
INTRODUCTION
Pancreatic ductal adenocarcinoma (PDA) is one of the most aggressive malignancies [1]. The poor prognosis is attributed to cancer stem cells (CSCs), which survive conventional cytotoxic therapy due to defense and survival mechanisms [2].
Viral treatment of cancer is an old concept that has been revisited in recent years using genetically modified viruses, including oncolytic adenoviruses (OAds). OAds are in clinical development for the treatment of various cancers because the replication of the viruses can be restricted to malignant cells [3–5]. OAds replicate in tumor cells; at the end of the viral replication cycle, the cell is lysed, and new infectious virus particles are released, which then infect and lyse neighboring tumor cells. The therapeutic potential involves the activation of endogenous tumor immune responses [6]. A major advantage of OAds is their excellent safety profile, which has been demonstrated in multiple studies of patients with ovarian cancer, melanoma, soft tissue or primary bone sarcoma and other neoplasms [3-5, 7-12]. In engineered OAds, the virus capsid is modified to improve binding to tumor cells [13]. Virus carriers have been studied to shield viruses against inactivation by neutralizing antibodies present in many patients from earlier adenovirus infections [14]. Mesenchymal stromal cells (MSCs) have been used as carriers due to their reported tumor-specific homing capacity, which enables the protected transfer and release of the viruses on site [14–17]. Castleton and colleagues [18] provided evidence that MSCs can be efficiently used as carriers for systemic delivery of oncolytic measles virus to treat acute lymphoblastic leukemia, even in the presence of high-titer neutralizing antibodies. Of particular interest is a small pilot study of OAd-delivery by MSCs in four children with metastatic neuroblastoma refractory to front-line therapies [19]. The patients received several doses of autologous MSCs carrying ICOVIR-5, an AdΔ24RGD-derivative OAd with tumor specificity that was designed to increase replication potency in tumor cells [20]. The tolerance to the treatment was excellent, and a complete clinical response was documented in one case. The child remained in complete remission 3 years after this therapy. Current efforts concentrate on increasing the efficacy of OAds, e.g., by inserting therapeutic genes [21] to support tumor lysis by inducing apoptosis or an anti-tumor immune response [6].
Recently, we genetically engineered adenoviruses for improved delivery by MSCs with the aim of enhancing oncolysis and improving virus production in PDA cells [22]. In this prior study, the capsid of adenovirus serotype 5, from which regular OAds are derived, was modified, resulting in consistently strong infection of PDA cells and MSCs [22]. The adenovirus was modified by inserting the chimeric Ad5/3 fiber, in which the knob domain with the HAdV-5 fiber has been replaced by the corresponding domain of the HAdV-3 fiber, binding to the widely expressed desmoglein-2 [22]. Thus, cell entry is independent of coxsackievirus-adenovirus receptor (CAR), which is poorly expressed on the cell surface of PDA cells and MSCs [22]. In addition, the replication and release of OAds in infected cells was improved by deleting the pro-apoptotic early adenoviral gene E1B19K or expression of the full-length human TRAIL gene from a viral transcription unit. The deletion of E1B19K has been reported to increase viral release and spread and, consequently, the therapeutic efficacy in experimental PDA models [23]. TRAIL was inserted because this tumor-specific death ligand induces paracrine apoptosis in neighboring tumor cells by eliminating non-infected tumor cells and autocrine effects presumably responsible for improved viral cell lysis and release in infected MSCs and PDA cells. Also, TRAIL exhibited low side effects in phase I/II clinical studies [24]. The resulting viruses Ad5/3-Δ19K-.Luc and Ad5/3-TRAIL demonstrated strongly increased total viral particle production and release from MSCs compared to the parental virus Ad5/3-Luc [22].
In the present study, we investigated the therapeutic efficacy of the improved OAds in established and primary CSC marker-enriched PDA cell lines after administration alone or via MSC carriers by in vitro invasion assays and xenotransplantation studies. The Ad5/3-TRAIL construct enabled effective tumor invasion by OAd-MSCs in spheroids and xenografts and significant in vivo elimination of tumorigenic cells.
RESULTS
Oncolytic adenoviruses infect primary pancreatic CSCs
To study the influence of OAd constructs on the potential for self-renewal, MIA-PaCa2 cells were infected, and live cells were re-seeded at clonogenic density 24 h later. After 2 weeks, the non-infected cells had formed colonies, but no colonies were detected in cells infected with the regular OAd construct Ad5/3-Luc or with the improved OAd constructs Ad5/3-Δ19K-.Luc or Ad5/3-TRAIL (Figure 1A, compare to Table 1), suggesting that the cells were already completely lysed by the parental virus. To evaluate the invasion potential of the OAd constructs in three-dimensional (3D), primary CSC spheroids, tumor cells were isolated from patient-derived PDA tissue by serial transplantation in mice and subsequent spheroidal culture (Figure 1B). These primary tumor spheroids are highly enriched in CSC markers [25]. The spheroids were infected, and 24 h later, the presence of adenoviral capsid protein, which reflects the amount and location of viruses, was detected by staining with a specific antibody. In addition, the expression of the CSC marker c-Met was detected by double immunofluorescence staining. Green-fluorescent-labeled adenoviral capsid protein was detected only in infected cells and not non-infected control cells (Figure 1C). By contrast, the red-fluorescent c-Met protein was present in infected and non-infected cells, demonstrating the CSC character of the spheroidal cell model. Double-stained, adenoviral capsid and c-Met-positive, yellow fluorescent cells were present in high amounts. These results indicate that all tested OAds successfully prevent colony formation and spread in 3D primary CSC spheroids, and no advantage of the improved OAds was detectable under these in vitro conditions.
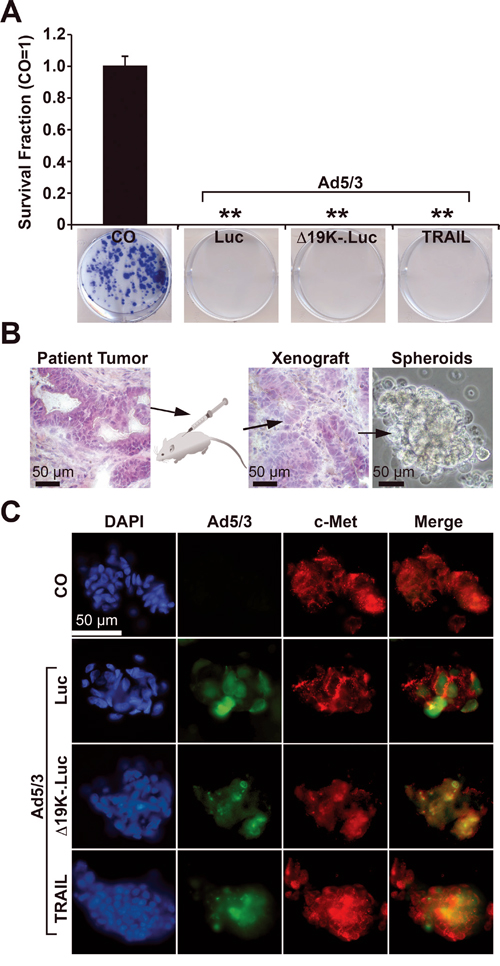
Figure 1: Infection of pancreatic CSCs by OAds. A. MIA-PaCa2 cells were infected at a titer of 200 TCID50 with Ad5/3-Luc (Luc), Ad5/3-Δ19K-.Luc (Δ19K-) or Ad5/3-TRAIL (TRAIL), or were left uninfected (CO). Twenty-four hours later, the cells were re-plated at a low density (400 cells/well) in 6-well plates. After two weeks, colonies containing more than 50 cells were counted using a dissecting microscope. The number of surviving colonies in the control was set to 1, and the survival fraction is presented. B. Tumor cells were freshly isolated from a mouse xenograft derived from the primary, low-passage, CSC-enriched human PDA xenograft cell line T30. The morphologies of the patient tumor and its mouse xenograft in passage 9 were detected by H&E staining and immunohistochemistry. The cells were cultured in an anchorage-independent manner as spheroids in vitro, which promotes the growth of CSCs (photograph on the right). C. Primary tumor spheroids were uninfected (CO) or infected at a titer of 200 TCID50 with Ad5/3-Luc (Luc), Ad5/3-Δ19K-.Luc (Δ19K-) or Ad5/3-TRAIL (TRAIL). Spheroids were fixed and centrifuged on slides 42 h after infection. Adenoviral capsid protein (green), expression of the CSC marker c-Met (red), or both together (merge, yellow) were detected using specific antibodies and double immunofluorescence staining. The cell nuclei were counterstained with DAPI (blue). Representative images at 400× magnification are shown.
