
Introduction: Landmark Improvements in HER2+ Disease
Since the approval of trastuzumab in the adjuvant setting by the FDA in 2006, most patients with HER2+ early BC are doing very well with standard trastuzumab therapy added to chemotherapy. Planned Joint Analysis of OS from NSABP B-31 and NCCTG N9831, demonstrated that the addition of trastuzumab to a taxane and anthracycline chemotherapy backbone resulted in a durable improvement in survival, after a median time on study of approximately eight and a half years. [11]
In fact, patients nearly diagnosed with localized HER2+ BC have an excellent prognosis, even if treated with less toxic adjuvant systemic therapy, as recently demonstrated by Dana Farber investigators [10]. This nonrandomized prospective trial evaluated an adjuvant chemotherapy regimen consisting of weekly paclitaxel at 80mg/m2 and trastuzumab at 2 mg/kg for 12 weeks, followed by 9 months of trastuzumab in 406 women with HER2-positive, node-negative tumors ≤ 3 cm. Disease-free survival achieved an impressive result at 3 years of 98.7% (P < .0001), and the regimen was associated with great cardiac safety. [12]
In the metastatic setting, Slamon et al. [8] evaluated the addition of trastuzumab to chemotherapy among women diagnosed with metastatic HER2+ BC in the landmark trial that lead to trastuzumab approval in the metastatic setting. The authors found that the addition of trastuzumab to chemotherapy was associated with a longer time to disease progression, higher rate of objective response, and a longer survival. Since this first trial, many others corroborated trastuzumab benefit in survival outcomes among women with metastatic HER2+ BC.
Incorporation of new agents, as evidenced by the CLEOPATRA trial, in which pertuzumab, a humanized monoclonal antibody that binds to HER2 at a different epitope than that at which trastuzumab binds, was added to the standard docetaxel and trastuzumab combination, and lead to striking improvements in PFS and OS in a cohort of advanced HER2+ BC patients, reaching the median OS boundary of almost 5 years. [13-15]
Despite this robust clinical benefit, anti-HER therapy resistance, either de novo or acquired, is an important clinical challenge in the management of BC patients. Research has been dedicated to a better understanding of the molecular mechanisms involved of trastuzumab resistance. [16]
Main Resistance Mechanisms Pathways to Trastuzumab
PIK3CA Pathway
Anti-HER2 Therapy Benefit and PIK3CA alterations
The PI3K/AKT/mTOR pathway is an important growth factor pathway and a key effector of HER2 signalling. HER2 phosphorylation may lead to pathway activation.[17] Constitutive activation of PI3K, either by PIK3CA mutation or PTEN loss, are associated with resistance to therapies targeting HER2, and possibly are able to identify a group of patients with poor prognosis after trastuzumab therapy. These alterations might result in continuous pathway signalling, despite HER2 blockage, priming a treatment escape mechanism. [18-20]
Many investigators evaluated trastuzumab benefit in patients enrolled in clinical trials in distinct disease scenarios, according to alterations in the PI3K pathway. Most of them failed to demonstrate a relationship between PIK3CA mutations and trastuzumab benefit. As an example, the FinHER adjuvant phase III trial genotyped 687 HER2+ BC patients. PIK3CA mutations were not statistically significantly associated with trastuzumab benefit, or survival outcomes. [21] Similarly, a recent metaanalysis also reached the conclusion that neither PTEN loss, nor PIK3CA mutation were associated with response rate of trastuzumab based neoadjuvant treatment. [22] Analysis of other trials also failed to demonstrate a relationship between PIK3CA or PTEN status and adjuvant trastuzumab benefit. [23, 24]
The EMILIA trial compared the effectiveness of TDM-1 versus lapatinib and capecitabine in patients previously treated with trastuzumab. Samples from patients were prospectively collected for PIK3CA mutation analysis. Patients in the lapatinib arm with PIK3CA mutations had worse outcomes than those with wild-type PIK3CA, but the presence of PIK3CA mutations had absolutely no effect on PFS or OS in patients treated with T-DM1, suggesting that this drug might be an attractive alternative for patients harbouring this alteration. [25]
The evidence described above is somehow contradictory to preclinical data. One important aspect to be taken into account is the fact that in vitro studies that initially identified PIK3CA mutation as a resistance factor for HER2-targeted treatment did not account for trastuzumab mediated antibody dependent cellular cytotoxicity (ADCC). Recognition of tumor cells opsonized by monoclonal antibodies, such as trastuzumab, is mediated through receptors expressed on effector cells, as well as monocytes, dendritic cells, and granulocytes. Upon recognition, these effectors induce tumor cell death. Dendritic cells capture the monoclonal antibody conjugated with tumor antigens released by dying cells in the form of immune complexes. Ultimately, these processed cells are presented to cytotoxic and helper T cells. As a consequence, both tumor-specific cytotoxic T cells and T-helper cells are activated, leading to tumor B-cell stimulation and expansion. The production of tumor-directed host antibodies is the final step in this important ADCC cytotoxic mechanism. [26] Also, trastuzumab enhances the ability of Natural Killer (NK) cells mediated ADCC, NK destruction of target cells through nonspecific mechanisms, ultimately leading to activation and expansion of tumor-specific cytotoxic T lymphocytes. [27] In fact, the importance of NK cell function is highlighted by the fact that there is a direct correlation between NK cell function and response to trastuzumab in metastatic HER2+ BC patients.[28] Preclinical models may not account for this, since many are conducted in vitro or in immunosuppressed models. Furthermore, trastuzumab is a human monoclonal antibody and therefore, unlikely to engender an immune reaction in a model system in vitro.
PIK3CA alterations as a prognostic marker
Survival outcomes of HER2+ BC patients might be influenced by PIK3CA status. Biomarker analysis from the CLEOPATRA trial demonstrated that PIK3CA mutation was associated with worse survival outcomes among patients with advanced HER2+ BC.[29] Nevertheless, the addition of pertuzumab benefited all patients, regardless of PIK3CA status.
Additionally, a group of researchers from Memorial Sloan-Kettering Cancer Center evaluated a group of 63 patients with HER2+ BC with disease recurrence after adjuvant trastuzumab treatment, or progressive metastatic disease on a trastuzumab-containing regimen.[20] All patients underwent biopsy of at least a single local or distant site to document progressive disease. PIK3CA and HER2 mutation status were analysed by Sequenon (genotyping DNA sequences), and HER2 and PTEN status were analysed by immunohistochemistry. Absent or significantly diminished PTEN expression was noted in 59%, and activating mutations in PIK3CA in 29% of cases evaluable for PTEN or PI3K mutation.
These authors also compared PTEN loss and PIK3CA mutation rate between 2 HER2+ BC cohorts of patients: one of them refractory to trastuzumab and the other unexposed to trastuzumab. They demonstrated that the combined rate of PTEN loss and PIK3CA mutation in the trastuzumab-refractory tumors was 71%, compared with 44% in the unexposed cohort, suggesting that trastuzumab exposure may lead to alterations in PIK3CA pathway.
Therefore, evidence supporting the hypothesis that PI3K alterations might lead to worse prognosis in HER2+ BC, as well as resistance to anti-HER2 therapy, is mostly based on small retrospective series. At the present time, the first (and still only) actionable genomic alteration in BC, is amplification of HER2, and anti-HER2 therapy is required in all disease scenarios where this alteration is observed.
Response to Neoadjuvant Chemotherapy combined with Anti-HER2 therapy according to PIK3CA status
Correlation between PI3K alterations and pCR achievement was extensively studied. Loibl and colleagues investigated whether the presence of a PIK3CA mutation affected the odds of achieving a pCR among patients enrolled in the GeparQuattro, GeparQuinto, and GeparSixto neoadjuvant clinical trials. [30] In these trials, HER2+ BC patients received either trastuzumab or lapatinib, or the combination plus anthracycline-taxane based chemotherapy.[31] The authors found that the pCR rate was lower among HER2+ women with at least one PIK3CA mutation in their tumor, compared with women without a PIK3CA mutation. In alignment with these results, investigators from the neoadjuvant TRYPHAENA study also found numerically lower pCR rates in patients with tumors carrying any PIK3CA mutation tested for, although this did not reach a statistically significant level, possibly due to small sample size. [32] Other neoadjuvant studies also found an association between PIK3CA mutation and lower rates of pCR, irrespectively of treatment arm.[33]
Of note, at the 2015 American Society of Clinical Oncology (ASCO) Meeting, investigators reported pCR rates according to PIK3CA status among HER2+ BC patients who received neoadjuvant trastuzumab, lapatinib, or both in addition to a taxane-based chemotherapy. Significantly lower rates of pCR were observed in PIK3CA mutant tumors after anti-HER2 treatment, namely 16.2% versus 29.6% (p < 0.001), for patients with and without a PIK3CA mutation, respectively. [34]
Table 1 summarizes differences in pCR rates according to PIK3CA mutation status.
