seems to be supported by a very recent clinical trial showing that TRAIL-R2 imaging with radioactively labelled tigatuzumab (CS-1008) is predictive of clinical benefit in the treatment of patients affected by metastatic colorectal cancer [35]. In addition, the existence of intracellular mechanisms of resistance to TRAIL are likely limiting the clinical efficacy of these agents [36].
BRCA2-mutations are known to confer chemosensitivity to ICL-agents. Although this effect is known to be mediated by cell cycle jamming in consequence of impaired DNA repair, apoptosis is eventually triggered in these cells. In the present paper we assessed the possibility that BRCA2-deficient cells could thus display enhanced sensitivity towards the action of TRAIL in order to define a subpopulation of tumors highly susceptible to TRAIL-induced apoptosis which would be highly likely to respond to the clinical application of TRAIL-R targeting compounds. This hypothesis was tested first in vitro applying a BRCA2 gene knockout (KO) model of the colorectal cancer cell line DLD1 [37] and a BRCA2 gene complementation model of the BRCA2-mutant pancreatic cancer cell line CAPAN1 [38]. The in vitro results were consecutively validated in vivo using LBY in a murine xenograft model of BRCA2-deficient cancer cells. In addition, the underlying mechanisms of the observed hypersensitivity of BRCA2-deficient cancer cells towards apoptosis-inducing agents were explored.
RESULTS
Genetic inactivation of BRCA2 enhances the susceptibility of cancer cells towards TRAIL-R-mediated apoptosis
To assess the effects of BRCA2 status on the sensitivity towards TRAIL-R-mediated apoptosis, proliferation assays were performed upon administration of recombinant human TRAIL in parental BRCA2 wild type DLD1 colon cancer cells (termed DLD1), heterozygote BRCA2+/− KO cells (clone termed A9A2) and homozygous BRCA2−/− KO cells (two independently established clones termed A10-1 and A10-3, respectively). As a control, the cells were treated with the ICL-agent MMC, for which the specific hypersensitivity of BRCA2-deficient cells is well-established.
Upon treatment with MMC, we expectedly observed a strongly reduced proliferation rate in the two BRCA2−/−cell clones A10-1 and A10-3 as compared to the parental BRCA2+/+ and heterozygote BRCA2+/− A9A2 cells (IC50 ratio approx. 10). A similar pattern was observed upon administration of TRAIL, which also caused a dose-dependent proliferation decrease in both BRCA2−/− clones as compared to control cells (IC50 ratio approx. 5) (Figure 1A). Similar results were achieved after treatment with the FAS-agonistic antibody anti-Apo-1, which shows that the effects of BRCA2-status are not limited to TRAIL-R stimulation, but affect mechanisms of the activation of apoptosis common to other receptors of the extrinsic signaling pathway (Figure S1).
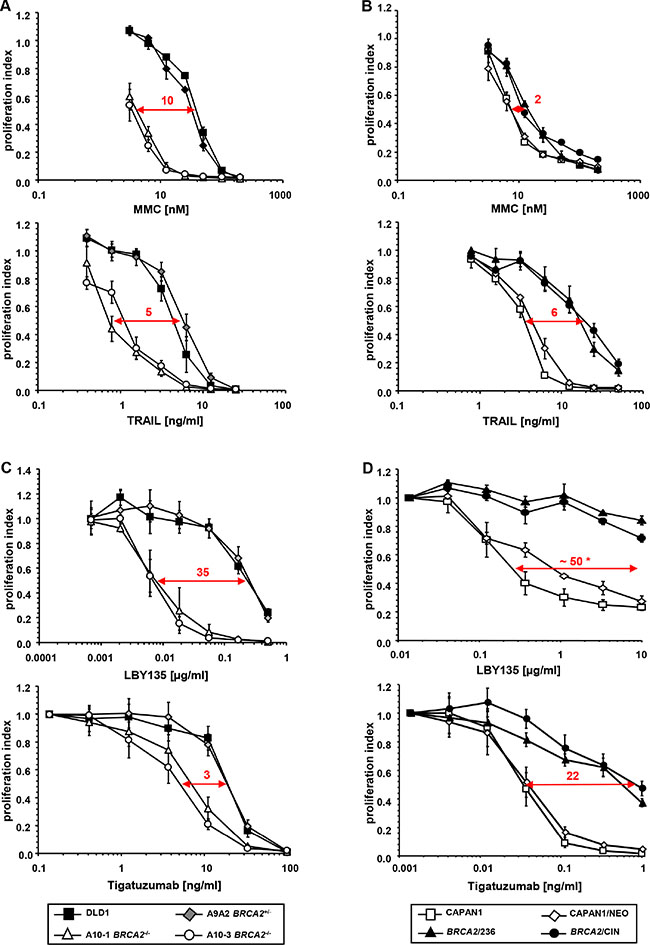
Figure 1: Genetic BRCA2 inactivation enhances the sensitivity of cancer cells towards TRAIL-R-mediated apoptosis. (A) Proliferation assays of BRCA2-proficient DLD1 and corresponding BRCA2+/− A9A2 cells versus two corresponding homozygously BRCA2-deleted clones (A10-1 BRCA2−/− and A10-3 BRCA2−/−) after treatment with the indicated agents. (B) Confirmatory proliferation assays of BRCA2-deficient parental (CAPAN1) and empty vector-transfected (CAPAN1/NEO) pancreatic cancer cells versus two BRCA2-complemented cell clones CAPAN1/CIN and CAPAN1/236 after treatment with the indicated agents. All experiments were performed in triplicate with error bars representing SEM from three independent experiments. * = extrapolated value. (C, D) Effects of BRCA2-status on cell viability upon administration of the clinically viable TRAIL-R2-targeting agonistic antibodies LBY-135 and tigatuzumab in BRCA2-proficient and BRCA2-deficient DLD1 cells (C) and in BRCA2-proficient and BRCA2-deficient CAPAN1 cells (D).
Susceptibility towards TRAIL-R-mediated apoptosis is decreased by re-expression of BRCA2 in BRCA2-deficient cancer cells
To rule out cell line- or method-dependent phenomena, the data obtained in DLD1 cells were confirmed in a BRCA2-complementation model established by Wang et al. [38], employing the pancreatic cancer cell line CAPAN1. This cell line harbors the naturally occurring, inactivating BRCA2 6174delT frameshift mutation accompanied by loss of the second allele, the severe impact of which on BRCA2 function has previously been extensively characterized [39]. In our experiments, parental CAPAN1 cells (termed CAPAN1) and empty-vector transfected cells (termed CAPAN1/NEO) were compared with two independently established CAPAN1 cell clones complemented by stably transfected BRCA2 (termed BRCA2/236 and BRCA2/CIN). In these cells, re-expression of BRCA2 decreased the sensitivity towards MMC as well as towards TRAIL (Figure 1B). However, the changes towards MMC were less obvious than those observed in the DLD1 model (IC50 ratio approx. 2), which might be attributable to the different experimental approaches (stable BRCA2 overexpression in the BRCA2-deficient CAPAN1 line versus complete BRCA2 KO in the BRCA2-proficient DLD1 line), a phenomenon previously observed in other studies [22, 23].
