
Introduction
RNA splicing is a crucial post-transcription process that regulates gene expression and increases genomic diversity.[1] Recently, somatic mutations involving core components of the RNA splicing machinery were detected in myelodysplastic syndrome (MDS).[2, 3] Mutations of the splicing factor (SF) genes occur most frequently in SRSF2, U2AF1, and SF3B1, but also ZRSR2, U2AF2, SF1, SF3A1, PRPF40B, PRPF8 and LUC7L2,[2] with a strong genotype and phenotype association.[4-6] Some of these mutations showed prognostic relevance in MDS, however, discrepancies exist among different studies.[7]
Although acute myeloid leukemia (AML) and MDS share some similar mutations in the pathogenesis, differences exist. For example, NPM1, CEBPA and FLT3 mutations that are common in AML occur infrequently in MDS, and the opposite is true for EZH2 and SF mutations. The reported incidence of SF mutations in AML varied from 4.5% to 12.5% depending on the patient population selected, the regions of SF genes screened, and the methods used.[2, 8-11] Due to lower incidence of SF mutations and small cohorts studied, the association of SF mutations with clinic-biologic features and their prognostic implication in de novo AML patients remain unclear. Further, there has been no report in literature concerning the stability of SF mutations in AML.
In this study, we assessed the clinical implication of SF mutations in 500 unselected adults with de novo AML and their interactions with other 18 genetic alterations. Longitudinal follow-ups of the status of SF mutations during the clinical course were also performed in 163 patients to investigate the stability and pathogenic role of these mutations in AML. To the best of our knowledge, this is the first study to address the prognostic implication of SF mutations in a large cohort of patients with de novo AML. We found that SF mutation was an independent poor-risk factor for overall survival (OS) and disease-free survival (DFS) in these patients.
Results
SF mutations in patients with de novo AML
Mutations of the RNA splicing machinery genes were identified in 54 (10.8%) of 500 patients, including 12 (2.4%) with SF3B1 mutations, 15 (3.0%) with U2AF1 mutations, and 27 (5.4%) with SRSF2 mutations, respectively (Table 1). SF mutations in all these patients were heterozygous. None had two of these three SF mutations at the same time, suggesting these three mutations were mutually exclusive (Table 1, Figure 1 and Supplementary Table 4).
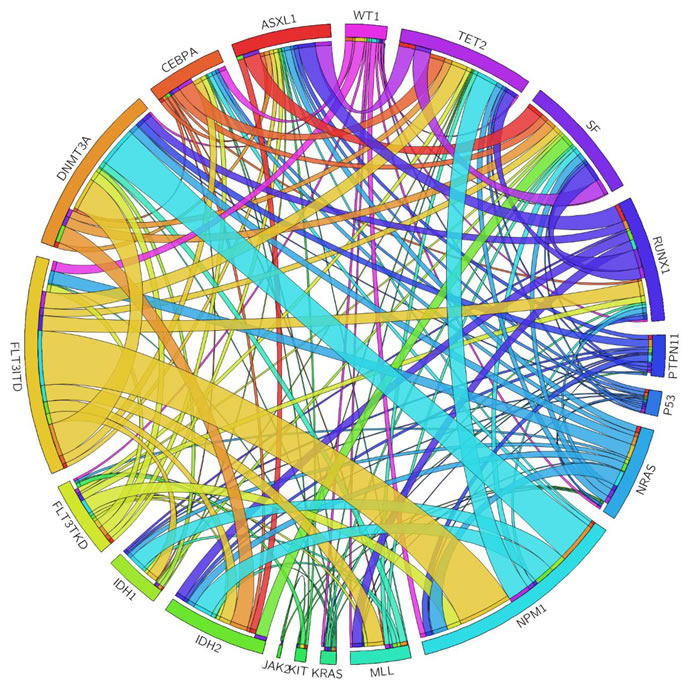
Figure 1: The Circos plots depicted the relative frequency and pairwise co-occurrence of genetic alterations. The length of the arc corresponded to the frequency of the first gene mutation, and the width of the ribbon corresponded to the proportion of the second gene mutation.
The most common SF3B1 mutation was K666M (n = 5), followed by K700E (n = 3) (Table 1). Ring sideroblasts could be detected in two (33%) of the six patients who had bone marrow smears for iron staining. Regarding U2AF1 mutations, ten patients had exon 2 mutations, including S34F in six patients, S34Y in three and S34T in one; six patients had exon 5 mutations, including Q157P in three patients, Q157R in two and E159_M160insYE in one. One patient (patient 18) had concurrent exon 2 S34F and exon 6 Q157R mutations. Among the 27 SRSF2-mutated patients, 24 patients had missense mutations, including P95H in 12 patients, P95L in 8 and P95R in 4. Two patients (patients 28 and 54) had P95_R102del (c.284_307del), a 24-base pair deletion, and the remaining one patient (patient 48) had R94_p95insR (c.283_284insGCC), a 3-base pair insertion (Table 1).
Table 1: The mutation patterns in 54 patients with SF3B1/U2AF1/SRSF2 mutations at diagnosis
UPN |
Age/Sex |
FAB |
RNA Splicing mutation |
Other accompanied gene mutations |
||
Location |
DNA change |
Protein change |
||||
| SF3B1 (n=12) | ||||||
| 1 | 65/F | 1 | Exon 15 | c.2112_2117dup | S705_A706dup | NRAS, ASXL1 |
| 2 | 77/M | 1 | Exon 14 | c.1998G>C | K666N | RUNX1, DNMT3A |
| 3 | 67/F | 1 | Exon 14 | c.1998G>C | K666N | RUNX1, TET2, P53 |
| 4 | 53/F | 1 | Exon 14 | c.1998G>C | K666N | CEBPA, RUNX1 |
| 5 | 73/F | 1 | Exon 15 | c.2098A>G | K700E | CEBPA, TET2, DNMT3A |
| 6 | 62/M | 2 | Exon 14 | c.1996A>C | K666Q | FLT3/ITD, MLL/PTD, RUNX1 |
| 7 | 70/M | 4 | Exon 14 | c.1988C>T | T663I | NPM1 |
| 8 | 31/M | 3 | Exon 14 | c.1998G>C | K666N | — |
| 9 | 43/F | 1 | Exon 15 | c.2098A>G | K700E | P53 |
| 10 | 82/M | 4 | Exon 14 | c.1998G>C | K666N | FLT3/ITD, MLL/PTD, RUNX1 |
| 11 | 86/M | 5 | Exon 14 | c.1873C>T | R625C | FLT3/ITD, DNMT3A |
| 12 | 70/M | 2 | Exon 15 | c.2098A>G | K700E | NPM1, FLT3/ITD, DNMT3A, IDH2 |
| U2AF1 (n=15) | ||||||
| 13 | 40/M | 1 | Exon 2 | c.101C>T | S34F | IDH2 |
| 14 | 22/M | 4 | Exon 6 | c.470A>G | Q157R | FLT3/TKD, ASXL1 |
| 15 | 54/M | 4 | Exon 2 | c.101C>A | S34Y | PTPN11, ASXL1, DNMT3A |
| 16 | 75/M | 4 | Exon 2 | c.101C>T | S34F | KRAS |
| 17 | 72/M | 1 | Exon 6 | c.470A>C | Q157P | ASXL1, IDH1, TET2 |
| 18 | 52/M | 0 | Exon 2 | c.101C>T | S34F | ASXL1 |
| Exon 6 | c.470A>G | Q157R | ||||
| 19 | 47/M | 4 | Exon 2 | c.101C>A | S34Y | PTPN11 |
| 20 | 43/M | 2 | Exon 2 | c.101C>T | S34F | WT1 |
| 21 | 71/F | 2 | Exon 2 | c.101C>T | S34F | CEBPA, NRAS, TET2 |
| 22 | 66/M | 2 | Exon 6 | c.476_477insGTATGA | E159_M160insYE | NRAS, IDH2 |
| 23 | 47/M | 1 | Exon 2 | c.101C>T | S34F | — |
| 24 | 48/M | 6 | Exon 6 | c.470A>C | Q157P | RUNX1 |
| 25 | 44/F | 0 | Exon 2 | c.101C>T | S34T | — |
| 26 | 46/M | 2 | Exon 2 | c.101C>A | S34Y | FLT3/ITD, MLL/PTD |
| 27 | 71/F | 4 | Exon 6 | c.470A>C | Q157P | NRAS, IDH2 |
| SRSF2 (n=27) | ||||||
28 |
89/M |
4 |
Exon 2 |
c.284_307del |
P95_R102del |
RUNX1, IDH2 |
| 29 | 80/M | U | Exon 2 | c.284C>T | P95L | — |
| 30 | 71/M | 4 | Exon 2 | c.284C>A | P95H | TET2 |
| 31 | 73/M | U | Exon 2 | c.284C>G | P95R | — |
| 32 | 67/M | U | Exon 2 | c.284C>A | P95H | ASXL1, IDH2 |
| 33 | 85/F | 2 | Exon 2 | c.284C>T | P95L | CEBPA, ASXL1, TET2 |
| 34 | 66/M | 2 | Exon 2 | c.284C>A | P95H | NPM1, RUNX1, ASXL1 |
| 35 | 70/M | 5 | Exon 2 | c.284C>A | P95H | ASXL1, TET2 |
| 36 | 65/M | 1 | Exon 2 | c.284C>G | P95R | IDH1 |
| 37 | 64/M | 2 | Exon 2 | c.284C>T | P95L | CEBPA, IDH2 |
| 38 | 42/F | 4 | Exon 2 | c.284C>A | P95H | FLT3/ITD, RUNX1, ASXL1, DNMT3A |
| 39 | 75/M | 5 | Exon 2 | c.284C>A | P95H | NPM1, ASXL1, TET2 |
| 40 | 84/M | 0 | Exon 2 | c.284C>T | P95L | RUNX1, IDH2, DNMT3A |
| 41 | 68/M | 4 | Exon 2 | c.284C>T | P95L | RUNX1, TET2 |
| 42 | 66/M | 4 | Exon 2 | c.284C>A | P95H | NRAS, ASXL1 |
| 43 | 63/M | 2 | Exon 2 | c.284C>A | P95H | NRAS, TET2 |
| 44 | 72/M | 1 | Exon 2 | c.284C>G | P95R | RUNX1, IDH1 |
| 45 | 82/M | 5 | Exon 2 | c.284C>T | P95L | RUNX1, ASXL1, TET2 |
| 46 | 70/M | 4 | Exon 2 | c.284C>A | P95H | KRAS, RUNX1 |
| 47 | 48/F | 1 | Exon 2 | c.284C>A | P95H | CEBPA, IDH2, DNMT3A |
| 48 | 71/M | 1 | Exon 2 | c.283_284insGCC | R94_p95insR | RUNX1, TET2 |
| 49 | 77/F | 4 | Exon 2 | c.284C>A | P95H | NPM1, TET2 |
| 50 | 87/M | 2 | Exon 2 | c.284C>T | P95L | NPM1, FLT3/ITD, TET2 |
| 51 | 63/F | 4 | Exon 2 | c.284C>G | P95R | PTPN11, IDH2 |
| 52 | 90/M | 2 | Exon 2 | c.284C>T | P95L | CEBPA, ASXL1, TET2, P53 |
| 53 | 69/M | 4 | Exon 2 | c.284C>A | P95H | NRAS, FLT3/TKD, RUNX1, IDH2, DNMT3A |
| 54 | 44/M | 1 | Exon 2 | c.284_307del | P95_R102del | RUNX1, IDH2 |
Abbreviations: UPN, unique patient number; FAB, French-American-British; U, undetermined.
Correlation of SF mutations with clinical and laboratory features
SF-mutated patients were older (median, 67.5 years vs. 49 years, P < 0.0001, Table 2), male predominant (14.4% in males vs. 6% in females, P = 0.0033) and had a lower incidence of FAB M2 subtype (P = 0.0499) than other patients. The SF mutations were positively associated with the expression of HLA-DR (P = 0.0156) and CD34 (P = 0.0131), but inversely associated with the expression of CD33 (P = 0.0379) and CD56 (P = 0.0493) on the leukemic cells (Supplementary Table 1). Correlation of the clinical and laboratory features with mutations in individual SF genes was shown in Supplementary Table 2.
Table 2: Comparison of clinical and laboratory features between AML patients with and without SF mutation
Variables |
Total (n = 500) |
SF-Mutated (n = 54, 10.8%) |
SF-Wild (n = 446, 89.2%) |
P value |
Sex† |
0.0033 |
|||
Male |
285 |
41 (14.4) |
244 (85.6) |
|
Female |
215 |
13 (6) |
202 (94) |
|
Age (year)‡ |
51 (15-90) |
67.5 (22-90) |
49 (15-90) |
<0.0001 |
Lab data‡ |
||||
WBC (/μL) |
19075 (120-627800) |
19865 (120-627800) |
19090 (300-42300) |
0.9837 |
Hb (g/dL) |
8 (2.9-16.2) |
8.2 (3.7-16.2) |
8 (2.9-14) |
0.5309 |
Platelet (×1,000 /μL) |
42 (2-802) |
36.5 (6-455) |
42 (2-802) |
0.6565 |
Blast (/μL) |
7401 (0-456725) |
6212 (14-456725) |
7479 (0-369070) |
0.953 |
LDH (U/L) |
889 (206-15000) |
821 (288-7930) |
856 (206-15000) |
0.8432 |
FAB† |
||||
M0 |
10 |
3 (30) |
7 (70) |
0.0827 |
M1 |
112 |
14 (12.5) |
98 (87.5) |
0.4933 |
M2 |
171 |
12 (7.0) |
159 (93.0) |
0.0499 |
M3 |
38 |
1 (2.6) |
37 (97.4) |
0.1058 |
M4 |
124 |
16 (12.9) |
108 (87.1) |
0.4052 |
M5 |
24 |
4 (16.7) |
20 (83.3) |
0.3139 |
M6 |
12 |
1 (8.3) |
11 (91.7) |
>0.9999 |
Undetermined |
9 |
3 (33.3) |
6 (66.7) |
0.0626 |
Induction response* |
363 |
32 |
331 |
|
CR |
284 |
11 (34.4) |
273 (82.5) |
<0.0001 |
PR/Refractory |
54 |
15 (46.9) |
39 (11.8) |
<0.0001 |
Induction death |
25 |
6 (18.7) |
19 (5.7) |
0.0153 |
Relapse* |
144 |
7 (63.6) |
137 (50.2) |
0.5412 |
† number of patients (%)
‡ median (range)
* only the 363 patients, including 32 with SF mutation and 331 without, who received conventional intensive induction chemotherapy and then consolidation chemotherapy if CR was achieved, as mentioned in the text, were included in the analysis.
Association of SF mutations with cytogenetic abnormalities
Chromosome data were available in 482 patients at diagnosis, including 51 SF-mutated and 431 SF-wild patients (Supplementary Table 3). SF mutations occurred more frequently in patients with intermediate-risk cytogenetics (13.2%) than in those with favorable- or unfavorable-risk cytogenetics (5.5%, P < 0.0001). None of the patients with t(8;21), inv(16), or t(7;11) showed SF mutation, but one patient with t(15;17) harbored this mutation concurrently. There was no association of SF mutations as a whole with other chromosomal abnormalities, including +8, +11, +13, +21, -5/del(5q), and -7/del(7q). Intriguingly, U2AF1 mutations occurred frequently in patients with -7/7q- (P = 0.0352)
Association of SF mutations with other molecular gene abnormalities
The interaction of SF mutations with mutations of 18 other genes was shown in Table 3. Among the 54 patients with SF mutations, 49 (90.7%) showed additional molecular abnormalities at diagnosis (Tables 1 and 3 and Figure 1). Eleven had one additional change, 21 had two, 12 had three, four had four and one had five. Patients with SF mutations had significantly higher incidences of RUNX1, ASXL1, IDH2 and TET2 mutations than those without the mutation (31.5% vs. 10.1%, P < 0.0001; 27.8% vs. 7.8%, P < 0.0001; 20.4% vs. 9.9%, P = 0.0344 and 27.8% vs. 11.4%; P = 0.0022, respectively). The interaction of mutations in each SF gene and other genetic alterations was shown in Supplementary Table 4.
Table 3: Association of SF mutation with other gene mutations
Variables |
No. of patients with alteration (%) |
P value |
||
Whole cohort (n = 500) |
SF-mutated patients (n = 54) |
SF-wild patients (n = 446) |
||
FLT3/ITD |
113 (22.6) |
7 (13.0) |
106 (22.7) |
0.0848 |
FLT3/TKD |
38 (7.6) |
2 (3.7) |
36 (8.1) |
0.4116 |
NRAS |
61 (12.2) |
7 (13.0) |
54 (12.1) |
0.8266 |
KRAS |
16 (3.2) |
2 (3.7) |
14 (3.1) |
0.6874 |
PTPN11 |
18 (3.6) |
3 (5.6) |
15 (3.3) |
0.4291 |
KIT |
15 (3.0) |
0 (0) |
15 (3.3) |
0.3891 |
JAK2 |
3 (0.6) |
0 (0) |
3 (0.7) |
>0.9999 |
WT1 |
33 (6.6) |
1 (1.9) |
32 (7.2) |
0.239 |
NPM1 |
103 (20.6) |
6 (11.1) |
97 (21.7) |
0.0753 |
CEBPA |
66 (13.2) |
7 (13.0) |
59 (13.2) |
>0.9999 |
RUNX1 |
62 (12.4) |
17 (31.5) |
45 (10.1) |
<0.0001 |
MLL/PTD |
27 (5.4) |
3 (5.6) |
24 (5.4) |
>0.9999 |
ASXL1 |
50 (10.0) |
15 (27.8) |
35 (7.8) |
<0.0001 |
IDH1 |
27 (5.4) |
3 (5.6) |
24 (5.4) |
>0.9999 |
IDH2 |
55 (11) |
11 (20.4) |
44 (9.9) |
0.0344 |
TET2 |
66 (13.2) |
15 (27.8) |
51 (11.4) |
0.0022 |
DNMT3A |
70 (14.0) |
9 (16.7) |
61 (13.7) |
0.5353 |
TP53 |
35 (7.0) |
2 (3.7) |
33 (7.4) |
0.409 |
Impact of SF mutation on response to therapy and clinical outcome
Of the 363 AML patients undergoing conventional intensive induction chemotherapy, 284 (78.5%) patients achieved a CR. Mutations in any of SF3B1, SRSF2 and U2AF1 were associated with lower CR rates (22.2% vs. 79.7%, P = 0.0005; 45.5% vs. 79.3%, P = 0.0162; 33.3% vs. 79.8%, P = 0.0009; respectively, Supplementary Table 2A, B, C). With a median follow-up of 55 months (ranges, 1.0 to 160), patients with mutations of either SF3B1 or U2AF1 had significantly shorter OS (2 months vs. 29.5 months, P < 0.001 and 4.5 months vs. 26 months, P = 0.001, respectively, Supplementary Figure 1A, E) and DFS (0 month vs. 9 months, P < 0.001 and 0 month vs. 9 months, P < 0.001, respectively, Supplementary Figure 1B, F), while patients with SRSF2 mutation had a significantly inferior OS (14.5 month vs. 29.5 months, P = 0.021, Supplementary Figure 1C) and a trend of shorter DFS than those without the mutation (0 month vs. 9 months, P = 0.172, Supplementary Figure 1D). As mutations of all three individual SF implicated a poor response to treatment and inferior outcome, we therefore analysed the clinical relevance of SF mutations as a whole. Patients with SF mutations had significantly poorer OS and DFS than those without SF mutation (median, 6 months vs. 38 months, P < 0.001, and median, 0 month vs. 10 months, P < 0.001, respectively, Figure 2A, 2B). The prognostic differences remained similar among the patients with non-M3 AML (median, 6 months vs. 25 months, P < 0.001 and median, 0 month vs. 9 months, < 0.001, respectively) and those with intermediate-risk cytogenetics (median, 7 months vs. 23.5 months, P < 0.001, Figure 2C and median, 0 month vs. 7.5 months, P < 0.001, Figure 2D, respectively). The same were also true for the subgroup of 161 patients with normal karyotype (median, 4.5 months vs. 61 months, P < 0.001, Figure 2E and median, 0 month vs. 10 months, P < 0.001, Figure 2F, respectively).
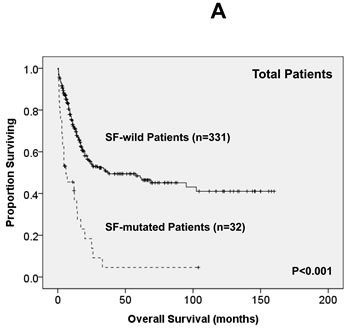
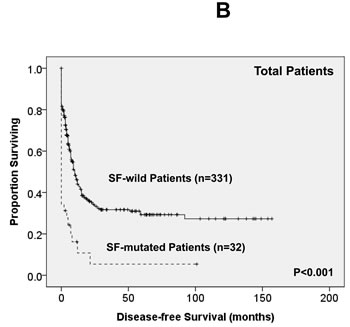
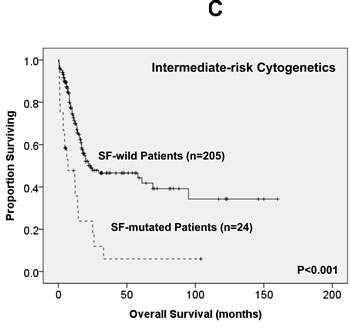
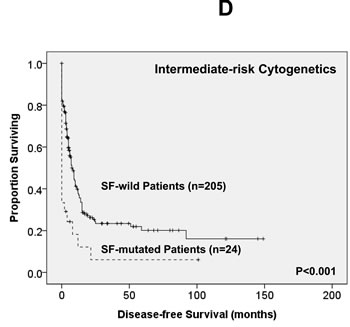
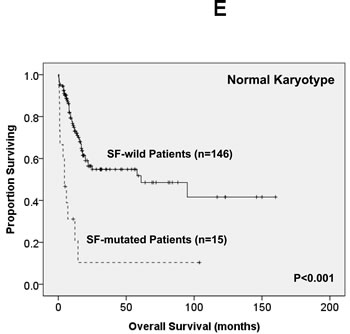
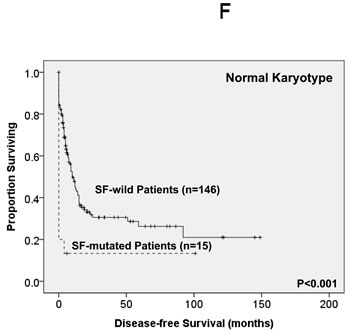
Figure 2: Kaplan-Meier survival curves for overall survival and disease-free survival stratified by the status of SF mutations in total 363 AML patients (A and B), 229 patients with intermediate-risk cytogenetics (C and D) and 161 patients with normal karyotype (E and F) who received standard intensive chemotherapy.
In multivariate analysis (Table 4), the independent poor risk factors for OS were older age >50 years, higher white blood cell (WBC) counts >50,000/μL, unfavorable-risk cytogenetics and mutations of SF (RR 2.243, 95% CI 1.380-3.647, P = 0.001), TP53, RUNX1, WT1 and DNMT3A. On the other hand, CEBPAdouble-mutation and NPM1 mutation in the absence of FLT3-ITD (NPM1+/FLT3-ITD-) were independent favorable prognostic factors. There was a trend of better OS in patients with IDH2 mutation (RR 0.539, 95% CI 0.284-1.020, P = 0.058). Similarly, the independent poor risk factors for DFS included older age > 50 years, higher WBC counts >50,000/μL, unfavorable-risk cytogenetics, and SF, TP53, RUNX1, WT1 and DNMT3A mutations. On the other hand, NPM1+/FLT3-ITD- was an independent favorable prognostic factor. In the 229 patients with intermediate-risk cytogenetics, the SF mutation was still an independent poor prognosis for OS and DFS (RR, 2.999; 95% CI, 1.002-2.999, P = 0.049 and RR, 1.705; 95% CI, 1.028-2.827, P = 0.039, respectively, Supplementary Table 5).
Intriguingly, among the 97 patients receiving allogeneic HSCT, either in first CR (n = 45) or beyond (n = 52), the poor prognostic impact of SF mutation on OS and DFS was lost (P = 0.439 and P = 0.348, respectively). It seems that HSCT may ameliorate the poor survival impact of SF mutations, similar to RUNX1 mutations.[12, 13] However, because the number of patients who had SF mutations and received HSCT was limited in our cohort, further studies in more patients are needed to clarify this point.
To better stratify the AML patients into different risk groups, a scoring system incorporating SF mutations with ten other prognostic factors, including age, WBC counts, cytogenetics at diagnosis, NPM1/FLT3-ITD, and mutations of CEBPA, IDH2, TP53, DNMT3A, RUNX1 and WT1, into survival analysis was formulated based on the results of our Cox proportional hazards model. The weight of the each variable was based on the value of relative risk (Table 4). To simplify the clinical utilization, a score of -3 was assigned for NPM1+/FLT3-ITD- and -2 for CEBPAdouble-mutation and IDH2 mutation whereas a score of +3 for TP53 mutation and +2 for other factors associated with an adverse outcome (SF, DNMT3A, WT1 and RUNX1 mutations, older age, higher WBC counts at diagnosis and unfavorable cytogenetics). The algebraic summation of these scores of each patient was the final score. This score system divided the AML patients into five groups with different clinical outcomes (P < 0.001 for both OS and DFS, Figure 3).
Table 4: Multivariate Analysis (Cox regression) on the Overall Survival and Disease-free Survival
Variables |
Overall Survival |
Disease-free Survival |
|||||||
95% CI |
95% CI |
||||||||
RR |
Lower |
Upper |
P |
RR |
Lower |
Upper |
P |
||
Age† |
2.228 |
1.598 |
3.106 |
<0.001* |
1.344 |
1.016 |
1.779 |
0.038* |
|
WBC§ |
2.192 |
1.539 |
3.123 |
<0.001* |
1.731 |
1.285 |
2.331 |
<0.001* |
|
KaryotypeΨ |
2.227 |
1.230 |
4.032 |
0.008* |
1.792 |
1.087 |
2.955 |
0.022* |
|
NPM1/FLT3-ITDζ |
0.343 |
0.171 |
0.686 |
0.002* |
0.304 |
0.163 |
0.567 |
<0.001* |
|
CEBPA‡ |
0.462 |
0.238 |
0.896 |
0.022* |
0.630 |
0.392 |
1.014 |
0.057 |
|
RUNX1 |
1.942 |
1.129 |
3.339 |
0.016* |
1.788 |
1.138 |
2.809 |
0.012* |
|
WT1 |
2.560 |
1.508 |
4.346 |
<0.001* |
2.469 |
1.614 |
3.778 |
<0.001* |
|
ASXL1 |
1.126 |
0.622 |
2.039 |
0.695 |
0.978 |
0.562 |
1.704 |
0.938 |
|
IDH2** |
0.539 |
0.284 |
1.020 |
0.058 |
0.840 |
0.530 |
1.333 |
0.459 |
|
DNMT3A |
1.919 |
1.166 |
3.158 |
0.010* |
2.130 |
1.400 |
3.241 |
<0.001* |
|
TP53 |
3.613 |
1.598 |
8.167 |
0.002* |
2.824 |
1.372 |
5.812 |
0.005* |
|
SF |
2.243 |
1.380 |
3.647 |
0.001* |
2.136 |
1.376 |
3.314 |
0.001* |
|
Abbreviation: RR, relative risk; CI, confidence interval, SF, splicing factor.
*Statistically significant (P < 0.05)
†Age > 50 relative to Age ≤50 (the reference)
§WBC greater than 50,000/µL vs. 50,000/µL or less
ζNPM1mut/FLT3-ITDneg vs. other subtypes
‡CEBPAdouble-mutation vs. others
Ψunfavorable cytogenetics vs. others
**IDH2 mutations included R140 and R172 mutations
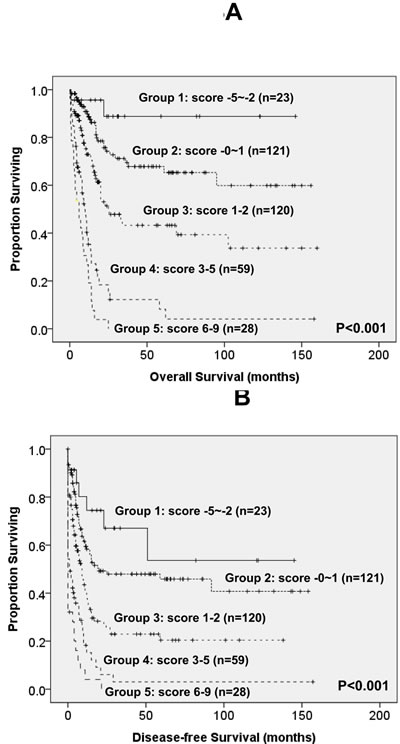
Figure 3: Kaplan-Meier survival curves for overall survival (A) and disease-free survival (B) in AML patients based on scoring system (P < 0.001 for both OS and DFS). AML patients were grouped according to scoring system based on SF mutation and 10 other prognostic markers (CEBPAdouble-mutation, NPM1/FLT3-ITD, IDH2, TP53, WT1, RUNX1 and DNMT3A mutations, cytogenetics, age and WBC counts at diagnosis). A score of -3 was assigned for NPM1+/FLT3-ITD- and -2 for CEBPAdouble-mutation and IDH2 mutation whereas a score of +3 for TP53 mutation and +2 for other factors associated with an adverse outcome (SF, DNMT3A, WT1 and RUNX1 mutations, older age, higher WBC counts at diagnosis and unfavorable cytogenetics). The algebraic summation of these scores of each patient was the final score. This score system divided the AML patients into five groups with different clinical outcomes (P < 0.001 for both OS and DFS). The 12 patients without chromosome data were not included in the analysis.
Sequential studies of SF mutations
SF mutations were serially studied in 489 samples from 163 patients, including 11 patients with SF mutations and 152 patients without the mutation at diagnosis (Table 5). Among the nine patients with SF mutations who obtained a CR and had available samples for study, eight lost the original mutation at remission status, but one (patient 22) retained it (Table 5). In addition to U2AF1 mutation, patient 22 also harbored concurrent mutations of NRAS and IDH2 at diagnosis and these two mutations disappeared at CR. The amplitude of the mutant sequence of U2AF1 in this patient was much lower at CR compared to that at diagnosis and relapse (Supplementary Figure 2). It implied that the cells with the mutation were present as a minor population at remission suggesting that the mutation was not hereditary, but acquired, and such residual leukemia cells would then cause relapse.
In the seven patients who had available samples for serial study at relapse, the original SF mutation could be detected at relapse in six patients (patients 3, 8, 14, 22, 26 and 34), but was lost in one (SRSF2 mutant in patient 36). Because direct sequencing might not be sensitive enough to detect low level of SF mutation signal, we therefore sequenced TA clones of the PCR product from patient 36 at relapse. The original SRSF2 mutant could be detected in one out of 45 clones. Interestingly, acquisition of novel mutations was noted at relapse in three SF-mutated patients (patients 3, 8 and 14, Table 5). On the other side, among the 152 patients who had no SF mutation at diagnosis, none acquired SF mutation at relapse.
Table 5: Sequential studies in the AML patients with SF mutations*
UPN |
Interval† (months) |
Status |
karyotype |
SF mutation |
Other mutations |
3 |
Initial |
46,XX |
SF3B1 (K666N) |
RUNX1, TET2, P53 |
|
4 |
CR |
ND |
— |
||
7.5 |
Relapse |
ND |
SF3B1 (K666N) |
RUNX1, TET2, P53, FLT3/ITD |
|
8 |
Initial |
46,XY,t(15;17)(q22;q21) |
SF3B1 (K666N) |
— |
|
12 |
Relapse |
46,XY,t(15;17)(q22;q21) |
SF3B1 (K666N) |
ASXL1 |
|
14 |
|
Initial |
45,XY,-7 |
U2AF1 (Q157R) |
FLT3/TKD, ASXL1 (P1377SfsX3) |
4 |
CR |
46,XY |
— |
— |
|
16.5 |
Relapse |
ND |
U2AF1 (Q157R) |
ASXL1 (S1255X) |
|
15 |
Initial |
46,XY |
U2AF1 (S34Y) |
PTPN11, ASXL1, DNMT3A |
|
5.4 |
CR |
ND |
— |
— |
|
22 |
Initial |
47,XY,+8 |
U2AF1 (E159_M160insYE) |
NRAS, IDH2 |
|
4.2 |
CR1 |
46,XY |
U2AF1 (E159_M160insYE) |
— |
|
11 |
Relapse 1 |
48,XY,+8,+15 |
U2AF1 (E159_M160insYE) |
NRAS, IDH2 |
|
2 |
CR2 |
ND |
U2AF1 (E159_M160insYE) |
— |
|
8 |
Relapse 2 |
46-48,XY,+X,+15 |
U2AF1 (E159_M160insYE) |
NRAS, IDH2 |
|
26 |
Initial |
47,XY,+11 |
U2AF1 (S34Y) |
FLT3/ITD, MLL/PTD |
|
8.7 |
Relapse |
ND |
U2AF1 (S34Y) |
FLT3/ITD, MLL/PTD |
|
34 |
Initial |
46,XY,del(7)(q22q36) |
SRSF2 (P95H) |
NPM1, RUNX1, ASXL1 |
|
5.5 |
CR1 |
46,XY |
— |
ASXL1 |
|
4 |
Relapse 1 |
46,XY |
SRSF2 (P95H) |
NPM1, RUNX1, ASXL1 |
|
36 |
Initial |
48,XY,+add(1)(p13),+8 |
SRSF2 (P95R) |
IDH1 |
|
1 |
CR1 |
46,XY |
— |
— |
|
7.5 |
Relapse 1 |
46,XY |
SRSF2 (P95R)†† |
IDH1 |
|
37 |
Initial |
46,XY |
SRSF2 (P95L) |
CEBPA, IDH2 |
|
2.5 |
CR1 |
ND |
— |
— |
|
47 |
Initial |
47,XX,+8 |
SRSF2 (P95H) |
CEBPA, IDH2, DNMT3A |
|
2 |
CR1 |
46,XX |
— |
— |
|
54 |
Initial |
46,XY |
SRSF2 (P95_R102del) |
RUNX1, IDH2 |
|
5 |
CR1 |
ND |
— |
— |
Abbreviations: UPN, unique patient number; CR, complete remission; ND, not done.
*The data of serial studies in other 152 patients, who did not have SF mutation both at diagnosis and relapse were not shown in this table
†Interval between the two successive status
††The SRSF2 (patient 36) mutation could be detected by TA cloning (one out of 45 clones), but not by direct sequencing, at relapse.
Discussion
Most studies on SF mutations in AML were focused on small patients cohorts.[2, 8, 9, 11] To the best of our knowledge, this study recruited the largest cohort of de novo AML. Patients with antecedent hematological diseases, family history of myeloid neoplasms or therapy-related AML were excluded the same way we did previously.[14, 15] We found that SF mutation was associated with distinct clinic-biological features and was a poor prognostic factor in AML patients, independent of age, WBC counts, karyotype and other genetic markers.
Mutations of the SF genes were identified in 54 (10.8%) patients, most commonly in those with intermediate-risk cytogenetics (13.2%). Similar to the data in MDS, the majority of mutations occurred in hotspot areas: K666N and K700E in SF3B1, S34 and Q157 in U2AF1 and P95 in SRSF2. The incidence of SF mutations in AML varied from 4.5%-12.5% in different reports.[2, 8-11] Yoshida et al found SF3B1, U2AF1 and SRSF2 mutations in 2.6%, 1.3% and 0.7%, respectively, of 151 AML patients.[2] Kihara et al reported 4.5% of 197 patients harbored SF mutations, including SF3B1 (1.5%), U2AF1 (1.5%), SRSF2 (1%) and ZRSR2 (0.5%) mutations. By analyzing the mutations in eight hotspots of SF genes in 325 patients, Taskesen et al showed 1.8% of AML patients had mutations in SF3B1, 1.2% in U2AF1 and 4.6% in SRSF2.[10] In a cohort of 200 adult AML patients reported by the Cancer Genome Atlas (TCGA), the incidence of mutations in 21 spliceosome genes detected by either whole-genome sequencing or whole-exome sequencing was 12.5%; among them, SF3B1 mutation was found in 0.5%, U2AF1 mutation in 4% and SRSF2 mutation in 0.5%.[11] Surprisingly, mutations in SRSF2 gene occurred in 81% of AML patients with isolated trisomy 13.[16] The reason of the variability in the incidence of SF mutations in different studies is unknown but may be due to differences in ethnic background, patient population selected (age range, FAB subtypes and karyotype, etc), the regions of SF genes screened, and the methods used. We analyzed exons 14-15 in SF3B1 genes, exons 2 and 6 in U2AF1 genes and exon 2 in SRSF2 gene to avoid missing some mutations outside hotspot regions. A higher frequency of SRSF2 mutations in this study might be partially due to age effect; elder patients were also enrolled in this cohort and SRSF2 mutation is closely associated with older age in myeloid neoplasm.[17]
Although a close association was observed between SF mutations and mutations in certain genes, especially those related to epigenetic modifications, in MDS (such as SF3B1 mutation with DNMT3A mutation, SRSF2 mutation with mutations of RUNX1, IDH and ASXL1 genes and U2AF1 mutation with mutations of ASXL1 and DNMT3A),[4, 6, 18, 19], little is known about the interaction between SF mutations and other molecular genetic alterations in AML patients. In a study of mutational status of three SF genes (SF3B1, U2AF1 and SRSF2), NPM1, FLT3, CEBPA, IDH1, DNMT3A, ASXL1 and NRAS/KRAS in 344 patients, including 47 refractory anemia with excess blasts (RAEB), 29 AML with low BM blast count and other AML patients, Taskesen et al could not find any molecular association.[10] However, with the help of combined genome-wide mRNA expression and DNA-methylation profiling they identified two distinct patient clusters highly enriched for SF-mutated RAEB/AML. One cluster was associated with erythroid phenotype; the other was correlated with NRAS/KRAS mutation (10 out of 25 patients, 40%). However, the reason why these two clusters were defined only by combined genome-wide mRNA expression and DNA-methylation profiling was unclear. In this study, we found SF mutations rarely occurred alone; 49 (90.7%) of 54 patients with SF mutations showed additional molecular abnormalities at diagnosis. This finding is in agreement with the concept that the development of AML requires concerted cooperation of different molecular genetic alterations.[11, 20] Intriguingly, patients with SF mutations had significantly higher incidences of RUNX1, ASXL1, IDH2 and TET2 mutations than those without the mutation, similar to the findings in MDS.[4, 6, 18, 19]
To the best of our knowledge, this study is the first to evaluate the dynamic change of SF mutation during disease progression in a large cohort of patients with de novo AML. In contrast to the instability of FLT3-ITD during disease evolution,[21] we found that the SF mutation seemed rather stable, analogous to DNMT3A mutations[14, 22] At relapse, the original SF mutations in all seven SF-mutated patients studied were retained, but the mutant level in one of them was much reduced at the time of AML relapse as it could only be detected by a sensitive cloning technique, but not by direct sequencing. (patient 36, Table 5) On the other side, among the 152 patients who had no SF mutation at diagnosis, all remained germline of the genes during clinical follow-ups. Taken together, these findings suggest that SF mutations were quite stable during disease evolution and may play an important role in development, but not progression of AML.
Few studies regarding the prognostic relevance of SF mutations in de novo AML have been reported. In a study of Taskesen et al, only one distinct SF-mutant patient cluster enriched for NRAS/KRAS mutation (cluster 3, 7.3% of 344 patients) had poorer prognosis. Patients with isolated trisomy 13 reported by Herold et al, in whom high frequencies of mutations in SRSF2 (81%) and RUNX1 (75%) were noted, had a dismal outcome.[16] In this study, we distinctly identified that SF mutation was an important prognostic factor, independent from all other variables in both total cohort and patients with intermediate-risk cytogenetics. Although SF3B1 mutations have been shown to predict better OS in MDS patients,[3, 19, 23, 24] we found the mutation was associated with a lower CR rate (Supplementary Table 2A) and shorter survival in de novo AML patients (Supplementary Figure 1A, B). The reason why SF3B1 mutation has different impact on clinical outcome between patients with MDS and AML remains to be explored. In fact, the reports concerning the prognostic impact of SF3B1 mutation in MDS showed inconsistent and conflicting results.[3, 19, 23, 24] The good prognostic impact of SF3B1 mutation could not be demonstrated in MDS patients in some studies.[3, 19, 23, 24] It was suggested the close association of SF3B1 mutation with old age and DNMT3A mutation and different treatment regimens might influence the implication of this mutation on survival of MDS patients.[19, 24] In AML, Lindsley et al[25] first showed that SF mutations as well as ASXL1, EZH2, BCOR, and STAG2 mutations were highly specific for secondary AML, and were secondary-type mutations in therapy-related AML and elderly de novo AML that defined a distinct subgroup of patients with poor outcome. In this study, we only recruited de novo AML patients, the same cohort as we reported previously.[14, 15] Secondary AML patients were carefully excluded and SF mutations in this study were closely associated with intermediate-risk cytogenetics, but not poor-risk cytogenetics or complex karyotype, which is frequently seen in secondary AML. The findings from this study reflected the poor prognostic implication of SF mutations in de novo AML patients.
Intriguingly, the poor prognostic impact of SF mutation in OS and DFS was lost if the patients received allogeneic HSCT. In other words, HSCT may ameliorate the poor survival impact of SF mutations. Further studies in more patients are needed to clarify this point. To better stratify AML patients into different risk groups, a survival scoring system incorporating SF mutation and ten other prognostic factors, including age, WBC counts, cytogenetics, NPM1/FLT3-ITD, CEBPA, IDH2, RUNX1, WT1, DNMT3A and TP53 mutations, into survival analysis was formulated. