
INTRODUCTION
Autophagy refers to an evolutionarily conserved, multi-step lysosomal degradation process, in which a cell degrades long-lived proteins and damaged organelles [1, 2]. Macroautophagy (hereafter referred to as autophagy) is a major, regulated catabolic mechanism that involves the delivery of cytoplasmic cargo sequestered inside double-membrane vesicles to the lysosome [3], and is linked to several pathological processes, including cancers and neurodegenerative diseases [4, 5]. Autophagy is considered as a physiological mechanism that may serve as a means for temporary survival and provide a way to recycle macromolecules as an alternative energy source. If cellular stress leads to continuous or excessively induced autophagy, cell death will ensue [6, 7]. Several studies have reconciled the opposing roles of autophagy in diseases and demonstrated that autophagy can act as either a guardian or executioner [8–11]. The different roles of autophagy depend on disease stages, surrounding cellular environment, and attempted therapeutic interventions [12–15].
Accordingly, targeting autophagy may be a promising therapeutic strategy for treatment of diseases. Recently, accumulating evidence has revealed many small-molecule compounds that can activate or inhibit autophagy and may therefore have remarkable therapeutic potential on diseases, such as cancers and neurodegenerative diseases [16, 17]. However, targeting autophagy for drug development remains in its infancy. Here, we designed a webserver called Autophagic Compound-Target Prediction (ACTP) (http://actp.liu-lab.com/) that can predict potential autophagic targets and relevant pathways for given compound (s). The ACTP webserver will help to explore more possible autophagy-activating or autophagy-inhibiting drugs for potential therapeutic purposes.
RESULTS
Potential autophagic targets in ACTP
We collected 430 target proteins (199 were reviewed, and 231 were unreviewed) from Uniprot. The basic protein information included the accession number, full name, and molecular function. We identified GO annotation terms and related diseases information from the Online Mendelian Inheritance in Man (OMIM) database. Crystal structures of 86 targets were downloaded from the Protein Data Bank (PDB) and saved as 948 PDB files. Six hundred and fifteen PDB structures were selected as available structures for docking, and their PDB codes were also saved (Table 1 and Supplementary Table S1). We prefer to retain PDBs that have both high resolution and complete amino acid motif covering active sites and compound-binding sites. For those PDBs have better resolution and worst coverage than a second one, we will firstly consider the sequence integrity (that means the PDB entry has a complete amino acid motif covering active sites and compound-binding sites) rather than resolution; thus, we will retain PDBs have complete amino acids motif even if they have relative lower resolution. For those PDBs have lower resolution and worst coverage, we will perform homology modeling instead of using these PDBs. These proteins were assigned to the following 9 functional target groups: antigen, enzyme, kinase, receptor, protein binding, nucleotide binding, transcription factor binding, tubulin binding, and others (Figure 1). For reviewed proteins without available crystal structures and the BLAST result with the template shown > 30% similarity, we performed homology modeling to generate predicted structures using Discovery Studio 3.5 (Supplementary Table S2 and Supplementary Table S3). 109 protein sequence files were downloaded from Uniprot and saved in FASTA format. Then, the templates were found using BLAST. Finally, the structures of 109 targets were generated and saved in PDB format. In addition, the PDB files were available from the corresponding PDB number hyperlink on the result page of the webserver. For example, the mTOR file contains the following information: the accession number, “P42345”; the name, “Serine/threonine-protein kinase mTOR (Mechanistic target of rapamycin)”; and the function, “Serine/threonine protein kinase is a central regulator of cellular metabolism, growth and survival in response to hormones, growth factors, nutrients, energy, and stress signals. mTOR can activate or inhibit the phosphorylation of at least 800 proteins directly or indirectly.” The PDB accession number for mTOR is 4dri, and the PDB file was downloaded from http://rcsb.org. Discovery Studio 3.5 was then used to prepare the PDB file for docking by deleting water, cleaning the protein, and detecting the interaction site.