Table 1: pCR Rates according to PIK3CA pathway alteration
%pCR |
PIK3CA WT |
PIK3CA MUT |
|
All treatments |
34.5% |
21.3% |
Majewski et al., 2015 |
29.6% |
16.2% |
Loibl et al., 2015 |
|
32.8% |
19.4% |
Loibl et al., 2014 |
|
%pCR |
PIK3CA WT |
PIK3CA MUT |
|
Dual Anti-HER2 Therapy |
55.8% |
28.6% |
Majewski et al., 2015 |
39.1% |
16.7% |
Loibl et al., 2015 |
|
37.1% |
17.4% |
Loibl et al., 2014 |
mTOR inhibitors combined with Anti-HER2 therapy
Therefore, much interest is focused on this important proliferative pathway that may contribute to trastuzumab resistance. Inhibition of mTOR has been shown to be an important therapeutic strategy in HER2+ BC. BOLERO-3 aimed to assess whether the addition of the mTOR inhibitor everolimus to trastuzumab might restore sensitivity to trastuzumab. Eligible patients for this trial had HER2+, trastuzumab-resistant, advanced BC who had previously received taxane therapy. Patients were randomized to receive daily everolimus (5 mg/day) plus weekly trastuzumab (2 mg/kg) and vinorelbine (25 mg/m2) or to placebo plus trastuzumab plus vinorelbine, in 3-week cycle. As a result, the addition of everolimus prolonged PFS from 5.78 months to 7.00 months (HR=0.78, p=0.0067).[35] In PFS subgroup analysis, most groups favoured the addition of everolimus, although women who had hormone receptor negative disease derived more benefit from adding the mTOR inhibitor to the standard therapy.
BOLERO-1 also focuses on the strategy of targeting the PI3K/AKT/mTOR pathway. In this trial, everolimus was added to paclitaxel and trastuzumab in patients with metastatic HER2+ BC patients who had not received previous trastuzumab or chemotherapy for advanced BC within 12 months of randomisation. Primary endpoint was PFS, and secondary endpoints included OS, response rate (RR), and clinical benefit rate. The results were presented at the 2014 San Antonio Breast Cancer Symposium (SABCS) and published in Lancet Oncology.[36] The study did not meet its primary objective in the full population, with comparable PFS among patients who received everolimus versus placebo. However, the authors found that in the hormone receptor (HR)-negative subpopulation, everolimus-treated patients achieved a median PFS of 20.27 months versus 13.08 months with placebo (HR = 0.66, P.0049).[37] Taken together, BOLERO-1 and 3 appear to demonstrate that the benefit of adding mTOR inhibitor to chemotherapy and trastuzumab in the metastatic disease setting is great in the HER2+ hormone receptor negative cohort.
Considering that mTOR inhibitors add a substantial amount of toxicity to therapy, biomarkers to identify patients who could derive more benefit from therapy are being studied. At the 2015 ASCO Meeting, researchers evaluated tumor tissue from patients recruited in BOLERO-1 and BOLERO-3. Exons of 282 cancer related genes were analyzed by next generation sequencing (NGS), and PTEN levels were evaluated by immunohistochemistry. Hyperactive PI3K pathway was defined as low PTEN levels or mutations in PIK3CA pathway. In both trials, patients with hyperactive PI3K derived more benefit from everolimus.[38]
Also, at 2013 ESMO (European Society of Medical Oncology) Conference, investigators presented data on patients from BOLERO-3 with low PTEN or high pS6 levels.[39] Those individuals were more likely to respond to the addition of everolimus to chemotherapy plus trastuzumab. In detail, patients with low PTEN levels achieved greater benefit with everolimus, with a median PFS absolute gain of about 18 weeks. Median PFS was 41.9 weeks with everolimus versus 23.1 weeks with placebo.
Contrasting with these results, biomarker analysis from BOLERO-2, which involved 3,230 exons of 182 oncogenes and tumor-suppressor genes that were sequenced using next-generation sequencing, on 309 tissue samples, demonstrated a positive treatment effect in favor of everolimus across the various key genetic marker subgroups.[40] In truth, a greater benefit from everolimus treatment was derived in patients with minimal genetic alterations in PIK3CA/PTEN/CCND1 or FGFR1/2 genes combined. Although BOLERO-2 [41] evaluated the benefit of adding everolimus in the treatment of a hormone receptor-positive, HER2-negative BC cohort, biomarker analysis evaluating alterations in PIK3CA pathway and mTOR inhibitors benefit, might provide some insights of everolimus benefit in other BC subtypes with specific somatic genomic alterations.
Other trials are currently being designed to specifically target the PI3K/AKT pathway in conjunction with HER2 itself, in order to hopefully achieve more durable and potent antitumor effects and overcome the resistant to treatment. These PI3K inhibitors are being introduced to the treatment of women with HER2+ BC in several stages of disease, from the neoadjuvant setting to the metastatic setting, after several lines of standard treatment.
As an example, the NeoPHOEBE study (NCT01816594), recruited women with HER2+ newly diagnosed early BC larger than 2cm. Patients were randomized to receive neoadjuvant weekly paclitaxel and Trastuzumab in combination with an oral PI3K inhibitor, BKM120, or placebo. The study has been completed, and was conducted in two separate cohorts (PIK3CA mutated and PI3K3CA wild-type) using a two-stage approach. NCT02152943 is evaluating everolimus, letrozole, and trastuzumab.
The combination of trastuzumab with a PI3K inhibitor was evaluated in a phase 1 clinical trial. Buparlisib plus trastuzumab was tested in a cohort of HER2+ metastatic BC patients resistant to trastuzumab-based therapy, with a manageable toxicity profile and preliminary evidence of clinical activity. Of note, inhibition of the PI3K/AKT/mTOR and RAS/MEK/ERK pathways was observed in paired tumor biopsies.[42] Pan PI3K inhibitors, such as pilaralisib (SAR245408), were tested in combination with trastuzumab and chemotherapy, with results showing objective evaluable responses.[43]
Recently, at ASCO 2015, an oral PI3Kα inhibitor, BYL719, was evaluated in combination with trastuzumab and a HER3 inhibitor, in a heavily pre treated HER2+ metastatic BC population (median number of previous therapy lines of 6). Clinical activity was observed in patients harbouring PIK3CA mutations. Nevertheless, gastrointestinal and metabolic toxicities were an important issue, warranting exploration of intermittent schedule regimens. [44]
The PIK3CA pathway may be targeted in other downstream points of the pathway, such as Akt. The Akt-inhibitor MK2206, was evaluated before surgery in patients with stage I, II or III BC with various combinations of ER, PR and HER2.
Many other trials are being performing with the important aim to target the PI3K/AKT/mTOR pathway in conjunction with HER2. Toxicity associated with PI3K/AKT/mTOR pathway inhibitors may be taken into account, as well as the added financial cost. Deaths attributable to everolimus were seen BOLERO-1, due to pneumonitis, pulmonary embolism, respiratory failure, pulmonary edema, pneumonia, and cardiorespiratory arrest. Management of adverse events related to these drugs, especially when combined with chemotherapy, is of extreme importance.
Therefore, the PI3K/AKT/mTOR pathway seems to play an important role in anti-HER2 therapy resistance. The optimal timing of targeting this pathway, either in the first line setting, or in a disease scenario where resistance is already established is yet to be determined. Also, the specific point of the pathway to be elected as the ideal target is still unclear. There are many options, ranging from mTOR-inhibitors to Akt-inhibitors and specific inhibitors of different isoforms of PI3K. Additionally, a balance between cost and side effects is of extreme importance in moving forward this strategy to clinical practice.
p95HER2
The aminoterminal-truncated form of the HER2 receptor, that undergoes a proteolytic cleavage generating a truncated fragment, is denominated p95HER2 and is constitutively active. Also, p95HER2 fragments may arise through translation of the mRNA encoding HER2 from internal initiation codons.[45] It has kinase activity, but lacks the extracellular domain and the binding site of trastuzumab, enabling activated signalling despite the presence of trastuzumab.[46]
The presence of p95HER2 has been associated with worse survival outcomes in patients with localized HER2+ BC. Primary breast tumor tissues were evaluated by Western blot analysis for HER-2 protein forms, including p185HER-2 and p95HER-2. Authors found that a high level of p95HER-2 in primary tumor tissue correlated with reduced 5-year disease-free survival (DFS) (HR 2.55; 95% IC, 2.13-8.01; P < 0.0001). Differences in DFS were quite impressive between the two groups, namely 139 versus 32 months, for low and high levels of p95HER2, respectively.[47]
Despite being an independent prognostic factor in breast cancer, defining a group of patients with HER2+ disease with significantly worse survival outcomes, p95HER2 is also a marker of resistance to trastuzumab. Previous studies demonstrated that metastatic BC patients overexpressing p95HER2 had considerably lower response rates to trastuzumab compared with patients expressing full-length HER2.[46] In this trial, Scaltriti et al. also evaluated the effect of lapatinib in a p95HER2 preclinical model of p95HER2-positive MCF-7 cells. Lapatinib, a small molecule that inhibits both HER2 and EGFR kinases, successfully inhibited p95HER2 phosphorylation, reducing downstream phosphorylation of Akt and MAPK, with cell growth inhibition.[46]
Other studies reported a correlation between elevated p95 expression and poor outcomes in response to trastuzumab treatment.[48, 49]
Interestingly, recent studies showed that tumors expressing the most active p95HER2 fragment, the so-called p95HER2/611CTF, indeed do respond to trastuzumab in combination with chemotherapy. The 611CTF fragment is highly oncogenic, because it spontaneously homodimerizes into a constitutively active form.[50] In vitro experiments observed that chemotherapy sensitizes p95HER2/611CTF-positive patient-derived xenografts to trastuzumab, which may be related to HER2 stabilization induced by chemotherapy.[51] Another analysis on the p95HER2/611CTF isoform on samples from the NeoALTTO trial also demonstrated better responses to trastuzumab in combination with paclitaxel in tumors that expressed high levels of this isoform.52
Larger studies are highly awaited for confirmation of p95HER2 as a biomarker of resistance or sensitivity to anti-HER2 therapy, enabling development of strategies to target this form of HER2. Evidence is accumulating indicating that trastuzumab alone is not effective in patients who have expression of p95HER2 isoforms. Figure 1 illustrates HER2 and p95HER2, along with their interactions with chemotherapy and trastuzumab, as well as proliferative molecular pathways.