BRCA2 inactivation enhances the sensitivity of cancer cells towards TRAIL-R agonistic antibodies
To assess the potential clinical relevance of TRAIL-R targeting in BRCA2-deficient tumors, experiments were conducted in both the BRCA2 KO DLD1 as well as the BRCA2-complementation CAPAN1 models using LBY135 [40] and tigatuzumab [41], two TRAIL-R2-targeting antibodies which have been recently made available for clinical application. These experiments clearly confirmed the enhanced susceptibility of BRCA2-deficient cells towards TRAIL-R-stimulation (Figure 1C and 1D). Of note, BRCA2−/− cell clones displayed a 35-fold increased sensitivity towards LBY135, an effect significantly exceeding the well-established effects of all common DNA-ICL agents [21, 37]. This agent was thus expected to exert particularly strong effects in the experimental in vivo setting. Consequently, LBY135 was used for subsequent experiments.
siRNA-mediated BRCA2 knockdown enhances the sensitivity of cancer cells towards TRAIL-R-mediated apoptosis
To rule out clonal artefacts potentially occurring in gene KO and gene complementation models, RNA-interference experiments using BRCA2-siRNA were conducted in DLD1 cells. At 72 h after transfection with BRCA2-siRNA at 25 or 50 nM, respectively, LBY135 treatment resulted in a small but significant decrease of cell viability as compared to control-transfected cells (IC50 ratio approx. 3 fold) (Figure 2). The quantitatively smaller effects observed in the BRCA2 siRNA model as compared to the BRCA2 KO model are likely attributable to the incomplete protein depletion upon siRNA, an effect previously observed by us [22, 23].
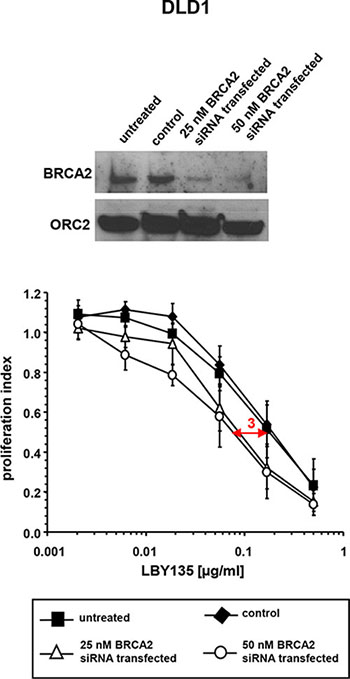
Figure 2: siRNA-mediated BRCA2 knockdown enhances the sensitivity of cancer cells towards TRAIL-R-mediated apoptosis. Western blot analysis of BRCA2 expression levels upon siRNA-mediated knockdown of untreated, control- and BRCA2-siRNA-transfected DLD1 cells (upper panel). Corresponding proliferation assays after treatment with LBY135 (lower panel). Experiments were performed in triplicate with error bars representing SEM from three independent experiments.
TRAIL-R stimulation causes early onset of apoptosis without preceding cell cycle arrest in BRCA2−/− cells
To analyze the mechanistic basis of the increased susceptibility of BRCA2-deficient cancer cells towards TRAIL-R-targeting agents, cell cycle arrest and apoptosis were determined upon treatment with LBY135 and the results compared to those observed upon treatment with MMC. First, cell cycle profiles were assessed by FACS analysis after administration of MMC or LBY135 in BRCA2-deficient vs. BRCA2-proficient cells. Then, apoptosis rates were assessed by measuring sub-G1 events, which correspond to the fraction of cells showing nuclear fragmentation in consequence of apoptotic death. Hypersensitivity of FA pathway-deficient cells towards ICL-agents is known to be associated with a late S-phase/G2/M arrest [7, 21]. Accordingly, MMC treatment of BRCA2−/− cells expectedly led to a dose- and time-dependent early increase of the cell fraction in G2/M at 24 and 48 h as compared to control DLD1 cells (Figure 3A, left panels). G2/M-arrest was followed by apoptosis at 48 h, which progressively increased and peaked at 72 h after treatment (Figure 3A, right panel). In contrast, LBY135 caused apoptosis as early as 24 h after treatment without cell cycle arrest specifically in BRCA2−/− cells (Figure 3B). Representative cell cycle profiles are shown (Figure 3C).
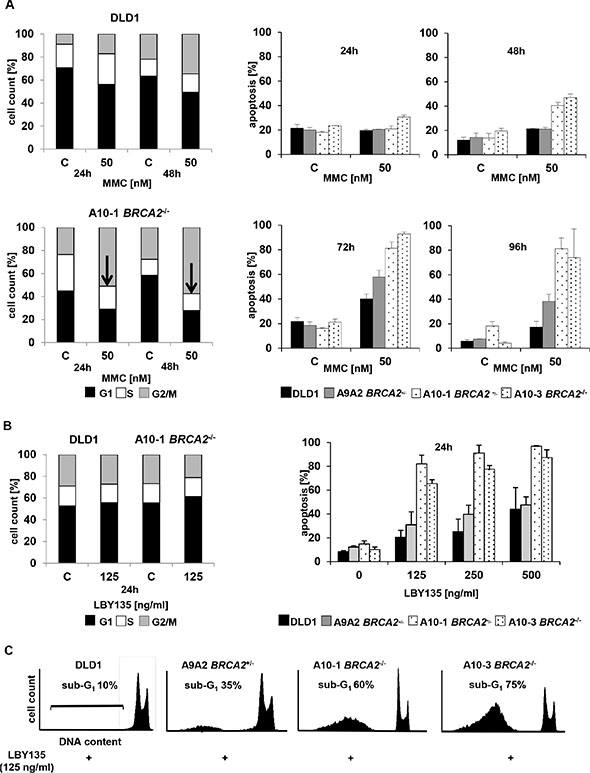
Figure 3: TRAIL-R stimulation causes early onset of apoptosis without concomitant cell cycle arrest in BRCA2−/− cells. (A) Cell cycle profiles (left panels) and subG1 fraction (right panels) of parental DLD1 cells (upper panels) versus BRCA2−/− A10-1 cells (lower panels), treated with MMC at 50 nM or left untreated and assessed at the indicated time points. Arrows indicate samples displaying a significant G2/M arrest. (B) Cell cycle distribution (left panel) and subG1 fraction (right panel) of parental DLD1 cells versus BRCA2−/− A10-1 cells, treated with LBY135 at the indicated concentrations or left untreated, assessed at 24 h after treatment. (C) Cell cycle profiles displaying representative results from one of at least three experiments derived from (B).
DNA fragmentation and caspase cleavage confirms increased apoptosis in BRCA2−/− cells
Apoptosis was validated by demonstration of massive DNA fragmentation as a typical apoptosis feature (Figure 4A) and increased cleavage of CASPASE3 (Figure 4B) in BRCA2−/− cells after treatment with LBY135. Additionally, increased cleavage was also observed for CASPASE8 in BRCA2−/− cells, suggesting that increased apoptosis occurs as consequence of the activation the death-inducing signaling complex (DISC) upon TRAIL-R-stimulation. This pattern of increased caspase cleavage was confirmed in the BRCA2-complementation CAPAN1 model (Figure 4C).
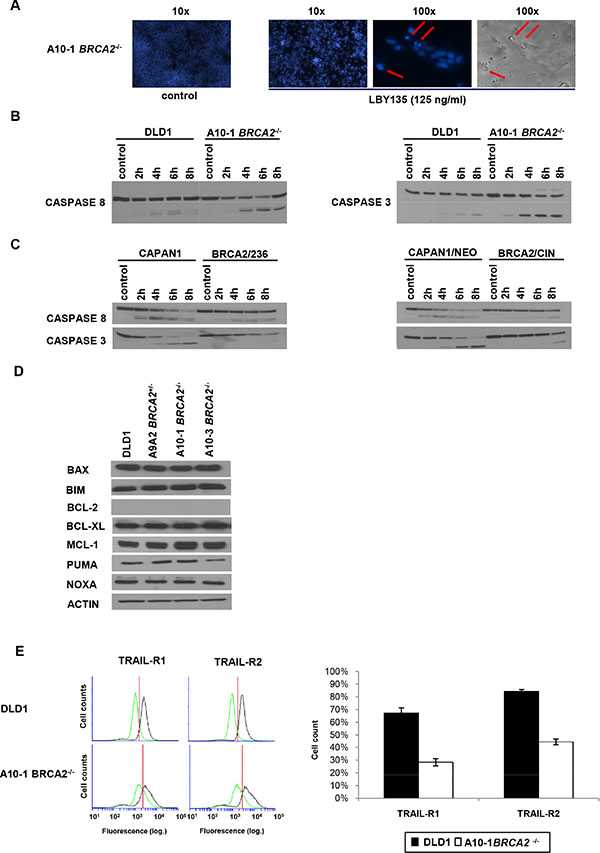
Figure 4: Apoptosis and CASPASE8 recruitment in BRCA2−/− cells are not dependent on the mitochondrial pathway or on the regulation of TRAIL-receptors. (A) Typical features of apoptosis (chromatin condensation and nuclear fragmentation) in DLD1 BRCA2−/− A10-1 cells 24 h after treatment with LBY135 at 125 ng/ml, as assessed by fluorescence microscopy after Hoechst staining. (B + C) Western blotting to detect caspase 8 and caspase 3 cleavage upon treatment with LBY at 125 ng/ml at the indicated time points in parental DLD1 versus BRCA2−/− A10-1 cells (B) and in BRCA2-deficient parental (CAPAN1) and empty vector-transfected (CAPAN1/NEO) cancer cells versus two BRCA2-complemented cell clones (CAPAN1/CIN and CAPAN1/236) (C). (D) Western blotting to assess the baseline expression levels of the indicated regulators of the mitochondrial pathway in BRCA2-proficient versus BRCA2-deficient DLD1 cells. BCL-2 was detectable in control cell lines (not shown) but not in DLD1 cells. (E) FACS analysis of surface receptor staining of TRAIL-R1 and TRAIL-R2 in BRCA2-proficient versus BRCA2-deficient DLD1 cells.
Since TRAIL-R-stimulation could result in necroptosis under certain circumstances [42], BRCA2−/− and control DLD1 cells were additionally incubated with LBY135 with or without the addition of necrostatin to block necrosis induction. However, addition of necrostatin did not affect cell morphology or apoptosis rates (data not shown).
TRAIL-R-induced apoptosis is not mediated by the mitochondrial apoptotic pathway in BRCA2-deficient cells
Since apoptosis triggered in response to DNA damage is typically triggered by alteration of the balance between pro- and anti-apoptotic factors that regulate mitochondrial polarisation [43], we next analyzed the baseline protein expression levels of principal mediators of the mitochondrial signaling pathway. However, except for a slight reduction of the expression of the pro-apoptotic molecule PUMA, which was observable only in the BRCA2−/− A10-3 but not the BRCA2−/− A10-1 clone (thus likely reflecting a clonal phenomenon), no significant changes in any of the tested regulators of the intrinsic apoptotic pathway were detected in BRCA2-proficient or -deficient cells, regarding both baseline expression (Figure 4D) and expression upon TRAIL-R stimulation (data not shown). Importantly, FACS analyses showed that membrane staining of TRAIL-R1 and –R2 did not differ in these cell lines (Figure 4E). These data suggest that the increased susceptibility of BRCA2-deficient cells towards TRAIL-R-targeting compounds specifically potentiates TRAIL-R-signaling and caspase 8 recruitment rather than enhancing downstream mitochondrial apoptotic signaling.
BRCA2-deficiency delays tumor growth upon administration of LBY135 in a murine tumor xenograft model in vivo
To assess whether the increased sensitivity of BRCA2-deficient cells towards TRAIL-R-targeting compounds observed in vitro was transferable to the in vivo setting, we applied a murine tumor xenograft model. Tumors from parental DLD1 or corresponding BRCA2−/− cells were induced in nude mice by subcutaneous injection. Animals exhibiting a solid palpable tumor mass at the injection site 5 days after tumor inoculation were randomized to receive intraperitoneally thrice a week LBY135 at 5 mg/kg or vehicle over a time period of 21 days (parental DLD1 cells: control group: N = 10, LBY135-treated group: N = 9. BRCA2−/− A10-3 cell clones: control group: N = 9; LBY135 group: N = 12). Tumor growth rate was assessed by repeated tumor size measurements of the lesions, using the initial tumor lesion as denominator. LBY135 treatment caused a strongly decreased tumor growth rate as compared to vehicle-treated tumors specifically in tumors originating from BRCA2−/− cells (LBY135-treated BRCA2−/− cells on d21: 130 ± 22 mm3 vs. baseline 46 ± 7 mm3 [3.7 ± 1.1 fold increase]; control-treated BRCA2−/− cells on d21: 308 ± 34 mm3 vs. baseline 36 ± 3.4 [9 ± 1.3 fold increase] – p < 0.001) (Figure 5A right panel), whereas no significant differences were observed in tumors from parental cells (LBY135-treated BRCA2+/+ cells on d21: 719 ± 146 mm3 vs. baseline 88 ± 9 mm3 [8.7 ± 1.8 fold increase]; control-treated BRCA2+/+ cells on d21: 663 ± 113 mm3 vs. baseline 75 ± 7 [8.8 ± 1.4 fold increase] – p = 0.70) (Figure 5A left panel).
