
Introduction
Infusion of in vitro expanded autologous tumor-infiltrating lymphocytes (TILs) following lymphodepletion has been shown to result in objective tumor regression in up to 70% of patients with metastatic melanoma, and almost a quarter of the treated patients achieved durable complete remission [1]. However, it is not always possible to obtain TILs with anti-melanoma activity and there has been limited success in obtaining TILs in other cancers. Thus, much effort has been devoted to develop efficient means of producing CTLs with antitumor activity. In addition, melanoma frequently relapses in the patients after a period of remission [1], and the relapse was found to be associated with a tumor immunosuppressive microenvironment that inhibits T cell function [2]. Emerging evidence indicates that the tumor-induced inhibition of T cell activation is largely attributed to the recruitment of regulatory T cells (Tregs) into the tumor and upregulation of immune inhibitory pathway signaling, which are both driven by T cell immune responses [3, 4]. These studies imply that, for achieving the desired therapeutic effects of adoptive immunotherapy, it is important to develop effective approaches overcoming these immunosuppressive pathways. However, such studies have mostly been performed in mice, and the limited availability of tumor-reactive human CTLs that resemble those from patients is one of the key impeding factors.
It has been shown first in mice [5, 6], and more recently in humans [7] that T cells expressing the transgenic TCR can be generated by introducing TCR genes into hematopoietic stem cells. We have previously shown that transplantation of human fetal thymus tissue (FTHY; under kidney capsule) and CD34+ fetal liver cells (FLCs; i.v.) in immunodeficient mice leads to the development of human lymphohematopoietic cells including T, B and dendritic cells, and the formation of secondary lymphoid organs consisting of human lymphohematopoietic cells [8-10]. Here, we investigate the possibility of using this humanized mouse (hu-mouse) model to generate melanoma antigen (MART-1)-specific human T cells for translational studies of adoptive cancer immunotherapies. We show that MART-1-specific human T cells can be generated efficiently in hu-mice made of CD34+ FLCs that were transduced with lentiviruses containing MART-1-specific TCR gene. Importantly, MART-1-specific human T cells developed in hu-mice are functional and capable of killing melanoma cells in an HLA/peptide-dependent manner. Furthermore, using hu-mouse-derived melanoma antigen-specific human T cells, we demonstrate that pretreatment of the T cells with rapamycin can significantly enhance the antitumor activity of adoptive T cell therapy in IL-15-treatted recipients.
Results
Development of melanoma antigen MART-1-specific human T cells in humanized mice made of TCR engineered CD34+ cells
A lentiviral vector encoding HLA-A*0201-restricted TCR (DMF5 clone) [11] specific for melanoma-associated antigen recognized by T cell-1 (MART-1) was used to engineer CD34+ FLCs. The hu-mice were made by intravenous injection of TCR-engineered HLA-A*0201+ CD34+ FLCs into NOD.Cg-Prkdcscid Il2rgtm1Wjl/SzJ (NSG) mice grafted with cryopreserved-thawed autologous FTHY (Figure 1A). We have shown that the use of cryopreserved-thawed FTHY may improve T cell development from virally-transduced CD34+ cells by eliminating preexisting T cell progenitors in the FTHY graft (Hu Z, Xia J, Yang YG. Unpublished data). In hu-mice that received HLA-A*0201+ FTHY and CD34+ FLCs transduced with lentiviral vectors containing MART-1-specific TCR gene, high levels of human T and B cell reconstitution were detected (Figure S1) and among human CD3+ T cells, a significant proportion was found to express MART-1-specific TCR, as identified by HLA-A2/MART-1 tetramer staining (Figure 1B). The majority of tetramer+ T cells had a naïve phenotype as shown by expression of CD45RA and CCR7 (Figure 1C). In concordance with the role of CD8 in recognition of MHC class I-restricted antigens, CD8+ T cells consisted of a significantly larger number of tetramer+ cells than CD4+ T cells (especially at the early time), and the percentage of CD8+ T cells in CD3+tetramer+ cells was significantly higher than found in CD3+tetramer- T cells (Figure 1D and 1E). All mice had at least more than a million (ranging from 1.1x106 and 12x106) of tetramer+ CD8+ T cells in the spleen, and a significant increase was seen in hu-mice receiving MART-1 peptide immunization (Table S1; also see Figure 2 below). Furthermore, most CD8 single positive (SP) and CD4+CD8+ double positive (DP) thymocytes expressed MART-1-specific TCR (Figure 1F). Although the frequency was lower, peripheral human CD4+ T cells and CD4SP thymocytes also contained a significant proportion of tetramer+ cells (with relatively lower levels of MART-1-TCR expression; Figure S2) in almost all mice examined (Figure 1D-1F).
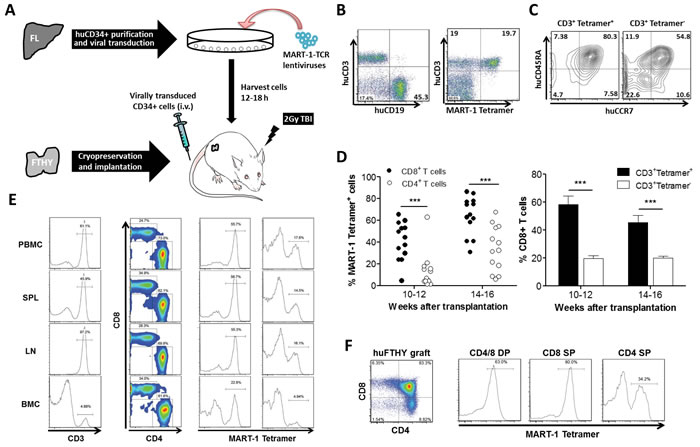
Figure 1: Generation of MART-1-specific T cells in humanized mice made by transplantation of human FTHY and TCR-engineered CD34+ FLCs. A. Schematic preparation of the humanized mouse model with human T cells expressing transgenic TCR specific for MART-1. B. Representative flow cytometric profiles showing reconstitution of human T and B cells (left) and MART-1 TCR+ T cells (right) in PBMCs of humanized mice. C. FACS analysis of MART-1 TCR+ and MART-1 TCR- T cells for CD45RA and CCR7 expression. D. Shown are percentages of MART-1 TCR+ T cells in CD8+ and CD4+ T cell compartments (left) and percentages (mean ± SEMs; n = 13) of CD8+ T cells in MART-1 TCR+ and MART-1 TCR- T cell population (right). E. FACS assessment of MART-1 TCR+ T cells in CD8+ and CD4+ T cells from the indicated tissues at week 22. F. FACS assessment of MART-1 TCR+ T cells in CD4+CD8+ (DP), CD8SP and CD4SP human thymocytes. ***, p < 0.001.