Table 1: Properties of the used OAd constructs
Virus name |
Abbreviation |
Properties |
Reference |
|---|---|---|---|
Ad5/3 |
- |
- |
present work |
Ad5/3-Luc |
Luc |
Luciferase gene fused via IRES to the fiber gene. |
[23] |
Ad5/3-Δ19K-.Luc |
Luc/19K- |
Luciferase gene fused via IRES to the fiber gene, deletion of E1B19K. |
[23] |
Ad5/3-TRAIL |
TRAIL |
Human TRAIL gene fused via IRES to the fiber gene. |
[23] |
All viruses have a 24 amino acid deletion in E1A for tumor-selectivity, an E3-deletion, an Ad5/3 chimeric fiber for enhanced cell entry and a GFP gene upstream of RITR, expressed from the major late promoter.
OAd-infected MSC carriers invade tumor spheroids
For protected and tumor-specific transfer, we used as carriers MSCs that were isolated from the bone marrow of different healthy donors, expanded, and selected. In passage 3, the progenitor cell character of the MSCs was confirmed according to the minimal criteria for defining multipotent mesenchymal stromal cells of the International Society for Cellular Therapy (ISCT). Thus, the MSC were plastic-adherent in standard culture conditions, they expressed a characteristic surface marker pattern of CD34-, CD45-, CD166-, CD44+, CD90+ and CD105+ and they differentiated to osteoblasts, adipocytes and chondroblasts in vitro (SFig. 1). Therefore, the MSCs were used for further experiments between passages 4 and 8.
To evaluate the attraction of OAd-infected MSCs to tumor spheroids in vitro, MSCs attached to the bottom of 24-well plates were marked with the red fluorescent dye PKH26. Twenty-four hours later, the MSCs were infected or left uninfected with parental Ad5/3-Luc or the improved Ad5/3-Δ19K-.Luc or Ad5/3-TRAIL viruses, and the cells were covered with a gel layer on which spheroids from PaCaDD183 or MIA-PaCa2 cells were seeded and co-incubated for 16 h (Figure 2). Forty-two hours after infection, that is, 6 h before viral lysis [22], the spheroids were separated, and the integration of red MSCs was analyzed by immunofluorescence microscopy. Red fluorescent MSCs were detected in all co-incubated spheroids, suggesting that infected- and non-infected MSCs were attracted. However, double-fluorescent, yellow, virus-expressing MSCs were only detected in spheroids co-incubated with infected MSCs, as expected. This result suggests the successful migration of MSCs in 3D spheroidal structures.
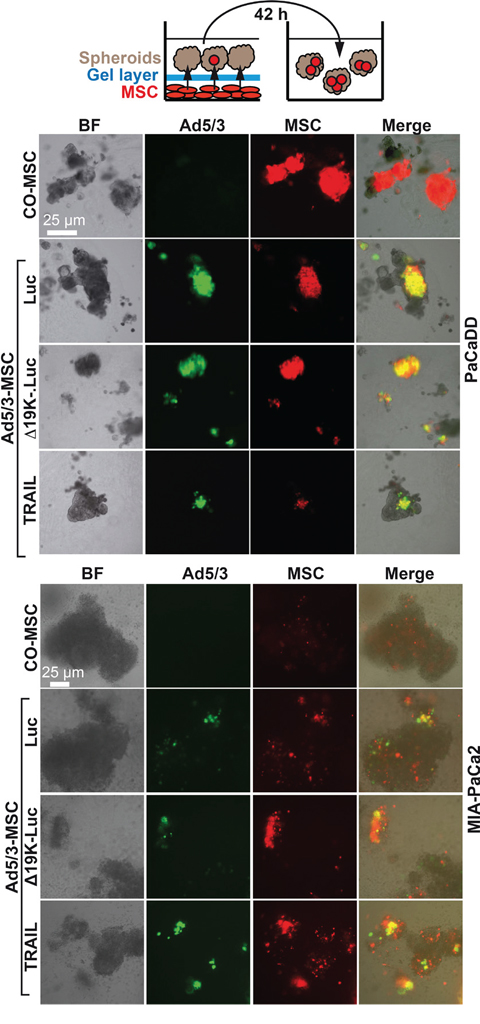
Figure 2: OAd-infected MSC carriers invade tumor spheroids. MSCs growing as a monolayer in 24-well plates were stained with the red fluorescent dye PKH26; 2 μM dye solution (provided by the manufacturer) was added per well. MSCs were infected with Ad5/3-Luc (Luc), Ad5/3-Δ19K-.Luc (Δ19K-) or Ad5/3-TRAIL (TRAIL) at a titer of 2000 TCID50 24 h later. Two hours after viral infection, the MSCs were covered with a 1:1:1 mixture of Matrigel, collagen and methylcellulose in DMEM medium supplemented with 2% FCS. Immediately afterward, tumor spheroids prepared from the primary, PDA cell line PaCaDD in passage 10 or from the established PDA cell line MIAPaCa-2 were seeded on top of the gel layer. The cells were co-incubated for 16 h to allow invasion of the MSCs into the spheroids through the gel layer. The spheroids were then transferred into new culture plates using a pipette and cultivated until 42 h after infection. Invasion was evaluated by detection of adenoviral capsid protein (green) by staining with a specific antibody and double immunofluorescence microscopy. Bright field (left), fluorescence (right: Adenoviral capsid protein: green; MSCs: red; Merge: yellow). Representative images at 400× magnification are shown.