Chemotherapy, in conjunction with trastuzumab, is essential to achieve response to therapy. Larger and well-controlled studies, using pre-defined clinical cutoffs for p95 expression and also blinded analyses are highly awaited.
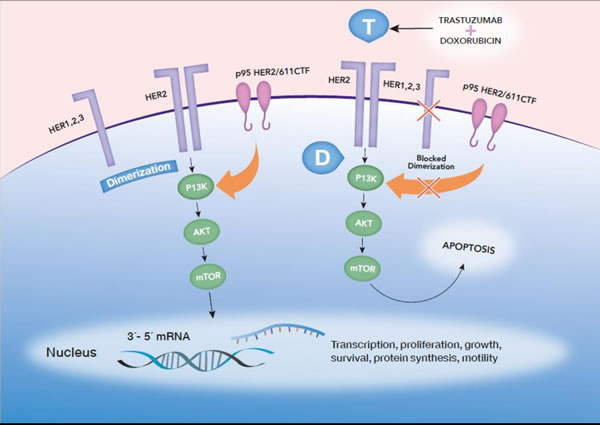
Figure 1: HER2 dimerizes with other partners from the HER2 family, activating intracellular proliferative pathways. Also, p95HER2/611CTF has intrinsic kinase activity, lacking the extracellular domain and the binding site of trastuzumab (T), enabling activated signaling, despite the presence of trastuzumab. Upon activation of proliferative pathways, such as PI3K/AKT/mTOR, the cell undergoes proliferation. Trastuzumab blocks HER2 dimerization and phosphorylation, but p95HER2/611CTF might still phosphorylate and enable cancer cell proliferation. In contrast, treatment with doxorubicin (D) and trastuzumab induces apoptosis more efficiently in p95HER2/611CTF-positive cells and, destabilizing phospho-HER2 and stabilizing HER2 in p95HER2/611CTF-positive cells. Therefore, cells are sensitized to trastuzumab and suffer apoptosis through trastuzumab therapy.
Insulin-like growth factor-I receptor (IGF-IR)
Insulin-like growth factor-I receptor (IGF-IR) is another pathway implicated in trastuzumab resistance.[53] This receptor tyrosine kinase plays an important role in tumor cell growth and survival. Upon ligand stimulation, IGF-IR generates signaling through the Ras/mitogen-activated protein kinase and phosphatidylinositol 3-kinase/AKT pathways, stimulating cell growth, proliferation and apoptosis inhibition.[54]
Important data demonstrated that interaction between HER2 and IGFR-IR occurs in trastuzumab-resistant cells and that inhibition of IGF-IR tyrosine kinase activity may lead to decreased HER-2 phosphorylation with consequent restoration of trastuzumab sensitivity. This important cross talk between HER-2 and IGF-IR could potentially lead to clinical use of IGF-IR-targeted agents to block this receptor.
In vitro studies demonstrated that inhibition of HER2 signaling using trastuzumab, and inhibition of IGF-IR signaling using a dominant negative construct produced synergistic growth inhibition of HER2-overexpressing breast cancer cells.[55, 56]
Of note, in experiments in which in vitro inhibition of IGF1R expression was made through small receptor tyrosine kinase inhibitors, response to trastuzumab was substantially improved, suggesting an interaction between HER2 and IGFR. [57]
Future clinical trials of HER2 and IGF1R-inhibitors are awaited to corroborate these preclinical findings. Analysis of potential predictive biomarkers to identify tumors that may achieve maximum benefit from this approach would be of great interest.
MET Aberrations
c-MET is a receptor tyrosine kinase (RTK) encoded by the MET proto-oncogene that is largely expressed in epithelial/endothelial cells. [58] Upon binding with the hepatocyte growth factor (HGF), the c-MET receptor dimerizes, autophosphorylates and activates downstream pathways, including mitogen-activated protein kinase (MAPK), phosphatidylinositol 3-kinase (PI3K) and signal transducer and activator of transcription.[59, 60]
Overexpression of c-MET has been shown to contribute to the development of the invasive phenotype during BC progression. Poorly differentiated and invasive cell lines may express high levels of the receptor and ligation by HGF is associated with increased motility and invasiness.[61]
Previous trials have shown that overexpression of c-MET in BC primary tumors is strongly associated with worse disease-free survival compared to tumors without c-MET overexpression.[62, 63]
MET expression might be associated with resistance to treatment in BC patients, even in a cohort of individuals co expressing ERBB2, in which a highly effective treatment, such as trastuzumab, is largely available.[64]
In fact, overexpression c-Met has emerged as a potential contributor to trastuzumab resistance and has been shown to be highly elevated in HER2-positive breast cancer cell lines and in 25% of HER2-positive breast cancer patients tissues.[62, 65] Loss of Met function is implicated in the development of trastuzumab response.[65]
Cell line experiments demonstrated that foretinib, a multikinase MET inhibitor, combined with erlotinib or lapatinib, was capable of cell growth inhibition of MET-amplified or overexpressing cell lines.[66] Additionally, in the presence of HGF, these cell lines demonstrated reduced sensitivity to lapatinib, which may indicate that MET activation can decrease the effectiveness of HER1/2 inhibitors.
Many therapeutic strategies to target Met are currently under clinical development, including tyrosine kinase inhibitors and monoclonal antibodies, which could potentially be associated with anti-HER2 therapy in patients with BC overexpressing HER2 to optimize treatment and avoid tumor resistance. As an example, cabozantinib, a small molecule inhibitor of the tyrosine kinases c-Met and VEGFR2, is currently being evaluated in combination with trastuzumab, in HER2+ BC patients who have brain metastasis (NCT02260531).
Src
The proto-oncogene Src encodes for the non-receptor protein tyrosine kinase Src, which is involved in many cellular events, mediating cell proliferation and survival. [67]
Src has extensive interaction with transmembrane receptor tyrosine kinases (RTKs) at the cell membrane, such as HER1 and HER2. Src activation confers resistance to trastuzumab, both acquired and de novo. In fact, data points to the fact that Src activation by itself is sufficient to confer trastuzumab resistance.[68]
The relationship between SRC activation and patient response to trastuzumab-based therapies was previously studied in a cohort of 57 patients who were evaluated for SRC activation in primary breast tumors. As featured, patients with high amounts of phosphorylated Src (pSrc) in tumors demonstrated a lower clinical response rate and a higher progressive disease rate after trastuzumab therapy. Additionally, the OS of patients with high pSrc tumors (median 34.2 months) was significantly lower than that of patients with low pSrc tumors (median 57.9 months).[68] In vitro targeting Src universally sensitized trastuzumab-resistant cells to trastuzumab treatment, and was able to suppress tumor growth in multiple preclinical resistance models. Other studies also demonstrated that Src activation is involved with trastuzumab mechanisms of resistance, and indicates poor prognosis in patients with HER2+ BC.[69]
Resistance to lapatinib was also demonstrated in Src activated cell lines. Saracatinib, a small molecule inhibitor of Src, was combined with lapatinib, and significantly prolonged survival of xenografted mice compared with saracatinib alone.[70]
Therefore, blocking Src interaction with HER2 is a promising strategy that might impact the management of HER2+ BC patients.
Immune Response and trastuzumab sensitivity
Trastuzumab mechanism of action is dependent on the host immune system. As a consequence, trastuzumab is only partially effective in mice defective for the receptor of immunoglobulins (gamma receptor knockout mice). Innate and adaptive immune mechanisms are emerging as key players in modulation of the effects of HER2-targeted drugs.[71]
Therefore, sensitivity to anti-HER2 therapy depends on a well functioning host immune system. Loi et al. evaluated tumor PIK3CA mutations, lymphocyte infiltration, and RFS in an early HER2+ BC cohort of patients enrolled on the Finher Trial. [72, 73] In this analysis, the presence of tumor lymphocyte infiltration (TIL) was associated with prediction of trastuzumab response.[73,74]
In the neoadjuvant setting, signals of immune system activation were associated with response to pertuzumab and trastuzumab in the Neosphere study. In this analysis, baseline core biopsies were collected from 387 out of 417 patients and evaluated by gene expression profiling. Patients identified as having low expression of PD-L1 had lower pathologic complete response rates (pCR). In contrast, individuals with high expression of STAT1 and IFNG, achieved high pCR rates, irrespective of estrogen receptor status.[75]
Tumor cells display genetic and epigenetic changes that provide plenty of tumor-associated antigens, which could potentially elicit host immune system recognition and tumor cell destruction. Avoidance of this host intrinsic defense mechanism is fundamental for tumor progression. As a consequence, immune-checkpoint proteins are frequently deregulated by tumors.[76] Cytotoxic T-lymphocyte antigen 4 (CTLA-4), which is a negative regulator of T cell activation, and PD-1 (programmed cell death protein 1), which is an immune checkpoint that limits the activity of T cells in peripheral tissues, are immune checkpoint proteins that are currently being targeted in a variety of solid tumors, such as melanoma, lung, renal and also breast cancer.
Among one of the first reports that recognized the importance of this rapidly evolving field in BC was presented at the 2013 SABCS.[77] Breast tumor samples from 156 patients with HER2-positive BC enrolled in the GeparQuattro trial were evaluated. In this cohort of patients, for every 10 percent increase in the levels of tumor-infiltrating lymphocytes there was a 16 percent increase in the number of patients who had a pCR. Furthermore, authors investigated samples from patients recruited in the FinHer trial, and found that high levels of CTLA-4 and PD-1 correlated with a survival benefit after trastuzumab treatment in a mouse model. Besides, treatment of mice with trastuzumab and either an agent that blocks PD-1 or PD-L1, resulted in greater tumor regression compared with trastuzumab alone, suggesting an important role of checkpoint-inhibitors in HER2+ BC.