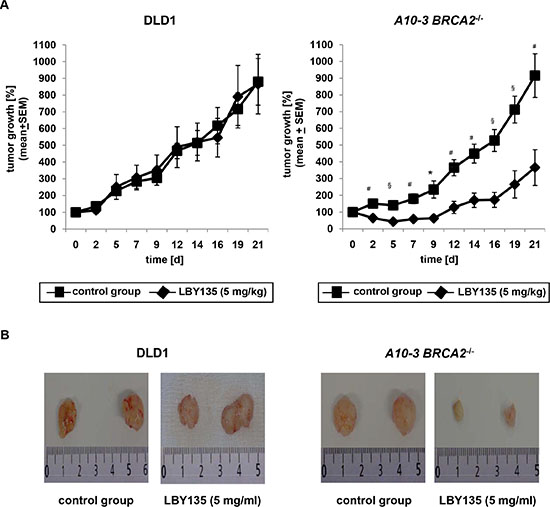
Figure 5: BRCA2-deficiency delays tumor growth upon administration of LBY135 in a murine tumor xenograft model in vivo. (A) Time course (21d) of tumor growth as assessed by repeated measurements of xenograft tumors in mice subcutaneously injected with parental DLD1 cells or A10-3 BRCA2−/− cell clones, respectively, which were consecutively treated intraperitoneally thrice a week with either LBY135 at 5 mg/kg or vehicle. Values are expressed as mean and standard error of the size of tumors at the indicated time points expressed as percentage to the baseline dimensions. t-test: *p < 0.01, #p < 0.005, §p < 0.001. (B) Representative pictures from excised tumors derived from parental and BRCA2−/− A10-3 DLD1 cells.
DISCUSSION
Aiming at expanding the therapeutic exploitation of BRCA2-deficiency for cancer therapy, we investigated here whether deleterious BRCA2 mutations might confer increased susceptibility towards TRAIL-R-targeting agents. Such compounds, which are known to induce apoptosis selectively in cancer cells, have been recently developed and are currently undergoing clinical investigation in multiple tumor entities [29, 44].
In agreement with previous studies [37], treatment of BRCA2-deficient cells with MMC caused early cell cycle arrest at 24 h after treatment, which was subsequently followed by apoptosis reaching a peak at 72 h. In contrast, apoptosis upon TRAIL-R stimulation was induced without preceding or concomitant cell cycle arrest already at 24 h, as demonstrated by caspase cleavage and early appearance of cellular apoptotic features. These data show that the increased susceptibility of BRCA2-deficient cancer cells towards TRAIL-R-mediated apoptosis occurs independently of a prior cell cycle arrest and can thus mechanistically be distinguished from the well-established effects of ICL-agents [37]. Of note, BRCA2-deficient cancer cells displayed a significantly stronger hypersensitivity towards TRAIL-R stimulation using LBY135 than towards the classical ICL-agent MMC in our studies.
Using a murine xenograft model, we were able to confirm these BRCA2 genotype-dependent effects of TRAIL-R stimulation in vivo: tumor growth in mice was significantly delayed upon intraperitoneal administration of LBY135 specifically in BRCA2−/− cells, while no significant differences in tumor growth kinetics were observed in parental DLD1 cells. Our in vitro- and in vivo-data thus clearly demonstrate a hypersensitivity phenotype of BRCA2-deficient cells towards TRAIL-R-stimulation, resembling and exceeding the effects of ICL-agents, and could serve as a platform for clinical trials applying TRAIL-R-agonistic antibodies specifically in patients with BRCA2-mutant cancers.
To assess the distinct mechanisms of apoptosis induction by MMC or TRAIL-R stimulation, respectively (i.e. delayed apoptosis as a consequence of DNA-damage and cell cycle arrest vs. direct apoptosis) we examined the two main mechanisms of apoptosis induction, namely the extrinsic (or receptor-mediated) and the intrinsic (or mitochondrial) apoptotic pathway. The extrinsic pathway is triggered in cancer cells by the stimulation of apoptotic membrane receptors like FAS and TRAIL-R [3] which lead to CASPASE8 activation and, in turn CASPASE3 cleavage.
In contrast, the intrinsic pathway is initiated by cell-internal stimuli including DNA repair errors, which can occur either spontaneously or upon administration of DNA-damaging agents [43]. This leads to a shift in the pro-/anti-apoptotic balance towards pro-apoptotic molecules (e.g. BAX, PUMA or NOXA), which permeabilize the mitochondrial membrane, or to the repression of anti-apoptotic molecules (e.g. BCL-2 and BCL-XL) which, conversely, lead to mitochondrial stabilization [36]. Characteristically, accumulation of unrepaired DNA damage can cause a cell cycle arrest in the G2/M phase and subsequent apoptosis. This is exemplified in our experiments by the observed early G2/M arrest followed by apoptosis in BRCA2-deficient cells upon MMC treatment. Similarly, a previous study has shown an increase in the BAX to BCL-2 ratio and elevated cytochrome c release from the mitochondria in doxorubicin-treated BRCA2-knockout mice [45]. Due to this role of the mitochondrial pathway in determining apoptosis in BRCA2-deficient cells, we hypothesized that the increased sensitivity of these cells towards TRAIL-R targeting agents could be due to the increased recruitment of the mitochondrial signalling pathway upon CASPASE8 cleavage. However, neither baseline- nor TRAIL-R stimulation-induced expression levels of principal regulators of the intrinsic apoptotic pathway (including BAX, BIM, BID, BCL2, BCL-XL, MCL-1, NOXA, PUMA) differed between BRCA2-proficient and BRCA2-deficient cells. The prompt increase of CASPASE8 cleavage upon stimulation of TRAIL-R-mediated apoptosis specifically in BRCA2-deficient cells, which do not differ in the baseline expression of membrane-bound TRAIL-receptors from their BRCA2-proficient counterparts, along with the lack of typical changes in the mitochondrial apoptosis pathway suggests that this hypersensitivity phenotype is mediated at the level of the formation of the death-inducing signaling complex (DISC) and CASPASE8 recruitment. Interestingly, as opposed to our results in BRCA2-deficient cells, a previous study showed that MMC sensitizes cancer cells to the pro-apoptotic effects of TRAIL by downregulating the antiapoptotic proteins BCL2, MCL-1 and BCL-XL while upregulating several pro-apoptotic proteins including BAX and BIM. In addition, MMC induced an increase of cell surface trafficking of both TRAIL receptors in this study [46]. Taken together, these data indicate that BRCA2 exerts antiapoptotic functions, which appear to be independent from its previously described role within the DNA repair system or mitochondrial pathway activation and suggest a role for BRCA2 as a regulator of DISC.