Tetramer+ CD8+, but not CD4+, T cells showed efficient in vivo responses following peptide immunization
Next we questioned if the tetramer+ T cells generated in hu-mice were functional. We immunized the hu-mice with MART-1 peptides emulsified by complete Freud’s adjuvant and measured the immune response 3 weeks later. Tetramer+ CD8+ T cells showed a MART-1-specific response following immunization, as shown by antigen-specific expansion (Figure 2A), conversion from a naïve to effector/memory phenotype (i.e., losing CCR7 and CD45RA expression; Figure 2B), and IFN-γ production (Figure 2C). In contrast to tetramer+ CD8+ T cells, tetramer- CD8+ T cells showed no detectable changes in either phenotypes or function after immunization with MART-1 peptides. However, unlike CD8+ T cells, neither tetramer+ nor tetramer- CD4+ T cells showed expansion (Figure 2D), conversion to an effector/memory phenotype (Figure 2E) or IFN-γ production (Figure 2F) following MART-1 peptide immunization, despite that the tetramer+ CD4+ T cells showed significant proliferation after in vitro stimulation with MART-1 peptides (Figure S3). These data indicate that tetramer+ CD8+, but not CD4+, T cells could mount MART-1-specific responses following in vivo peptide immunization.
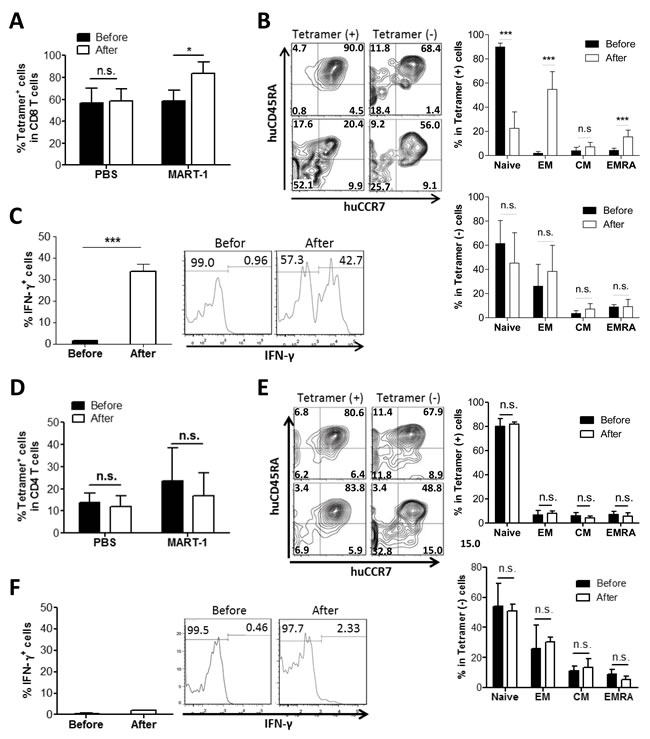
Figure 2: MART-1 TCR+ CD8, but not CD4, T cells show antigen-specific responses in humanized mice following MART-1 peptide immunization. Hu-mice were immunized with MART-1 peptide or PBS as control 15 weeks after humanization, and CD8+ A.-C. and CD4+ D.-F. T cell responses were assessed by flow cytometry 1 week prior to (before) and 3 weeks after immunization. A. and D. Percentages of tetramer+ cells in peripheral blood CD8+ A. and CD4+ D. T cells (mean±SEM; n = 3). B. and E. Expression of CD45RA and CCR7 on tetramer+ vs. tetramer- CD8+ B. and CD4+ E. T cells (mean ± SEMs; n = 7). Left, representative staining profiles of the cells prepared before (top) and after (bottom) immunization; Right, percentages of T cell subsets in tetramer+ (top) and tetramer- (bottom) T cells prepared before and after immunization. Naïve, CD45RA+CCR7+ naïve T cells; EM, CD45RA-CCR7- effector memory T cells; CM, CD45RA-CCR7+ central memory T cells; EM/RA, CD45RA+CCR7- effector memory T cells. C. and F. Percentages of IFN-γ producing tetramer+ CD8+ C. and CD4+ F. T cells prepared before and after immunization (mean ± SEMs; n = 5). *, p < 0.05; **, p < 0.01; ***, p < 0.001; n.s., not significant.
MART-1-specific human T cells developed in humanized mice induce efficient killing of melanoma cells in an antigen-specific manner, and retain their antigen-specific cytotoxicity after extensive in vitro expansion
We then measured the killing of melanoma cells by tetramer+ T cells isolated from hu-mice. Tetramer+ human T cells purified from hu-mice were stimulated for 5 days with anti-huCD3/CD28 microbeads, and their cytotoxicity against melanoma cells was assessed by 51Cr-release assay. As shown in Figure 3A, tetramer+ T cells induced significant killing of HLA-A2+MART-1+ melanoma cells (Mel 624), providing further evidence that human T cells developing from the engineered CD34+ cells in hu-mice were functional. Moreover, these T cells failed to produce cytotoxicity against HLA-A2-MART-1+ (Mel 888) and HLA-A2+MART-1- (Mel A375) melanoma cells (Figure 3A), demonstrating an HLA-A2-restricted, MART-1-specific cytotoxic activity of tetramer+ T cells developed in the hu-mice.
