
INTRODUCTION
Wnt/β-catenin signaling is important for cancer progression, including tumor initiation, tumor growth, cell death, and metastasis [1-5]. β-Catenin is the central player in signal transduction of this pathway. In the absence of Wnt ligands, β-catenin levels are efficiently regulated by a supramolecular complex containing adenomatous polyposis coli (APC), axin, and glycogen synthetase kinase 3β (GSK3β). This complex promotes β-catenin phosphorylation and subsequent β-catenin degradation. Several components of the Wnt/β-catenin pathway have been identified as oncogenes or tumor suppressors (showing gain-of-function or loss-of-function mutations, respectively) in many types of human cancers [1-3]. Mutations in these genes are most evident in colorectal cancers. About 80% of all colorectal cancers contain loss-of function mutations in the tumor suppressor gene APC. Gain-of-function mutations in the oncogene CTNNB1 (β-catenin encoding gene) are present in approximately 10% of the colorectal cancers. The consequence of either APC inactivation or CTNNB1 activation mutations is the failure of proper β-catenin degradation leading to its cytosolic accumulation. The β-catenin then translocates into the nucleus where it interacts with T-cell factor/lymphoid enhancing factor (TCF/LEF) to induce the expression of downstream target genes [6, 7]. It is well established that aberrant activation of Wnt/β-catenin signaling is a necessary initiating event in the genesis of most colorectal cancers [6-8], and that the Wnt/β-catenin pathway has emerged as one of the most promising targets for colorectal cancer chemoprevention and treatment [1, 3, 9-11].
Among heterocyclic compounds, libraries of compounds possessing the quinazoline template in particular have attracted considerable attention in lead discovery efforts. Contributing factors for their desirability include: (a) facile synthetic access, (b) lead-like and drug-like attributes and (c) opportunities that the template provides for molecular manipulations for lead optimizations. The introduction of quinazolines such as lapatinib and gefitinib as kinase inhibitory drugs for cancer treatment exemplifies recent successes in optimization of quinazoline lead compounds into drugs [12-15].
In our efforts to discover novel inhibitors of Wnt/β-catenin signaling, we have identified a structurally related series of quinazolines as potent inhibitors in colorectal cancer cells harboring mutations in CTNNB1 or APC. Although a few quinazoline analogs have been reported in the literature as inhibitors of the Wnt/β-catenin signaling pathway [16-19], the set of compounds that we have identified is structurally distinct. Moreover, the quinazoline leads displayed potent anticancer activities with IC50 values between 4.9 and 17.4 μM for colorectal cancer cells, and the IC50 values are comparable to their inhibitory activities on Wnt/β-catenin signaling in colorectal cancer cells.
RESULTS
Quinazoline leads suppress Wnt/β-catenin signaling in colorectal cancer cells and inhibit cell proliferation
In support of lead discovery efforts on various biological targets, we synthesized a library of ~ 500 compounds containing the quinazoline template. From this library of quinazolines, we screened a set of 40 compounds using the Super8XTOPFlash Wnt reporter assay in colorectal cancer HCT116 cells (Supplementary Table S1). Interestingly, results from this screen led to the identification of two sets of structurally related quinazolines as inhibitors of Wnt/β-catenin signaling (Figure 1A and Table 1). Axin2 is a specific transcriptional target of Wnt/β-catenin signaling, and the expression level of axin2 is the signature of the activation of the Wnt/β-catenin pathway [20-23]. In addition, cyclin D1 and survivin have been identified as two key transcriptional targets of the Wnt/β-catenin pathway, and are critical for cancer cell proliferation and cell death [24-27]. Additional studies with these compounds revealed that they suppressed the expression of Wnt/β-catenin signaling targets axin2, cyclin D1 and survivin in both colorectal cancer HCT116 cells (harboring a CTNNB1 mutation) and SW480 cells (harboring an APC mutation) (Figure 1B), and displayed potent anticancer activities with IC50 values between 4.9 and 17.4 μM for HCT116, SW480 and SW620 cells (Table 1). The IC50 values of these quinazoline inhibitors are comparable to their inhibitory activities on Wnt/β-catenin in colorectal cancer cells (Table 1), suggesting that the anticancer activities of the quinazoline leads are associated with their inhibitory effects on Wnt/β-catenin signaling in colorectal cancer cells.