OAd in vitro infection prevents tumorigenicity
To assess the influence of OAd infection on tumorigenicity, we first used a control Ad5/3 construct to establish the detection of viral spread in xenograft tumors. MIA-PaCa2 cells were xenotransplanted in fertilized chick eggs. This method is an in vivo replacement method for mouse studies. Chick embryos are naturally immunodeficient because full immunocompetence in birds develops only after hatching at day 21 of development [26]. Xenografted tumors are well supplied by blood vessels from the CAM, and chick fibroblasts participate in the formation of the tumor stroma. Most importantly, the CAM of the chick embryo is not innervated; therefore, unlike mice, the embryo does not feel pain during transplantation and tumor growth. Favorably, this system has no administrative barriers, it is inexpensive and well suited for short-term in vivo xenograft studies. MIA-PaCa2 cells were transplanted in the CAM of fertilized chick eggs, either as uninfected cells or in cell mixtures containing 1% or 5% infected cells. Tumor xenografts developed rapidly from uninfected control cells 9 days after transplantation (Figure 3A). The embryos were humanely euthanized, followed by tumor resection and calculation of the percentage of engrafted xenografts and tumor volumes. The tumor take rate was 86% in the control group and 90% for tumor cells containing 1% infected MIA-PaCa2 cells but decreased to 58% in the presence of 5% infected cells. The adenoviral capsid protein was present in tumor samples of infected xenografts, as shown by immunohistochemistry (Figure 3B). Simultaneously, the progression marker CD24 was reduced in infected tumors compared to uninfected controls, demonstrating the reduction of CD24-expressing tumor cells by the OAd virus. In addition, proliferation was strongly reduced in 1% infected cells and nearly undetectable in 5% infected cells, as concluded from double immunofluorescence microscopy with the human-specific markers Ki67 and cytokeratin 19 (Figure 3C). Moreover, DAPI staining of the cell nuclei revealed strongly shriveled nuclei in tumors containing 5% infected cells, indicating complete elimination of tumorigenic cells. Interestingly, the morphology of xenograft tissue from infected cell mixtures differed from that of uninfected control xenografts and contained densely packed, small cells located in cluster-like structures (Figure 4), suggesting that these are the regions in which the virus spread and lysed the cells.
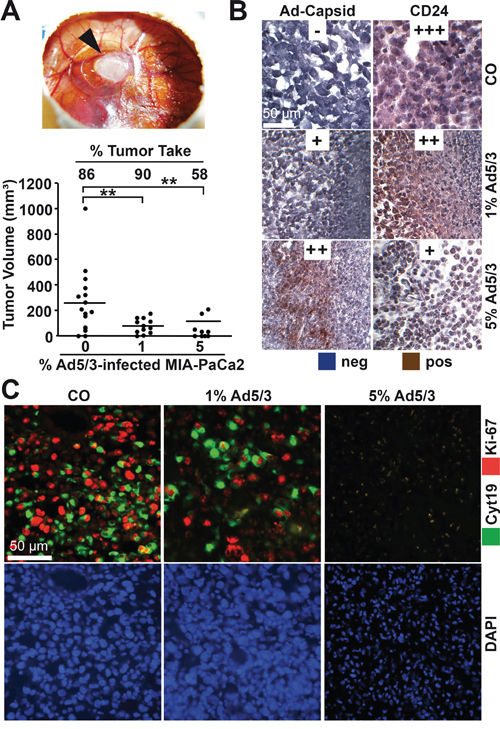
Figure 3: OAd-infected PDA cells exhibit reduced tumorigenicity. A. MIA-PaCa2 cells were uninfected or infected with Ad5/3. Two hours later, 5 × 105 uninfected cells (CO) or ratios of 99:1 (1) or 95:5 (5) 5 × 105 uninfected:infected MIA-PaCa2 cells were transplanted to the CAM of fertilized chick eggs at day 9 of development (15 eggs per group). The results for xenografted eggs with live chick embryos were evaluated. The black arrow marks tumor xenograft formed by uninfected MIA-PaCa-2 cells at day 18 of development. The engraftment efficiency expressed as the % tumor take of transplanted tumors and the tumor volumes were determined, and the single data points and the means of each group are shown (**P < 0.01). B. The presence of adenoviral capsid protein (Ad-Capsid) and of the CSC marker CD24 was evaluated by staining with specific antibodies and immunohistochemistry. To evaluate expression levels, a semi-quantitative scoring system was used based on the determination of the percentage of positive cells by visual inspection: high (+++), medium (++), low (+) and absent (-). C. Double immunofluorescence staining of xenograft tissue with a human-specific antibody against cytokeratin 19 (Cyt19, green) and the proliferation marker Ki67 (red). Cell nuclei were counterstained with DAPI (blue). Representative images at 400× magnification are shown.
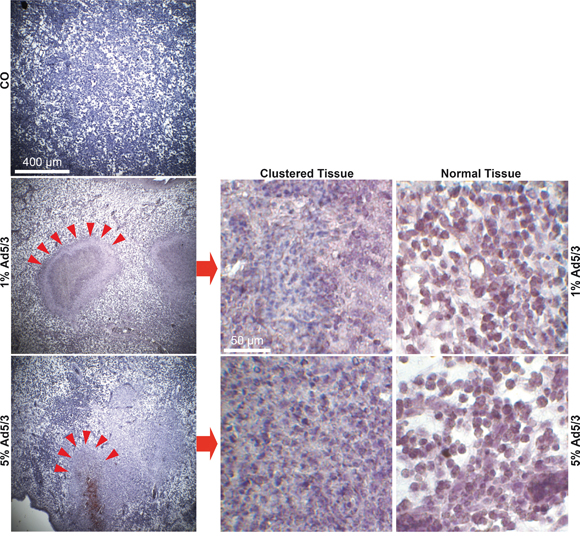
Figure 4: OAd treatment leads to clustered cell regions. MIA-PaCa2 cells were uninfected or infected with Ad5/3. Two hours later, 5×105 uninfected cells (CO) or ratios of 99:1 (1) or 95:5 (5) 5×105 uninfected:infected MIA-PaCa2 cells were transplanted to the CAM of fertilized chick eggs at day 9 of development (15 eggs per group). Tissue sections from the resected xenograft tumors were prepared and stained with a human-specific antibody against cytokeratin 19 (Cyt19). Representative images are shown at 40× magnification, and compact, cluster-like structures are marked with red arrows. Details of cluster-like structures (Clustered Tissue) and normal tissue structures (Normal Tissue) are shown at 400× magnification.
OAd-TRAIL is most therapeutically significant virus in vivo
To compare the invasion, spread and lysis mediated by the systemic delivery of infected MSCs, Mia-PaCa2 cells were transplanted to the CAM of fertilized chick eggs. Seven days later, the xenografted eggs were left untreated, or the CAM vessels were injected with virus-infected MSCs or the Ad5/3-TRAIL virus without carrier (Figure 5). Two days later, the tumors were resected to examine the virus distribution in the xenograft tissue. Tissue sections were prepared and stained with a specific antibody to detect adenoviral capsid protein. Positive signals were detected in xenograft tissue from eggs injected with virus-infected MSCs but not in xenografts from eggs injected with Ad5/3-TRAIL virus alone or in control xenografts. By contrast, adenoviral capsid protein was not detectable in the xenografts of non-injected eggs, as expected, or in xenografts from eggs injected with the Ad5/3-TRAIL-GFP virus alone without MSC carriers. This result suggests that the delivery of virus with MSCs was more efficient, and most likely several particles were present in the same tumor region and thereby enabled the detection by immunohistochemistry. We cannot rule out that virus particles were also present in tumor tissue after transfer of the virus alone, but the isolated particles may have been too tiny and thus under the detection limit of immunohistochemistry. Because significant oncolysis occurred after delivery of virus without MSCs, the virus must have been replicated and spread with a time delay compared to MSC-delivered virus (Figure 6A). As a control for tumor-specific virus distribution, we stained normal chick embryonal tissue with an antibody for adenoviral capsid protein, but we were not able to detect virus particles in liver, heart and lung after virus delivery with or without MSCs (data not shown), suggesting that the delivery was tumor-specific. Also, we were not able to prove the presence of MSCs in tumor tissue, due to a lack of a specific single marker that would distinguish MSCs from tumor fibroblasts. However, the fact that adenoviral capsid protein was detectable in tumor tissue after transfer by MSCs only strongly supports the conclusion that virus-infected MSCs migrated into the tumors.
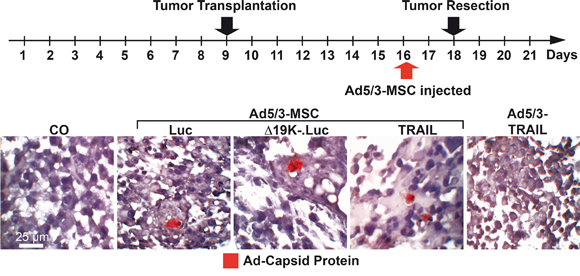
Figure 5: OAd-infected MSCs invade tumors in vivo. At developmental day 9, 5×105 MIA-PaCa2 cells were transplanted to the CAM of chick embryos (18 eggs per group). At day 16, the CAM was injected with 100 μL of cell culture medium (CO), 1×108 TCID50 Ad5/3-TRAIL in 100 μL of cell culture medium, or 5×104 MSCs in 100 μL of cell culture medium 2 h after infection with 2000 TCID50 of Ad5/3-Luc (Luc), Ad5/3-Δ19K-.Luc (Δ19K-) or Ad5/3-TRAIL (TRAIL). The tumor xenografts were resected at day 18. Tissue sections were prepared and paraffin-embedded, and the expression of adenoviral capsid protein (red) was detected by immunohistochemistry. Representative images at 400× magnification are shown.
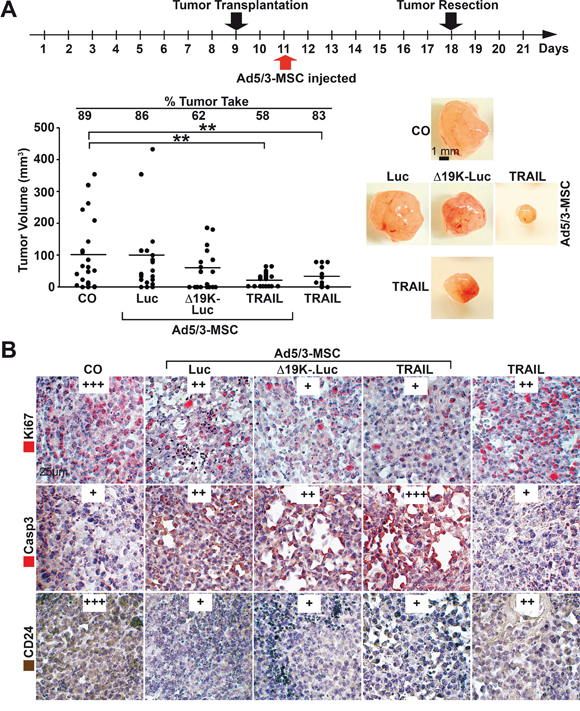
Figure 6: MSC-delivered OAds reduce tumor growth in vivo. A. Experimental procedure: MIA-PaCa2 cells (5×105) were transplanted to the CAM of chick embryos at developmental day 9 (20 eggs per group), followed by therapeutic injection at day 11, as described in Figure 5, and tumor resection at day 18. The tumor take rate and the volume of the resected xenografts were determined, and the single data points and the means of each group are shown (**P<0.01) along with representative images of tumor xenografts of each group. B. Sections were prepared from the xenograft tumors and paraffin-embedded, and the expression of Ki67, cleaved fragment of Caspase 3 (Casp3) and CD24 were examined by immunohistochemistry. Representative images are shown at 400× magnification. The expression levels were evaluated by a semi-quantitative scoring system as described in Figure 4B.
To assess tumor inhibition, MIA-PaCa2 cells were transplanted, but the therapy was injected later than before, at day 11, in CAM blood vessels to allow virus spread and lysis until the tumors were resected at day 18 (Figure 6A). This strategy resulted in a strong reduction of the engraftment rate from 89% in the control group to 62% and 58% in the groups that received MSC carriers with Ad5/3-Δ19K-.Luc or Ad5/3-TRAIL, respectively. However, MSC carriers with the parental Ad5/3-Luc virus or application of the Ad5/3-TRAIL virus alone reduced the tumor take rate only minimally. The Ad5/3-TRAIL virus effectively reduced the tumor volume whether transferred with or without MSCs, suggesting that the virus alone also homed to the tumor xenografts, but viral spread and lysis occurred with a time delay. Toxicity to the chick embryos was not observed, as expected due to the human-specific nature of the OAds. Immunohistochemistry of tumor sections demonstrated that all groups treated with OAds had reduced viability, as determined by Ki67 staining, and the strongest inhibition was mediated by the two improved OAds Ad5/3-Δ19K-.Luc and Ad5/3-TRAIL transferred by MSCs (Figure 6B). Similarly, the Ad5/3-TRAIL virus transferred by MSCs was superior at inducing apoptosis, as shown by staining of the active fragment of caspase-3. Additionally, all virus-treated groups had reduced expression of the progression marker CD24, with strongest and comparable effects in xenografts of eggs injected with OAds in MSC carriers. Taken together, our findings reveal that MSC delivery of OAds is superior to the delivery of OAds alone in tumor therapy. Among the two improved OAds tested, the Ad5/3-TRAIL virus was superior, most likely due to the paracrine induction of apoptosis in parallel to virus-induced tumor cell lysis.
DISCUSSION
In the present preclinical study, we investigated the therapeutic suitability of improved OAds with the aim of targeting highly therapy-resistant pancreatic stem-like tumor cells. The genetic engineering of OAds specific for PDA and MSC carriers and their infection and lysis efficiency were recently described by us [22]. The present study focused on the elimination of tumorigenicity, tumor growth and progression markers in vivo after administration of the improved OAds alone or via MSC carriers. We examined the Ad5/3-Δ19K-.Luc construct, which features a deletion of the viral gene E1B19K to permit increased viral release and spread [23], and the Ad5/3-TRAIL construct to exploit viral lysis by autocrine TRAIL activity and complement it with inducing paracrine apoptosis in neighboring, non-infected tumor cells.
Whereas our former publication demonstrated the successful infection of spheroidal growing established PDA cell lines [22], the present study showed the successful OAd infection of primary CSC spheroids from PDA patient tumors. The OAd constructs completely prevented the self-renewal potential, which is further underlined by our observation that no colonies were formed after infection of the established PDA cell line MIA-PaCa2. No obvious differences among the three viruses used were detectable under these in vitro conditions. The present data extend our prior findings demonstrating the successful infection of spheroidal cultures formed by MIA-PaCa2 cells [22]. Moreover, these data confirm recent reports in which an effective adenoviral lysis of primary CSCs derived from breast cancer was observed both in vitro and in orthotopic tumors in mice [27]. These results support the suitability of OAds for future therapeutic targeting of CSCs, in agreement with a recent review article suggesting oncolytic viral therapy for the targeting of CSCs [28]. Because CSCs are suggested to be the root of cancer growth, spread and recurrence [29], only the elimination of these tumor progenitors may provide a cure for patients with PDA.
Recent clinical studies have demonstrated inefficient delivery of oncolytic viruses by systemic application. This inefficiency is due to inactivation of viruses by the immune system and clearance from the blood by liver macrophages [30]. To circumvent this problem, we transferred the engineered OAds via MSC carriers because these progenitor cells have tumor tropism [31], sustain the replication of OAds [22], especially of the improved OAds used here [22], and were recently shown to release infectious particles in orthotopic lung and breast tumors [32]. The positive results of our invasion assays, in which infected MSC virus carriers successfully integrated through a gel layer into 3D spheroidal cultures of primary and established PDA cell lines, demonstrate that MSCs retain their tumor tropism and migration activity to overcome connective tissue-like structures despite OAd infection. This finding is particularly important because the extremely dense fibrotic matrix of PDA prevents even small therapeutic molecules from entering and perfusing [33] and is likely responsible for the marked resistance to chemo- and radiotherapy [34]. Another in vitro study has highlighted the advantage of adenoviral vector delivery by MSCs and observed increased delivery to breast and ovarian tumor xenografts in mice, with negligible levels of systemic toxicity compared with administration of adenovirus alone [35]. In preparation for a phase I clinical trial in ovarian cancer, significantly prolonged survival was observed in ovarian cancer-bearing mice treated with measles virus-infected MSCs from the adipose tissue of healthy donors and ovarian cancer patients but not those treated with the virus alone [36].
To detect differences between our OAd constructs, we examined the viral efficiency in a chick egg tumor xenograft model. Compared to mice, the egg system has several advantages, when used for short-time transplantation of 10 days. The tumor cells are transplanted to the chorioallantoic membrane (CAM), which is a well-vascularized extra-embryonic tissue located underneath the eggshell. According to our experience, the chick blood vessels quickly supply the tumor cells with blood. The chick embryo is naturally immunodeficient and the CAM readily supports the engraftment of tumor cells. Up to day 18 of chick development an animal application is not required, and the costs for the chick system are much lower compared to the mouse system. The experiments can be performed in a normal laboratory by the use of inexpensive egg incubators. Tumor xenografts growing on eggs have a pronounced tumor stroma, which consists of human tumor cells, chick fibroblasts, chick vessels and other host cells and thus provide the advantage of a tumor stroma, similar to the mouse xenotransplantation system. In a first approach, we evaluated the effect of a regular OAd virus infected in vitro, followed by xenotransplantation of a small amount of infected cells mixed with uninfected cells. This strategy was chosen because the uninfected cells can form tumors until the virus from the infected cells starts to replicate, lyse and spread to neighboring tumor cells embedded in the protective tissue structure. Indeed, we observed strongly decreased tumor engraftment and tumor volume as well as morphological changes in the virus-bearing tumor tissues, characterized by clusters of dense tissue. These dense clusters are likely the result of a cytopathic virus effect, which has been reported in response to adenovirus infection [37]. Consistent with these effects, the proliferation and expression of the CSC marker CD24 was decreased, and hardly any CD24-positive or proliferating cells were found in the dense clusters. We proceeded to transfer the OAd viruses, either alone or in MSCs, by systemic injection into blood vessels a few days after the tumor xenografts were transplanted. Viral capsid protein was detected in tumor tissue two days after injection, suggesting successful transfer by MSCs. By contrast, adenoviral capsid protein could not be detected in tumor tissue after the transfer of virus alone, or in embryonic tissue, suggesting that the virus transfer without MSC was less efficient and the delivery by MSC was specific for tumor tissue. Similar observations were made in a previous study demonstrating homing of virus-infected MSCs to different tumor types, including tumors with poor accessibility, such as glioma [15, 38]. Our results indicate that both of the improved OAds strongly reduced the expression of markers for stemness (CD24) and progression (Ki67) and enhanced the presence of the apoptosis marker “cleaved fragment of active caspase-3” after delivery by MSCs in vivo. Although our recent in vitro data suggested that the improved 19K-depleted OAd was superior regarding virus replication and release [22], the present in vivo data show that the OAd-TRAIL exhibited a superior effect after MSC delivery compared to delivery of OAd-TRAIL alone or compared to MSCs infected with the 19K-depleted OAd or with regular OAds. This conclusion was based on the strong and significant potential to reduce the tumor take rate and tumor volume and demonstrates the prominent bystander effect mediated by OAd-TRAIL. The enhanced efficiency of OAds expressing TRAIL is supported by other studies in which the viruses were not transferred by MSCs [39, 40]. Another advantage of TRAIL transfer by OAds is the known potential of TRAIL to induce apoptosis in senescent and malignant cells while showing no cytotoxicity in untransformed cells [41]. This finding is important because prior studies have suggested a tumorigenic potential of MSCs [42, 43]. However, this can be prevented by our strategy: MSCs are lysed by OADs after tumor-specific transfer. To address the toxicity and virus distribution in individuals with advanced PDA, an initial phase I dose escalation study using intratumoral injections of the OAd VCN-01 with or without gemcitabine has just started in Spain and is recruiting patients (NCT02045589). Similar to our OAd-TRAIL, the VCN-01 construct combines selective tumor replication with an improved tropism by insertion of the integrin-binding RGD peptide into the fiber. Instead of TRAIL, it expresses the hyaluronidase gene to degrade the dense extracellular tumor matrix and belongs to human adenovirus serotype 5 [44].
Taken together, our results demonstrate that delivery of the new OAd Ad5/3-TRAIL construct by MSC carriers induces a strong anti-tumor response in our experimental system. This therapeutic approach was even effective in tumorigenic cells with CSC characteristics, which do not respond to conventional chemotherapy. An important advantage of OAd Ad5/3-TRAIL is the selective replication in MSCs and PDA cells and the expression of TRAIL to induce tumor-specific apoptosis in non-infected cells. Oncolysis and the induction of apoptosis may induce an advanced inflammatory response and thus “open” the tumor microenvironment for conventional cytotoxic therapy or innate immune attacks. The clinical feasibility of such systemic MSC-OAd therapy warrants clinical investigation and may be understood as an initial aid to disrupt the dense tumor stroma for tumor cell eradication by innate immune responses and/or cytotoxic tumor therapy.
MATERIALS AND METHODS
Cell lines
The established human PDA cell line MIA-PaCa2 was obtained from American Type Culture Collection (ATCC, LGC Standards, Molsheim Cedex, France) and cultured in DMEM (18 mmol/l glucose) supplemented with 10% FCS and 5% HEPES. Human primary bone marrow-derived MSCs and the primary PDA cell line PaCaDD183 were isolated and cultured as previously described [45, 46]. The primary PDA xenograft line T30 was isolated from surgical non-diagnostic specimens by serial transplantation in mice and subsequent anchorage-independent in vitro culture, resulting in the selection of a CSC population as previously described [26]. All cells were grown in a humidified incubator at 37°C and 5% CO2 and authenticated throughout culture by their typical morphology. To maintain the authenticity of the cell lines, frozen stocks were prepared initially, and every three months a new frozen stock was used for experiments. MIA-PaCa2 cells were recently authenticated by a commercial service (Multiplexion, Heidelberg, Germany). Mycoplasm-negative cultures were ensured by performing monthly mycoplasm tests.
Isolation of human MSCs from bone marrow and culture
Human bone marrow was harvested from the iliac crest of 2 young healthy donors between 20 and 30 years old. 40 mL bone marrow aspirate were collected in a syringe containing 10,000 IU heparin to prevent coagulation. The mononuclear cell fraction was isolated by Biocoll density-gradient centrifugation (d = 1.077 g/cm3; Biochrom, Berlin, Germany). Cells were plated on fibronectin (10 ng/mL, Sigma, Taufkirchen, Germany)-coated cell culture flasks and selected by plastic adherence upon culture in MSC-NH expansion medium (Miltenyi Biotec, Bergisch Gladbach, Germany).
Flow cytometry (FACS) analysis of MSC marker expression
FACS analysis of marker expression was performed as described [45].
Recombinant oncolytic adenoviruses
Improved OAds were constructed as described [22]. The new OAd construct Ad-5/3 was cloned in the present study by inserting a GFP gene (from plasmid pEGFP-N1, Clontech, Saint-Germain-en-Laye, France) into the plasmid pSΔ24 [47], followed by homologous recombination with plasmids containing the Ad5/3ΔE3 genome. In all virus strains, the capsid contains a fiber 5/3 chimera with the shaft of serotype 5 and the knob domain of serotype 3. All constructs contain a GFP gene in the E4 locus. Other properties of the virus constructs are described in Table 1.
Colony-forming assay
Twenty-four hours after OAd infection of MIA-PaCa2 cells with 200 TCID50, 400 cells/well in 6-well tissue culture plates were re-seeded in complete medium and incubated for 2 weeks without changing the medium. The cells were then fixed with 3.7% PFA for 10 min and 70% ethanol for 10 min. After washing with distilled water, the cells were stained with 0.05% Coomassie Blue for 5 min. Then, the cells were washed with distilled water and dried overnight. Colonies with more than 50 cells were counted under a dissecting microscope.
Establishment and infection of primary tumor spheroids
Spheroids obtained from the primary xenograft line T30 were cultured in NeuroCult NS-A basal serum-free medium (human) (StemCell Technologies, Vancouver, Canada) supplemented with 2 μg/ml heparin (StemCell Technologies, Vancouver, Canada), 20 ng/ml hEGF (R&D Systems, Wiesbaden-Nordenstadt, Germany), 10 ng/ml hFGF-b (PeproTech, Hamburg, Germany) and 10% NeuroCult NS-A Proliferation Supplements (StemCell Technologies, Vancouver, Canada) and were used for experiments within 7 d after isolation. The cells were resuspended in DMEM medium containing 2% FCS for infection and incubated with 200 TCID50 (50% tissue culture infective dose) for 2 h before use in experiments.
Invasion of MSC-delivered OAds into spheroidal tumor cells
PaCaDD or MIA-PaCa2 cells were seeded in DMEM cell culture medium containing 2% heat-inactivated FCS and 0.25% methylcellulose (PureCol, Biomaterials, Fremont, USA) in a 96-well suspension culture plate (Greiner Bio-One, Frickenhausen, Germany) with a density of 10,000 cells/well for spheroid formation. The cells were used for co-incubation experiments with infected MSCs 72 h later. MSCs were stained with the PKH26 red fluorescent cell linker kit for general cell membrane labeling (Sigma-Aldrich, Munich, Germany), and 5 × 104 MSCs/well were seeded in a 24-well plate in DMEM cell culture medium supplemented with 2% FCS. The MSCs were infected 24 h later with a virus titer of 2000 TCID50. Two hours after infection, the MSCs were overlaid with a gel layer of methylcellulose, Matrigel and collagen (PureCol, Biomaterials, Fremont, USA) at a ratio of 1:1:1 in DMEM supplemented with 2% FCS. PaCaDD183 or MIA-PaCa2 spheroids were seeded on top of the gel layer and co-incubated with MSCs for 16 h. Next, the spheroids were transferred with a cut tip of a 10 μl pipette into new 96-well suspension culture plates. Forty-two hours after infection, images of non-fixed spheroids were acquired under a BIOREVO BZ-9000 microscope (Keyence, Neu-Isenburg, Germany).
Immunohistochemistry and immunofluorescence staining
Paraffin-embedded 6-μm xenograft sections or spheroids fixed in 4% PFA and centrifuged in suspension onto slides in a CytoSpin 4 cytocentrifuge (Thermo Scientific, Schwerte, Germany) were stained as previously described [26]. Samples were stored at −20°C prior to further analysis. The primary antibodies were rabbit polyclonal antibodies against human c-Met (Enzo Life Science, Lörrach, Germany), cytokeratin 19, Ki67 (both from Abcam, Cambridge, UK), and the cleaved fragment of active human caspase-3 (R&D Systems, Abingdon, UK) and mouse monoclonal antibodies against human CD24 (SW11 hybridoma, kindly provided by Dr. P. Altevogt) and adenovirus-capsid (Merck Millipore, Darmstadt, Germany). Biotinylated goat anti-rabbit or anti-mouse IgG (Vector Laboratories, Peterborough, UK) was used as the secondary antibody for immunohistochemistry. The primary antibodies were omitted in the negative control. Goat anti-mouse Alexa Fluor 488 IgG and goat anti-rabbit Alexa Fluor 594 (Molecular Probes, Karlsruhe, Germany) were used as the secondary antibodies for immunofluorescence staining. The signals were detected with a Leica DMRB fluorescence microscope (Leica, Wetzlar; Germany). Images of representative fields were captured with a SPOT™ FLEX 15.2 64 Mp shifting pixel digital color camera (Diagnostic Instruments, Inc. USA) and analyzed with SPOT Basic/Advanced 4.6 software.
Tumor xenotransplantation to the CAM of fertilized chick eggs
Tumor cells were transplanted to the chorioallantoic membrane (CAM) of fertilized eggs from genetically identical hybrid LB chicks, followed by treatment and humane euthanasia of the chick embryo as described [26, 48]. After resection, the tumor take rates were calculated from the percent of transplanted tumors that engrafted and the tumor volumes according to the formula Volume = 4/3 × π × r3 (r = 1/2 × square root of diameter 1 × diameter 2) [49]. Tumor tissue was fixed in Roti Histofix (Carl Roth, Karlsruhe, Germany) for 2-3 days and embedded in paraffin for further analysis.
Statistical analysis
Experiments with established and primary PDA cell lines were performed three times with similar outcomes. In vivo transplantation experiments in fertilized chick eggs were performed twice with similar outcomes, and representative experiments are shown (15 to 20 eggs/group). The significance of the differences between the tumor volumes of OAd-infected tumors or tumors treated with adenovirus-infected MSCs were evaluated by Student’s t test, or as shown in Figure 4, with a non-parametric Mann-Whitney test with Bonferroni-Holm correction for multiple testing. P<0.05 (*) and P<0.01 (**) were deemed statistically significant.
ACKNOWLEDGMENTS
We thank Dr. P. Schemmer for providing bone marrow form healthy donors and E. Aleksandrowicz for helpful discussions of the in vivo experiments. This study was supported by grants from the German Research Community (DFG HE 3186/11-1, DFG NE 832/4-1), Federal Ministry of Education and Research (BMBF 031A213), Heidelberger Surgery Foundation, Foundation for Cancer- and Scarlet-Research, Dietmar Hopp-Foundation and the Hanns A. Pielenz-Foundation.
CONFLICTS OF INTEREST
The authors declare no conflicts of interest.
Author contributions
IH, DN: Concept and design; AK, KH: Development of methodology; AK, KH, SV, CN, PF, ZZ, JG: Acquisition of data; AK, KH, WG, DN, IH: Analysis and interpretation of data; IH, LL, AK, KH, DN: Writing, review, and/or revision of the manuscript.
Abbreviations
Cancer stem cells (CSCs); Chorioallantoic membrane (CAM); Epithelial mesenchymal transition (EMT); Mesenchymal stromal cells (MSCs); Oncolytic adenoviruses (AOds); Pancreatic ductal adenocarcinoma (PDA).
REFERENCES
1. Oberstein PE, Saif MW. First-Line Treatment for Advanced Pancreatic Cancer. Highlights from the “2011 ASCO Gastrointestinal Cancers Symposium”. San Francisco, CA, USA. January 20-22, 2011. Jop. 2011; 12: 96-100.
2. Rasheed ZA, Kowalski J, Smith BD, Matsui W. Concise review: Emerging concepts in clinical targeting of cancer stem cells. Stem Cells. 2011; 29: 883-887.
3. Alemany R, Balague C, Curiel DT. Replicative adenoviruses for cancer therapy. Nat Biotechnol. 2000; 18: 723-727.
4. Nettelbeck DM. Virotherapeutics: conditionally replicative adenoviruses for viral oncolysis. Anticancer Drugs. 2003; 14: 577-584.
5. Yamamoto M, Curiel DT. Current issues and future directions of oncolytic adenoviruses. Mol Ther. 2010; 18: 243-250.
6. Pol J, Bloy N, Obrist F, Eggermont A, Galon J, Cremer I, Erbs P, Limacher JM, Preville X, Zitvogel L, Kroemer G, Galluzzi L. Trial Watch:: Oncolytic viruses for cancer therapy. Oncoimmunology. 2014; 3: e28694.
7. Bramante S, Koski A, Kipar A, Diaconu I, Liikanen I, Hemminki O, Vassilev L, Parviainen S, Cerullo V, Pesonen SK, Oksanen M, Heiskanen R, Rouvinen-Lagerstrom N, et al. Serotype chimeric oncolytic adenovirus coding for GM-CSF for treatment of sarcoma in rodents and humans. Int J Cancer. 2014; 135: 720-730.
8. Liikanen I, Ahtiainen L, Hirvinen ML, Bramante S, Cerullo V, Nokisalmi P, Hemminki O, Diaconu I, Pesonen S, Koski A, Kangasniemi L, Pesonen SK, Oksanen M, et al. Oncolytic adenovirus with temozolomide induces autophagy and antitumor immune responses in cancer patients. Mol Ther. 2013; 21: 1212-1223.
9. Kanerva A, Nokisalmi P, Diaconu I, Koski A, Cerullo V, Liikanen I, Tahtinen S, Oksanen M, Heiskanen R, Pesonen S, Joensuu T, Alanko T, Partanen K, et al. Antiviral and antitumor T-cell immunity in patients treated with GM-CSF-coding oncolytic adenovirus. Clin Cancer Res. 2013; 19: 2734-2744.
10. Kim KH, Dmitriev IP, Saddekni S, Kashentseva EA, Harris RD, Aurigemma R, Bae S, Singh KP, Siegal GP, Curiel DT, Alvarez RD. A phase I clinical trial of Ad5/3-Delta24, a novel serotype-chimeric, infectivity-enhanced, conditionally-replicative adenovirus (CRAd), in patients with recurrent ovarian cancer. Gynecol Oncol. 2013; 130: 518-524.
11. Bramante S, Kaufmann JK, Veckman V, Liikanen I, Nettelbeck DM, Hemminki O, Vassilev L, Cerullo V, Oksanen M, Heiskanen R, Joensuu T, Kanerva A, Pesonen S, et al. Treatment of melanoma with a serotype 5/3 chimeric oncolytic adenovirus coding for GM-CSF: Results in vitro, in rodents and in humans. Int J Cancer. 2015.
12. Liu TC, Galanis E, Kirn D. Clinical trial results with oncolytic virotherapy: a century of promise, a decade of progress. Nat Clin Pract Oncol. 2007; 4: 101-117.
13. Glasgow JN, Everts M, Curiel DT. Transductional targeting of adenovirus vectors for gene therapy. Cancer Gene Ther. 2006; 13: 830-844.
14. Willmon C, Harrington K, Kottke T, Prestwich R, Melcher A, Vile R. Cell carriers for oncolytic viruses: Fed Ex for cancer therapy. Mol Ther. 2009; 17: 1667-1676.
15. Xia X, Ji T, Chen P, Li X, Fang Y, Gao Q, Liao S, You L, Xu H, Ma Q, Wu P, Hu W, Wu M, et al. Mesenchymal stem cells as carriers and amplifiers in CRAd delivery to tumors. Mol Cancer. 2011; 10: 134.
16. Beckermann BM, Kallifatidis G, Groth A, Frommhold D, Apel A, Mattern J, Salnikov AV, Moldenhauer G, Wagner W, Diehlmann A, Saffrich R, Schubert M, Ho AD, et al. VEGF expression by mesenchymal stem cells contributes to angiogenesis in pancreatic carcinoma. Br J Cancer. 2008; 99: 622-631.
17. Kallifatidis G, Beckermann BM, Groth A, Schubert M, Apel A, Khamidjanov A, Ryschich E, Wenger T, Wagner W, Diehlmann A, Saffrich R, Krause U, Eckstein V, et al. Improved lentiviral transduction of human mesenchymal stem cells for therapeutic intervention in pancreatic cancer. Cancer Gene Ther. 2008; 15: 231-240.
18. Castleton A, Dey A, Beaton B, Patel B, Aucher A, Davis DM, Fielding AK. Human mesenchymal stromal cells deliver systemic oncolytic measles virus to treat acute lymphoblastic leukemia in the presence of humoral immunity. Blood. 2014; 123: 1327-1335.
19. Garcia-Castro J, Alemany R, Cascallo M, Martinez-Quintanilla J, Arriero Mdel M, Lassaletta A, Madero L, Ramirez M. Treatment of metastatic neuroblastoma with systemic oncolytic virotherapy delivered by autologous mesenchymal stem cells: an exploratory study. Cancer Gene Ther. 2010; 17: 476-483.
20. Cascallo M, Alonso MM, Rojas JJ, Perez-Gimenez A, Fueyo J, Alemany R. Systemic toxicity-efficacy profile of ICOVIR-5, a potent and selective oncolytic adenovirus based on the pRB pathway. Mol Ther. 2007; 15: 1607-1615.
21. Cody JJ, Douglas JT. Armed replicating adenoviruses for cancer virotherapy. Cancer Gene Ther. 2009; 16: 473-488.
22. Hammer K, Kazcorowski A, Liu L, Behr M, Schemmer P, Herr I, Nettelbeck DM. Engineered adenoviruses combine enhanced oncolysis with improved virus production by mesenchymal stromal carrier cells. Int J Cancer. 2015; 137: 978-990.
23. Oberg D, Yanover E, Adam V, Sweeney K, Costas C, Lemoine NR, Hallden G. Improved potency and selectivity of an oncolytic E1ACR2 and E1B19K deleted adenoviral mutant in prostate and pancreatic cancers. Clin Cancer Res. 2010; 16: 541-553.
24. Tolcher AW, Mita M, Meropol NJ, von Mehren M, Patnaik A, Padavic K, Hill M, Mays T, McCoy T, Fox NL, Halpern W, Corey A, Cohen RB. Phase I pharmacokinetic and biologic correlative study of mapatumumab, a fully human monoclonal antibody with agonist activity to tumor necrosis factor-related apoptosis-inducing ligand receptor-1. J Clin Oncol. 2007; 25: 1390-1395.
25. Forster T, Rausch V, Zhang Y, Isayev O, Heilmann K, Schoensiegel F, Liu L, Nessling M, Richter K, Labsch S, Nwaeburu CC, Mattern J, Gladkich J, et al. Sulforaphane counteracts aggressiveness of pancreatic cancer driven by dysregulated Cx43-mediated gap junctional intercellular communication. Oncotarget. 2014; 5: 1621-1634. doi: 10.18632/oncotarget.1764.
26. Zhang Y, Liu L, Fan P, Bauer N, Gladkich J, Ryschich E, Bazhin AV, Giese NA, Strobel O, Hackert T, Hinz U, Gross W, Fortunato F, et al. Aspirin counteracts cancer stem cell features, desmoplasia and gemcitabine resistance in pancreatic cancer. Oncotarget. 2015; 6: 9999-10015. doi: 10.18632/oncotarget.3171.
27. Eriksson M, Guse K, Bauerschmitz G, Virkkunen P, Tarkkanen M, Tanner M, Hakkarainen T, Kanerva A, Desmond RA, Pesonen S, Hemminki A. Oncolytic adenoviruses kill breast cancer initiating CD44+CD24-/low cells. Mol Ther. 2007; 15: 2088-2093.
28. Smith TT, Roth JC, Friedman GK, Gillespie GY. Oncolytic viral therapy: targeting cancer stem cells. Oncolytic Virother. 2014; 2014: 21-33.
29. Abbott A. Cancer: the root of the problem. Nature. 2006; 442: 742-743.
30. Ferguson MS, Lemoine NR, Wang Y. Systemic delivery of oncolytic viruses: hopes and hurdles. Adv Virol. 2012; 2012: 805629.
31. Brennen WN, Denmeade SR, Isaacs JT. Mesenchymal stem cells as a vector for the inflammatory prostate microenvironment. Endocr Relat Cancer. 2013; 20: R269-290.
32. Hakkarainen T, Sarkioja M, Lehenkari P, Miettinen S, Ylikomi T, Suuronen R, Desmond RA, Kanerva A, Hemminki A. Human mesenchymal stem cells lack tumor tropism but enhance the antitumor activity of oncolytic adenoviruses in orthotopic lung and breast tumors. Hum Gene Ther. 2007; 18: 627-641.
33. Olive KP, Jacobetz MA, Davidson CJ, Gopinathan A, McIntyre D, Honess D, Madhu B, Goldgraben MA, Caldwell ME, Allard D, Frese KK, Denicola G, Feig C, et al. Inhibition of Hedgehog signaling enhances delivery of chemotherapy in a mouse model of pancreatic cancer. Science. 2009; 324: 1457-1461.
34. Gukovskaya AS, Pandol SJ. Cell death pathways in pancreatitis and pancreatic cancer. Pancreatology. 2004; 4: 567-586.
35. Dembinski JL, Spaeth EL, Fueyo J, Gomez-Manzano C, Studeny M, Andreeff M, Marini FC. Reduction of nontarget infection and systemic toxicity by targeted delivery of conditionally replicating viruses transported in mesenchymal stem cells. Cancer Gene Ther. 2010; 17: 289-297.
36. Mader EK, Butler G, Dowdy SC, Mariani A, Knutson KL, Federspiel MJ, Russell SJ, Galanis E, Dietz AB, Peng KW. Optimizing patient derived mesenchymal stem cells as virus carriers for a phase I clinical trial in ovarian cancer. J Transl Med. 2013; 11: 20.
37. White E, Stillman B. Expression of adenovirus E1B mutant phenotypes is dependent on the host cell and on synthesis of E1A proteins. J Virol. 1987; 61: 426-435.
38. Sonabend AM, Ulasov IV, Tyler MA, Rivera AA, Mathis JM, Lesniak MS. Mesenchymal stem cells effectively deliver an oncolytic adenovirus to intracranial glioma. Stem Cells. 2008; 26: 831-841.
39. Cao X, Yang M, Wei RC, Zeng Y, Gu JF, Huang WD, Yang DQ, Li HL, Ding M, Wei N, Zhang KJ, Xu B, Liu XR, Qian QJ, Liu XY. Cancer targeting Gene-Viro-Therapy of liver carcinoma by dual-regulated oncolytic adenovirus armed with TRAIL gene. Gene Ther. 2011; 18: 765-777.
40. Li X, Mao Q, Wang D, Zhang W, Xia H. A fiber chimeric CRAd vector Ad5/11-D24 double-armed with TRAIL and arresten for enhanced glioblastoma therapy. Hum Gene Ther. 2012; 23: 589-596.
41. Stuckey DW, Shah K. TRAIL on trial: preclinical advances in cancer therapy. Trends Mol Med. 2013; 19: 685-694.
42. Spaeth EL, Dembinski JL, Sasser AK, Watson K, Klopp A, Hall B, Andreeff M, Marini F. Mesenchymal stem cell transition to tumor-associated fibroblasts contributes to fibrovascular network expansion and tumor progression. PLoS ONE. 2009; 4: e4992.
43. Wang Y, Huso DL, Harrington J, Kellner J, Jeong DK, Turney J, McNiece IK. Outgrowth of a transformed cell population derived from normal human BM mesenchymal stem cell culture. Cytotherapy. 2005; 7: 509-519.
44. Rodriguez-Garcia A, Gimenez-Alejandre M, Rojas JJ, Moreno R, Bazan-Peregrino M, Cascallo M, Alemany R. Safety and efficacy of VCN-01, an oncolytic adenovirus combining fiber HSG-binding domain replacement with RGD and hyaluronidase expression. Clin Cancer Res. 2015; 21: 1406-1418.
45. Apel A, Groth A, Schlesinger S, Bruns H, Schemmer P, Buchler MW, Herr I. Suitability of human mesenchymal stem cells for gene therapy depends on the expansion medium. Exp Cell Res. 2009; 315: 498-507.
46. Ruckert F, Aust D, Bohme I, Werner K, Brandt A, Diamandis EP, Krautz C, Hering S, Saeger HD, Grutzmann R, Pilarsky C. Five primary human pancreatic adenocarcinoma cell lines established by the outgrowth method. J Surg Res. 2012; 172: 29-39.
47. Suzuki K, Alemany R, Yamamoto M, Curiel DT. The presence of the adenovirus E3 region improves the oncolytic potency of conditionally replicative adenoviruses. Clin Cancer Res. 2002; 8: 3348-3359.
48. Aleksandrowicz E, Herr I. Ethical euthanasia and short-term anesthesia of the chick embryo. ALTEX. 2015; 32: 143-147.
49. Balke M, Neumann A, Szuhai K, Agelopoulos K, August C, Gosheger G, Hogendoorn PC, Athanasou N, Buerger H, Hagedorn M. A short-term in vivo model for giant cell tumor of bone. BMC Cancer. 2011; 11: 241.