Table 1: Structure-based autophagic targets (Reviewed)
Uniprot ID |
Uniprot Accession |
Number of PDBs |
Target type |
Uniprot ID |
Uniprot Accession |
Number of PDBs |
Target type |
|---|---|---|---|---|---|---|---|
P53_HUMAN |
P04637 |
74 |
antigen |
5HT2B_HUMAN |
P41595 |
2 |
receptor |
DRA_HUMAN |
P01903 |
55 |
antigen |
NR1D1_HUMAN |
P20393 |
5 |
receptor |
2B11_HUMAN |
P04229 |
34 |
antigen |
B2CL1_HUMAN |
Q07817 |
38 |
protein binding |
2B14_HUMAN |
P13760 |
9 |
antigen |
RGS19_HUMAN |
P49795 |
1 |
others |
DRB5_HUMAN |
Q30154 |
4 |
antigen |
BAD_HUMAN |
Q92934 |
1 |
protein binding |
ITB4_HUMAN |
P16144 |
8 |
antigen |
BCL2_HUMAN |
P10415 |
11 |
protein binding |
DRB3_HUMAN |
P79483 |
3 |
antigen |
TAU_HUMAN |
P10636 |
6 |
protein binding |
DPB1_HUMAN |
P04440 |
3 |
antigen |
S100A9_HUMAN |
P06702 |
3 |
others |
DPA1_HUMAN |
P20036 |
3 |
antigen |
PARK7_HUMAN |
Q99497 |
26 |
others |
PRKN2_HUMAN |
O60260 |
4 |
enzyme |
S100A8_HUMAN |
P05109 |
3 |
others |
SIR2_HUMAN |
Q8IXJ6 |
5 |
enzyme |
SQSTM_HUMAN |
Q13501 |
5 |
protein binding |
CATD_HUMAN |
P07339 |
4 |
enzyme |
MLP3B_HUMAN |
Q9GZQ8 |
6 |
others |
ATG4B_HUMAN |
Q9Y4P1 |
4 |
enzyme |
PA1B2_HUMAN |
P68402 |
1 |
others |
ATG4A_HUMAN |
Q8WYN0 |
2 |
enzyme |
NBR1_HUMAN |
Q14596 |
8 |
others |
UBP13_HUMAN |
Q92995 |
2 |
enzyme |
GOPC_HUMAN |
Q9HD26 |
8 |
others |
HDAC6_HUMAN |
Q9UBN7 |
3 |
enzyme |
SPT5H_HUMAN |
O00267 |
5 |
protein binding |
TIGAR_HUMAN |
Q9NQ88 |
1 |
enzyme |
MT3_HUMAN |
P25713 |
2 |
others |
SIR1_HUMAN |
Q96EB6 |
4 |
enzyme |
RAB1A_HUMAN |
P62820 |
8 |
nucleotide binding |
ATG3_HUMAN |
Q9NT62 |
1 |
enzyme |
BNIP3_HUMAN |
Q12983 |
2 |
protein binding |
HERC1_HUMAN |
Q15751 |
2 |
enzyme |
PSN1_HUMAN |
P49768 |
1 |
protein binding |
EPM2A_HUMAN |
O95278 |
2 |
enzyme |
GBRL1_HUMAN |
Q9H0R8 |
2 |
tubulin binding |
MK14_HUMAN |
Q16539 |
103 |
kinase |
GATA4_HUMAN |
P43694 |
1 |
transcription factor binding |
AKT1_HUMAN |
P31749 |
13 |
kinase |
IF16_HUMAN |
Q16666 |
4 |
others |
CDK5_HUMAN |
Q00535 |
5 |
kinase |
BECN1_HUMAN |
Q14457 |
5 |
others |
DAPK1_HUMAN |
P53355 |
21 |
kinase |
TBC14_HUMAN |
Q9P2M4 |
1 |
others |
KPYM_HUMAN |
P14618 |
16 |
kinase |
FOXO1_HUMAN |
Q12778 |
2 |
others |
MK08_HUMAN |
P45983 |
20 |
kinase |
MLP3A_HUMAN |
Q9H492 |
2 |
others |
DAPK2_HUMAN |
Q9UIK4 |
7 |
kinase |
ACBD5_HUMAN |
Q5T8D3 |
1 |
others |
DAPK3_HUMAN |
O43293 |
4 |
kinase |
CISD2_HUMAN |
Q8N5K1 |
2 |
others |
ABL2_HUMAN |
P42684 |
7 |
kinase |
NPC1_HUMAN |
O15118 |
1 |
others |
AAKB2_HUMAN |
O43741 |
8 |
kinase |
ZC12A_HUMAN |
Q5D1E8 |
1 |
ion binding |
AAPK2_HUMAN |
P54646 |
5 |
kinase |
MLP3C_HUMAN |
Q9BXW4 |
2 |
others |
PIM2_HUMAN |
Q9P1W9 |
2 |
kinase |
ATG13_HUMAN |
O75143 |
1 |