Recently, Perez et al. defined a group with enriched immune functions in patients enrolled in the N9831 trastuzumab adjuvant trial, based on the expression of 87 genes.[78] Using genomic technology, the authors identified significantly enriched biological processes associated with increased RFS that were linked to immune functions. This group was denominated immune group and experienced more favorable outcomes when treated with trastuzumab. Of note, patients who did not exhibit immune function enrichment and were treated with trastuzumab did not experience better RFS compared to patients with immune function enrichment who were treated with chemotherapy alone (HR=0.93, p=0.72). Therefore, a state of immunological function appears to be associated with HER2-targeted therapy efficacy and warrants further evaluation in future trials.
Analysis of immune markers in HER2+ BC indicates that host immune system integrity is extremely important to trastuzumab response. Future clinical trials exploring patient immune system, with the ultimate goal of maximizing immune response, might result in great improvement in trastuzumab benefit and sensitivity.
Table 2 summarizes the main resistance mechanisms to trastuzumab therapy, correlating with associated strategies to overcome it.
Table 2: IGFR Insulin growth factor receptor; TIL tumor infiltrating lymphocytes
Mechanisms of Trastuzumab resistance and strategies to target it |
|
Resistance Mechanism |
Strategy to Overcome resistance Anti-HER2 Therapy plus |
PIK3CA Alterations PIK3CA Mutation PIK3CA Copy Number gain PIK3CA Amplification PIK3CA Increased Expression PTEN loss |
Pan PI3K Inhibitor Specific PIK3CA Inhibitors AKT Inhibitors mTOR Inhibitors |
High Levels of p95HER2 |
Chemotherapy Lapatinib |
IGFR1 tyrosine kinase receptor activation |
IGF1R monoclonal antibodies or tyrosine kinase inhibitors |
MET Aberrations MET Amplification MET Mutation |
MET inhibitors |
Low Immune Response Low TIL levels Lack of immune genes expression |
Immune checkpoint inhibitors |
Main Resistance Mechanisms Pathways to other anti-HER2 Therapies
Lapatinib and Other Tyrosine Kinase Inhibitors (TKIs)
Lapatinib is a synthetic, orally active TKI that reversibly blocks phosphorylation of the epidermal growth factor receptor (EGFR), HER2, Erk-1 and-2 and AKT kinases [79]. Other TKIs that irreversibly block phosphorylation of these receptors include neratinib, afatinib (BIBW2992), CUDC-101, and canertinib (CI-1033), among others. Irreversible TKIs, such as neratinib, could have the theoretical advantage to potentially revert first-generation HER receptor tyrosine kinase inhibitors resistance [80].
Lapatinib in combination with capecitabine is FDA-approved for the treatment of HER2+ metastatic BC that have received prior therapy with trastuzumab [7]. Also, it is approved in combination with letrozole for the treatment of postmenopausal women with HER2+ and hormone receptor-positive metastatic BC, for whom hormonal therapy is indicated [81].
Combining trastuzumab with lapatinib is an attractive strategy, since both intracellular and extracellular HER2 domains would be targeted. Indeed, in vitro synergistic interaction was demonstrated between those two drugs [82]. Dual anti-HER2 blockade with lapatinib and trastuzumab improved PFS and clinical benefit rate in women with metastatic HER2+ BC [83], and also improved pathologic complete response rate in locally advanced HER2+ BC patients who received neoadjuvant chemotherapy [84, 85]. Nevertheless, lapatinib in combination with trastuzumab did not improve survival outcomes in the adjuvant setting [86].
Unfortunately, acquired resistance to lapatinib often develops in patients who initially responded to therapy. Chronic exposure to lapatinib may convert HER2-over-expressing BC cells that are initially sensitive to lapatinib-induced apoptosis to resistant cells [87]. Importantly, in cell lines that display overexpression of both estrogen receptor (ER) and HER2, FOXO3a and ER-regulated gene products are actually induced by lapatinib. Also, lapatinib modulates the expression of proteins that promote transcription of ER-regulated genes. Therefore, it is hypothesized that regulation of cell survival switches from HER2 to ER during the development of acquired resistance to lapatinib [88]. In cell lines that are ER-, resistance to lapatinib appears to be an ER-independent process.
Small molecule TKIs are being studied in combination with drugs that target the PIK3CA/AKT pathway, which was previously discussed as being implicated in trastuzumab resistance. PI3K inhibitors in monotherapy might display modest clinical activity, and a potential resistance mechanism frequently involves increased expression and phosphorylation of the HER family of receptors, particularly HER2 and HER3 [89, 90]. Therapeutic combinations to target both pathways could prevent or delay the development of resistance. Actually, combining a PI3K inhibitor with a small molecule inhibitor of EGFR/HER2/HER3 signaling was shown to have synergistic growth inhibition in breast cancer cell lines in vitro, suggesting that this is a strategy that might be a possible treatment option to be tested in future clinical trials [91].
Another potential mechanism to lapatinib resistance is activation of AXL, which is a membrane-bound receptor tyrosine kinase [92]. In a lapatinib-resistant BC cell clone model, AXL expression was increased and treatment with GSK1363089, a potent inhibitor of AXL, MET, and VEGFR, restored drug sensitivity [93]. Also, phosphorylation of AXL is reported to be associated with HER2 signaling, leading to downstream AKT activation, and generating activation of the AKT/mTOR intracellular proliferative pathway [94].
Pertuzumab
This humanized monoclonal antibody that binds to HER2 is an effective inhibitor of EGFR/HER2 driven signaling pathway. Compared to trastuzumab, much less is known about development of resistance to this targeted agent. Of note, in a tamoxifen-resistant BC cell line, pertuzumab promoted rapid formation of HER3/EGFR heterodimers, with subsequent phosphorylation of AKT and ERK1/2 [95]. In this cell line model, the rapid formation of HER3/EGFR heterodimers provided a mechanism whereby epidermal growth factor stimulation could overcome the growth inhibitory effect of this agent. Moreover, HER3/EGFR heterodimer levels were increased in tamoxifen/pertuzumab resistant cell lines, further suggesting that pertuzumab-induced EGFR/HER3 heterodimerization might play a role in the rapid acquisition of resistance to this agent in tamoxifen-resistant cells.
In ovarian cancer, microRNA-150 (miR-150) expression was induced by pertuzumab in a cell line model, and suppression of miRNA-150 resulted in decreased drug sensitivity to pertuzumab and cell apoptosis [96]. miRNAs are small noncoding, single-stranded RNAs that regulate several biological processes, controlling gene expression at a posttranscriptional level [97]. miRNA-150 was found to be a negative PI3K-AKT pathway regulator. Also, an AKT inhibitor, was used to treat pertuzumab resistant cancer cells, and was capable of achieving anti tumor effects [96]. Although this model was performed in an ovarian cancer cell line, this might provide some insights to breast cancer as well.
The development of resistance to anti-HER2 therapies seems complex, and is probably associated with multiple mechanisms. A generated transgenic mouse model of HER2+, PIK3CA mutant BC acquired resistance to trastuzumab, pertuzumab in combination with the pan-PI3K inhibitor BKM120 after treatment exposure. Initially, tumor regression occurred after 6 weeks of therapy. Nevertheless, within two months, all tumors relapsed. Of note, p95 HER2, which was not detected in untreated tumors, was expressed in resistant clones, and HER2 expression was significantly reduced [98]. This data suggests that a wide range of heterogeneity of mechanisms of acquired resistance may occur in different HER2+/PIK3CA-mutant BC patients.
T-DM1
Trastuzumab emtansine (T-DM1) is an antibody-drug conjugate that is active in second line, as well as later lines in the treatment of advanced HER2+ BC that has progressed on trastuzumab therapy [99, 100]. Although most patients will develop resistance to this agent, the mechanisms associated with this phenomenon are incompletely understood.
Primary resistance to T-DM1 may be relatively infrequent, particularly in patients who have no prior exposure to trastuzumab. The most common scenario is found in patients who initially responded, but eventually ceased to respond, despite continued treatment with T-DM1. It is important to know that DM1 and its metabolites need to accumulate in cell cytoplasm to reach a concentration that exceeds the threshold needed to evoke cell death [101]. Consequently, factors that affect the ability of the drug to reach a satisfactory intracellular level might be implicated in lack of clinical efficacy. Low tumor HER2 expression, poor internalization of the HER2-T-DM1complexes, defective intracellular and endosomal trafficking of the HER2-T-DM1 complex and defective lysosomal degradation of T-DM1 are all associated with low intracellular DM1 levels [102]. Also, drug efflux pumps might be implicated in resistance to therapy and expression of multi-drug resistance transporters may play a role in T-DM1 resistance.
Another important resistance mechanism of T-DM1 is mediated by neuregulin b1 (NRG) [103], the ligand protein of HER3, encoded by the NRG1 gene [104]. The presence of NRG can suppress the cytotoxic activity of T-DM1 in BC cell lines. This effect was reversed when cells were treated with pertuzumab. NRG can trigger the formation of HER2-HER3 heterodimers, a potent activating mechanism of the PI3K pathway, which was previously described in this review as a proliferative pathway involved with anti-HER2 therapy resistance [105].
Other HER2-targeting antibody-drug conjugate (ADC) are currently under development, such as SYD985. In the ongoing phase 1 clinical trial, safety and efficacy of SYD985 have been evaluated in European Oncology Centers, where patients with locally advanced or metastatic solid tumors of any origin were treated with SYD985. Of note, objective responses were seen in metastatic HER2+ BC patients who were refractory to previous HER2-targeted treatments, including T-DM1 [106].