Tetramer+ human T cells were prepared from the hu-mice and expanded for 4-7 weeks (approximately 2x103 to 1x105 fold increase in number) in media containing anti-huCD3 (OTK3), rhIL-2 and feeder cells (Figure 3B). CD8+ and CD4+ T cells were then sorted out from the expanded tetramer+ T cells and evaluated for antitumor activity. As shown in Figure 3C (left panel), tetramer+ CD8 T cells mediated efficient killing of HLA-A2+MART-1+ (Mel 624), but not HLA-A2-MART-1+ (Mel 888), melanoma cells. Furthermore, IFN-γ secretion was detected in tetramer+ CD8+ T cells cocultured with HLA-A2+MART-1+ melanoma (Mel 624) cells, but not in those cocultured with HLA-A2-MART-1+ melanoma (Mel 888) cells (Figure 3C, right panel). Similarly, high levels of granzyme B were detected in tetramer+ CD8+ T cells cocultured with Mel 624 cells, but not in those cocultured with Mel 888 cells (Figure S4). However, perforin production was not induced in tetramer+ CD8+ T cells after stimulation with either Mel 624 or Mel 888 cells (Figure S4), suggesting that perforin is not a major effector molecule for these T cells. Although to a markedly lower extend compared to tetramer+ CD8+ T cells, tetramer+ CD4+ T cells were also capable of mediating specific cytotoxicity and producing IFN-γ when cocultured with Mel 624, but not Mel 888, melanoma cells (Figure 3D). Taken together, these data demonstrate that tetramer+ human T cells (especially CD8 T cells) that developed in hu-mice are capable of killing melanoma cells in an HLA-A2/MART-1 peptide complex-dependent manner. Furthermore, these tetramer+ T cells can be substantially expanded in vitro without significantly compromising their cytotoxic activity.
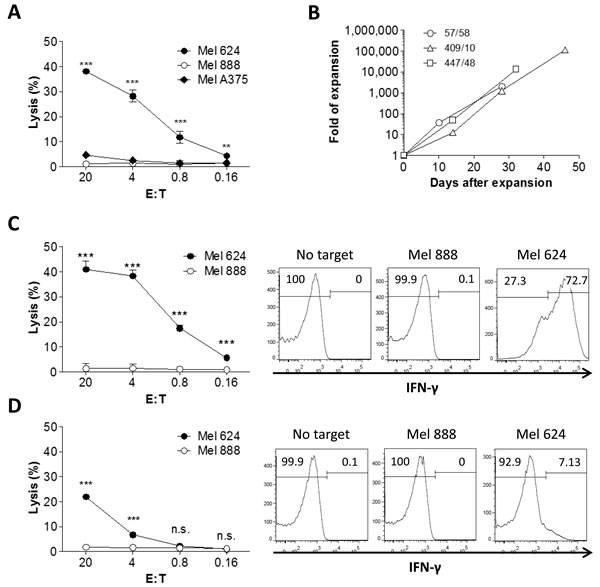
Figure 3: In vitro expansion and cytotoxicity of MART-1 TCR+ T cells isolated from humanized mice. A. Tetramer+ T cells were purified from spleen and bone marrow of hu-mice and their cytotoxicity against Mel 624 (HLA-A2+MART-1+), Mel 888 (HLA-A2-MART-1+), Mel A375 (HLA-A2+MART-1-) melanoma cells was measured after a short period (5 days) of in vitro stimulation with anti-huCD3/CD28 microbeads. B. In vitro expansion of tetramer+ T cells in the presence of OTK3, rhIL-12 and irradiated feeder cells. Tetramer+ T cells from 3 representative hu-mice are shown and data are presented as fold of expansion in cell numbers. C. and D. Tetramer+ CD8+ C. and CD4+ D. T cells sorted from in vitro expanded tetramer+ cells (shown in B.) were examined for cytotoxicity against melanoma cells using 51Cr release assay (left panel) and for IFN-γ production after cocultured with melanoma cells by flow cytometry (right panel). *, p < 0.05; **, p < 0.01; ***, p < 0.001; n.s., not significant.
Adoptive immunotherapy using in vitro-expanded MART-1-specific T cells generated in humanized mice
Tetramer+ CD8+ T cells were sorted out from hu-mouse splenocytes and expanded in vitro for approximately 30 days (Figure 4A). At the end of expansion, the majority of expanded cells were tetramer+ CD8+ T cells with a effector/memory (i.e., CCR7-CD45RA-) phenotype (Figure 4B), and capable of mediating efficient killing of HLA-A2+MART-1+ (Mel 624) melanoma cells (Figure 4C). We then assessed the antitumor activity of the expanded MART-1-specific CD8 T cells in melanoma-bearing mice. In NSG mice that were subcutaneously inoculated with Mel 624 melanoma cells (1x106 per mouse), tumor growth was significantly inhibited by adoptive transfer of in vitro expanded tetramer+ CD8+ T cells (Figure 4D, left panel). Flow cytometric analysis revealed that most TILs were tetramer+ CD8+ T cells with a CD45RA-CCR7- effector/memory phenotype (Figure 4D, right panel). Adoptive transfer of in vitro expanded tetramer+ CD8 T cells also mediated a significant protection against metastatic melanoma and markedly prolonged the survival in mice that were injected i.v. with Mel 624 melanoma cells (2x105 per mouse; Figure 4E). These data provide direct in vivo evidence that tetramer+ CD8+ T cells developed in hu-mice are capable of infiltrating into tumors and killing tumor cells after adoptive transfer into tumor-bearing recipients.