Interestingly, preliminary data on isogenic RKO cancer cells harbouring defects in the proximal FA pathway revealed increased TRAIL-sensitivity only upon FANCG inactivation, while FANCC deficient cells remained unaffected (our own unpublished results). These distinct and gene-specific functions of individual proximal FA genes [47] have previously been observed by us in regard to ionizing radiation [21] and clearly warrant further exploration.
Our study has also certain limitations. Despite providing mechanistic insights, particularly by confining the influence of BRCA2 on apoptosis to the level of CASPASE8 recruitment or cleavage, the present work does not clarify in-depth how exactly BRCA2 mediates the sensitivity towards TRAIL-R stimulation. Specifically, the mechanisms that lead to increased CASPASE8 recruitment need to be further elucidated, including the involvement of the caspase-antagonist FLIP [48], the mechanisms controlling TRAIL-R clustering (e.g. O-glycosylation) [49] or the roles of certain kinases such as PKC, as recently suggested [50]. Furthermore, future proof-of-principle ex-vivo experiments using primary tumor samples could help to further strengthen the translational relevance of our findings as a precondition prior to their clinical application.
In conclusion, our data demonstrate that BRCA2-deficiency - modeled by two independent experimental techniques - confers increased sensitivity towards TRAIL-R-stimulation in cancer cell lines in vitro and in vivo. These effects are mechanistically characterized through early onset of apoptosis without concomitant cell cycle arrest and appear not be related to the well-known functions of BRCA2 during DNA-repair. Importantly, while FA pathway- and particularly BRCA2-dependent hypersensitivities have previously been convincingly shown to be highly specific, reliably reproducible in different model systems and restricted mainly to ICL-agents [21, 37] and PARP inhibitors [51, 52], our data now expand the therapeutically exploitable spectrum to also include TRAIL R-targeting agents, providing a pre-clinical basis for clinical trials of newly developed TRAIL-R-agonistic antibodies in genetically defined subsets of patients with BRCA2-deficient tumors. On the one hand, patients developing resistance towards ICL-agents could benefit from a second-line treatment with TRAIL-R targeting compounds. On the other hand, the combination of TRAIL-R targeting compounds with ICL-agents is expected to either enhance the effects of ICL-agents or facilitate a dose-reduction, thus mitigating their side effects [23, 53]. Therefore, future pre-clinical studies will need to assess whether ICL-agents and TRAIL R-targeting drugs used in combination will cause synergistic, additive or antagonistic effects in BRCA2-deficient tumors [53]. Finally, recent studies in pancreatic and other cancer tissue samples indicate that the sub-cellular localization of TRAIL receptors influences the cellular sensitivity towards TRAIL [31–33]. As the recent clinical investigation of radio-labelled tigatuzumab demonstrates [35], these drug combinations might be thus particularly effective in those subsets of patients with BRCA2-deficient tumors that display an uptake for TRAIL-R-targeting antibodies.
MATERIALS AND METHODS
Cell lines and reagents
LBY135 was kindly provided by Novartis (Nuernberg, Germany), tigatuzumab (CS-1008) by Daiichi-Sankyo Pharma Development (Edison NJ, USA). Protein A was added at 0.01 times the concentration of LBY135 or tigatuzumab to facilitate antibody crosslinking. Recombinant human TRAIL/TNFSF10, was purchased from R & D Systems (Minneapolis, USA), Anti-Apo 1–3 from Alexis/ENZO Life Sciences (Lörrach, Germany). Parental DLD1 cells along with the corresponding heterozygote BRCA2+/− (termed A9A2) and homozygote BRCA2−/− (termed A10-1, A10-3) were kindly provided by Thomas Hucl and originally developed by him and Eike Gallmeier in Scott Kern’s laboratory (Johns Hopkins University, Baltimore/MD, USA) [37]. CAPAN1 cells along with the corresponding empty vector-transfected CAPAN1/NEO and two BRCA2-complemented CAPAN1/236 and CAPAN1/CIN cell clones were kindly provided by Mien-Chie Hung in July 2010 (M.D. Anderson Cancer Center, Houston, Texas) [38]. BRCA2 gene knockout was tested and confirmed in DLD1 cells on the genetic level by direct sequencing directly upon arrival, on the protein level by western blotting and on the functional level by assessment of sensitivity towards MMC and RAD51 focus formation. Likewise, BRCA2 gene complementation of CAPAN1 cells was confirmed on the genetic level by direct sequencing, on the protein level by western blotting and on the functional level by assessment of RAD51 focus formation. Extensive screening efforts earlier conducted by us and others have previously illustrated the specific hypersensitivity of BRCA2, FANCG- and/or FANCC-deficient cells towards ICL-agents [21, 37] and PARP inhibitors [51, 52] and demonstrated that these gene defects did not have any effects towards most other therapeutic agents, including gemcitabine [7, 37, 54], which was therefore used among several other agents as negative control in our experiments (data not shown). All cell lines were grown in DMEM supplemented with 10% fetal calf serum, L-glutamine and penicillin/streptomycin (PAA, Cölbe, Germany).
Cell proliferation assays
1,000–1,500 cells were plated in 96-well plates, allowed to adhere and then treated with the indicated agents at the indicated concentrations. After 6 days, cells underwent osmotic lysis in 100 μl H2O. After addition of 0.5% Picogreen (Molecular Probes, Invitrogen, Karlsruhe, Germany), fluorescence was measured (Cytofluor Series 4000, Applied Biosystems, Darmstadt, Germany) and the proliferation index calculated, defining the untreated samples as 1. Three independent experiments were performed per agent, with each data point reflecting triplicate wells. Error bars represent standard error of the mean (SEM) from three experiments.