Conclusions
Resistance to trastuzumab and other anti-HER2 therapies is an event that may occur during the course of therapy or de novo. In either case, this is a potentially life-threatening event, which may lead to rapidly progressive disease, or can result in important quality of life impairment for patients, due to disease related symptoms. A better knowledge of these mechanisms is of extreme importance for the development of effective strategies to overcome resistance.
In developing more effective and durable treatment strategies, multitargeted therapy should be considered. The recognition of specific molecular predictors of response to emerging therapies will allow a more personalized approach to the treatment of HER2-amplified BC.
Enrolling patients into clinical trials, with the purpose of understand and target the molecular mechanisms involved in HER2 therapy resistance is of crucial importance. Laboratory, pre clinical and clinical findings, such as seen at the BOLERO-1 and 3 trials, are very promising in this field, but have not yet been incorporated in clinical practice. Also, a better characterization between the relationship of tumor inflammatory infiltrate or molecular inflammatory signature, and trastuzumab benefit is warranted, before its incorporation in clinical practice.
Nowadays, the focus on HER2 expression/amplification status alone is not a realistic approach to understand the underlying mechanisms of disease progression and resistance. Improvement of patient outcomes in this disease setting will only be clinically meaningful if resistance pathways involved in disease progression are better recognized, understood and targeted.
Financial Support
The original figure was designed by authors and electronically illustrated with financial support from Roche Pharmaceuticals.
conflicts of interests
Dr. Debora de Melo Gagliato, Dr. Denis Leonardo Fontes Jardim and Dr. Gabriel Hortobagyi report no conflict of interest. Mario Marchesi works for Roche Pharmaceuticals. The content of this review was not influenced by this relationship.
References
1. DeSantis C, Ma J, Bryan L and Jemal A. Breast cancer statistics, 2013. CA Cancer J Clin. 2014; 64(1):52-62.
2. Siegel R, DeSantis C, Virgo K, Stein K, Mariotto A, Smith T, Cooper D, Gansler T, Lerro C, Fedewa S, Lin C, Leach C, Cannady RS, et al. Cancer treatment and survivorship statistics, 2012. CA Cancer J Clin. 2012; 62(4):220-241.
3. Slamon DJ, Clark GM, Wong SG, Levin WJ, Ullrich A and McGuire WL. Human breast cancer: correlation of relapse and survival with amplification of the HER-2/neu oncogene. Science. 1987; 235(4785):177-182.
4. Olayioye MA. Update on HER-2 as a target for cancer therapy: intracellular signaling pathways of ErbB2/HER-2 and family members. Breast Cancer Res. 2001; 3(6):385-389.
5. Rubin I and Yarden Y. The basic biology of HER2. Ann Oncol. 2001; 12 Suppl 1:S3-8.
6. Hudis CA. Trastuzumab--mechanism of action and use in clinical practice. N Engl J Med. 2007; 357(1):39-51.
7. Geyer CE, Forster J, Lindquist D, Chan S, Romieu CG, Pienkowski T, Jagiello-Gruszfeld A, Crown J, Chan A, Kaufman B, Skarlos D, Campone M, Davidson N, et al. Lapatinib plus capecitabine for HER2-positive advanced breast cancer. N Engl J Med. 2006; 355(26):2733-2743.
8. Slamon DJ, Leyland-Jones B, Shak S, Fuchs H, Paton V, Bajamonde A, Fleming T, Eiermann W, Wolter J, Pegram M, Baselga J and Norton L. Use of chemotherapy plus a monoclonal antibody against HER2 for metastatic breast cancer that overexpresses HER2. N Engl J Med. 2001; 344(11):783-792.
9. Cobleigh MA, Vogel CL, Tripathy D, Robert NJ, Scholl S, Fehrenbacher L, Wolter JM, Paton V, Shak S, Lieberman G and Slamon DJ. Multinational study of the efficacy and safety of humanized anti-HER2 monoclonal antibody in women who have HER2-overexpressing metastatic breast cancer that has progressed after chemotherapy for metastatic disease. J Clin Oncol. 1999; 17(9):2639-2648.
10. Moja L, Tagliabue L, Balduzzi S, Parmelli E, Pistotti V, Guarneri V and D’Amico R. Trastuzumab containing regimens for early breast cancer. Cochrane Database Syst Rev. 2012; 4:CD006243.
11. Perez EA, Romond EH, Suman VJ, Jeong JH, Sledge G, Geyer CE, Jr., Martino S, Rastogi P, Gralow J, Swain SM, Winer EP, Colon-Otero G, Davidson NE, et al. Trastuzumab plus adjuvant chemotherapy for human epidermal growth factor receptor 2-positive breast cancer: planned joint analysis of overall survival from NSABP B-31 and NCCTG N9831. J Clin Oncol. 2014; 32(33):3744-3752.
12. Tolaney SM BW, Dang CT, Yardley DA, Moy B, Marcom PK, Albain KS, Rugo HS, Ellis M, Shapira I, Wolff AC, Carey LA, Overmoyer BA, Partridge AH, Guo H, Hudis CA, Krop IE, Burstein HJ, Winer EP. (2013). A phase II study of adjuvant paclitaxel and trastuzumab (APT) trial for node-negative, HER2-positive breast cancer. 2013 San Antonio Breast Cancer Symposium.
13. Baselga J, Cortes J, Kim SB, Im SA, Hegg R, Im YH, Roman L, Pedrini JL, Pienkowski T, Knott A, Clark E, Benyunes MC, Ross G, et al. Pertuzumab plus trastuzumab plus docetaxel for metastatic breast cancer. N Engl J Med. 2012; 366(2):109-119.
14. Swain SM, Kim SB, Cortes J, Ro J, Semiglazov V, Campone M, Ciruelos E, Ferrero JM, Schneeweiss A, Knott A, Clark E, Ross G, Benyunes MC, et al. Pertuzumab, trastuzumab, and docetaxel for HER2-positive metastatic breast cancer (CLEOPATRA study): overall survival results from a randomised, double-blind, placebo-controlled, phase 3 study. Lancet Oncol. 2013; 14(6):461-471.
15. Swain SM, Baselga J, Kim SB, Ro J, Semiglazov V, Campone M, Ciruelos E, Ferrero JM, Schneeweiss A, Heeson S, Clark E, Ross G, Benyunes MC, et al. Pertuzumab, trastuzumab, and docetaxel in HER2-positive metastatic breast cancer. N Engl J Med. 2015; 372(8):724-734.
16. Pohlmann PR, Mayer IA and Mernaugh R. Resistance to Trastuzumab in Breast Cancer. Clin Cancer Res. 2009; 15(24):7479-7491.
17. Isakoff SJ, Engelman JA, Irie HY, Luo J, Brachmann SM, Pearline RV, Cantley LC and Brugge JS. Breast cancer-associated PIK3CA mutations are oncogenic in mammary epithelial cells. Cancer Res. 2005; 65(23):10992-11000.
18. Nagata Y, Lan KH, Zhou X, Tan M, Esteva FJ, Sahin AA, Klos KS, Li P, Monia BP, Nguyen NT, Hortobagyi GN, Hung MC and Yu D. PTEN activation contributes to tumor inhibition by trastuzumab, and loss of PTEN predicts trastuzumab resistance in patients. Cancer Cell. 2004; 6(2):117-127.
19. Berns K, Horlings HM, Hennessy BT, Madiredjo M, Hijmans EM, Beelen K, Linn SC, Gonzalez-Angulo AM, Stemke-Hale K, Hauptmann M, Beijersbergen RL, Mills GB, van de Vijver MJ, et al. A functional genetic approach identifies the PI3K pathway as a major determinant of trastuzumab resistance in breast cancer. Cancer Cell. 2007; 12(4):395-402.
20. Chandarlapaty S, Sakr RA, Giri D, Patil S, Heguy A, Morrow M, Modi S, Norton L, Rosen N, Hudis C and King TA. Frequent mutational activation of the PI3K-AKT pathway in trastuzumab-resistant breast cancer. Clin Cancer Res. 2012; 18(24):6784-6791.
21. Loi S, Michiels S, Lambrechts D, Fumagalli D, Claes B, Kellokumpu-Lehtinen PL, Bono P, Kataja V, Piccart MJ, Joensuu H and Sotiriou C. Somatic mutation profiling and associations with prognosis and trastuzumab benefit in early breast cancer. J Natl Cancer Inst. 2013; 105(13):960-967.
22. Wang Y, Liu Y, Du Y, Yin W and Lu J. The predictive role of phosphatase and tensin homolog (PTEN) loss, phosphoinositol-3 (PI3) kinase (PIK3CA) mutation, and PI3K pathway activation in sensitivity to trastuzumab in HER2-positive breast cancer: a meta-analysis. Curr Med Res Opin. 2013; 29(6):633-642.
23. Pogue-Geile KL, Song N, Jeong JH, Gavin PG, Kim SR, Blackmon NL, Finnigan M, Rastogi P, Fehrenbacher L, Mamounas EP, Swain SM, Wickerham DL, Geyer CE, Jr., et al. Intrinsic Subtypes, PIK3CA Mutation, and the Degree of Benefit From Adjuvant Trastuzumab in the NSABP B-31 Trial. J Clin Oncol. 2015; 33(12):1340-1347.
24. Perez EA, Dueck AC, McCullough AE, Chen B, Geiger XJ, Jenkins RB, Lingle WL, Davidson NE, Martino S, Kaufman PA, Kutteh LA, Sledge GW, Harris LN, et al. Impact of PTEN protein expression on benefit from adjuvant trastuzumab in early-stage human epidermal growth factor receptor 2-positive breast cancer in the North Central Cancer Treatment Group N9831 trial. J Clin Oncol. 2013; 31(17):2115-2122.