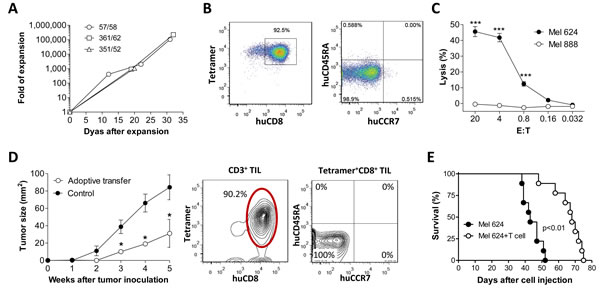
Figure 4: Antitumor effect by in vitro expanded MART-1-specific human CD8 T cells from humanized mice. Tetramer+ CD8+ T cells purified from hu-mice (n = 3) were expanded in cultures as shown in Fig 3b A.-C., and the anti-melanoma activity of in vitro expanded CD8+ T cells were assessed by adoptive transfer into melanoma-bearing recipients D.-E. A. Human T cell expansion in cultures at the indicated time points. B. Expression of MART-1-specific TCR, CD8, CD45RA and CCR7 on expanded human T cells. C. Cytotoxicity of the expanded human CD8+ T cells against melanoma cells. D. Left panel, tumor burden in mice receiving 1×106 of Mel 624 (HLA-A2+MART-1+; s.c.) cells with or without (control) adoptive transfer of in vitro-expanded tetramer+ CD8 T cells (1x107 per mouse ; n = 5 per group); Right panel, phenotypic analysis of CD3+ TILs at week 5 post-transfer of in vitro-expanded tetramer+ CD8 T cells. E. Survival of mice that received 2×105 Mel 624 cells (i.v.) with or without adoptive transfer of 1×107 in vitro-expanded tetramer+ CD8 T cells (n = 9 per group). *, p < 0.05; **, p < 0.01; ***, p < 0.001.
IL-15 promotes survival and antitumor responses of in vitro expanded MART-1 TCR+ T cells
We then assessed the effect of IL-15 on the survival and antitumor activity of adoptively transferred tetramer+ T cells in a metastatic melanoma model, in which mice were injected i.v. with Mel 624 melanoma cells (2x105 per mouse). IL-15 treatment was given by hydrodynamic injection of IL-15-expressing plasmids [12] one day prior to T cell transfer. At week 1 post-T cell transfer, the ratio of tetramer+ CD8+ T cells in peripheral blood was significantly (approximately 3-fold) greater in IL-15-treated than in the control group (Figure 5A). Importantly, this increase in tetramer+ CD8+ T cells in IL-15-treated mice was associated with a significantly enhanced antitumor response. As shown in Figure 5B, adoptive transfer of tetramer+ CD8+ T cells was significantly more effective in prolonging survival in IL-15-treated than in control mice bearing metastatic melanoma. These data indicate that IL-15 treatment has the potential to improve both the survival and antitumor activity of tumor antigen-specific CTLs. Because treatment with IL-15 alone did not induce detectable antitumor responses (Figure 5B), we conclude that the enhancement of antitumor effects by IL-15 was mediated by the adoptively transferred tetramer+ T cells.
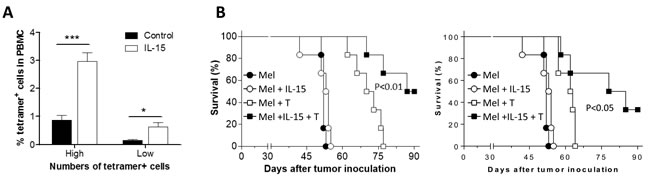
Figure 5: Improved antitumor effect by in vitro expanded MART-1-specific human CD8 T cells in IL-15-treated mice. A. Survival of adoptively transferred in vitro-expanded tetramer+ CD8+ T cells (T cells were expanded as described in Figure 4A) in mice with or without IL-15 treatment at day 7 post-cell transfer (‘High’ and ‘Low’ represent mice receiving 1x107 or 5x106 tetramer+ CD8+ T cells per mouse, respectively; n = 6 per group). B. Survival of mice that received 2×105 Mel 624 cells (i.v.) alone or together with 1x107 (left) or 5x106 (right) in vitro-expanded tetramer+ CD8 T cells with or without IL-15 treatment (n = 6 per group).*, p < 0.05; **, p < 0.01; ***, p < 0.001.
A short treatment of the expanded MART-1 TCR+ T cells with rapamycin immediately prior to adoptive therapy leads to markedly improved protection against metastatic melanoma
We further explore the potential of treatment with rapamycin prior to adoptive transfer to improve the survival and antitumor activity of tumor antigen-specific T cells. In vitro expanded tetramer+ CD8+ T cells were cultured with rapamycin (100nM) for 3 days immediately before transfer into IL-15-treated or non-treated melanoma-bearing recipients that were injected i.v. with Mel 624 melanoma cells (1x105 per mouse). A short treatment with rapamycin significantly improved T cell survival in both non-treated and IL-15-treated recipients (Figure 6A). In the recipients without IL-15 treatment, the percentage of rapamycin-treated tetramer+ CD8+ T (Rapa-T) cells measured at week 1 (0.86±0.09%) was significantly (approximately 3 fold) greater than that of non-treated tetramer+ T (Ctrl-T) cells (0.30±0.06%; Figure 6A). Treatment of the recipients with IL-15 synergistically enhanced the effect of rapamycin in improving T cell survival. The percentage of surviving Rapa-T cells at week 1 in IL-15-treated mice (Rapa-T/IL-15; 3.26±0.46%) was more than 10-fold and about 4-fold greater than that of Ctrl-T and Rapa-T cells, respectively, in mice that did not receive IL-15 treatment, and approximately 4-fold greater than that of Ctrl-T cells in IL-15-treated mice (Ctrl-T/IL-15; 0.84±0.14%; Figure 6A).
Rapa-T cells also mediated significantly stronger antitumor responses than Ctrl-T cells. Consistent with the results shown in Figure 5B, adoptive transfer of Ctrl-T cells induced significant protection against metastatic melanoma, which was further enhanced by IL-15 treatment (Figure 6B). Compared to Ctrl-T cells, Rapa-T cells were significantly more effective in eradicating melanoma, leading to improved survival rates in mice with metastatic melanoma (Figure 6B). Notably, a powerful antitumor response leading to 100% survival for more than 300 days was seen in IL-15-treated mice infused with Rapa-T cells, while most of the mice receiving Rapa-T cells without IL-15 and half of the IL-15-treated mice receiving Ctrl-T cells succumbed to metastatic melanoma. All surviving mice were sacrificed at day 320 post-infusion and none showed detectable tumors at autopsy.
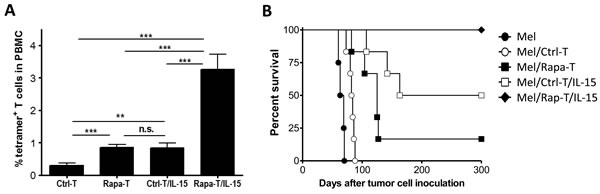
Figure 6: A short in vitro treatment with rapamycin significantly improves the survival and antitumor activity of in vitro expanded tetramer+ CD8+ T cells. A. Survival of adoptively transferred rapamycin-treated (Rap-T) and non-treated (Ctrl-T) tetramer+ CD8+ T cells at day 7 in mice with or without IL-15 treatment. B. Survival of IL-15-treated and non-treated mice that received injection i.v. of 1×105 Mel 624 cells plus 5x106 Ctrl-T (Mel/Ctrl-T and Mel/Ctrl-T/IL-15) or Rapa-T (Mel/Rapa-T and Mel/Rapa-T/IL-15) cells (n = 6 per group), and non-treated mice receiving 1×105 Mel 624 cells only (Mel; n = 4). **, p < 0.01; ***, p < 0.001; n.s., not significant
Discussion
In this study, we developed a hu-mouse model that permits efficient generation of tumor antigen-specific human T cells from genetically engineered CD34+ cells. Using this model in combination with in vitro cell expansion, we were able to produce large quantities of melanoma antigen MART-1-specific T cells with antigen-specific antitumor activity. This approach provides a practical means of producing potentially unlimited source of tumor-specific human T cells, which can be used in the development and testing of new cancer immunotherapy protocols in the preclinical settings. The recent development of “personalized immune (PI)” hu-mice demonstrates the feasibility of generating patient-specific human T cells [13]. Thus, the combination of the strategy described in the current study with the “PI” hu-mouse model may permit the production of tumor-reactive human T cells for preclinical exploration of individualized cancer immunotherapy.
Clinical trials using autologous peripheral lymphocytes genetically engineered by retroviral vector containing tumor antigen specific TCR gene have shown objective cancer regression in 10-30% patients with metastatic melanoma [14-16]. However, a challenge for this strategy is to prevent misparing of the endogenous TCR subunits with introduced TCR chains in the genetically-manipulated T cells, which may potentially alter TCR specificity leading to not only the loss of anti-tumor responses, but also formation of autoreactive T cells [17-19]. Unlike genetically engineered mature T cells, MART-1-TCR+ human T cells in the hu-mouse are developed from TCR gene-transduced hematopoietic stem cells, so that most of these cells should express only the tumor antigen-specific TCR.
As expected, the majority of human T cells expressing the HLA-A2-restricted MART-1-TCR+ developing in hu-mice expressed CD8. However, these hu-mice also had a significant population of tetramer+ CD4+ T cells including FoxP3+CD4+ Tregs (Figure S5). Similar to our results, the presence of CD4+ [20] and CD4-CD8- [21] human T cells that recognize melanoma antigens in an HLA class I-restricted manner was also found in humans. Previous studies on the ability of MHC class I-restricted CD4 T cells to generate antitumor effects have been contradictory [22-25]. A recent study showed that the antitumor efficacy of CD4+ T cells expressing MHC class I-restricted TCR depends on TCR affinity, and only those with high affinity TCR may mediate CD8-independent cytotoxicity [26]. In the present study, we showed that, although to a significantly lower extent than tetramer+ CD8+ T cells, tetramer+ CD4+ T cells developing in the hu-mice were also functional and capable of mediating antigen-specific responses (i.e., cytotoxicity and IFN-γ production) in vitro when cocultured with the target cells. However, we failed to detect antigen-specific responses in tetramer+ CD4+ T cells from MART-1 peptide-immunized hu-mice, despite that tetramer+ CD8+ T cells from the same hu-mice responded robustly to immunization. Clearly, further studies are needed to determine whether the tetramer+ CD4+ T cells may provide help to CD8 T cells expressing the same TCR. Nonetheless, our results implicate that in vitro analysis may not accurately predicate the in vivo antitumor effect of tumor antigens-specific T cells.
IL-15 is an immune regulatory cytokine with broad activities that include inducing differentiation and proliferation of T, B and NK cells, enhancing the cytolytic activity of CD8+ T cells and contributing to long term survival of memory T cells [27]. The potential of IL-15 to promote the survival and antitumor activity of adoptively transferred tumor-specific CD8+ T cells has been demonstrated in mice [28, 29], but remains poorly understood in humans. In the present study, we observed that in vivo treatment with IL-15 dramatically promoted the survival and/or proliferation of activated MART-1-TCR+ CD8 T cells, which was associated with a significantly increased antitumor effect, in melanoma-bearing hosts. The data support the idea of using IL-15 to enhance antitumor responses of adoptive T-cell transfer therapy, and a clinical trial launched recently to test the effectiveness of TIL administration combined with IL-15 in metastatic melanoma [27].
Another novel observation made in this study is that IL-15 administration combined with infusion of rapamycin-treated MART-1-TCR+ human CD8+ T cells produced a strong synergistic effect in promoting the survival and antitumor responses of adoptively transferred tumor antigen-specific human T cells. It has been shown that inhibition of mTORC1 by rapamycin promotes memory CD8+ T cell generation and function [30, 31] through down-regulation of T-bet and upregulation of Eomes in CD8+ T cells [32]. Using TCR transgenic mouse models, it was recently reported that a short course of injection with rapamycin enhances vaccine-induced CD8+ T cell memory and antitumor responses in mice [33, 34]. In this study, we observed that a brief culture of human CD8+ T cells with rapamycin significantly improved their survival and antitumor responses in melanoma-bearing recipients, particularly in mice treated IL-15. Since enhanced CD8+ T cell memory responses by mTOR inhibition is independent of IL-15 [32, 33], rapamycin-treated CD8 T cells should retain the ability to respond to the survival signaling provided by IL-15, which may explain why IL-15 and rapamycin can act synergistically to improve the survival and antitumor responses of tumor antigen-specific CD8+ T cells. The combinatorial treatment of the tumor-reactive CD8 T cells with rapamycin and of the recipient with IL-15 may provide a novel protocol for potentiating adoptive T cell immunotherapy.