Immunoblotting
Proteins were loaded on 10% SDS PAGE gels and separated for 15 minutes at 80 V and for 60 minutes at 140 V and then transferred onto PVDF membranes. After blocking for 1 h in TBST (tris-buffered saline with Tween-20) containing 5% milk or 1% milk and 1% bovine serum albumin, membranes were incubated overnight with the following primary antibodies: CASPASE3 (#9662), CASPASE8 (#9508), CASPASE 9 (#9508) and BCL-XL (#2764), purchased from Cell Signaling/New England Biolabs GmbH (Frankfurt am Main, Germany), β-ACTIN (#A5441) from Sigma Aldrich (Munich, Germany), Bcl-2 (#610539) and BCL-XL (#610746) from BD Biosciences (Heidelberg, Germany), MCL-1 (#sc-12756), BAD (#sc-8044), BAK (#sc-7873), PUMA (#sc-374223), and NOXA (#sc-30209) from Santa Cruz Biotechnology (Heidelberg, Germany), BAX (#ab7977) from Abcam (Cambridge, UK), BIM (#559685) from BD Pharmingen (Heidelberg, Germany), XIAP (#IMG-5770) from Novus Biologicals LLC (Littleton, USA). Corresponding secondary antibodies were used at concentrations of 1:10.000. Detection was performed using the Super Signal West Pico Chemiluminescent Substrate (Thermo Scientific #34077 – Schwerte, Germany). BRCA2 immunoblotting was performed using anti-BRCA2 antibody (#OP95, Merck Millipore, Schwalbach, Germany) and ORC-2 (rabbit anti-ORC-2, #559266, purchased by BD Pharmingen) as a loading control [55].
FACS analysis of surface receptors
DLD1 and A10-1 BRCA2−/− cell clones were detached from the plates by 1% Trypsin-EDTA, washed and incubated for 30 min. with the following monoclonal FITC-coupled antibodies: TRAIL-R1 (ALX-804-297F-T100), TRAIL-R2 (ALX-804-298F-T100) from Enzo Life Science (Loerach, Germany) and control IgG1 (BD Bioscience, Heidelberg, Germany).
Apoptosis and cell cycle assays
For cell cycle and apoptosis analyses, 8.0 × 104–1.5 × 105 cells were seeded in 12-well plates and, after overnight incubation, treated with the indicated agents. After propidium iodide staining, fluorescence-activated cell sorting (FACS – Accuri C6 flow cytometer, BD Biosciences San Jose, CA USA) was performed as previously described [9]. Apoptosis was quantified by the fraction of cells with sub-diploid DNA content (Sub-G1) and confirmed morphologically by Hoechst 33342 staining and fluorescence microscopy (Zeiss, Jena, Germany).
siRNA-mediated BRCA2 knockdown
Parental DLD1 cells were allowed to reach 30%–50% confluence and then incubated with oligofectamine (Invitrogen) and siRNA directed against BRCA2 (Hs BRCA2 7, Qiagen, Hilden, Germany) or with oligofectamine alone, which served as control. siRNA were used at final concentrations of 25 nM and 50 nM, respectively. 72 h after transfection, the cells were used for proliferation assays and immunoblotting.
In vivo-studies
For xenograft tumor induction, 8 week old nu/nu nude mice were injected in both flanks with either 106 isolated DLD1 parental cells or with the corresponding BRCA2−/− KO cells (A10-3 clone). Each group consisted of 12 animals. After 5 days, animals displaying tumor engraftment, i.e. solid palpable lesions at either one or both of the injection sites, were randomized to receive a solution of LBY135 at 5 mg/kg or vehicle intraperitoneally thrice a week. Tumor growth was monitored daily over a time period of 21 days through repeated assessment of tumor volume, determined through measurement of the two major tumor diameters with a caliper. These data were expressed for each cell line as the ratio of the tumor size to the initial tumor size (measured at day 5 after injection of tumor cells). All animal experiments were performed according to the guidelines of the German law for animal life protection and approved by the local ethics committee.
ACKNOWLEDGMENTS
We are grateful to Mrs. Stephanie Ochs for expert technical assistance.
FINANCIAL SUPPORT
This work was supported by the Deutsche Forschungsgemeinschaft (DFG) with the grant DFG GA762/3-2 to EG and by the Else Kröner-Fresenius Stiftung with the grant 2011_A226 to EDT.
CONFLICTS OF INTEREST
The authors have no conflicts of interest to disclose.
REFERENCES
1. Hucl T, Gallmeier E. DNA repair: exploiting the Fanconi anemia pathway as a potential therapeutic target. Physiol Res. 2011; 60:453–465.
2. Branzei D, Foiani M. Regulation of DNA repair throughout the cell cycle. Nat Rev Mol Cell Biol. 2008; 9:297–308.
3. Papenfuss K, Cordier SM, Walczak H. Death receptors as targets for anti-cancer therapy. Journal of cellular and molecular medicine. 2008; 12:2566–2585.
4. D’Andrea AD. Susceptibility pathways in Fanconi’s anemia and breast cancer. The New England journal of medicine. 2010; 362:1909–1919.
5. Kee Y, D’Andrea AD. Expanded roles of the Fanconi anemia pathway in preserving genomic stability. Genes & development. 24:1680–1694.
6. Kennedy RD, D’Andrea AD. The Fanconi Anemia/BRCA pathway: new faces in the crowd. Genes & development. 2005; 19:2925–2940.
7. van der Heijden MS, Brody JR, Gallmeier E, Cunningham SC, Dezentje DA, Shen D, Hruban RH, Kern SE. Functional defects in the fanconi anemia pathway in pancreatic cancer cells. The American journal of pathology. 2004; 165:651–657.
8. van der Heijden MS, Yeo CJ, Hruban RH, Kern SE. Fanconi anemia gene mutations in young-onset pancreatic cancer. Cancer research. 2003; 63:2585–2588.
9. Neveling K, Kalb R, Florl AR, Herterich S, Friedl R, Hoehn H, Hader C, Hartmann FH, Nanda I, Steinlein C, Schmid M, Tonnies H, Hurst CD, et al. Disruption of the FA/BRCA pathway in bladder cancer. Cytogenetic and genome research. 2007; 118:166–176.
10. Tokunaga E, Okada S, Kitao H, Shiotani S, Saeki H, Endo K, Morita M, Kakeji Y, Maehara Y. Low incidence of methylation of the promoter region of the FANCF gene in Japanese primary breast cancer. Breast Cancer. 2011; 18:120–123.