25. José Baselga SV, Jungsil Ro, Jens Huober, Ellie Guardino, Liang Fang, Steven Olsen, Gail Lewis Phillips, Sanne de Haas, and Mark Pegram. (2013). Relationship between tumor biomarkers and efficacy in EMILIA, a phase III study of trastuzumab emtansine (T-DM1) in HER2-positive metastatic breast cancer. AACR Annual Meeting.
26. Park S, Jiang Z, Mortenson ED, Deng L, Radkevich-Brown O, Yang X, Sattar H, Wang Y, Brown NK, Greene M, Liu Y, Tang J, Wang S, et al. The therapeutic effect of anti-HER2/neu antibody depends on both innate and adaptive immunity. Cancer Cell. 2010; 18(2):160-170.
27. Collins DM, O’Donovan N, McGowan PM, O’Sullivan F, Duffy MJ and Crown J. Trastuzumab induces antibody-dependent cell-mediated cytotoxicity (ADCC) in HER-2-non-amplified breast cancer cell lines. Ann Oncol. 2012; 23(7):1788-1795.
28. Beano A, Signorino E, Evangelista A, Brusa D, Mistrangelo M, Polimeni MA, Spadi R, Donadio M, Ciuffreda L and Matera L. Correlation between NK function and response to trastuzumab in metastatic breast cancer patients. J Transl Med. 2008; 6:25.
29. Baselga J, Cortes J, Im SA, Clark E, Ross G, Kiermaier A and Swain SM. Biomarker analyses in CLEOPATRA: a phase III, placebo-controlled study of pertuzumab in human epidermal growth factor receptor 2-positive, first-line metastatic breast cancer. J Clin Oncol. 2014; 32(33):3753-3761.
30. Loibl S DC, Schneeweiss A, Paepke S, Lehmann A, Rezai M, Zahm P, Sinn P, Khandan F, Eidtmann H, Dohnal K, Huober J, Loi S, Pfitzner B, Fasching PA, André F, Lindner J, Sotiriou C, Guo S, Gade S, Nekljudova V, Untch M, von Minckwitz G. (2013). PIK3CA Mutation Predicts Resistance to Anti-HER2/Chemotherapy in Primary HER2-Positive/Hormone-Receptor-Positive Breast Cancer – Prospective Analysis of 737 Participants of the GeparSixto and GeparQuinto Studies. San Antonio Breast Cancer Annual Meeting. (San Antonio, Tx.
31. Loibl S, von Minckwitz G, Schneeweiss A, Paepke S, Lehmann A, Rezai M, Zahm DM, Sinn P, Khandan F, Eidtmann H, Dohnal K, Heinrichs C, Huober J, et al. PIK3CA mutations are associated with lower rates of pathologic complete response to anti-human epidermal growth factor receptor 2 (her2) therapy in primary HER2-overexpressing breast cancer. J Clin Oncol. 2014; 32(29):3212-3220.
32. Schneeweiss A, Chia S, Hegg R, Tausch C, Deb R, Ratnayake J, McNally V, Ross G, Kiermaier A and Cortes J. Evaluating the predictive value of biomarkers for efficacy outcomes in response to pertuzumab- and trastuzumab-based therapy: an exploratory analysis of the TRYPHAENA study. Breast Cancer Res. 2014; 16(4):R73.
33. Majewski IJ, Nuciforo P, Mittempergher L, Bosma AJ, Eidtmann H, Holmes E, Sotiriou C, Fumagalli D, Jimenez J, Aura C, Prudkin L, Diaz-Delgado MC, de la Pena L, et al. PIK3CA Mutations Are Associated With Decreased Benefit to Neoadjuvant Human Epidermal Growth Factor Receptor 2-Targeted Therapies in Breast Cancer. J Clin Oncol. 2015; 33(12):1334-1339.
34. Loibl S, Majewski I, Guarneri V, Nekljudova V, Holmes EM, Bria E, Denkert C, Eidtmann H, Sotiriou C, Loi S, Andre F, Untch M, Conte PF, et al. Correlation of PIK3CA mutation with pathological complete response in primary HER2-positive breast cancer: Combined analysis of 967 patients from three prospective clinical trials. ASCO Meeting Abstracts. 2015; 33(15_suppl):511.
35. Andre F, O’Regan R, Ozguroglu M, Toi M, Xu B, Jerusalem G, Masuda N, Wilks S, Arena F, Isaacs C, Yap YS, Papai Z, Lang I, et al. Everolimus for women with trastuzumab-resistant, HER2-positive, advanced breast cancer (BOLERO-3): a randomised, double-blind, placebo-controlled phase 3 trial. Lancet Oncol. 2014; 15(6):580-591.
36. Hurvitz SA, Andre F, Jiang Z, Shao Z, Mano MS, Neciosup SP, Tseng LM, Zhang Q, Shen K, Liu D, Dreosti LM, Burris HA, Toi M, et al. Combination of everolimus with trastuzumab plus paclitaxel as first-line treatment for patients with HER2-positive advanced breast cancer (BOLERO-1): a phase 3, randomised, double-blind, multicentre trial. Lancet Oncol. 2015; 16(7):816-829.
37. Hurvitz SA AF, Jiang Z, Shao Z, Neciosup SP, Mano MS, Tseng L-M, Zhang Q, Shen K, Liu D, Dreosti LM, Feng J, Burris HA, Toi M, Buyse ME, Cabaribere D, Lindsay M-A, Kunz T, Rao S, Pacaud LB, Taran T, Slamon D. (2014). Phase 3, randomized, double-blind, placebo-controlled multicenter trial of daily everolimus plus weekly trastuzumab and paclitaxel as first-line therapy in women with HER2+ advanced breast cancer. San Antonio Breast Cancer Symposium. (San Antonio, Tx.
38. Slamon DJ, Hurvitz SA, Chen D, Andre F, Tseng L-M, Jerusalem GHM, Wilks S, O’Regan R, Isaacs C, Toi M, Burris HA, He W, Riester M, et al. Predictive biomarkers of everolimus efficacy in HER2+ advanced breast cancer: Combined exploratory analysis from BOLERO-1 and BOLERO-3. ASCO Meeting Abstracts. 2015; 33(15_suppl):512.
39. Jerusalem G AF, Chen D. (2013). Evaluation of everolimus (EVE) in HER2+ advanced breast cancer (BC) with activated PI3K/mTOR pathway: Exploratory biomarker observations from the BOLERO -3 trial. . ESMO 2013. (Amsterdam, Netherlands.
40. Hortobagyi GN, Piccart-Gebhart MJ, Rugo HS, Burris HA, Campone M, Noguchi S, Perez AT, Deleu I, Shtivelband M, Provencher L, Masuda N, Dakhil SR, Anderson I, et al. Correlation of molecular alterations with efficacy of everolimus in hormone-receptor-positive (HR+), HER2-negative advanced breast cancer: Preliminary results from BOLERO-2. ASCO Meeting Abstracts. 2013; 31(15_suppl):LBA509.
41. Baselga J, Campone M, Piccart M, Burris HA, 3rd, Rugo HS, Sahmoud T, Noguchi S, Gnant M, Pritchard KI, Lebrun F, Beck JT, Ito Y, Yardley D, et al. Everolimus in postmenopausal hormone-receptor-positive advanced breast cancer. N Engl J Med. 2012; 366(6):520-529.
42. Saura C, Bendell J, Jerusalem G, Su S, Ru Q, De Buck S, Mills D, Ruquet S, Bosch A, Urruticoechea A, Beck JT, Di Tomaso E, Sternberg DW, et al. Phase Ib study of Buparlisib plus Trastuzumab in patients with HER2-positive advanced or metastatic breast cancer that has progressed on Trastuzumab-based therapy. Clin Cancer Res. 2014; 20(7):1935-1945.
43. Tolaney S, Burris H, Gartner E, Mayer IA, Saura C, Maurer M, Ciruelos E, Garcia AA, Campana F, Wu B, Xu Y, Jiang J, Winer E, et al. Phase I/II study of pilaralisib (SAR245408) in combination with trastuzumab or trastuzumab plus paclitaxel in trastuzumab-refractory HER2-positive metastatic breast cancer. Breast Cancer Res Treat. 2015; 149(1):151-161.
44. Shah PD, Chandarlapaty S, Dickler MN, Ulaner G, Zamora SJ, Sterlin V, Iasonos A, Coughlin CM, Morozov A, Ero J, Rosen N, Gilewski T, Fornier MN, et al. Phase I study of LJM716, BYL719, and trastuzumab in patients (pts) with HER2-amplified (HER2+) metastatic breast cancer (MBC). ASCO Meeting Abstracts. 2015; 33(15_suppl):590.
45. Arribas J, Baselga J, Pedersen K and Parra-Palau JL. p95HER2 and breast cancer. Cancer Res. 2011; 71(5):1515-1519.
46. Scaltriti M, Rojo F, Ocana A, Anido J, Guzman M, Cortes J, Di Cosimo S, Matias-Guiu X, Ramon y Cajal S, Arribas J and Baselga J. Expression of p95HER2, a truncated form of the HER2 receptor, and response to anti-HER2 therapies in breast cancer. J Natl Cancer Inst. 2007; 99(8):628-638.
47. Saez R, Molina MA, Ramsey EE, Rojo F, Keenan EJ, Albanell J, Lluch A, Garcia-Conde J, Baselga J and Clinton GM. p95HER-2 predicts worse outcome in patients with HER-2-positive breast cancer. Clin Cancer Res. 2006; 12(2):424-431.
48. Sperinde J, Jin X, Banerjee J, Penuel E, Saha A, Diedrich G, Huang W, Leitzel K, Weidler J, Ali SM, Fuchs EM, Singer CF, Kostler WJ, et al. Quantitation of p95HER2 in paraffin sections by using a p95-specific antibody and correlation with outcome in a cohort of trastuzumab-treated breast cancer patients. Clin Cancer Res. 2010; 16(16):4226-4235.