Materials and Methods
Animals and human tissues and cells
NOD.Cg-Prkdcscid Il2rgtm1Wjl/SzJ (NOD/SCID/c-/- or NSG) mice were purchased from the Jackson Laboratory (Bar Harbor, ME), housed in a specific pathogen-free micro-isolator environment and used in experiments at 6 to 8 weeks of age. Human fetal thymus and liver tissues of gestational age of 17 to 20 weeks were obtained from Advanced Bioscience Resource (Alameda, CA). Protocols involving the use of discarded human tissues and animals were approved by the Institutional Review Board (IRB) and Institutional Animal Care and Use Committee (IACUC) of Columbia University Medical Center (New York, NY).
Humanized mouse preparation
NSG mice were conditioned with sublethal (2 Gy) total body irradiation, and received lentiviral vector transduced human CD34+ FLCs (2×105/mouse, i.v.) with a cryopreservation treated FTHY fragment measuring about 1mm3 (under the recipient kidney capsule) from the same fetal donor, as previously described [9, 10]. CD34+ FLCs were isolated by a magnetic-activated cell sorter (MACS) separation system using anti-CD34 microbeads (Miltenyi Biotec, Auburn, CA). Levels of human hematopoietic cells in humanized mice were determined by flow cytometric (FCM) analysis using various combinations of the following mAbs: anti-human CD45, CD19, CD3, CD4, CD8, CD45RA, CCR7; anti-mouse CD45 and Ter119; and isotype control mAbs (all antibodies were purchased from BD PharMingen, San Diego, CA). MART-1-TCR+ T cells were identified by HLA-A*0201/MART-1 (ELAGIGILTV) Tetramer (Beckman Coulter Immunotech). Mononuclear cells were prepared using density gradient centrifugation with Histopaque 1077 (Sigma-Aldrich, St. Louis, MO). Analysis was performed on a FACSCanto or LSR II (Becton Dickinson, Mountain View, CA). Dead cells were excluded from the analysis by gating out lower forward scatter and high propidium iodide-retaining cells.
Lentiviral production and human CD34+ FLC transduction
Pseudotyped lentiviral vectors were produced by transfection using Lipofectamine 2000 (Invitrogen, San Diego, CA) of 293FT cells in 10-cm plates. a 4-plasmid system consisting of the transfer vector (MART-1 antigen specific TCR, DMF5 clone; 10µg) and 3 packaging plasmids (VSV-G 3.6µg, pMDLg/pRRE 6.7 µg, and pRSV-Rev 6.7 µg) was used for MART-1 TCR-lentivirus production. The supernatant was collected 48 hour post-transfection and concentrated by ultracentrifugation at 50,000 g for 2 hours. Lentiviruses were stored at -80°C until use. Human CD34+ FLCs were stimulated overnight in media containing 50ng/mL rhSCF (R&D, Minneapolis, MN), 50ng/mL Flt3L (ebioscience, San Diego, CA), 25 ng/mL TPO (R&D), and 10ng/mL IL-3 (R&D), in a 24-well plate pre-coated with retronectin (Takara Bio Inc), followed by transduction with lentiviral vectors for 12 hours. Cells were washed twice and intravenously injected into sub-lethal irradiated mice as described above.
MART-1 peptide immunization
MART-1 peptides (ELAGIGILTV, Biosynthesis Inc, Lewisville, Texas) were emulsified with Freud’s Complete Adjuvant (CFA) with 1:1 ratio and subcutaneously injected into humanized mice. Immunized mice were analyzed for MART-1-specific responses 3 weeks after immunization.
CTL assay
Purified MART-1 tetramer+ human T cells were co-incubated with 51Cr-labeled melanoma cells at the indicated effector:target (E:T) ratios in 96-well plates at 37°C for 4-6 hours. Lysis of tumor cells was measured by 51Cr release in the supernatant counted using a Perkin Elmer 1450 microbeta liquid scintillation & luminescence counter. Specific lysis (%) of target cells was calculated as: = [(experimental release (cpm) - spontaneous release (cpm)]/[(maximum release (cpm) - spontaneous release (cpm)] ×100%.
In vitro expansion of MART-1 antigen specific T cells
MART-1 tetramer+ T cells were purified from lymphoid organs of humanized mice by cell sorting using an Influx cell sorter (BD Biosciences) and expanded in vitro using a previously described protocol with some modification [35, 36]. Briefly, purified T cells were stimulated with pooled allogeneic PBMCs (1.5×107/mL, 35 Gy irradiated) and Epstein-Barr virus-transformed lymphoblastoid cell lines (EBV-LCL, 4.3×105/mL, 60 Gy irradiated), 30 ng/mL anti-huCD3 (OKT3), and 100U/mL recombinant human IL-2 (rhIL-2) in RMPI 1640 medium supplemented with 10% human AB serum. One third of the culture supernatant was replaced with fresh media containing 300U/mL rhIL-2 every 2-3 days. The cells were restimulated with anti-human CD3 and freshly irradiated feeder cells (i.e., allogeneic PBMCs and EBV-LCL) every 2 weeks.
Assessment of antitumor effects of in vitro-expanded MART-1 TCR+ T cells in melanoma-bearing mice
In vitro expanded CD8 MART-1 TCR+ T cells were adoptively transferred (i.v.) into NSG mice that were inoculated with Mel 624 cells (s.c. or i.v.) on the same day, and the antitumor effect was evaluated by measuring tumor mass or recipient survival time in mice receiving s.c. and i.v. injection of melanoma cells, respectively. Tumor mass was measured using fine calipers (Marathon, Richmond Hill, Canada) and calculated by the product of the two largest perpendicular diameters a×b (mm2), as previously described [6]. To assess the effect of IL-15 on antitumor responses, some recipient mice were given hydrodynamic injection (50μg/2mL/mouse via tail vein) of plasmid containing rhIL-15 gene one day prior to injection of melanoma cells, as previously described [12]. In some experiments, rapamycin (100nM; Sigma Aldrich; St. Louis, Missouri) was added into the T cell expansion culture for 3 days immediately before adoptive transfer.