others |
AAKG1_HUMAN |
P54619 |
3 |
kinase |
WDFY3_HUMAN |
Q8IZQ1 |
1 |
others |
STK11_HUMAN |
Q15831 |
1 |
kinase |
GBRL2_HUMAN |
P60520 |
1 |
protein binding |
LRRK2_HUMAN |
Q5S007 |
2 |
kinase |
ATG12_HUMAN |
O94817 |
2 |
others |
PK3C3_HUMAN |
Q8NEB9 |
5 |
kinase |
A16L1_HUMAN |
Q676U5 |
3 |
others |
AAKB1_HUMAN |
Q9Y478 |
1 |
kinase |
ATG5_HUMAN |
Q9H1Y0 |
3 |
others |
TBK1_HUMAN |
Q9UHD2 |
4 |
kinase |
VPS51_HUMAN |
Q9UID3 |
1 |
others |
AAPK1_HUMAN |
Q13131 |
1 |
kinase |
FBX7_HUMAN |
Q9Y3I1 |
1 |
others |
ULK1_HUMAN |
O75385 |
1 |
kinase |
LYRIC_HUMAN |
Q86UE4 |
1 |
transcription factor binding |
ABL1_HUMAN |
P00519 |
30 |
kinase |
TCPR1_HUMAN |
Q7Z6L1 |
1 |
others |
MTOR_HUMAN |
P42345 |
12 |
kinase |
STX17_HUMAN |
P56962 |
1 |
others |
GBRAP_HUMAN |
O95166 |
7 |
receptor |
VAMP8_HUMAN |
Q9BV40 |
1 |
others |
OPTN_HUMAN |
Q96CV9 |
3 |
receptor |
SNP29_HUMAN |
O95721 |
1 |
others |
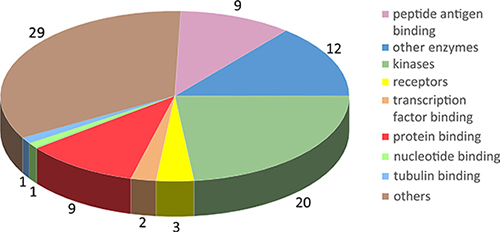
Figure 1: Molecular classification of potential autophagic targets. Herein, 86 targets with crystal structures were assigned to the following groups: peptide antigen binding, other enzymes, kinases, receptors, transcription factor binding, protein binding, nucleotide binding, tubulin binding and others. Groups are marked with different colors. The number of targets contained in each group is displayed in the pie chart.
Target prediction and pathways for autophagy-activating or autophagy- inhibiting compounds
The docking results were shown in a table of target proteins and include the top 10 docking scores and the P-value of the score. In this study, we used rapamycin and LY294002 as an example. We found that mTOR has the best binding score with rapamycin, 151.062; while PI3K has the best binding score with LY294002, 162.157 (Figure 2A). Rapamycin and LY294002 bound perfectly in the mTOR and PI3K inhibitor pocket, respectively. Moreover both of them had a similar conformation in different docking algorithms (Figure 2B).
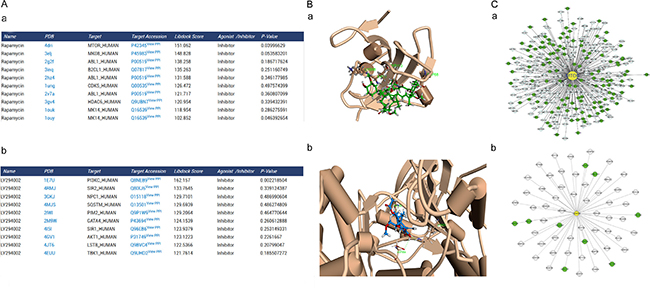
Figure 2: Predicted autophagic targets and related pathways from ACTP result page. (A) The output pages for (a) rapamycin (CAS number: 53123–88–9) and (b) LY294002 (CAS number: 154447–36–6) were displayed. The dock scoring table displayed on the page shows the top 10 possible targets according to the dock score. (B) Snapshots of (a) rapamycin docked with mTOR and (b) LY294002 docked with PI3K (the highest scored target in the result table) were also shown. (C) Users can also see the target PPI network graphically by clicking the view PPI hyperlink in the superscript of the target Uniprot AC, (a) mTOR, (b) PI3K. The PPI network is displayed by the cytoscape web plugin.