49. Lipton A, Goodman L, Leitzel K, Cook J, Sperinde J, Haddad M, Kostler WJ, Huang W, Weidler JM, Ali S, Newton A, Fuchs EM, Paquet A, et al. HER3, p95HER2, and HER2 protein expression levels define multiple subtypes of HER2-positive metastatic breast cancer. Breast Cancer Res Treat. 2013; 141(1):43-53.
50. Pedersen K, Angelini PD, Laos S, Bach-Faig A, Cunningham MP, Ferrer-Ramon C, Luque-Garcia A, Garcia-Castillo J, Parra-Palau JL, Scaltriti M, Ramon y Cajal S, Baselga J and Arribas J. A naturally occurring HER2 carboxy-terminal fragment promotes mammary tumor growth and metastasis. Mol Cell Biol. 2009; 29(12):3319-3331.
51. Parra-Palau JL, Morancho B, Peg V, Escorihuela M, Scaltriti M, Vicario R, Zacarias-Fluck M, Pedersen K, Pandiella A, Nuciforo P, Serra V, Cortes J, Baselga J, et al. Effect of p95HER2/611CTF on the response to trastuzumab and chemotherapy. J Natl Cancer Inst. 2014; 106(11).
52. Scaltriti M NP, Bradbury I, Sperinde J, Serra V, Parra JL, Prudkin L, Harbeck N, Pusztai L, Ellis C, Eidtmann H, Arribas J, Piccart M, Baselga J. (2013). High HER2 expression correlates with response to trastuzumab and the combination of trastuzumab and lapatinib. 36th San Antonio Breast Cancer Symposium. (San Antonio, TX.
53. Lu Y, Zi X, Zhao Y, Mascarenhas D and Pollak M. Insulin-like growth factor-I receptor signaling and resistance to trastuzumab (Herceptin). J Natl Cancer Inst. 2001; 93(24):1852-1857.
54. Wang Y, Hailey J, Williams D, Wang Y, Lipari P, Malkowski M, Wang X, Xie L, Li G, Saha D, Ling WL, Cannon-Carlson S, Greenberg R, et al. Inhibition of insulin-like growth factor-I receptor (IGF-IR) signaling and tumor cell growth by a fully human neutralizing anti-IGF-IR antibody. Mol Cancer Ther. 2005; 4(8):1214-1221.
55. Nahta R, Yuan LX, Zhang B, Kobayashi R and Esteva FJ. Insulin-like growth factor-I receptor/human epidermal growth factor receptor 2 heterodimerization contributes to trastuzumab resistance of breast cancer cells. Cancer Res. 2005; 65(23):11118-11128.
56. Camirand A, Lu Y and Pollak M. Co-targeting HER2/ErbB2 and insulin-like growth factor-1 receptors causes synergistic inhibition of growth in HER2-overexpressing breast cancer cells. Med Sci Monit. 2002; 8(12):BR521-526.
57. Browne BC, Crown J, Venkatesan N, Duffy MJ, Clynes M, Slamon D and O’Donovan N. Inhibition of IGF1R activity enhances response to trastuzumab in HER-2-positive breast cancer cells. Ann Oncol. 2011; 22(1):68-73.
58. Bottaro DP, Rubin JS, Faletto DL, Chan AM, Kmiecik TE, Vande Woude GF and Aaronson SA. Identification of the hepatocyte growth factor receptor as the c-met proto-oncogene product. Science. 1991; 251(4995):802-804.
59. Boccaccio C and Comoglio PM. Invasive growth: a MET-driven genetic programme for cancer and stem cells. Nat Rev Cancer. 2006; 6(8):637-645.
60. Blumenschein GR, Jr., Mills GB and Gonzalez-Angulo AM. Targeting the hepatocyte growth factor-cMET axis in cancer therapy. J Clin Oncol. 2012; 30(26):3287-3296.
61. Beviglia L, Matsumoto K, Lin CS, Ziober BL and Kramer RH. Expression of the c-Met/HGF receptor in human breast carcinoma: correlation with tumor progression. Int J Cancer. 1997; 74(3):301-309.
62. Lengyel E, Prechtel D, Resau JH, Gauger K, Welk A, Lindemann K, Salanti G, Richter T, Knudsen B, Vande Woude GF and Harbeck N. C-Met overexpression in node-positive breast cancer identifies patients with poor clinical outcome independent of Her2/neu. Int J Cancer. 2005; 113(4):678-682.
63. Raghav KP, Wang W, Liu S, Chavez-MacGregor M, Meng X, Hortobagyi GN, Mills GB, Meric-Bernstam F, Blumenschein GR, Jr. and Gonzalez-Angulo AM. cMET and phospho-cMET protein levels in breast cancers and survival outcomes. Clin Cancer Res. 2012; 18(8):2269-2277.
64. Minuti G, Cappuzzo F, Duchnowska R, Jassem J, Fabi A, O’Brien T, Mendoza AD, Landi L, Biernat W, Czartoryska-Arlukowicz B, Jankowski T, Zuziak D, Zok J, et al. Increased MET and HGF gene copy numbers are associated with trastuzumab failure in HER2-positive metastatic breast cancer. Br J Cancer. 2012; 107(5):793-799.
65. Shattuck DL, Miller JK, Carraway KL, 3rd and Sweeney C. Met receptor contributes to trastuzumab resistance of Her2-overexpressing breast cancer cells. Cancer Res. 2008; 68(5):1471-1477.
66. Liu L, Shi H, Liu Y, Anderson A, Peterson J, Greger J, Martin AM and Gilmer TM. Synergistic effects of foretinib with HER-targeted agents in MET and HER1- or HER2-coactivated tumor cells. Mol Cancer Ther. 2011; 10(3):518-530.
67. Yeatman TJ. A renaissance for SRC. Nat Rev Cancer. 2004; 4(6):470-480.
68. Zhang S, Huang WC, Li P, Guo H, Poh SB, Brady SW, Xiong Y, Tseng LM, Li SH, Ding Z, Sahin AA, Esteva FJ, Hortobagyi GN, et al. Combating trastuzumab resistance by targeting SRC, a common node downstream of multiple resistance pathways. Nat Med. 2011; 17(4):461-469.
69. Peiro G, Ortiz-Martinez F, Gallardo A, Perez-Balaguer A, Sanchez-Paya J, Ponce JJ, Tibau A, Lopez-Vilaro L, Escuin D, Adrover E, Barnadas A and Lerma E. Src, a potential target for overcoming trastuzumab resistance in HER2-positive breast carcinoma. Br J Cancer. 2014; 111(4):689-695.
70. Formisano L, Nappi L, Rosa R, Marciano R, D’Amato C, D’Amato V, Damiano V, Raimondo L, Iommelli F, Scorziello A, Troncone G, Veneziani B, Parsons SJ, et al. Epidermal growth factor-receptor activation modulates Src-dependent resistance to lapatinib in breast cancer models. Breast Cancer Res. 2014; 16(3):R45.
71. Bellati F, Napoletano C, Ruscito I, Liberati M, Panici PB and Nuti M. Cellular adaptive immune system plays a crucial role in trastuzumab clinical efficacy. J Clin Oncol. 2010; 28(21):e369-370; author reply e371.
72. Joensuu H, Bono P, Kataja V, Alanko T, Kokko R, Asola R, Utriainen T, Turpeenniemi-Hujanen T, Jyrkkio S, Moykkynen K, Helle L, Ingalsuo S, Pajunen M, et al. Fluorouracil, epirubicin, and cyclophosphamide with either docetaxel or vinorelbine, with or without trastuzumab, as adjuvant treatments of breast cancer: final results of the FinHer Trial. J Clin Oncol. 2009; 27(34):5685-5692.
73. Loi S, Michiels S, Salgado R, Sirtaine N, Jose V, Fumagalli D, Kellokumpu-Lehtinen PL, Bono P, Kataja V, Desmedt C, Piccart MJ, Loibl S, Denkert C, et al. Tumor infiltrating lymphocytes are prognostic in triple negative breast cancer and predictive for trastuzumab benefit in early breast cancer: results from the FinHER trial. Ann Oncol. 2014; 25(8):1544-1550.
74. Loi S, Michiels S, Lambrechts D, Salgado R, Sirtaine N, Fumagalli D, Claes B, Kellokumpu-Lehtinen P-L, Bono P, Kataja VV, Larsimont D, Piccart-Gebhart MJ, Joensuu H, et al. Tumor PIK3CA mutations, lymphocyte infiltration, and recurrence-free survival (RFS) in early breast cancer (BC): Results from the FinHER trial. ASCO Meeting Abstracts. 2012; 30(15_suppl):507.
75. Gianni L BG, Valagussa P, Belousov A, Thomas M, Ross G, Pusztai L. Adaptive immune system and immune checkpoints are associated with response to pertuzumab (P) and trastuzumab (H) in the NeoSphere study. SABCS Annual Meeting Abstracts 2012.
76. Pardoll DM. The blockade of immune checkpoints in cancer immunotherapy. Nat Rev Cancer. 2012; 12(4):252-264.
77. al SLe. (2013). Tumor infiltrating lymphocytes (TILs) indicate trastuzumab benefit in early-stage HER2- positive breast cancer (HER2+ BC). 2013 San Antonio Breast Cancer Symposium.
78. Perez EA, Thompson EA, Ballman KV, Anderson SK, Asmann YW, Kalari KR, Eckel-Passow JE, Dueck AC, Tenner KS, Jen J, Fan JB, Geiger XJ, McCullough AE, et al. Genomic analysis reveals that immune function genes are strongly linked to clinical outcome in the North Central Cancer Treatment Group n9831 Adjuvant Trastuzumab Trial. J Clin Oncol. 2015; 33(7):701-708.