11. Narayan G, Arias-Pulido H, Nandula SV, Basso K, Sugirtharaj DD, Vargas H, Mansukhani M, Villella J, Meyer L, Schneider A, Gissmann L, Durst M, Pothuri B, et al. Promoter hypermethylation of FANCF: disruption of Fanconi Anemia-BRCA pathway in cervical cancer. Cancer research. 2004; 64:2994–2997.
12. Marsit CJ, Liu M, Nelson HH, Posner M, Suzuki M, Kelsey KT. Inactivation of the Fanconi anemia/BRCA pathway in lung and oral cancers: implications for treatment and survival. Oncogene. 2004; 23:1000–1004.
13. Taniguchi T, Tischkowitz M, Ameziane N, Hodgson SV, Mathew CG, Joenje H, Mok SC, D’Andrea AD. Disruption of the Fanconi anemia-BRCA pathway in cisplatin-sensitive ovarian tumors. Nat Med. 2003; 9:568–574.
14. Fackenthal JD, Olopade OI. Breast cancer risk associated with BRCA1 and BRCA2 in diverse populations. Nature reviews. 2007; 7:937–948.
15. Szabo CI, King MC. Population genetics of BRCA1 and BRCA2. Am J Hum Genet. 1997; 60:1013–1020.
16. Ford D, Easton DF, Stratton M, Narod S, Goldgar D, Devilee P, Bishop DT, Weber B, Lenoir G, Chang-Claude J, Sobol H, Teare MD, Struewing J, et al. Genetic heterogeneity and penetrance analysis of the BRCA1 and BRCA2 genes in breast cancer families. The Breast Cancer Linkage Consortium. Am J Hum Genet. 1998; 62:676–689.
17. Vaziri SA, Krumroy LM, Rostai M, Casey G. Frequency of BRCA1 and BRCA2 mutations in a clinic-based series of breast and ovarian cancer families. Hum Mutat. 2001; 17:74.
18. Goggins M, Schutte M, Lu J, Moskaluk CA, Weinstein CL, Petersen GM, Yeo CJ, Jackson CE, Lynch HT, Hruban RH, Kern SE. Germline BRCA2 gene mutations in patients with apparently sporadic pancreatic carcinomas. Cancer research. 1996; 56:5360–5364.
19. Ozcelik H, Schmocker B, Di Nicola N, Shi XH, Langer B, Moore M, Taylor BR, Narod SA, Darlington G, Andrulis IL, Gallinger S, Redston M. Germline BRCA2 6174delT mutations in Ashkenazi Jewish pancreatic cancer patients. Nat Genet. 1997; 16:17–8.
20. Ramus SJ, Gayther SA. The contribution of BRCA1 and BRCA2 to ovarian cancer. Molecular oncology. 2009; 3:138–150.
21. Gallmeier E, Calhoun ES, Rago C, Brody JR, Cunningham SC, Hucl T, Gorospe M, Kohli M, Lengauer C, Kern SE. Targeted disruption of FANCC and FANCG in human cancer provides a preclinical model for specific therapeutic options. Gastroenterology. 2006; 130:2145–2154.
22. Gallmeier E, Hucl T, Brody JR, Dezentje DA, Tahir K, Kasparkova J, Brabec V, Bachman KE, Kern SE. High-throughput screening identifies novel agents eliciting hypersensitivity in Fanconi pathway-deficient cancer cells. Cancer research. 2007; 67:2169–2177.
23. Gallmeier E, Kern SE. Targeting Fanconi anemia/BRCA2 pathway defects in cancer: the significance of preclinical pharmacogenomic models. Clin Cancer Res. 2007; 13:4–10.
24. Chalasani P, Kurtin S, Dragovich T. Response to a third-line mitomycin C (MMC)-based chemotherapy in a patient with metastatic pancreatic adenocarcinoma carrying germline BRCA2 mutation. JOP. 2008; 9:305–308.
25. James E, Waldron-Lynch MG, Saif MW. Prolonged survival in a patient with BRCA2 associated metastatic pancreatic cancer after exposure to camptothecin: a case report and review of literature. Anticancer Drugs. 2009; 20:634–638.
26. Villarroel MC, Rajeshkumar NV, Garrido-Laguna I, De Jesus-Acosta A, Jones S, Maitra A, Hruban RH, Eshleman JR, Klein A, Laheru D, Donehower R, Hidalgo M. Personalizing cancer treatment in the age of global genomic analyses: PALB2 gene mutations and the response to DNA damaging agents in pancreatic cancer. Mol Cancer Ther. 2011; 10:3–8.
27. Sonnenblick A, Kadouri L, Appelbaum L, Peretz T, Sagi M, Goldberg Y, Hubert A. Complete remission, in BRCA2 mutation carrier with metastatic pancreatic adenocarcinoma, treated with cisplatin based therapy. Cancer Biol Ther. 2011; 12.
28. Kaufman B, Shapira-Frommer R, Schmutzler RK, Audeh MW, Friedlander M, Balmana J, Mitchell G, Fried G, Stemmer SM, Hubert A, Rosengarten O, Steiner M, Loman N, et al. Olaparib monotherapy in patients with advanced cancer and a germline BRCA1/2 mutation. Journal of clinical oncology. 2015; 33:244–250.
29. Yang A, Wilson NS, Ashkenazi A. Proapoptotic DR4 and DR5 signaling in cancer cells: toward clinical translation. CurrOpinCell Biol. 2010; 22:837–844.
30. De Toni EN, Thieme SE, Herbst A, Behrens A, Stieber P, Jung A, Blum H, Goke B, Kolligs FT. OPG is regulated by beta-catenin and mediates resistance to TRAIL-induced apoptosis in colon cancer. ClinCancer Res. 2008; 14:4713–4718.
31. Gallmeier E, Bader DC, Kriegl L, Berezowska S, Seeliger H, Göke B, Kirchner T, Bruns CJ, De Toni EN. Loss of TRAIL-receptors is a recurrent feature in pancreatic cancer and determines the prognosis of patients with no nodal metastasis after surgery. PLoSOne. 2013.
32. Kriegl L, Jung A, Engel J, Jackstadt R, Gerbes AL, Gallmeier E, Reiche JA, Hermeking H, Rizzani A, Bruns CJ, Kolligs FT, Kirchner T, Goke B, et al. Expression, cellular distribution, and prognostic relevance of TRAIL receptors in hepatocellular carcinoma. ClinCancer Res. 2010; 16:5529–5538.
33. Kriegl L, Jung A, Horst D, Rizzani A, Jackstadt R, Hermeking H, Gallmeier E, Gerbes AL, Kirchner T, Goke B, De Toni EN. Microsatellite Instability, KRAS Mutations and Cellular Distribution of TRAIL-Receptors in Early Stage Colorectal Cancer. PLoSOne. 2012; 7:e51654.