Statistical analysis
The level of significant differences in group means was determined by the student’s t test. Graft survival data were presented as Kaplan-Meier survival curves and differences between groups were analyzed by logrank test using GraphPad Prism (San Diego, CA). All statistical analysis was performed using Prism 4 (GraphPad Software, San Diego, CA). A p value of ≤ .05 was considered significant in all analyses herein.
Acknowledgments
The authors thank Dr. Kang Liu (Columbia University) for critical review of this manuscript, Dr. Steven A. Rosenberg for providing the MART-1-specific lentiviral vector and human melanoma cell lines, and Dr. Jianzhu Chen at MIT for providing huIL-15 plasmid. This study was supported by grants from Chinese MOST (2015CB964400, 2013CB966900, and 2014AA021601), NSFC (81273334, 81200397 and 81570145), the Project of Science and Technology of Jilin Province (20140520013JH), and NIH (P01AI045897 and R01AI064569).
Conflicts of Interest
The authors declare no competing financial interest.
References
1. Rosenberg SA, Yang JC, Sherry RM, Kammula US, Hughes MS, Phan GQ, Citrin DE, Restifo NP, Robbins PF, Wunderlich JR, Morton KE, Laurencot CM, Steinberg SM, et al. Durable Complete Responses in Heavily Pretreated Patients with Metastatic Melanoma Using T-Cell Transfer Immunotherapy. Clinical Cancer Research. 2011; 17:4550-4557.
2. Alexandrescu DT, Ichim TE, Riordan NH, Marincola FM, Di Nardo A, Kabigting FD and Dasanu CA. Immunotherapy for Melanoma: Current Status and Perspectives. Journal of Immunotherapy. 2010; 33:570-590 510.1097/CJI.1090b1013e3181e1032e1098.
3. Wolchok JD, Kluger H, Callahan MK, Postow MA, Rizvi NA, Lesokhin AM, Segal NH, Ariyan CE, Gordon R-A, Reed K, Burke MM, Caldwell A, Kronenberg SA, et al. Nivolumab plus Ipilimumab in Advanced Melanoma. New England Journal of Medicine. 2013; 369:122-133.
4. Spranger S, Spaapen RM, Zha Y, Williams J, Meng Y, Ha TT and Gajewski TF. Up-Regulation of PD-L1, IDO, and Tregs in the Melanoma Tumor Microenvironment Is Driven by CD8+ T Cells. Science Translational Medicine. 2013; 5:200ra116.
5. Yang L, Qin XF, Baltimore D and Van Parijs L. Generation of functional antigen-specific T cells in defined genetic backgrounds by retrovirus-mediated expression of TCR cDNAs in hematopoietic precursor cells. Proceedings of the National Academy of Sciences of the United States of America. 2002; 99:6204-6209.
6. Yang L and Baltimore D. Long-term in vivo provision of antigen-specific T cell immunity by programming hematopoietic stem cells. Proceedings of the National Academy of Sciences of the United States of America. 2005; 102:4518-4523.
7. Vatakis DN, Koya RC, Nixon CC, Wei L, Kim SG, Avancena P, Bristol G, Baltimore D, Kohn DB, Ribas A, Radu CG, Galic Z and Zack JA. Antitumor activity from antigen-specific CD8 T cells generated in vivo from genetically engineered human hematopoietic stem cells. Proceedings of the National Academy of Sciences. 2011; 108:E1408-E1416.
8. Lan P, Wang L, Diouf D, Eguchi H, Bronson R, Sachs DH, Sykes M and Yang YG. Induction of human T cell tolerance to porcine xenoantigens through mixed hematopoietic chimerism. Blood. 2004; 103:3964-3969.
9. Lan P, Tonomura N, Shimizu A, Wang S and Yang YG. Reconstitution of a functional human immune system in immunodeficient mice through combined human fetal thymus/liver and CD34+ cell transplantation. Blood. 2006; 108:487-492 [Epub 2006 Jan 2012].
10. Tonomura N, Habiro K, Shimizu A, Sykes M and Yang YG. Antigen-specific human T-cell responses and T cell-dependent production of human antibodies in a humanized mouse model. Blood. 2008; 111:4293-4296.
11. Johnson LA, Morgan RA, Dudley ME, Cassard L, Yang JC, Hughes MS, Kammula US, Royal RE, Sherry RM, Wunderlich JR, Lee C-CR, Restifo NP, Schwarz SL, et al. Gene therapy with human and mouse T-cell receptors mediates cancer regression and targets normal tissues expressing cognate antigen. Blood. 2009; 114:535-546.
12. Chen Q, Khoury M and Chen J. Expression of human cytokines dramatically improves reconstitution of specific human-blood lineage cells in humanized mice. Proceedings of the National Academy of Sciences. 2009; 106:21783-21788.
13. Kalscheuer H, Danzl N, Onoe T, Faust T, Winchester R, Goland R, Greenberg E, Spitzer TR, Savage DG, Tahara H, Choi G, Yang Y-G and Sykes M. A model for personalized in vivo analysis of human immune responsiveness. Science Translational Medicine. 2012; 4:125ra130.
14. Rosenberg SA, Restifo NP, Yang JC, Morgan RA and Dudley ME. Adoptive cell transfer: a clinical path to effective cancer immunotherapy. NatRevCancer. 2008; 8:299-308.
15. Morgan RA, Dudley ME, Wunderlich JR, Hughes MS, Yang JC, Sherry RM, Royal RE, Topalian SL, Kammula US, Restifo NP, Zheng Z, Nahvi A, de Vries CR, et al. Cancer regression in patients after transfer of genetically engineered lymphocytes. Science. 2006; 314:126-129.
16. Johnson LA, Morgan RA, Dudley ME, Cassard L, Yang JC, Hughes MS, Kammula US, Royal RE, Sherry RM, Wunderlich JR, Lee CC, Restifo NP, Schwarz SL, et al. Gene therapy with human and mouse T-cell receptors mediates cancer regression and targets normal tissues expressing cognate antigen. Blood. 2009; 114:535-546.
17. Cohen CJ, Zhao Y, Zheng Z, Rosenberg SA and Morgan RA. Enhanced antitumor activity of murine-human hybrid T-cell receptor (TCR) in human lymphocytes is associated with improved pairing and TCR/CD3 stability. Cancer research. 2006; 66:8878-8886.
