
INTRODUCTION
Breast cancer is the leading cause of cancer-related death in women worldwide [1]. Previous twin studies suggest that 12–30% of breast cancers are primarily genetic in origin and result from autosomal dominant inheritance of a single gene mutation [2, 3]. The best-known genes are BRCA1 and BRCA2, which cause hereditary breast and ovarian cancer syndrome (HBOC). Genetic counseling and a BRCA gene test is recommended for breast cancer patients with early-onset or a significant family history. This strategy significantly reduces cancer-related mortality in BRCA mutation carriers, who receive regular cancer screening or prophylactic mastectomy and oophorectomy. However, pathogenic mutations of BRCA1 and BRCA2 only explain 10–20% of breast cancers in patients with early-onset or a significant family history. Of the non-BRCA genes, ATM, BRIP1, PALB2, PTEN and CHEK2, are reported to be medium-to-high penetrance genes that cause hereditary breast cancer [4–6]. A longitudinal study showed that, by 70 years of age, the absolute breast-cancer risk for female PALB2 mutation carriers ranges from 33% (95% CI, 25 to 44) for those with no family history of breast cancer to 58% (95% CI, 50 to 66) for those with a family history [7]. Comprehensive multiple gene sequencing is therefore necessary to understand the predisposing genetic factors in development of breast cancer.
Next-generation sequencing (NGS) technology makes it possible to sequence large numbers of target genes and is now used not only in research, but for multiple gene testing for clinical application. Although multiple predisposing genes can be sequenced in parallel, several points have not been answered about the application of it into clinical practice. First, it is not known how many predisposing genes need to be tested in these patients and whether there is an association between gene and tumor phenotype (pathology). Second, there is no consensus on the best approach to genetic counseling, cancer-risk assessment, and intervention for patients with non-BRCA mutations. Third, it is difficult to distinguish genetic variants of uncertain significance (VUS), especially for non-BRCA1/2 predisposing genes, in clinical patients.
In this study, we developed a customized sequencing panel containing 68 genes with a known and potential association with hereditary cancer syndromes (Table 1). Using this panel, we aimed to assess the clinical value of multiple predisposing genes in breast cancer patients with an early-onset or a significant family history.
Table 1: List of genes for sequencing and reasons for their inclusion
Gene |
Hereditary syndrome or increased breast cancer risk (BC risk↑) |
DNA repair |
Gene |
Hereditary syndrome or increased breast cancer risk (BC risk↑) |
DNA repair |
|---|---|---|---|---|---|
APC |
FAP |
|
GT198 |
BC risk↑ |
|
ARLTS1 |
BC risk↑ |
|
ku70 |
|
NHEJ |
ATM |
BC risk↑ |
|
ku80/XRCC5 |
BC risk↑ |
NHEJ |
BARD1 |
BC risk↑ |
|
MAP3K1 |
BC risk↑ |
|
BMPR1A |
JPS |
|
MDM4 |
BC risk↑ |
|
BRCA1 |
HBOC |
|
MLH1 |
Lynch syndrome |
MMR |
CDH1 |
Gastric/breast cancer |
|
MLH3 |
Lynch syndrome |
MMR |
CHEK2 |
BC risk↑ |
|
MRE11 |
BC risk↑ |
HR, NHEJ |
DDB1/XPE |
CS |
NER |
MSH2 |
Lynch syndrome |
MMR |
DDB2/XPE |
CS |
NER |
MSH3 |
Lynch syndrome |
MMR |
EPCAM |
Lynch syndrome |
|
MSH6 |
Lynch syndrome |
MMR |
ERCC1 |
XP |
NER |
MUTYH |
MYH-polyposis |
BER |
ERCC2/XPD |
XP, CS, TTD |
NER |
NBN |
BC risk↑ |
|
ERCC3/XPB |
XP, CS, TTD |
NER |
NBS1 |
BC risk↑ |
HR, NHEJ |
ERCC4 |
XP |
NER |
OGG1 |
BC risk↑ |
BER |
ERCC5 |
XP, CS |
NER |
PMS1 |
Lynch syndrome |
MMR |
ERCC6/CSB |
CS, |
NER |
PMS2 |
Lynch syndrome |
MMR |
ERCC8/CSA |
CS |
NER |
polymerase delta1 |
|
TLS |
FANCA |
Fanconi anemia |
HR |
polymerase epsilon |
|
TLS |
FANCB |
Fanconi anemia |
HR |
polymerase beta |
|
TLS |
FANCC |
Fanconi anemia |
HR |
polymerase eta |
|
TLS |
FANCD1/BRCA2 |
Fanconi anemia, HBOC |
HR |
polymerase kappa |
|
TLS |
FANCD2 |
Fanconi anemia |
HR |
PTEN |
Cowden syndrome |
|
FANCE |
Fanconi anemia |
HR |
RAD50 |
BC risk↑ |
HR, NHEJ |
FANCF |
Fanconi anemia |
HR |
RAD51 |
BC risk↑ |
HR |
FANCG/XRCC9 |
Fanconi anemia |
HR |
RAD51D |
BC risk↑ |
HR |
FANCI |
Fanconi anemia |
HR |
SMAD4 |
JPS |
|
FANCJ/BRIP1 |
Fanconi anemia, BC risk↑ |
HR |
STK11 |
Peutz-Jeghers syndrome |
|
FANCL/PHF9 |
Fanconi anemia |
HR |
TP53 |
Li frenmanii |
|
FANCM |
Fanconi anemia |
HR |
XPA |
XP |
NER |
FANCN/PALB2 |
Fanconi anemia, BC risk↑ |
HR |
XPC |
XP, CS, TTD |
NER |
FANCO/RAD51C |
Fanconi anemia, BC risk↑ |
HR |
XRCC2 |
BC risk↑ |
NHEJ |
FANCP/SLX4 |
Fanconi anemia, BC risk↑ |
HR |
XRCC3 |
BC risk↑ |
NHEJ |
FGFR2 |
BC risk↑ |
|
XRCC4 |
BC risk↑ |
NHEJ |
CS: Cockayne syndrome; FAP: familial adenomatous polyposis; HBOC: hereditary breast and ovarian cancer syndrome; JPS: juvenile polyposis syndrome; TTD: trichothiodystrophy; XP: xeroderma pigmentosum; BER: base excision repair; HR: homologous recombination; NER: nucleotide excision repair; NHEJ: nonhomologous DNA end joining; MMR: mismatch repair; TLS: translesion synthesis
RESULTS
Patients’ characteristics and performance of the illumina DNA sequencing
A total of 133 breast cancer patients were enrolled in this study. Their median age at diagnosis was 44 years; 41 were aged £ 35 years, 56 were 35–50 years, and 36 were > 50 years. Thirteen patients had metachronous breast cancer, five had a history of ovarian cancer, two had a history of colon cancer, and one each with a history of gastric cancer, nasopharyngeal cancer and multiple myeloma. A family history of breast cancer, ovarian cancer, prostate cancer, male breast cancer, or other malignancies was found in 97, 15, 7, 2, and 42 patients, respectively. The clinical characteristics of the patients are listed in Table 2.
Table 2: Characteristics of the study participants and comparison of patients with and without a pathogenic mutation
Variants |
Without |
With |
P value |
|---|---|---|---|
Patient number |
103 |
30 |
|
Median (range) |
44 (25–75) |
41 (29–60) |
0.309 |
≤ 35 years (patient no.) |
30 |
11 |
|
> 35–50 years (patient no.) |
45 |
11 |
|
> 50 years (patient no.) |
28 |
8 |
|
Personal history |
|||
Single/Metachronous breast cancer |
94/9 |
26/4 |
0.456 |
Ovarian cancer |
4 |
1 |
|
Other cancer |
4 |
1 |
|
Molecular type |
|
|
0.025 |
HR(+)Her2(−) |
68 |
16 |
|
HR(+)Her2(+) |
16 |
4 |
|
HR(−)Her2(+) |
7 |
0 |
|
TNBC |
12 |
10 |
|
Family cancer history |
|||
Breast cancer |
74 |
23 |
0.650 |
Ovarian cancer |
10 |
5 |
0.327 |
Prostate cancer |
3 |
4 |
0.024 |
Male breast cancer |
0 |
2 |
0.008 |
Other cancers |
31 |
11 |
0.510 |
Criteria of enrollment |
|
|
0.304 |
(1) |
23 |
6 |
|
(2) |
45 |
10 |
|
(3) |
34 |
15 |
|
(1) Early-onset breast cancer (age ≤ 35 years) or bilateral breast cancer (without family hsitory); (2) Breast cancer onset age ≤ 50 years and at least one first or second-degree relative with breast cancer or ovarian cancer; (3) Breast cancer onset after the age of 50 years, but with two relatives with breast cancer or one with ovarian cancer.
The average mean depth in the coding exons of the 68 genes was 195X (range: 2–348), respectively. The coding sequencing exons covered by at least 50 reads were 90.8% (range of depth per sample: 82.0%–94.5%).
Deleterious mutations
As shown in Figure 1A and 1B, 30 patients (22.6%) were found to have germline heterozygous deleterious mutations of known cancer susceptibility genes, 9 in BRCA1, 11 in BRCA2, 2 in RAD50, 2 in TP53 and one each in ATM, BRIP1, FANCI, MSH2, MUTYH, and RAD51C. The mutation prevalence for BRCA1 and BRCA2 in this cohort was 15.0%, indicating multiple gene sequencing increasing about 7.5% of the detection rate.
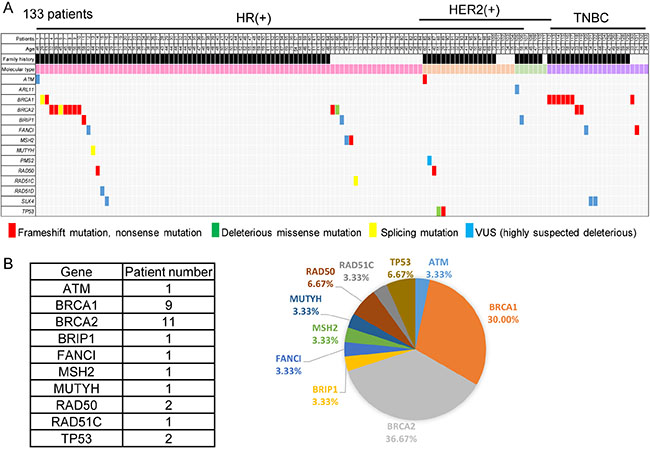
Figure 1: Mutation of predisposing genes in breast cancer patients with early-onset or a family history. (A) Each of the predisposing genes identified in the patients is listed on the left. A family history is shown in black. For the molecular types, HR(+)Her2(−) breast cancer is colored pink, HR(+)Her2(+) orange, HR(−)Her2(+) light green, and TNBC purple. (B) Twenty-five mutations of a predisposing gene were identified, 1 (4.0%) in ATM, 8 (32.0%) in BRCA1, 10 (40.0%) in BRCA2, and 1 each (4.0%) in BRIP1, FANCI, MSH2, RAD50, RAD51C, and TP53.
As shown in Table 3, analysis of mutation type showed that nonsense mutations were found in 8 patients, frameshift mutations in 15, missense mutations in 3, and mutations involving uncorrected splicing in 4. All of the nonsense and frameshift mutations were located in exons. One patient had a missense mutation of BRCA2 p.G2748D, which is reported to result in defective homologous recombination [8, 9], while others carried the heterozygous TP53 p.G245S or p.R248Q mutation, which result in a defective function of TP53 protein [10]. The intronic deletion of chr17:.g.41251910_41251919delGTAAAGAACA leads to deletion of a branch site in BRCA1 intron 6 [11], while the BRCA2 c.G631C mutation affects the donor site for splicing and the RAD51C c.905–2A > C and MUTYH c.934–2A > G mutation affects the recipient site; these three mutations, each found in 1 patient, are considered to cause uncorrected splicing and to be deleterious.
Table 3: Deleterious mutations identified in this cohort
Gene |
Mutation |
Transcript |
gDNA/cDNA |
Amino acid |
Reported/novel |
|---|---|---|---|---|---|
ATM |
frameshift deletion |
NM_000051 |
c.8434_8435delTC |
p.2812del |
Novel |
BRCA1 |
frameshift deletion |
NM_007294 |
c.1934delC |
p.S645fs |
Novel |
BRCA1 |
frameshift deletion |
NM_007294 |
c.1361delG |
p.S454fs |
Novel |
BRCA1 |
frameshift deletion |
NM_007294 |
c.470_471delCT |
p.S157fs |
Reported, rs80357887 |
BRCA1 |
splicing |
NG_005905 |
g.41251910_41251919delGTAAAGAACA |
|
Reported [30] |
BRCA1 |
frameshift deletion |
NM_007294 |
c.5470_5477delATTG GGCA |
p.I1824Dfs |
Reported, rs80357973 |
BRCA1 |
frameshift deletion |
NM_007294 |
c.3770_3771delAG |
p.E1257Gfs |
Reported, rs80357993 |
BRCA1 |
frameshift deletion |
NM_007294 |
c.3228_3229delAG |
p.G1077Afs |
Reported, rs80357635 |
BRCA1 |
nonsense mutation |
NM_007294 |
c.3607C > T |
p.R1203Ter |
Reported, rs62625308 |
BRCA1 |
frameshift deletion |
NM_007294 |
c.2679_2682delGAAA |
p.K893fs |
Reported, rs80357596 |
BRCA2 |
nonsense mutation |
NM_000059 |
c.8934delA |
p.E2981KfsTer7 |
Novel |
BRCA2 |
nonsense mutation |
NM_000059 |
c.6645delC |
pS2216PfsTer13 |
Novel |
BRCA2 |
nonsense mutation |
NM_000059 |
c.5574_5577delAATT |
p.I1859KfsTer3 |
Novel |
BRCA2 |
nonsense mutation |
NM_000059 |
c.5164_5165delAG |
p.S1722YfsTer4 |
Reported, rs80359490 |
BRCA2 |
missense mutation |
NM_000059 |
c.G8243A |
p.G2748D |
Reported, rs80359071 |
BRCA2 |
splicing |
NM_000059 |
c.G631C |
|
Reported, rs80358871 |
BRCA2 |
frameshift deletion |
NM_000059 |
c.6448delA |
p.K2150fs |
Novel |
BRCA2 |
frameshift deletion |
NM_000059 |
c.2806_2809del |
p.A938Pfs |
Reported, rs80359351 |
BRCA2 |
frameshift deletion |
NM_000059 |
c.8531_8532del |
p.E2844fs |
Novel |
BRCA2 |
frameshift insertion |
NM_000059 |
c.7407dupT |
p.T2469fs |
Novel |
BRCA2 |
frameshift deletion |
NM_000059 |
c.8323delA |
p.M2775CfsTer2 |
Novel |
BRIP1 |
nonsense mutation |
NM_032043 |
c.G1343A |
p.W448X |
Novel |
FANCI |
nonsense mutation |
NM_001113378 |
c.G568T |
p.E190X |
Novel |
MSH2 |
nonsense mutation |
NM_000251 |
c.C2785T |
p.R929X |
Reported, |
MUTYH |
splicing |
NM_001128425 |
c.934–2A > G |
|
Reported, rs77542170 |
RAD50 |
frameshift insertion |
NM_005732 |
c.2157dupA |
p.L719fs |
Novel |
RAD50 |
frameshift deletion |
NM_005732 |
c.2498_2499del |
p.Q833fs |
Novel |
RAD51C |
splicing |
NM_058216 |
c.905–2A > C |
|
Novel |
TP53 |
missense mutation |
NM_000546.5 |
c.733G > A |
p.G245S |
Reported, rs28934575 |
TP53 |
missense mutation |
NM_000546.5 |
c.743G > A |
p.R248Q |
Reported, rs11540652 |
Missense mutations and variants of uncertain significance
A total of 14717 missense mutations were identified among the 68 genes in the 133 patients. After searching for the database (http://www.ncbi.nlm.nih.gov/snp/) and bioinformatics analyses to evaluate the pathogenicity, most of the missense mutations were considered as benign variants and 12 missense mutations were classified as VUS with suspicion of being deleterious, averaging 0.09 VUS per participant (Table 4).
Table 4: Variants of uncertain significance strongly suspected of being deleterious mutations
gene |
rs number |
cDNA position |
amino acid |
ESP6500 |
1000 Genomes |
Patient number |
Polyphen2 |
SIFT |
GVGD align |
|---|---|---|---|---|---|---|---|---|---|
ARL11 |
- |
C467T |
A156V |
- |
- |
1 |
1 |
0 |
- |
ATM |
- |
A8450G |
Y2817C |
- |
- |
1 |
1 |
0.004 |
C65 |
BRIP1 |
- |
A2324G |
N775S |
- |
- |
1 |
1 |
0 |
- |
BRIP1 |
rs201869624 |
C2440T |
R814C |
- |
0.0005 |
1 |
1 |
0.019 |
- |
FANCI |
rs149243307 |
G286A |
E96K |
0.000154 |
0.00279553 |
2 |
1 |
0.004 |
- |
MSH2 |
- |
G1601T |
R534L |
- |
- |
1 |
1 |
0 |
C65 |
PMS2 |
rs182246929 |
C883T |
R295W |
- |
0.000199681 |
1 |
1 |
0 |
C65 |
RAD51D |
rs145309168 |
T932A |
I311N |
0.000231 |
0.0009 |
1 |
0.998 |
0 |
- |
SLX4 |
- |
G1457A |
R486H |
- |
- |
1 |
1 |
0 |
- |
SLX4 |
- |
T2453C |
L818P |
- |
- |
1 |
1 |
0 |
- |
SLX4 |
rs201622632 |
A2381T |
D794V |
- |
0.000199681 |
1 |
0.993 |
0 |
|
For variants identified by above programs, we search for the protein database to simulate the mutant structure for visually checking the potential deleterious impact to the protein. Three missense mutations, FANCI p.E96K, MSH2 p.R534L, and PMS2 p.R295W, were further evaluated by simulation using known protein templates [12–14]. As shown in Figure 2 and Supplementary Figure 4, the missense mutation FANCD2 E96K may disrupt the FANCI-FANCD2 complex and inability to carry out DNA interstrand cross-linking (Figure 2A and Supplementary Figure 4A and 4B). The MSH2 p. R534L may affect the mismatch repair by influencing the DNA attraction and interaction with MSH6 protein. The other missense mutation PMS2 p. R295W changes the polarity of the amino acid position 295 and may affect the ATP entry to the ATP binding pocket. However, whether these causes a functional defect requires further functional assays.
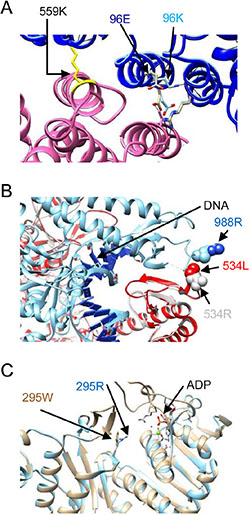
Figure 2: Structural analyses of three mutations. (A) Ribbon presentation of the FANCI E96K mutant structure (dark blue) superimposed on the structure of the wild type FANCI interacting with FANCD2 (FANCI, light blue; FANCD2, purple; pdb 3S4W) to form the ssDNA groove. This interface is maintained by Van Der Walls forces between FANCD2 HD2 (Leu614) and FANCI solenoid 1 (Leu92, Met94, and Leu95). The lysine substitution, with a longer sidechain, may disrupt the FANCI cap-solenoid 1 structure, leading to disturbance of the binding affinity for FANCD2 and influencing the ssDNA groove. This may result in defective function of the FANCI-FANCD2 complex and inability to carry out DNA interstrand cross-linking. FANCD2 Lys559 (yellow) is a mono-ubiquitination site. (B) Ribbon presentation of the MSH2 R534L structure (red) superimposed on that for the wild type MSH2-MSH6 complex (pdb: 2O8E, light gray; MSH6, light blue; DNA helix, dark blue), showing that MSH2 Arg534 is located at the clamping region, which is involved in DNA contact and matching of MSH6. Substitution of the arginine with leucine reduces the basic nature of this region, which may alter the attraction of DNA. This structure change also affects the interaction with MSH6. MSH2 p. R534L is highly suspected to have decreased function. (C) PMS2 p. R295W (khaki color) superimposed on the wild type PMS2 structure (pdb: 1EA6, light blue). This missense mutation changes the polar amino acid arginine to the non-polar tryptophan and is located nearby the entrance to the ATP binding pocket. However, whether this causes a functional defect requires further functional assays.
Association between genetic mutations and clinicopathologic characteristics
The mutation prevalence was 26.8% in the group aged £ 35 years, 19.6% in the group aged 35–50 years, and 22.2% in the group aged more than 50 years. Two patients with the deleterious mutation BRCA2 or RAD50 had male family members with breast cancer. There was no significant difference in the incidence of mutations between patients with a family history of female breast/ovarian cancer and early-onset breast cancer patients without a family history (23.1% vs. 20.7%, Pearson’s Chi-squared test p = 0.786). However, a significantly higher incidence of deleterious mutations was found in patients with a family history of prostate cancer (p = 0.024) or male breast cancer (p = 0.008) compared to those without such a family history.
When assessed by the molecular subtype of breast cancer, the mutation prevalence was 19.0% in patients with hormone receptor(HR, +)Her2(−) breast cancer, 20.0% in patients with HR(+)Her2(+) breast cancer, 0% in patients with only Her2(+) cancer, and 45.5% with TNBC (p = 0.024). Seven of the 9 BRCA1 mutations and the single FANCI mutation were in the TNBC group; 9 of the 11 BRCA2, 1 of the 2 RAD50 as well as the BRIP1, MSH2, MUTYH, and RAD51C mutations were in the hormone receptor (HR)(+)Her2(−) group, and the other RAD50, ATM, and TP53 mutations were in the HR(+)Her2(+) group.
Clinical relevance of the genetic results
Given the clinical significance of the pathogenic variants, patients carrying these pathogenic mutations are considered as high risk to develop cancer. Not only for BRCA mutation carriers, but we also provided genetic counseling for carriers of other 10 actionable mutations according to the NCCN guidelines and their cancer risk [15] (Supplementary Table 1).
One TNBC patient, who had received right breast conserving surgery 9 years previously, was referred for genetic tests due to developing left site metachronous breast cancer, which confirmed her germline BRCA1 mutation, so she decided to undergo bilateral mastectomy with reconstruction and one occult malignancy was found in the resected right breast tissue. In patients with a BRCA mutation, one received prophylactic contralateral mastectomy, two decided to undergo prophylactic oophorectomy, and all other BRCA mutation carriers preferred not to undergo prophylactic surgery and were advised to have an annual mammography with MRI of the breasts and transvaginal examination and the CA-125 test for prevention of gynecologic malignancy. Two patients with a TP53 mutation diagnosed as Li-Fraumeni syndrome were advised to have an annual mammography and a comprehensive whole body physical examination. Patients with the pathogenic mutation of MSH2 and MUTYH were advised to consider annual colonoscopy examination for patients and family members. The participant who had breast cancer carrying MUTYH mutation underwent colonoscopy, identifying five tubular adenomas that were removed. All patients with deleterious variants involving double-strand DNA repair (ATM, BRIP1, FANCI, RAD50, and RAD51C) were advised to have an annual screening of the breasts. In addition to the patients themselves, their family members were suggested to test whether they carrying deleterious mutations and mutation carriers were advised to receive screening.
DISCUSSION
Our study demonstrates a high value of a large gene panel for cancer-risk assessment using the NGS and is the first report of the use of this technique in the Asian-Pacific region. We found 30 (22.6%) pathogenic variants; 9 in BRCA1, 11 in BRCA2, and 9 in other genes. The mutation prevalence of BRCA1/2 (15.0%) in patients (Han Chinese) with early-onset or with a significant family history was similar to that reported in Western countries [16] and we found a 7.5% mutation rate of non-BRCA genes in women who tested negative for BRCA1/2 mutation. These data show that multiple gene sequencing increases the mutation detection rate compared to BRCA testing alone and that there is no ethnic difference in its application.
In this study, multiple gene testing identify 10 non-BRCA mutation carriers. This result is compatible with the suggestion from NCCN guideline that multiple gene sequencing may be more efficient and cost-effective for cancer-risk assessment for patients with a high probability of hereditary breast cancer [15]. Participants found to carry deleterious mutation are considered as high-risk cases to develop malignancy and targeted organ screening are advised for reducing cancer related-mortality. However, the cancer penetrance of non-BRCA genes may be intermediate, and there are no standardized screening guidelines. To manage the potential actionable mutations, we provide suggestions based on the biologic functions of these genes. For example, since mutations of ATM, BRIP1, FANCI, RAD50, and RAD51C affect double-strand DNA repair and may have a similar carcinogenic effect to BRCA genes [17, 18], we suggest screening should be started for these carriers. This would provide valuable information about screening for non-BRCA mutations and help with future genetic counseling and provide a rationale for a prospective study to elucidate the effect of this policy.
There are still unanswered questions about clinical multiple gene sequencing, such as the design of the gene panel and VUS interpretation. First, it is uncertain how many genes need to survey for testing hereditary breast cancer syndromes so that we do not know if this panel is suitable. The concept of the panel design was that gene mutations in the homologous recombination pathway may have a similar carcinogenic effect to BRCA mutations. A recent large study which investigated 17 breast cancer susceptibility genes in 1824 TNBC patients confirmed this rationale, as it showed deleterious mutations in 14 genes, mainly in genes involved in homologous recombination [19]. In addition, mutations in genes for other DNA repair pathway proteins, such as MSH2 (mismatch DNA repair) and XRCC1 (base excision repair) are reported to increase breast cancer risk [20, 21]. Germline mutation of tumor suppressor genes, for example TP53 and PTEN, causes hereditary cancers, including breast cancer [22]. Of the 68 genes selected for our panel, 8 were found to be deleterious mutations of non-BRCA predisposing genes. A previous large scale study evaluated 42 cancer predisposing genes in 198 patients who met the criteria for BRCA testing and found 16 pathogenic variants in 9 non-BRCA genes [23]. Combining the results of the above two studies and our own, deleterious mutations have been found in 21 non-BRCA genes (Supplementary Table 2). This suggests more studies are warranted to evaluate the selection of predisposing genes for clinical patients.
The increased numbers of VUS identified by multiple gene sequencing is another problem because they cause difficulty in risk assessment and may prompt anxiety and overtreatment for patients. It is therefore important to establish a rapid and robust method for reducing the number of VUS in clinical practice. It is efficient to use bioinformatics analysis to pre-screen the VUS to exclude obvious non-deleterious VUS and select possible deleterious VUSs for functional evaluation [24]. Potential deleterious VUS were selected by mutation frequency analysis (less than 1% in the general population) and a high score using mathematical prediction software (Polyphen2, SIFT and GVGD align). We also performed structural analysis to view whether the mutation affected the protein function. This strategy efficiently reduced the number of VUS, and only 12 strongly suspected of being deleterious were identified in the 68 sequenced genes. However, the result of bioinformatics analysis cannot be used in clinical diagnosis. In order to ensure that the uncertainty did not cause excessive anxiety for these patients, while, at the same time, informing them of the possible risk, they were well-informed and further functional assays are planned. With widespread use of multiple gene sequencing and the sharing of results in an open database (such dbSNP/clinvar at NCBI), the incidence of VUS will decline.
In summary, the 22% prevalence of mutations of cancer predisposing genes is a strong incentive to perform gene testing in these high-risk patients in early cancer screening. We demonstrates that multiple gene sequencing using the NGS is clinically applicable and is an effective method to increase detected rate of high-risk cases, rather than simply testing for BRCA1/2. Adequate targeted organ screening may help them to reduce the cancer-related mortality. However, a suitable guide for genetic counseling and better VUS interpretation of non-BRCA genes are needed.
PATIENTS AND METHODS
Patients
Patients had early-onset breast cancer or bilateral breast cancer or had a family history of breast or ovarian cancer. All patients had to meet one of the following criteria: (1) Early-onset breast cancer (age ≤ 35 years) or bilateral breast cancer; (2) Breast cancer onset age ≤ 50 years and at least one first or second-degree relative with breast cancer or ovarian cancer; (3) Breast cancer onset after the age of 50 years, but with two relatives with breast cancer or one with ovarian cancer [24]. The study was approved by the Institutional Review Board of the National Taiwan University Hospital (201308077RINA).
Designing of the gene panel
Most predisposing genes in hereditary cancer syndromes are tumor suppressor genes and DNA repair genes [25]. For example, the molecular mechanism of Lynch syndrome is a genetic defect in mismatch repair genes (MLH1, MLH3, MSH2, MSH3, MSH6, PMS1, PMS2, and EPCAM) [26]. Pathogenic mutations of BRCA1, BRCA2, and PALB2, which involved in homologous recombination for double-strand DNA repair, are linked to hereditary breast cancer, ovarian cancer, and prostate cancer [17]. Defects of the nucleotide excision repair genes cause xeroderma pigmentosum and predispose to skin cancer and lung cancer [27]. Mutations of the PTEN gene, a tumor suppressor gene, cause Cowden syndrome and predispose to breast cancer, follicular thyroid cancer, and endometrial cancer [22]. Following a literature review, we hypothesized that germline mutations of DNA repair genes and tumor suppressor genes might predispose to development of breast cancer. Based on this hypothesis, we selected 68 genes for the sequencing panel; these consisted of (i) DNA repair genes involved in homologous recombination, base excision repair, nucleotide excision repair, mismatch repair, nonhomologous DNA end joining, and translesion DNA synthesis; (ii) tumor suppressor genes, such as PTEN and TP53; and (iii) other genes predisposing to cancer development (Table 1). The overall region of the 68 gene was 4967005 bp (Supplementary Table 3).
Library preparation, NGS, and sequence mapping
After the patient had given written informed consent, genomic DNA (gDNA) was isolated from peripheral blood mononuclear cells using QIAGEN Genomic DNA extraction kits (Qiagen Inc., Valencia, CA USA) and its purity and concentration checked by agarose gel electrophoresis and the OD260/280 ratio, followed by Covaris fragmentation (Covaris, Inc., Woburn, MA, USA) and checking of the size of the fragmented gDNA using an Agilent Bioanalyzer 2100 (Agilent Technologies, Inc., Santa Clara, CA, USA) and a NanoDrop spectrophotometer (Thermo Fisher Scientific, Inc., Wilmington, DE, USA). Finally, the target gene library was generated using NimblGen capture kits (Roche NimblGen, INC.). The samples were then sequenced on an Illumina MiSeq that generated paired-end reads of 300 nucleotides.
The analysis algorithm is shown as Supplementary Figure 1. The raw sequencing data was aligned with the reference human genome (Feb. 2009, GRCh37/hg19) using Burrows-Wheeler Aligner software (version 0.5.9) [28]. SAMtools (version 0.1.18) was used to perform the necessary data conversion, sorting, and indexing [29]. For single nucleotide polymorphisms (SNPs) and small insertion/deletions (indels), Genome Analysis Toolkit (GATK; version 2.7) was used for variant calling by using Base/indel-calibrator and HaplotypeCaller. Genetic variants larger than 100 bp cannot be identified by GATK, so Pindel or Breakdancer software were used to find structural variants, such as large deletions, insertions, and duplications [30]. After variant calling, ANNOVAR was used for annotation of the genetic variants [31, 32]. Filtering of common variants of sequencing results was performed using dbSNP (version138), Exome sequencing Project 6500 (ESP6500), and the 1000 Genomes variant dataset (2014 Sep). Finally, all potential genetic variants identified in patients were confirmed by repeated PCR amplification of the indicated gene region(s) and direct Sanger sequencing.
In order to check the sensitivity and specificity of the NGS platform and bioinformatics algorithm, we checked the concordance of the results with prior 10 clinical sequencing, which contained large scale deletion and known BRCA1 genetic variants. The NGS results were fully concordant with the previous sequencing results (Supplementary Methods and Supplementary Figures 1–3).
Variant classification
The sequence variants were classified according to the IARC variant classification [33]. Large-scale deletion, frame-shift mutation, nonsense mutation, genetic variants associated with uncorrected splicing, and mutations affecting protein function demonstrated by functional analyses are considered as deleterious or pathogenic mutations. An allele frequency greater than 0.01 in the general population in the 1000 Genomes variant dataset (2014Sep) or ESP6500 database suggests a benign or likely benign genetic variant. Silent and intronic variants that do not affect splicing are also considered as benign or probably benign. Other variants, mainly missense mutations without known functional data, are considered to be VUS.
In order to reduce the number of VUS, we used the bioinformatics analysis to evaluate the potential pathogenicity, including PolyPhen2 [34], SIFT [35], and Align GVGD [36], as well as structural analysis. The mathematical prediction is mainly based on an evolutional approach examining the degree of cross-species amino acid conservation by sequence alignment and the properties of the amino acids. After the bioinformatics analysis, we defined VUSs that were suspected of being deleterious mutations as those that met the following two criteria: (1) a population frequency of less than 0.01 in the 1000 Genomes and ESP6500 databases and (2) a bioinformatics analysis result with a SIFT score less than 0.05 and a polyphen2 score greater than 0.95. Several variants were also analyzed using align GVGD software, the results had to be C65 (most likely to interfere with function).
For comparative structural modeling, the variant was simulated based on a known protein structure in the RCSB protein database (http://www.rcsb.org) [24]. For example, a BRCA1 mutant can be created using the SWISS-MODEL program based on the template of the human wild-type BRCA1 BRCT domain interacting with a BACH1 phosphorylated peptide (PDB code: 1T29) [37]. The 3-dimensional structure of the mutation was constructed using the UCSF Chimera program [38].
Statistical analysis
Descriptive statistics included medians, means, and standard deviations for continuous data. The X2 test and Fisher’s exact test were used to calculate the significance of differences between the means for two groups. All p values were 2-sided and p values less than 0.05 were considered significant.
ACKNOWLEDGMENTS AND FUNDING
This work was supported, in part, by research grants from the National Taiwan University Hospital (NTUH. 103-N2524 and NTUH. 104-N2901), the Ministry of Science and Technology (MOST 103–2314–B–002–005-MY2), and the Ministry of Health and Welfare (MOHW103–TDU–212–114002). The authors thank the A1 laboratory of the National Taiwan University Hospital for providing access to the Illumina Miseq instrument and the National Applied Research Laboratories for providing access to their high-performance computer to analyze the post-NGS data.
Authors’ contributions
P.-H. Lin, W.-H. Kuo, Y.-H. Ni, and C.-S. Huang conceived and designed the study. P.-H. Lin, H.-Y. Li, Y.-H. Yang, Y.-H. Hsu, and S.-C. Fan performed the experiments. W.-H. Kuo, A.-C. Huang, Y.-S. Lu, C.-H. Lin, S.-H. Kuo, M.-Y. Wang, C.-Y. Liu, Fiona T.-F. Cheng, M.-H. Yeh, K.-J. Chang, and C.-S. Huang collected samples and analyzed the clinical data. P.-H. Lin, H.-Y. Li, L.-Y. Li, P.-L. Chen and T.-Z. Yuan analyzed and interpreted the sequencing results. P.-H. Lin, Y.-H. Ni, C.-S. Huang performed writing, review, and/or revision of the manuscript. P.-H. Lin, A.-C. Huang, H.-Y. Li, Y.-H. Yang, Y.-H. Hsu and S.-C. Fan provided Administrative, technical, or material support. All authors contributed to the final version of the paper.
CONFLICTS OF INTEREST
The authors declare they have no known conflicts of interest in this work.
REFERENCES
1. DeSantis C, Naishadham D, Jemal A. Cancer statistics for African Americans, 2013. CA Cancer J Clin. 2013; 63:151–166.
2. Euhus D. Genetic Testing Today. Annals of surgical oncology. 2014; 21:3209–15.
3. Locatelli I, Lichtenstein P, Yashin AI. The heritability of breast cancer: a Bayesian correlated frailty model applied to Swedish twins data. Twin Res. 2004; 7:182–191.
4. Prokopcova J, Kleibl Z, Banwell CM, Pohlreich P. The role of ATM in breast cancer development. Breast Cancer Res Treat. 2007; 104:121–128.
5. Zheng Y, Zhang J, Niu Q, Huo D, Olopade OI. Novel germline PALB2 truncating mutations in African American breast cancer patients. Cancer. 2012; 118:1362–1370.
6. Han FF, Guo CL, Liu LH. The effect of CHEK2 variant I157T on cancer susceptibility: evidence from a meta-analysis. DNA Cell Biol. 2013; 32:329–335.
7. Antoniou AC, Casadei S, Heikkinen T, Barrowdale D, Pylkas K, Roberts J, Lee A, Subramanian D, De Leeneer K, Fostira F, Tomiak E, Neuhausen SL, Teo ZL, et al. Breast-cancer risk in families with mutations in PALB2. The New England journal of medicine. 2014; 371:497–506.
8. Spugnesi L, Balia C, Collavoli A, Falaschi E, Quercioli V, Caligo MA, Galli A. Effect of the expression of BRCA2 on spontaneous homologous recombination and DNA damage-induced nuclear foci in Saccharomyces cerevisiae. Mutagenesis. 2013; 28:187–195.
9. Farrugia DJ, Agarwal MK, Pankratz VS, Deffenbaugh AM, Pruss D, Frye C, Wadum L, Johnson K, Mentlick J, Tavtigian SV, Goldgar DE, Couch FJ. Functional assays for classification of BRCA2 variants of uncertain significance. Cancer research. 2008; 68:3523–3531.
10. Petitjean A, Achatz MI, Borresen-Dale AL, Hainaut P, Olivier M. TP53 mutations in human cancers: functional selection and impact on cancer prognosis and outcomes. Oncogene. 2007; 26:2157–2165.
11. Li SS, Tseng HM, Yang TP, Liu CH, Teng SJ, Huang HW, Chen LM, Kao HW, Chen JH, Tseng JN, Chen A, Hou MF, Huang TJ, et al. Molecular characterization of germline mutations in the BRCA1 and BRCA2 genes from breast cancer families in Taiwan. Human genetics. 1999; 104:201–204.
12. Joo W, Xu G, Persky NS, Smogorzewska A, Rudge DG, Buzovetsky O, Elledge SJ, Pavletich NP. Structure of the FANCI-FANCD2 complex: insights into the Fanconi anemia DNA repair pathway. Science. 2011; 333:312–316.
13. Edelbrock MA, Kaliyaperumal S, Williams KJ. Structural, molecular and cellular functions of MSH2 and MSH6 during DNA mismatch repair, damage signaling and other noncanonical activities. Mutation research. 2013; 743–744:53–66.
14. Guarne A, Junop MS, Yang W. Structure and function of the N-terminal 40 kDa fragment of human PMS2: a monomeric GHL ATPase. The EMBO journal. 2001; 20:5521–5531.
15. Daly MB, Pilarski R, Axilbund JE, Buys SS, Crawford B, Friedman S, Garber JE, Horton C, Kaklamani V, Klein C, Kohlmann W, Kurian A, Litton J, et al. Genetic/familial high-risk assessment: breast and ovarian, version 1.2014. J Natl Compr Canc Netw. 2014; 12:1326–1338.
16. Hall MJ, Reid JE, Burbidge LA, Pruss D, Deffenbaugh AM, Frye C, Wenstrup RJ, Ward BE, Scholl TA, Noll WW. BRCA1 and BRCA2 mutations in women of different ethnicities undergoing testing for hereditary breast-ovarian cancer. Cancer. 2009; 115:2222–2233.
17. Pennington KP, Swisher EM. Hereditary ovarian cancer: beyond the usual suspects. Gynecologic oncology. 2012; 124:347–353.
18. Jalal S, Earley JN, Turchi JJ. DNA repair: from genome maintenance to biomarker and therapeutic target. Clin Cancer Res. 2011; 17:6973–6984.
19. Couch FJ, Hart SN, Sharma P, Toland AE, Wang X, Miron P, Olson JE, Godwin AK, Pankratz VS, Olswold C, Slettedahl S, Hallberg E, Guidugli L, et al. Inherited mutations in 17 breast cancer susceptibility genes among a large triple-negative breast cancer cohort unselected for family history of breast cancer. J Clin Oncol. 2015; 33:304–311.
20. Engel C, Loeffler M, Steinke V, Rahner N, Holinski-Feder E, Dietmaier W, Schackert HK, Goergens H, von Knebel Doeberitz M, Goecke TO, Schmiegel W, Buettner R, et al. Risks of less common cancers in proven mutation carriers with lynch syndrome. J Clin Oncol. 2012; 30:4409–4415.
21. Smith TR, Liu-Mares W, Van Emburgh BO, Levine EA, Allen GO, Hill JW, Reis IM, Kresty LA, Pegram MD, Miller MS, Hu JJ. Genetic polymorphisms of multiple DNA repair pathways impact age at diagnosis and TP53 mutations in breast cancer. Carcinogenesis. 2011; 32:1354–1360.
22. Mester JL, Moore RA, Eng C. PTEN germline mutations in patients initially tested for other hereditary cancer syndromes: would use of risk assessment tools reduce genetic testing? The oncologist. 2013; 18:1083–1090.
23. Kurian AW, Hare EE, Mills MA, Kingham KE, McPherson L, Whittemore AS, McGuire V, Ladabaum U, Kobayashi Y, Lincoln SE, Cargill M, Ford JM. Clinical evaluation of a multiple-gene sequencing panel for hereditary cancer risk assessment. J Clin Oncol. 2014; 32:2001–2009.
24. Kuo WH, Lin PH, Huang AC, Chien YH, Liu TP, Lu YS, Bai LY, Sargeant AM, Lin CH, Cheng AL, Hsieh FJ, Hwu WL, Chang KJ. Multimodel assessment of BRCA1 mutations in Taiwanese (ethnic Chinese) women with early-onset, bilateral or familial breast cancer. Journal of human genetics. 2012; 57:130–138.
25. Knudson AG. Cancer genetics. American journal of medical genetics. 2002; 111:96–102.
26. Martin-Lopez JV, Fishel R. The mechanism of mismatch repair and the functional analysis of mismatch repair defects in Lynch syndrome. Familial cancer. 2013; 12:159–168.
27. Matakidou A, Eisen T, Fleischmann C, Bridle H, Houlston RS, Consortium G. Evaluation of xeroderma pigmentosum XPA, XPC, XPD, XPF, XPB, XPG and DDB2 genes in familial early-onset lung cancer predisposition. International journal of cancer Journal international du cancer. 2006; 119:964–967.
28. Li H, Durbin R. Fast, accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009; 25:1754–1760.
29. Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R, Genome Project Data Processing S. The Sequence Alignment/Map format and SAMtools. Bioinformatics. 2009; 25:2078–2079.
30. Mimori T, Nariai N, Kojima K, Takahashi M, Ono A, SatoY, Yamaguchi-Kabata Y, Nagasaki M. iSVP: an integrated structural variant calling pipeline from high-throughput sequencing data. BMC systems biology. 2013; 7 Suppl 6:S8.
31. McKenna A, Hanna M, Banks E, Sivachenko A, Cibulskis K, Kernytsky A, Garimella K, Altshuler D, Gabriel S, Daly M, DePristo MA. The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome research. 2010; 20:1297–1303.
32. Wang K, Li M, Hakonarson H. ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic acids research. 2010; 38:e164.
33. Plon SE, Eccles DM, Easton D, Foulkes WD, Genuardi M, Greenblatt MS, Hogervorst FB, Hoogerbrugge N, Spurdle AB, Tavtigian SV, Group IUGVW. Sequence variant classification and reporting: recommendations for improving the interpretation of cancer susceptibility genetic test results. Human mutation. 2008; 29:1282–1291.
34. Adzhubei IA, Schmidt S, Peshkin L, Ramensky VE, Gerasimova A, Bork P, Kondrashov AS, Sunyaev SR. A method and server for predicting damaging missense mutations. Nature methods. 2010; 7:248–249.
35. Kumar P, Henikoff S, Ng PC. Predicting the effects of coding non-synonymous variants on protein function using the SIFT algorithm. Nature protocols. 2009; 4:1073–1081.
36. Mathe E, Olivier M, Kato S, Ishioka C, Hainaut P, Tavtigian SV. Computational approaches for predicting the biological effect of p53 missense mutations: a comparison of three sequence analysis based methods. Nucleic acids research. 2006; 34:1317–1325.
37. Arnold K, Bordoli L, Kopp J, Schwede T. The SWISS-MODEL workspace: a web-based environment for protein structure homology modelling. Bioinformatics. 2006; 22:195–201.
38. Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, Ferrin TE. UCSF Chimera—a visualization system for exploratory research and analysis. Journal of computational chemistry. 2004; 25:1605–1612.