79. Burris HA, 3rd. Dual kinase inhibition in the treatment of breast cancer: initial experience with the EGFR/ErbB-2 inhibitor lapatinib. Oncologist. 2004; 9 Suppl 3:10-15.
80. Kwak EL, Sordella R, Bell DW, Godin-Heymann N, Okimoto RA, Brannigan BW, Harris PL, Driscoll DR, Fidias P, Lynch TJ, Rabindran SK, McGinnis JP, Wissner A, et al. Irreversible inhibitors of the EGF receptor may circumvent acquired resistance to gefitinib. Proc Natl Acad Sci U S A. 2005; 102(21):7665-7670.
81. Johnston S, Pippen J, Jr., Pivot X, Lichinitser M, Sadeghi S, Dieras V, Gomez HL, Romieu G, Manikhas A, Kennedy MJ, Press MF, Maltzman J, Florance A, et al. Lapatinib combined with letrozole versus letrozole and placebo as first-line therapy for postmenopausal hormone receptor-positive metastatic breast cancer. J Clin Oncol. 2009; 27(33):5538-5546.
82. Konecny GE, Pegram MD, Venkatesan N, Finn R, Yang G, Rahmeh M, Untch M, Rusnak DW, Spehar G, Mullin RJ, Keith BR, Gilmer TM, Berger M, et al. Activity of the dual kinase inhibitor lapatinib (GW572016) against HER-2-overexpressing and trastuzumab-treated breast cancer cells. Cancer Res. 2006; 66(3):1630-1639.
83. Blackwell KL, Burstein HJ, Storniolo AM, Rugo H, Sledge G, Koehler M, Ellis C, Casey M, Vukelja S, Bischoff J, Baselga J and O’Shaughnessy J. Randomized study of Lapatinib alone or in combination with trastuzumab in women with ErbB2-positive, trastuzumab-refractory metastatic breast cancer. J Clin Oncol. 2010; 28(7):1124-1130.
84. Baselga J, Bradbury I, Eidtmann H, Di Cosimo S, de Azambuja E, Aura C, Gomez H, Dinh P, Fauria K, Van Dooren V, Aktan G, Goldhirsch A, Chang TW, et al. Lapatinib with trastuzumab for HER2-positive early breast cancer (NeoALTTO): a randomised, open-label, multicentre, phase 3 trial. Lancet. 2012; 379(9816):633-640.
85. Robidoux A, Tang G, Rastogi P, Geyer CE, Jr., Azar CA, Atkins JN, Fehrenbacher L, Bear HD, Baez-Diaz L, Sarwar S, Margolese RG, Farrar WB, Brufsky AM, et al. Lapatinib as a component of neoadjuvant therapy for HER2-positive operable breast cancer (NSABP protocol B-41): an open-label, randomised phase 3 trial. Lancet Oncol. 2013; 14(12):1183-1192.
86. Piccart-Gebhart MJ, Holmes AP, Baselga J, De Azambuja E, Dueck AC, Viale G, Zujewski JA, Goldhirsch A, Santillana S, Pritchard KI, Wolff AC, Jackisch C, Lang I, et al. First results from the phase III ALTTO trial (BIG 2-06; NCCTG [Alliance] N063D) comparing one year of anti-HER2 therapy with lapatinib alone (L), trastuzumab alone (T), their sequence (T->L), or their combination (T+L) in the adjuvant treatment of HER2-positive early breast cancer (EBC). ASCO Meeting Abstracts. 2014; 32(15_suppl):LBA4.
87. Vicario R, Peg V, Morancho B, Zacarias-Fluck M, Zhang J, Martinez-Barriocanal A, Navarro Jimenez A, Aura C, Burgues O, Lluch A, Cortes J, Nuciforo P, Rubio IT, et al. Patterns of HER2 Gene Amplification and Response to Anti-HER2 Therapies. PLoS One. 2015; 10(6):e0129876.
88. Xia W, Bacus S, Hegde P, Husain I, Strum J, Liu L, Paulazzo G, Lyass L, Trusk P, Hill J, Harris J and Spector NL. A model of acquired autoresistance to a potent ErbB2 tyrosine kinase inhibitor and a therapeutic strategy to prevent its onset in breast cancer. Proc Natl Acad Sci U S A. 2006; 103(20):7795-7800.
89. Serra V, Scaltriti M, Prudkin L, Eichhorn PJ, Ibrahim YH, Chandarlapaty S, Markman B, Rodriguez O, Guzman M, Rodriguez S, Gili M, Russillo M, Parra JL, et al. PI3K inhibition results in enhanced HER signaling and acquired ERK dependency in HER2-overexpressing breast cancer. Oncogene. 2011; 30(22):2547-2557.
90. Chakrabarty A, Sanchez V, Kuba MG, Rinehart C and Arteaga CL. Feedback upregulation of HER3 (ErbB3) expression and activity attenuates antitumor effect of PI3K inhibitors. Proc Natl Acad Sci U S A. 2012; 109(8):2718-2723.
91. Crafter C, Vincent JP, Tang E, Dudley P, James NH, Klinowska T and Davies BR. Combining AZD8931, a novel EGFR/HER2/HER3 signalling inhibitor, with AZD5363 limits AKT inhibitor induced feedback and enhances antitumour efficacy in HER2-amplified breast cancer models. Int J Oncol. 2015.
92. Hafizi S and Dahlback B. Signalling and functional diversity within the Axl subfamily of receptor tyrosine kinases. Cytokine Growth Factor Rev. 2006; 17(4):295-304.
93. Liu L, Greger J, Shi H, Liu Y, Greshock J, Annan R, Halsey W, Sathe GM, Martin AM and Gilmer TM. Novel mechanism of lapatinib resistance in HER2-positive breast tumor cells: activation of AXL. Cancer Res. 2009; 69(17):6871-6878.
94. Bose R, Molina H, Patterson AS, Bitok JK, Periaswamy B, Bader JS, Pandey A and Cole PA. Phosphoproteomic analysis of Her2/neu signaling and inhibition. Proc Natl Acad Sci U S A. 2006; 103(26):9773-9778.
95. Iain Hutcheson DB, Max Hasmann and Robert Nicholson. (2007). Induction of erbB3/EGFR heterodimers mediates resistance to pertuzumab in a tamoxifen-resistant MCF-7 breast cancer cell line. AACR-NCI-EORTC International Conference. (San Francisco, CA.
96. Wuerkenbieke D, Wang J, Li Y and Ma C. miRNA-150 downregulation promotes pertuzumab resistance in ovarian cancer cells via AKT activation. Arch Gynecol Obstet. 2015.
97. He L and Hannon GJ. MicroRNAs: small RNAs with a big role in gene regulation. Nat Rev Genet. 2004; 5(7):522-531.
98. Ariella B. Hanker CDY, Thomas P. Stricker, Rebecca S. Cook, and Carlos L. Arteaga. (2014). HER2/PIK3CAH1047R transgenic tumors develop acquired resistance to triple therapy with trastuzumab, pertuzumab, and PI3K inhibitors via multiple mechanisms. AACR Annual Meeting 2014; April 5-9, 2014. (San Diego, CA.
99. Verma S, Miles D, Gianni L, Krop IE, Welslau M, Baselga J, Pegram M, Oh DY, Dieras V, Guardino E, Fang L, Lu MW, Olsen S, et al. Trastuzumab emtansine for HER2-positive advanced breast cancer. N Engl J Med. 2012; 367(19):1783-1791.
100. Krop IE, Kim SB, Gonzalez-Martin A, LoRusso PM, Ferrero JM, Smitt M, Yu R, Leung AC, Wildiers H and collaborators TRs. Trastuzumab emtansine versus treatment of physician’s choice for pretreated HER2-positive advanced breast cancer (TH3RESA): a randomised, open-label, phase 3 trial. Lancet Oncol. 2014; 15(7):689-699.
101. Kovtun YV and Goldmacher VS. Cell killing by antibody-drug conjugates. Cancer Lett. 2007; 255(2):232-240.
102. Barok M, Joensuu H and Isola J. Trastuzumab emtansine: mechanisms of action and drug resistance. Breast Cancer Res. 2014; 16(2):209.
103. Phillips GD, Fields CT, Li G, Dowbenko D, Schaefer G, Miller K, Andre F, Burris HA, 3rd, Albain KS, Harbeck N, Dieras V, Crivellari D, Fang L, et al. Dual targeting of HER2-positive cancer with trastuzumab emtansine and pertuzumab: critical role for neuregulin blockade in antitumor response to combination therapy. Clin Cancer Res. 2014; 20(2):456-468.
104. Holmes WE, Sliwkowski MX, Akita RW, Henzel WJ, Lee J, Park JW, Yansura D, Abadi N, Raab H, Lewis GD and et al. Identification of heregulin, a specific activator of p185erbB2. Science. 1992; 256(5060):1205-1210.
105. Holbro T, Beerli RR, Maurer F, Koziczak M, Barbas CF, 3rd and Hynes NE. The ErbB2/ErbB3 heterodimer functions as an oncogenic unit: ErbB2 requires ErbB3 to drive breast tumor cell proliferation. Proc Natl Acad Sci U S A. 2003; 100(15):8933-8938.
106. C.M.L. van Herpen UB, E.C.M. Mommers, N.P. Koper, P. Goedings, J. Lopez, A. Awada, H.B. Fiebrich, P.G. Aftimos. (2015). Phase I dose-escalation trial with the DNA-alkylating anti-HER2 antibody-drug conjugate SYD985. ESMO 2015. (Vienna, Austria).