To construct the global human PPI network based on PrePPI, we collected 24,035 human protein accession numbers from Uniprot and saved them in a text file. The results page was created using PHP with accession numbers from the text file and request interaction data. All the information were imported into MySQL database. As a result, 1.1 million PPIs were collected to construct the global network. We generated the ARP subnetwork and created the autophagy subnetwork, which contains 93,532 PPIs. The autophagy subnetwork data were also available on the “download” page. It was combined with PHP and MySQL web2.0 to generate a dynamic graphical network. For example, mTOR has 308 PPIs and PI3K has 60 PPIs in their cytoscape visual network. The target was in the center and was marked in yellow. The targets of high and low credible level ARP were displayed in green and gray, respectively (Figure 2C). Moreover we carried out an additional blind-test with 15 compounds with known targets (Supplementary Table S4). And, the result showed the predict targets with the significant Libdock score of 14 compounds contained the real target of the compound (10 compounds’ real target had the top Libdock score). Only one compound’s predicted targets result did not contain the real target. Thus, it suggests that ACTP has a reliable accuracy for predict the target of autophagy-activating or autophagy-inhibiting compounds.
Webserver development
Based upon the above-mentioned results, we developed the ACTP webserver to offer a simple interface for users to submit compounds and predict their potential targets. For the first time, a user should create an account to submit compounds and view the results though the user interface. We support either CAS number or mol/mol2 files as the submission format. When submitted, the compound is sent to the Discovery Studio to perform virtual screening with ARPs. We would notify the users when the job is complete. If a user has submitted any compound previously, the webserver will display the results directly. The results page includes not only the docking scores and a snap-shot, but important information about the target proteins. For example, if rapamycin was submitted, the input can be either 53123–88–9 or a mol/mol2 file. Then, the task and process stage are shown on a user dashboard. When the task is complete, the user can click “VIEW” to see the score table, target information and PPIs (Figure 3). Currently, a due to the limitation of server is that a user could only submit 5 tasks per day.

Figure 3: The ACTP user interface. The simple user interface enables task submitting by inputting the compound name, CAS number, or by uploading a mol/mol2 formatted file. The pre-input example and tips help users become accustomed to the input format.
DISCUSSION
Autophagy may possess the contradictory functions because in addition to being primarily a survival mechanism, it can also lead to type II programmed cell death (type II PCD) under certain conditions [18, 19]. Our understanding of the relationship between autophagy and diseases has benefited from the availability of rapamycin and other autophagy-activating or autophagy-inhibiting agents, such as tamoxifen, chloroquine and resveratrol, which have been approved for potential clinical use [20, 21]. Several small-molecule compounds have been reported to activate or inhibit autophagy in different diseases. However, few of them has been purposefully designed as autophagic activators or inhibitors. Thus, it is urgent to find an avenue for rapidly screening and identifying a wealth of possible autophagy-activating or autophagy-inhibiting compounds without labor-intensive experiments.
Herein, we designed the Autophagic Compound-Target Prediction (ACTP) (http://actp.liu-lab.com/) webserver, which can predict a specific compound’s autophagic targets and relevant pathways. We used a series of bioinformatics methods to assemble together for solving only one problem. When a given compound has been submitted, we could correspondingly predict its potential autophagic targets and relevant pathways for therapeutic purposes. There are some key points for our methods to construct the ACTP webserver. Firstly, the autophagy-related protein (ARP) data were collected and classified into different subclasses for accurate target identification. Secondly, autophagic targets and their relevant pathways were provided for possible mechanism analysis. Lastly but most importantly, autophagic targets and relevant pathways could be predicted according to given compounds by structure-based docking technique. Interestingly, the ACTP could provide a clue for themselves prone to activators or inhibitors of these predicted autophagic targets.