34. Cretney E, Takeda K, Yagita H, Glaccum M, Peschon JJ, Smyth MJ. Increased susceptibility to tumor initiation and metastasis in TNF-related apoptosis-inducing ligand-deficient mice. JImmunol. 2002; 168:1356–1361.
35. Ciprotti M, Tebbutt NC, Lee FT, Lee ST, Gan HK, McKee DC, O‘Keefe GJ, Gong SJ, Chong G, Hopkins W, Chappell B, Scott FE, Brechbiel MW, et al. Phase I Imaging and Pharmacodynamic Trial of CS-1008 in Patients With Metastatic Colorectal Cancer. Journal of clinical oncology. 2015.
36. Fulda S. Targeting extrinsic apoptosis in cancer: Challenges and opportunities. Seminars in cell & developmental biology. 2015; 39:20–25.
37. Hucl T, Rago C, Gallmeier E, Brody JR, Gorospe M, Kern SE. A syngeneic variance library for functional annotation of human variation: application to BRCA2. Cancer research. 2008; 68:5023–5030.
38. Wang SC, Shao R, Pao AY, Zhang S, Hung MC, Su LK. Inhibition of cancer cell growth by BRCA2. Cancer research. 2002; 62:1311–1314.
39. Gallmeier E, Kern SE. Absence of specific cell killing of the BRCA2-deficient human cancer cell line CAPAN1 by poly(ADP-ribose) polymerase inhibition. Cancer Biol Ther. 2005; 4:703–706.
40. Sharma S, de Wries EG, Infante JR, Oldenhuis C, Chiang L, Bilic S, Goldbrunner M, Scott JW, Burris HA. Phase I trial of LBY135, a monoclonal antibody agonist to DR5, alone and in combination with capecitabine in advanced solid tumors. Journal of Clinical Oncology. 2008; 26.
41. Forero-Torres A, Shah J, Wood T, Posey J, Carlisle R, Copigneaux C, Luo FR, Wojtowicz-Praga S, Percent I, Saleh M. Phase I trial of weekly tigatuzumab, an agonistic humanized monoclonal antibody targeting death receptor 5 (DR5). Cancer BiotherRadiopharm. 2010; 25:13–19.
42. Fulda S. The mechanism of necroptosis in normal and cancer cells. Cancer Biol Ther. 2013; 14.
43. Nowsheen S, Yang ES. The intersection between DNA damage response and cell death pathways. Experimental oncology. 2012; 34:243–254.
44. Sharma S, de Vries EG, Infante JR, Oldenhuis CN, Gietema JA, Yang L, Bilic S, Parker K, Goldbrunner M, Scott JW, Burris HA, 3rd. Safety, pharmacokinetics, and pharmacodynamics of the DR5 antibody LBY135 alone and in combination with capecitabine in patients with advanced solid tumors. Investigational new drugs. 2013.
45. Singh KK, Shukla PC, Quan A, Desjardins JF, Lovren F, Pan Y, Garg V, Gosal S, Garg A, Szmitko PE, Schneider MD, Parker TG, Stanford WL, et al. BRCA2 protein deficiency exaggerates doxorubicin-induced cardiomyocyte apoptosis and cardiac failure. The Journal of biological chemistry. 2012; 287:6604–6614.
46. Cheng H, Hong B, Zhou L, Allen JE, Tai G, Humphreys R, Dicker DT, Liu YY, El-Deiry WS. Mitomycin C potentiates TRAIL-induced apoptosis through p53-independent upregulation of death receptors: evidence for the role of c-Jun N-terminal kinase activation. Cell cycle. 2012; 11:3312–3323.
47. Gallmeier E, Hucl T, Calhoun ES, Cunningham SC, Bunz F, Brody JR, Kern SE. Gene-specific selection against experimental fanconi anemia gene inactivation in human cancer. Cancer Biol Ther. 2007; 6:654–660.
48. Haag C, Stadel D, Zhou S, Bachem MG, Moller P, Debatin KM, Fulda S. Identification of c-FLIP(L) and c-FLIP(S) as critical regulators of death receptor-induced apoptosis in pancreatic cancer cells. Gut. 2011; 60:225–237.
49. Wagner KW, Punnoose EA, Januario T, Lawrence DA, Pitti RM, Lancaster K, Lee D, von Goetz M, Yee SF, Totpal K, Huw L, Katta V, Cavet G, et al. Death-receptor O-glycosylation controls tumor-cell sensitivity to the proapoptotic ligand Apo2L/TRAIL. NatMed. 2007; 13:1070–1077.
50. Lemke J, Noack A, Adam D, Tchikov V, Bertsch U, Roder C, Schutze S, Wajant H, Kalthoff H, Trauzold A. TRAIL signaling is mediated by DR4 in pancreatic tumor cells despite the expression of functional DR5. JMolMed(Berl). 2010; 88:729–740.
51. Bryant HE, Schultz N, Thomas HD, Parker KM, Flower D, Lopez E, Kyle S, Meuth M, Curtin NJ, Helleday T. Specific killing of BRCA2-deficient tumours with inhibitors of poly(ADP-ribose) polymerase. Nature. 2005; 434:913–917.
52. Farmer H, McCabe N, Lord CJ, Tutt AN, Johnson DA, Richardson TB, Santarosa M, Dillon KJ, Hickson I, Knights C, Martin NM, Jackson SP, Smith GC, et al. Targeting the DNA repair defect in BRCA mutant cells as a therapeutic strategy. Nature. 2005; 434:917–921.
53. Hucl T, Gallmeier E, Kern SE. Distinguishing rational from irrational applications of pharmacogenetic synergies from the bench to clinical trials. Cell cycle. 2007; 6:1336–1341.
54. van der Heijden MS, Brody JR, Dezentje DA, Gallmeier E, Cunningham SC, Swartz MJ, DeMarzo AM, Offerhaus GJ, Isacoff WH, Hruban RH, Kern SE. In vivo therapeutic responses contingent on Fanconi anemia/BRCA2 status of the tumor. Clin Cancer Res. 2005; 11:7508–7515.
55. Krawczyk PM, Eppink B, Essers J, Stap J, Rodermond H, Odijk H, Zelensky A, van Bree C, Stalpers LJ, Buist MR, Soullie T, Rens J, Verhagen HJ, et al. Mild hyperthermia inhibits homologous recombination, induces BRCA2 degradation, and sensitizes cancer cells to poly (ADP-ribose) polymerase-1 inhibition. Proceedings of the National Academy of Sciences of the United States of America. 2011; 108:9851–9856.