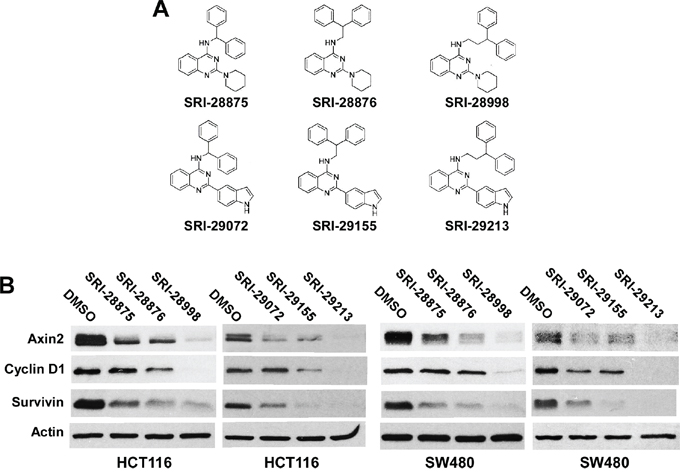
Figure 1: Effects of the six lead compounds on Wnt/β-catenin signaling in colorectal cancer cells. A. Structures of the six lead quinazolines. B. Cancer cells in 6-well plates were treated with test compounds at 10 μM (HCT116) or 20 μM (SW480) for 24 h. The levels of axin2, cyclin D1 and survivin were examined by Western blotting. All the samples were also probed with anti-actin antibody to verify equal loading.
Table 1: Activity data on the lead compounds
ID |
MWT |
CLogPa |
PSAa |
Wnt reporter activity in HCT116 cells, IC50 (μM) |
Cell viability, IC50 (μM) |
||
|---|---|---|---|---|---|---|---|
HCT116 |
SW480 |
SW620 |
|||||
SRI-28875 |
395 |
6.2 |
39.9 |
14.7 |
11.9 |
17.4 |
13.6 |
SRI-28876 |
409 |
6.9 |
39.9 |
6.6 |
7.9 |
13.5 |
10.2 |
SRI-28998 |
423 |
7.3 |
39.9 |
6.0 |
5.2 |
7.0 |
8.2 |
SRI-29072 |
427 |
6.7 |
48.7 |
22.3 |
8.8 |
12.3 |
6.0 |
SRI-29155 |
441 |
7.5 |
48.7 |
9.7 |
8.5 |
8.9 |
9.2 |
SRI-29213 |
455 |
7.8 |
48.7 |
8.9 |
4.9 |
5.8 |
7.1 |
SRI-31230 |
332 |
5.5 |
39.9 |
12.7 |
6.2 |
5.9 |
6.7 |
aCLogP and Polar Surface Area (PSA) were calculated using ChemBioDraw Ultra 2010.
The presence of diaryl group in SRI-28876 is not essential for its inhibitory effect on Wnt/β-catenin signaling in colorectal cancer cells
Compound SRI-31230 is a structurally simplified analog of SRI-28876 (Figure 2A). We found that SRI-31230, similarly to SRI-28876, was able to suppress the Super8XTOPFlash Wnt reporter activity in HCT116 cells (Figure 2B), and inhibited the expression of Wnt/β-catenin signaling targets axin2, cyclin D1 and survivin in HCT116 cells and SW480 cells (Figure 2C). Moreover, SRI-31230 exhibited similar potent anticancer activity with IC50 values between 5.9 and 6.7 μM for HCT116, SW480 and SW620 cells (Figure 2D and Table 1). Together, these results indicate that the presence of diaryl group such as the diphenylethylamino moiety present in compound SRI-28876 may not be essential for the inhibitory effect of the quinazoline leads on Wnt/β-catenin signaling in colorectal cancer cells.