Of course, there are some limitations for ACTP. The binding sites of the reviewed targets are directly imported from PDB files; thus, ACTP cannot predict the binding of compounds to other pockets. Moreover, for many proteins, the structures are not available yet, and the homology modeling is not sufficiently accurate for prediction. Therefore, ACTP cannot currently confirm the results for these proteins. However, with a growing number of protein structures to be analyzed, we will continue to add some new protein structures, which could be used for accurate target prediction. Moreover, we plan to update the latest data every two months, enabling continuous improvement of the webserver and processes.
In summary, Autophagic Compound-Target Prediction (ACTP) may provide a basis for the rapid prediction of potential targets and relevant pathways for a given autophagy-modulating compound. These results will help a user to assess whether the submitted compound can activate or inhibit autophagy by targeting which kind of key autophagic proteins and also has a therapeutic potential on diseases. Importantly, ACTP will also provide a clue to guide further experimental validation on one or more autophagy-activating or autophagy-inhibiting compounds for future drug discovery.
MATERIALS AND METHODS
Target protein information collection and preprocessing
Autophagy-related proteins (ARPs) included genes or proteins that are associated with the Gene Ontology (GO) term “autophagy” (http://www.geneontology.org) [22]. The useful information on ARPs was extracted from Uniprot database (http://www.uniprot.org). Autophagic targets were classified based on their molecular functions. Targets were assigned to 9 functional target groups. Cluster analysis was deemed to be relevant if the over-represented functional groups contained at least 5 targets. Moreover, functional clustering was performed by the DAVID functional annotation tool (http://david.abcc.ncifcrf.gov/). The functional categories were GO terms that is related to molecular function (MF). Specific docking strategies were employed for different groups. For instance, kinase binding pockets were focused on the active sites, while antigens were focused on their interaction surfaces with other proteins. It may reduce the number of false positive results in in silico analysis [23, 24]. Also, the active sites were divided into two groups by their position for predicting if a compound is an inhibitor or agonist of the target [25, 26]. Taken a kinase as an example, inhibitors targeting active sites for kinases, the agonists were chose screening sites for according to the different regulation mechanism of kinases. For example, the AMPK agonist named compound 991 is envisaged to strengthen the interaction between the kinase and carbohydrate-binding module (CBM) to protect a major proportion of the active enzyme against dephosphorylation [25]. If available, ARP crystal structures were downloaded from the Protein Data Bank (PDB) website (www.rcsb.org) [27]. For proteins that have more than one PDB entry, we screened the PDB files by resolution and sequence length until only one PDB entry remained. For proteins without crystal structure, we created homology modeling from sequences using Discovery Studio 3.5 (Accelrys, San Diego, California, United States). Sequence data were downloaded from Uniprot in FASTA format, and the templates were identified using BLASTP (Basic Local Alignment Search Tool) (http://blast.ncbi.nlm.nih.gov). ARPs were divided into two credibility levels (high and low) according to their review status in Uniprot.
Protein-protein interaction (PPI) network construction
The cellular biological processes of specific targets were predicted based on the global architecture of PPI network. We used an in-house PHP script to construct Autophagy interaction networks (AINs) based on the global PPI network were from PrePPI database (https://bhapp.c2b2.columbia.edu/PrePPI) [28] and Uniprot accession numbers. The ARP accession numbers were used to generate an AIN subnetwork. PPIs with different credible levels were marked in ACTP. The interactions were recorded in SQL format, which could be imported into MySQL database. The Cytoscape web plug-in was used to visualize the interactions [29].
Webserver generation
The ACTP webserver was generated with Linux, Apache, MySQL and PHP. Users can inquiry the database with their private data through the web interface. Currently, all major web browsers are supported. The processed results will be returned to the website. Web 2.0 technologies (i.e., JavaScript/AJAX and CSS functionalities) enables interactive data analysis. For example, based on AJAX and flash, ARP interaction networks can be indexed by accession numbers and visualized on the web page with Cytoscape web.