18. Kuball J, Dossett ML, Wolfl M, Ho WY, Voss RH, Fowler C and Greenberg PD. Facilitating matched pairing and expression of TCR chains introduced into human T cells. Blood. 2007; 109:2331-2338.
19. Bendle GM, Linnemann C, Hooijkaas AI, Bies L, de Witte MA, Jorritsma A, Kaiser AD, Pouw N, Debets R, Kieback E, Uckert W, Song JY, Haanen JB, et al. Lethal graft-versus-host disease in mouse models of T cell receptor gene therapy. Nature medicine. 2010; 16:565-570, 561p following 570.
20. Nishimura MI, Avichezer D, Custer MC, Lee CS, Chen C, Parkhurst MR, Diamond RA, Robbins PF, Schwartzentruber DJ and Rosenberg SA. MHC Class I-restricted Recognition of a Melanoma Antigen by a Human CD4+ Tumor Infiltrating Lymphocyte. Cancer research. 1999; 59:6230-6238.
21. Voelkl S, Moore T, Rehli M, Nishimura M, Mackensen A and Fischer K. Characterization of MHC class-I restricted TCRαβ+ CD4− CD8− double negative T cells recognizing the gp100 antigen from a melanoma patient after gp100 vaccination. Cancer Immunol Immunother. 2009; 58:709-718.
22. Johnson LA, Heemskerk B, Powell DJ, Jr., Cohen CJ, Morgan RA, Dudley ME, Robbins PF and Rosenberg SA. Gene Transfer of Tumor-Reactive TCR Confers Both High Avidity and Tumor Reactivity to Nonreactive Peripheral Blood Mononuclear Cells and Tumor-Infiltrating Lymphocytes. The Journal of Immunology. 2006; 177:6548-6559.
23. Frankel TL, Burns WR, Peng PD, Yu Z, Chinnasamy D, Wargo JA, Zheng Z, Restifo NP, Rosenberg SA and Morgan RA. Both CD4 and CD8 T Cells Mediate Equally Effective In Vivo Tumor Treatment When Engineered with a Highly Avid TCR Targeting Tyrosinase. The Journal of Immunology. 2010; 184:5988-5998.
24. Xue S-A, Gao L, Ahmadi M, Ghorashian S, Barros RD, Pospori C, Holler A, Wright G, Thomas S, Topp M, Morris EC and Stauss HJ. Human MHC Class I-restricted high avidity CD4+ T cells generated by co-transfer of TCR and CD8 mediate efficient tumor rejection in vivo. OncoImmunology. 2013; 2:e22590.
25. Willemsen R, Ronteltap C, Heuveling M, Debets R and Bolhuis R. Redirecting human CD4+ T lymphocytes to the MHC class I-restricted melanoma antigen MAGE-A1 by TCR [alpha][beta] gene transfer requires CD8[alpha]. Gene Ther. 2005; 12:140-146.
26. Soto C, Stone J, Chervin A, Engels B, Schreiber H, Roy E and Kranz D. MHC-class I-restricted CD4 T cells: a nanomolar affinity TCR has improved anti-tumor efficacy in vivo compared to the micromolar wild-type TCR. Cancer Immunol Immunother. 2013; 62:359-369.
27. Steel JC, Waldmann TA and Morris JC. Interleukin-15 biology and its therapeutic implications in cancer. Trends in Pharmacological Sciences. 2012; 33:35-41.
28. Melchionda F, Fry TJ, Milliron MJ, McKirdy MA, Tagaya Y and Mackall CL. Adjuvant IL-7 or IL-15 overcomes immunodominance and improves survival of the CD8+ memory cell pool. The Journal of Clinical Investigation. 2005; 115:1177-1187.
29. Klebanoff CA, Finkelstein SE, Surman DR, Lichtman MK, Gattinoni L, Theoret MR, Grewal N, Spiess PJ, Antony PA, Palmer DC, Tagaya Y, Rosenberg SA, Waldmann TA and Restifo NP. IL-15 enhances the in vivo antitumor activity of tumor-reactive CD8+ T Cells. Proceedings of the National Academy of Sciences of the United States of America. 2004; 101:1969-1974.
30. Araki K, Turner AP, Shaffer VO, Gangappa S, Keller SA, Bachmann MF, Larsen CP and Ahmed R. mTOR regulates memory CD8 T-cell differentiation. Nature. 2009; 460:108-112.
31. Pearce EL, Walsh MC, Cejas PJ, Harms GM, Shen H, Wang L-S, Jones RG and Choi Y. Enhancing CD8 T-cell memory by modulating fatty acid metabolism. Nature. 2009; 460:103-107.
32. Rao Rajesh R, Li Q, Bupp Melanie RG and Shrikant Protul A. Transcription Factor Foxo1 Represses T-bet-Mediated Effector Functions and Promotes Memory CD8+ T Cell Differentiation. Immunity. 2012; 36:374-387.
33. Li Q, Rao R, Vazzana J, Goedegebuure P, Odunsi K, Gillanders W and Shrikant PA. Regulating Mammalian Target of Rapamycin To Tune Vaccination-Induced CD8+ T Cell Responses for Tumor Immunity. The Journal of Immunology. 2012; 188:3080-3087.
34. Thomas D, Doty R, Tosic V, Liu J, Kranz D, McFadden G, MacNeill A and Roy E. Myxoma virus combined with rapamycin treatment enhances adoptive T cell therapy for murine melanoma brain tumors. Cancer Immunol Immunother. 2011; 60:1461-1472.
35. Dudley ME, Wunderlich JR, Robbins PF, Yang JC, Hwu P, Schwartzentruber DJ, Topalian SL, Sherry R, Restifo NP, Hubicki AM, Robinson MR, Raffeld M, Duray P, et al. Cancer Regression and Autoimmunity in Patients After Clonal Repopulation with Antitumor Lymphocytes. Science. 2002; 298:850-854.
36. Riddell SR and Greenberg PD. The use of anti-CD3 and anti-CD28 monoclonal antibodies to clone and expand human antigen-specific T cells. JImmunolMethods. 1990; 128:189-201.