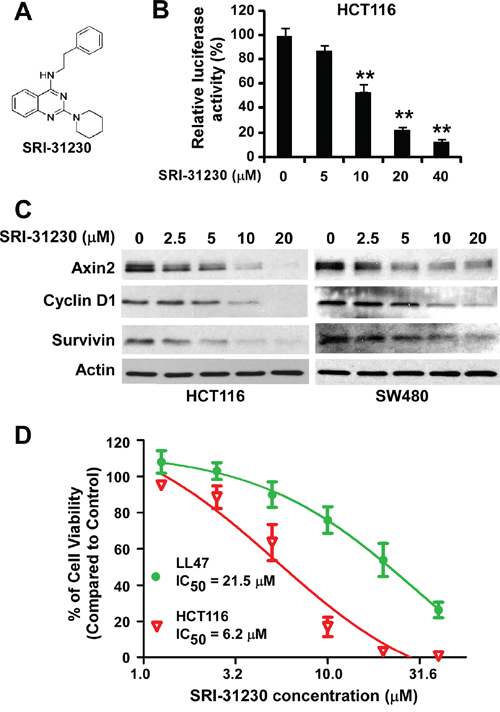
Figure 2: Effects of the lead compound SRI-31230 on Wnt/β-catenin signaling in colorectal cancer cells. A. The structure of SRI-31230. B. Cancer cells in 24-well plates were transiently transfected with Super8XTOPFlash construct and β-galactosidase-expressing vector in each well. After being incubated for 24 h, cells were treated with SRI-31230 at the indicated concentrations for 24 h. The luciferase activity was then measured 24 h later with normalization to the activity of the β-galactosidase. Values are averages of three determinations with the SD indicated by error bars. *P < 0.05, **P < 0.01 versus corresponding control cells without SRI-31230 treatment. C. Colorectal cancer HCT116 and SW480 cells in 6-well plates were treated with SRI-31230 at the indicated concentrations for 24 h. The levels of axin2, cyclin D1 and survivin were examined by Western blotting. All the samples were also probed with anti-actin antibody to verify equal loading. D. HCT116 and LL47 cells in 96-well plates were treated with SRI-31230 for 72 h. Cell viability was measured by the Cell TiterGlo assay. All the values are the average of triplicate determinations with the SD indicated by error bars.
SRI-31230 is able to selectively inhibit colorectal cancer cell proliferation
To further characterize our quinazoline lead SRI-31230, we performed cell proliferation/viability assay in normal lung fibroblast line LL47, and found that SRI-31230 was less cytotoxic against LL47 cells than colorectal cancer cells (Figure 2D), suggesting that SRI-31230 is able to selectively kill Wnt-dependent cancer cells.
Quinazoline inhibitors suppress Wnt/β-catenin signaling by acting on the downstream elements of the pathway
Uncomplexed cytosolic β-catenin (free β-catenin) can translocate to the cell nucleus to activate the transcription factors of the TCF/LEF family, leading to the transcription of Wnt target genes. To identify the site of action of the quinazoline leads in Wnt/β-catenin signaling, we determined the β-catenin levels after the treatment with the lead compounds. It was found that the levels of cytosolic β-catenin and total cellular β-catenin were not significantly changed in response to treatment with the six quinazoline leads at 20 μM for 24 h (Figure 3A) or SRI-31230 at 2.5 to 20 μM for 24 h (Figure 3B). These results indicate that these quinazoline inhibitors suppress Wnt/β-catenin signaling by acting on the downstream elements of the pathway.
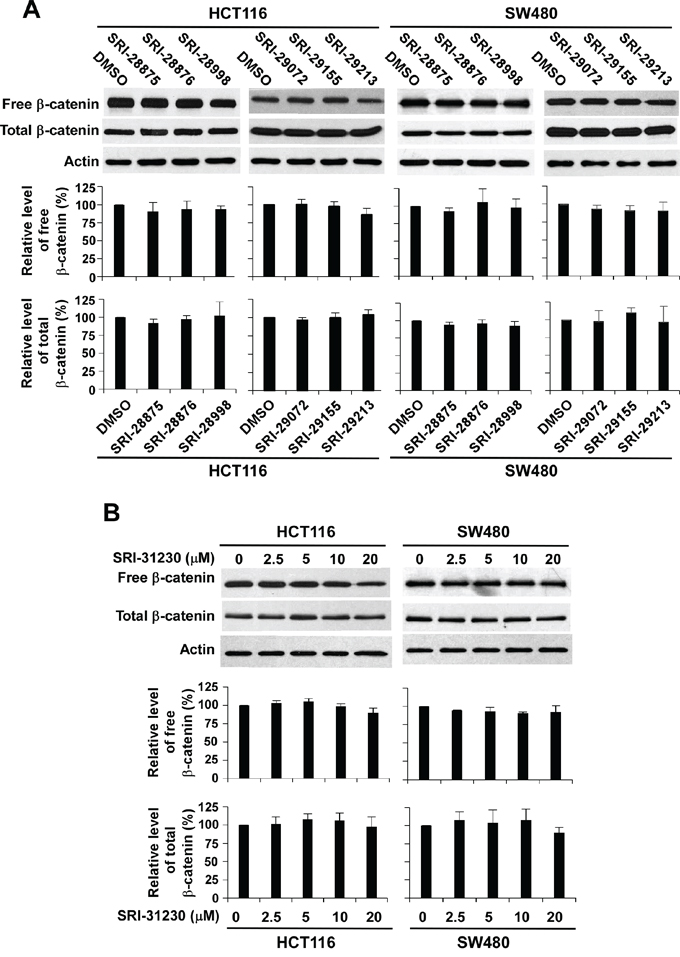
Figure 3: Effect of the seven lead compounds on levels of cytosolic free β-catenin and total β-catenin signaling in colorectal cancer cells. A. Cancer cells in 6-well plates were treated with the test compounds at 10 μM (HCT116) or 20 μM (SW480) for 24 h. B. Cancer cells in 6-well plates were treated with SRI-31230 at the indicated concentrations for 24 h. The levels of cytosolic free β-catenin and total β-catenin signaling were examined by Western blotting. All the samples were also probed with anti-actin antibody to verify equal loading. The bands were quantified by densitometry using the Image J software. The intensity of the total cellular β-catenin bands was quantified relative to actin. The results represented in the histograms are shown as the mean ± SD and are the average of three independent experiments.
Inhibition of Wnt/β-catenin signaling by the quinazoline lead compounds are responsible for their anticancer activity
Compound SRI-20039 is a 2-phenylquinazoline derivative that was ineffective in blocking Wnt/β-catenin signaling in colorectal cancer cells. As a negative control, SRI-20039 displayed very weak activity against colorectal cancer cell proliferation (Figure 4). These results further suggest that inhibition of Wnt/β-catenin signaling by the quinazoline lead compounds are responsible for their anticancer activity.
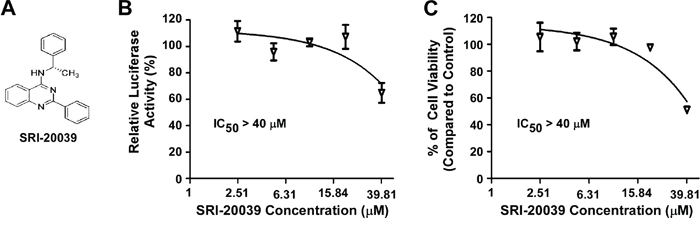
Figure 4: Effect of the compound SRI-20039 on Wnt/β-catenin signaling in colorectal cancer cells and cancer cell viability. A. The structure of SRI-20039. B. HCT116 cells in 24-well plates were transiently transfected with Super8XTOPFlash construct and β-galactosidase-expressing vector in each well. After being incubated for 24 h, cells were treated with SRI-20039 at the indicated concentrations for 24 h. The luciferase activity was then measured 24 h later with normalization to the activity of the β-galactosidase. Values are averages of three determinations with the SD indicated by error bars. C. HCT116 cells in 96-well plates were treated with SRI-20039 for 72 h. Cell viability was measured by the Cell TiterGlo assay. All the values are the average of triplicate determinations with the SD indicated by error bars.
DISCUSSION
Aberrant activation of the Wnt/β-catenin signaling pathway is a necessary initiating event in the genesis of most colorectal cancers [6-8]. Accumulating evidence strongly suggests that novel inhibitors of Wnt/β-catenin signaling could emerge as chemotherapeutic agents for treatment of colorectal cancer [1, 3-5, 9-11]. Despite tremendous efforts in the past decade, most of the compounds reported in the literature including the clinical candidate LGK974 [28, 29], target upstream components of Wnt/β-catenin signaling [4, 5]. Such compounds may not be effective against colorectal cancers in which the Wnt/β-catenin pathway is deregulated by mutations in downstream components APC and CTNNB1. Recent studies suggest that such targeting of downstream signaling could provide therapeutic advantages over targeting upstream signaling components [30, 31].
Among heterocyclic compounds, libraries of compounds possessing the quinazoline template in particular have attracted considerable attention in lead discovery efforts [12-15]. In the present study, we describe the discovery of a series of novel quinazolines possessing the drug-like quinazoline framework that inhibit the Wnt/β-catenin pathway in colorectal cancer cells. Our studies revealed that the quinazoline compounds repress Wnt/β-catenin signaling without altering the level of β-catenin protein in colorectal cancer cells harboring mutations in CTNNB1 or APC, suggesting that they act on the downstream elements of the pathway. Importantly, we also found that a structurally related quinazoline lacking inhibitory effect on Wnt/β-catenin signaling was unable to suppress colorectal cancer cell proliferation, suggesting that the anticancer activity of the quinazoline lead compounds is associated with their inhibitory effect on Wnt/β-catenin signaling. However, additional studies are needed to dissect molecular mechanisms underlying the quinazoline-mediated inhibition of Wnt/β-catenin signaling in detail.
In summary, we have discovered a structurally related series of quinazolines as potent inhibitors of Wnt/β-catenin signaling in colorectal cancer cells. Of interest is the fact that structure–activity relationships (SAR) are discernible in the profile of this initial set of compounds, thus providing an opportunity to follow-up the SAR trend in a systematic way to enhance potency, selectivity, and drug-like characteristics in the future. The Wnt/β-catenin pathway is critical to uncontrolled cell proliferation in a number of tumor types, particularly colorectal cancer. Therefore, the quinazoline leads identified in the current effort are potentially promising candidates for development as novel chemotherapeutic agents for colorectal cancer.
MATERIALS AND METHODS
Materials
All of the test compounds were synthesized at Southern Research Institute. Details of the chemical synthesis will be published elsewhere. Plasmid pGST-E-cadherin was provided by Dr. Gail Johnson (University of Rochester, Rochester, NY). The Super8XTOPFlash luciferase construct was provided by Dr. Randall T. Moon (University of Washington, Seattle, WA). The β-galactosidase-expressing vector was obtained from Promega. Rabbit monoclonal anti-axin2 (#2151) was purchased from Cell Signaling Technology. Rabbit polyclonal anti-cyclin D1 (#04-221) was from EMD Millipore. Mouse monoclonal anti-survivin (D-8) (sc-17779) was from Santa Cruz Biotechnology. Mouse monoclonal anti-β-catenin (#61054) was from BD Biosciences. Mouse monoclonal anti-actin (#A2228) was from Sigma. Peroxidase labeled anti-mouse and anti-rabbit secondary antibodies and ECL system were purchased from Amersham Life Science. The luciferase and β-galactosidase assay systems were from Promega. Tissue culture media, fetal bovine serum (FBS), and plastic-ware were obtained from Life Technologies, Inc. Proteinase inhibitor cocktail Complete™ was obtained from Boehringer Mannheim.
Cell culture
All cell lines were obtained from ATCC and grown under standard cell culture conditions at 37°C in a humidified atmosphere with 5% CO2. The cells were cultured in DMEM medium containing 10% of FBS, 2 mM of L-glutamine, 100 units/ml of penicillin, and 100 μg/ml of streptomycin.
Western blotting
Colorectal cancer cells in 6-well plates were lysed in 0.5 ml of lysis buffer (phosphate-buffered saline containing 1% Triton X-100 and 1 mM PMSF) at 4°C for 10 min. Equal quantities of protein were subjected to SDS-PAGE under reducing conditions. Following transfer to immobilon-P transfer membrane, successive incubations with a primary antibody, and a horseradish peroxidase-conjugated secondary antibody were carried out for 60-120 min at room temperature. The immunoreactive proteins were then detected using the ECL system. Films showing immunoreactive bands were scanned by HP Scanjet 5590. Blots were quantitated by densitometry using Image J Software (NIH, Bethesda, MD, USA) and normalized to a housekeeper marker β-actin.
Cytosolic free β-catenin analysis with GST-E-cadherin binding assay
The GST-E-cadherin binding assay was carried out exactly as previously described [32, 33]. Briefly, cells in six-well plates were lysed in 0.5 ml of lysis buffer at 4°C for 10 min, and extracts were clarified by centrifugation at 18,000×g for 2 min. One hundred micrograms of total cell extracts were incubated with Sepahrose beads bound to the GST-E-cadherin. The GST-E-cadherin Sepahrose beads were prepared as described [34]. After 1 h of incubation at 4°C, the Sepharose beads were collected by centrifugation at 10,000×g for 1 min, washed 2 times with lysis buffer containing 0.1% SDS and 0.5% bovine serum albumin and 2 times with PBS buffer, and boiled in SDS sample buffer containing β-mercaptoethanol. The supernatants were subjected to SDS–PAGE and Western blotting with the β-catenin antibody.
Luciferase reporter assay for Wnt/β-catenin signaling
Colorectal cancer cells were plated into 24-well plates. After overnight culture, the cells were transiently transfected with the Super8XTOPFlash luciferase construct and β-galactosidase-expressing vector. After 24 h incubation, cells were treated with the test compounds at the indicated concentrations. Cells were then lysed 24 h later and both luciferase and β-galactosidase activities were determined. The luciferase activity was normalized to the β-galactosidase activity.
Cell viability assay
Colorectal cancer cells were seeded into 96-well tissue culture treated microtiter plates at a density of 5000 cells/well. DMEM containing 10% FBS was used as assay media. After 24 h incubation, the cells were treated with the test compounds at the indicated concentrations for 72 h. Cell viability was measured by the Cell Titer Glo Assay, which is a luminescent assay that is an indicator of live cells as a function of metabolic activity and ATP content.
Statistics
Statistical analyses were performed using Student’s unpaired t-test. Data were presented as mean ± SD. Differences at P < 0.05 were considered statistically significant.
ACKNOWLEDGMENTS
We are grateful to Dr. Gail Johnson (University of Rochester) for providing GST-E Cadherin cDNA, and Dr. Randall T. Moon (University of Washington) for providing the Super8XTOPFlash luciferase construct. This work was supported by grants from the National Institutes of Health RO1CA124531 and R21CA182056 and Alabama Innovation Fund.
CONFLICTS OF INTEREST
The authors declare no conflict of interest.
REFERENCES
1. Anastas JN, Moon RT. WNT signalling pathways as therapeutic targets in cancer. Nat Rev Cancer. 2013; 13:11-26.
2. Clevers H, Nusse R. Wnt/beta-catenin signaling and disease. Cell. 2012; 149:1192-1205.
3. Polakis P. Drugging Wnt signalling in cancer. EMBO J. 2012; 31:2737-2746.
4. Kahn M. Can we safely target the WNT pathway? Nat Rev Drug Discov. 2012; 13:513-532.
5. Madan B, Virshup DM. Targeting Wnts at the source—new mechanisms, new biomarkers, new drugs. Mol Cancer Ther. 2015; 14:1087-1094.
6. Morin PJ, Sparks AB, Korinek V, Barker N, Clevers H, Vogelstein B, Kinzler KW. Activation of beta-catenin-Tcf signaling in colon cancer by mutations in beta-catenin or APC. Science. 1997; 275:1787-1790.
7. Kinzler KW, Vogelstein B. Lessons from hereditary colorectal cancer. Cell. 1996; 87:159-170.
8. Dow LE, O’Rourke KP, Simon J, Tschaharganeh DF, van Es JH, Clevers H, Lowe SW. Apc Restoration Promotes Cellular Differentiation and Reestablishes Crypt Homeostasis in Colorectal Cancer. Cell. 2015; 161:1539-1552.
9. de Sousa EM, Vermeulen L, Richel D, Medema JP. Targeting Wnt signaling in colon cancer stem cells. Clin Cancer Res. 2011; 17:647-653.
10. Burgess AW, Faux MC, Layton MJ, Ramsay RG. Wnt signaling and colon tumorigenesis—a view from the periphery. Exp Cell Res. 2011; 317:2748-2758.
11. Serafino A, Moroni N, Zonfrillo M, Andreola F, Mercuri L, Nicotera G, Nunziata J, Ricci R, Antinori A, Rasi G, Pierimarchi P. WNT-pathway components as predictive markers useful for diagnosis, prevention and therapy in inflammatory bowel disease and sporadic colorectal cancer. Oncotarget. 2014; 5:978-992. doi: 10.18632/oncotarget.1571.
12. Petrov KG, Zhang YM, Carter M, Cockerill GS, Dickerson S, Gauthier CA, Guo Y, Mook RA, Jr., Rusnak DW, Walker AL, Wood ER, Lackey KE. Optimization and SAR for dual ErbB-1/ErbB-2 tyrosine kinase inhibition in the 6-furanylquinazoline series. Bioorg Med Chem Lett. 2006; 16:4686-4691.
13. Morphy R. Selectively nonselective kinase inhibition: striking the right balance. J Med Chem. 2010; 53:1413-1437.
14. Barker AJ, Gibson KH, Grundy W, Godfrey AA, Barlow JJ, Healy MP, Woodburn JR, Ashton SE, Curry BJ, Scarlett L, Henthorn L, Richards L. Studies leading to the identification of ZD1839 (IRESSA): an orally active, selective epidermal growth factor receptor tyrosine kinase inhibitor targeted to the treatment of cancer. Bioorg Med Chem Lett. 2001; 11:1911-1914.
15. Wu X, Li M, Qu Y, Tang W, Zheng Y, Lian J, Ji M, Xu L. Design and synthesis of novel Gefitinib analogues with improved anti-tumor activity. Bioorg Med Chem. 2010; 18:3812-3822.
16. Chen Z, Venkatesan AM, Dehnhardt CM, Dos Santos O, Delos Santos E, Ayral-Kaloustian S, Chen L, Geng Y, Arndt KT, Lucas J, Chaudhary I, Mansour TS. 2,4-Diamino-quinazolines as inhibitors of beta-catenin/Tcf-4 pathway: Potential treatment for colorectal cancer. Bioorg Med Chem Lett. 2009; 19:4980-4983.
17. Mao Y, Lin N, Tian W, Han X, Han X, Huang Z, An J. Design, synthesis, and biological evaluation of new diaminoquinazolines as beta-catenin/Tcf4 pathway inhibitors. J Med Chem. 2012; 55:1346-1359.
18. Dehnhardt CM, Venkatesan AM, Chen Z, Ayral-Kaloustian S, Dos Santos O, Delos Santos E, Curran K, Follettie MT, Diesl V, Lucas J, Geng Y, Dejoy SQ, Petersen R, et al. Design and synthesis of novel diaminoquinazolines with in vivo efficacy for beta-catenin/T-cell transcriptional factor 4 pathway inhibition. J Med Chem. 2010; 53:897-910.
19. Nencini A, Pratelli C, Quinn JM, Salerno M, Tunici P, De Robertis A, Valensin S, Mennillo F, Rossi M, Bakker A, Benicchi T, Cappelli F, Turlizzi E, et al. Structure-activity relationship and properties optimization of a series of quinazoline-2,4-diones as inhibitors of the canonical Wnt pathway. Eur J Med Chem. 2015; 95:526-545.
20. Yan D, Wiesmann M, Rohan M, Chan V, Jefferson AB, Guo L, Sakamoto D, Caothien RH, Fuller JH, Reinhard C, Garcia PD, Randazzo FM, Escobedo J, et al. Elevated expression of axin2 and hnkd mRNA provides evidence that Wnt/beta-catenin signaling is activated in human colon tumors. Proc Natl Acad Sci U S A. 2001; 98:14973-14978.
21. Jho EH, Zhang T, Domon C, Joo CK, Freund JN, Costantini F. Wnt/beta-catenin/Tcf signaling induces the transcription of Axin2, a negative regulator of the signaling pathway. Mol Cell Biol. 2001; 22:1172-1183.
22. Leung JY, Kolligs FT, Wu R, Zhai Y, Kuick R, Hanash S, Cho KR, Fearon ER. Activation of AXIN2 expression by beta-catenin-T cell factor. A feedback repressor pathway regulating Wnt signaling. J Biol Chem. 2002; 277:21657-21665.
23. Lustig B, Jerchow B, Sachs M, Weiler S, Pietsch T, Karsten U, van de Wetering M, Clevers H, Schlag PM, Birchmeier W, Behrens J. Negative feedback loop of Wnt signaling through upregulation of conductin/axin2 in colorectal and liver tumors. Mol Cell Biol. 2002; 22:1184-1193.
24. Shtutman M, Zhurinsky J, Simcha I, Albanese C, D’Amico M, Pestell R, Ben-Ze’ev A. The cyclin D1 gene is a target of the beta-catenin/LEF-1 pathway. Proc Natl Acad Sci U S A. 1999; 96:5522-5527.
25. Tetsu O, McCormick F. Beta-catenin regulates expression of cyclin D1 in colon carcinoma cells. Nature. 1999; 398:422-426.
26. Kim PJ, Plescia J, Clevers H, Fearon ER, Altieri DC. Survivin and molecular pathogenesis of colorectal cancer. Lancet. 2003; 362:205-209.
27. Ma H, Nguyen C, Lee KS, Kahn M. Differential roles for the coactivators CBP and p300 on TCF/beta-catenin-mediated survivin gene expression. Oncogene. 2005; 24:3619-3631.
28. Liu J, Pan S, Hsieh MH, Ng N, Sun F, Wang T, Kasibhatla S, Schuller AG, Li AG, Cheng D, Li J, Tompkins C, Pferdekamper A, et al. Targeting Wnt-driven cancer through the inhibition of Porcupine by LGK974. Proc Natl Acad Sci U S A. 2013; 110:20224-20229.
29. Jiang X, Hao HX, Growney JD, Woolfenden S, Bottiglio C, Ng N, Lu B, Hsieh MH, Bagdasarian L, Meyer R, Smith TR, Avello M, Charlat O, et al. Inactivating mutations of RNF43 confer Wnt dependency in pancreatic ductal adenocarcinoma. Proc Natl Acad Sci U S A. 2013; 110:12649-12654.
30. Gonsalves FC, Klein K, Carson BB, Katz S, Ekas LA, Evans S, Nagourney R, Cardozo T, Brown AM, DasGupta R. An RNAi-based chemical genetic screen identifies three small-molecule inhibitors of the Wnt/wingless signaling pathway. Proc Natl Acad Sci U S A. 2011; 108:5954-5963.
31. Watanabe K, Dai X. Winning WNT: race to Wnt signaling inhibitors. Proc Natl Acad Sci U S A. 2011; 108:5929-5930.
32. Lu W, Kim KA, Liu J, Abo A, Feng X, Cao X, Li Y. R-spondin1 synergizes with Wnt3A in inducing osteoblast differentiation and osteoprotegerin expression. FEBS Lett. 2008; 582:643-650.
33. Lu W, Lin C, Roberts MJ, Waud WR, Piazza GA, Li Y. Niclosamide suppresses cancer cell growth by inducing Wnt co-receptor LRP6 degradation and inhibiting the Wnt/beta-catenin pathway. PLoS One. 2011; 6:e29290.
34. Bafico A, Gazit A, Wu-Morgan SS, Yaniv A, Aaronson SA. Characterization of Wnt-1 and Wnt-2 induced growth alterations and signaling pathways in NIH3T3 fibroblasts. Oncogene. 1998; 16:2819-2825.