Reverse docking
Reverse docking is the virtual screening of targets by given compounds based on various scoring functions. Reverse docking allows a user to find the protein targets which can bind to a particular ligand [30]. We performed reverse docking with Libdock protocol [31], which is a high-throughput docking algorithm that positions catalyst-generated compound conformations in protein hotspots. Before docking, force fields including energies and forces on each particle in a system were applied with CHARMM [32] to define the positional relationships among atoms and to detect their energy. The binding site image consists of a list of non-polar hot spots, and positions in the binding site that were favorable for a non-polar atom to bind. Polar hot spot positions in the binding site were favorable for the binding of a hydrogen bond donor or acceptor. For Libdock algorithm, a given ligand conformation was put into the binding site as a rigid body and the atoms of the ligand were matched to the appropriate hot spots. The conformations were ranked using the following score:
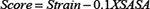
where SASA is the solvent accessible surface area of a particular conformation measured in Å2 and the strain is in units of kcal/mol. A match then determines the unique rigid body transformation that minimizes the following equation:
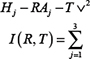
where R is a 3 × 3 rotation matrix and T is a translation vector. A single conformation can produce up to 10,000 matches. Thus, in the final stage, the matches were clustered after ranking, and only the top 25–100 entries were chosen for the next stage. Two values were reported as the measures of success of the two scores in pulling out active compounds. The first step of these measurements is the enrichment factor and is given by the following equation:
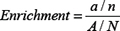
N is the number of compounds in the library; A is the number of active compounds; and a is the number of active compounds in the top n compounds. The second value is the statistical significance of the enrichment and is given by the following equation:
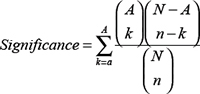
N, A, n, and a are defined as the enrichment. Comparing with other dock algorithm Libdock is quicker and support concurrent computation as well. Moreover protocol in Discovery Studio can be used to perform the Libdock algorithm for a series of proteins. Thus, Libdock is a suitable algorithm for high throughput identifying the various conformations of compounds within a receptor. Target-compound interactions were further optimized by molecular dynamics using CHARMM and Clean Geometry function of Discovery Studio. A T-test was also add to analyze the significance of the Libdock score of each target.
ACKNOWLEDGMENTS AND FUNDING
We are grateful to Prof. Canhua Huang (Sichuan University) and Prof. Yan Cheng (Central South University) for their critical reviews on this manuscript. This work was supported by grants from the National 973 Basic Research Program of China (No. 2013CB911300), and the National Natural Science Foundation of China (Nos. 81402496, 81473091 and 81260628).
CONFLICTS OF INTEREST
We declare that we have no conflicts of interest.
REFERENCES
1. Klionsky DJ, Emr SD. Autophagy as a regulated pathway of cellular degradation. Science. 2000; 290:1717–1721.
2. Klionsky DJ. Autophagy: from phenomenology to molecular understanding in less than a decade. Nat Rev Mol Cell Biol. 2007; 8:931–937.
3. Levine B, Kroemer G. Autophagy in the pathogenesis of disease. Cell. 2008; 132:27–42.
4. Wang SY, Yu QJ, Zhang RD, Liu B. Core signaling pathways of survival/death in autophagy-related cancer networks. Int J Biochem Cell Biol. 2011; 43:1263–1266.
5. Liu B, Wen X, Cheng Y. Survival or death: disequilibrating the oncogenic and tumor suppressive autophagy in cancer. Cell Death Dis. 2013; 4:e892.
6. Ylä-Anttila P, Vihinen H, Jokitalo E, Eskelinen EL. Monitoring autophagy by electron microscopy in Mammalian cells. Methods Enzymol. 2009; 452:143–164.
7. Wen X, Wu JM, Wang FT, Liu B, Huang CH. Deconvoluting the role of reactive oxygen species and autophagy in human diseases. Free Radical Bio Med. 2013; 65:402–410.
8. Rabinowitz JD, White E. Autophagy and metabolism. Science. 2010; 330:1344–1348.
9. Yang Z, Klionsky DJ. Eaten alive: a history of macroautophagy. Nat Cell Biol. 2010; 12:814–822.
10. Mizushima N, Levine B, Cuervo AM, Klionsky DJ. Autophagy fights disease through cellular self-digestion. Nature. 2008; 451:1069–1075.
11. Huang J, Klionsky DJ. Autophagy and human disease. Cell. 2007; 6:1837–1849.
12. Rubinsztein DC, Gestwicki JE, Murphy LO, Klionsky DJ. Potential therapeutic applications of autophagy. Nat Rev Drug Discov. 2007; 6:304–312.
13. Levine B, Klionsky DJ. Development by self-digestion: molecular mechanisms and biological functions of autophagy. Dev Cell. 2004; 6:463–477.
14. Hannigan AM, Gorski SM. Macroautophagy: the key ingredient to a healthy diet? Autophagy. 2009; 5:140–151.
15. Liu JJ, Mou L, Yu JY, Liu B, Bao JK. Targeting apoptotic and autophagic pathways for cancer therapeutics. Cancer Lett. 2011; 300:105–114.
16. Tong XP, Chen Y, Zhang SY, Xie T, Tian M, Guo MR, Kasimu R, Ouyang L, Wang JH. Key autophagic targets and relevant small-molecule compounds in cancer therapy. Cell Prolif. 2015; 48:7–16.
17. Sarkar S, Rubinsztein DC. Small molecule enhancers of autophagy for neurodegenerative diseases. Mol Biosyst. 2008; 4:895–901.
18. Cheng Y, Ren X, Hait WN, Yang JM, Therapeutic targeting of autophagy in disease: biology and pharmacology. Pharmacol Rev. 2013; 65:1162–1197.
19. Ouyang L, Shi Z, Zhao S, Wang FT, Zhou TT, Liu B, Bao JK. Programmed cell death pathways in cancer: a review of apoptosis, autophagy and programmed necrosis. Cell Prolif. 2012; 45:487–498.
20. Liu B, Cheng Y, Yang JM. Autophagy in Health and Disease, Chapter 10, 161–180. Drug Discovery in the Autophagy Pathways. 2013. Roberta Gottlieb (Ed.), Academic Press, Elsevier.
21. Li J, Li S, Zhang L, Ouyang L, Liu B. Deconvoluting the complexity of autophagy and Parkinson›s disease for potential therapeutic purpose. Oncotarget. 2015; 6:40480–40495. doi: 10.18632/oncotarget.5803.
22. The Gene Ontology (GO) project. Gene Ontology Consortium Nucleic Acids Res. 2006; 34:D322–D326.
23. Huang da W, Sherman BT, Lempicki RA. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc. 2009; 4:44–57.
24. Huang da W, Sherman BT, Lempicki RA. Bioinformatics enrichment tools: paths toward the comprehensive functional analysis of large gene lists. Nucleic Acids Res. 2009; 37:1–13.
25. Bing X, Matthew JS, David C, Nicola JB, Lesley FH, Elizabeth U, Bhakti RP, Richard B, Philip AW, Stefan H, Fabrizio G, Stephen RM, DC, Steven JG. Structural basis of AMPK regulation by small molecule activators. Nat Commun. 2013; 4:3017.
26. März AM, Fabian AK, Kozany C, Bracher A, Hausch F. Large FK506-Binding Proteins Shape the Pharmacology of Rapamycin. Mol Cell Biol. 2013; 33:1357–1367.
27. Parasuraman S. Protein data bank. J Pharmacol Pharmacother. 2012; 3:351–352.
28. Zhang C, Donald P, Lei D, Li Q, Yu S, Chan AT, Brygida B, Celine L, Domenico A, Hunter T, Maniatis T, Califano A. Structure-based prediction of protein-protein interactions on a genome-wide scale Barry Honig. Nature. 2013; 490:556–560.
29. Keiichiro O, Barry D, Trey I. Cytoscape tools for the web age: D3.js and Cytoscape.js exporters Version 2. F1000 Res. 2014; 3:143.
30. Kothiwale, Jens M, Edward WL. Computational Methods in Drug Discovery Gregory Sliwoski. Sandeepkumar Pharmacol Rev. 2014; 66:334–395.
31. Diller DJ, Merz KM. High throughput docking for library design and library prioritization. Jr Proteins 2001; 43:113–124.
32. Brooks BR, Brooks CL, MacKerell AD III, Nilsson L, Petrella RJ, Roux B, Won Y, Archontis G, Bartels CS. CHARMM: The Biomolecular Simulation Program, J Comput Chem. 2010; 30:1545–1614.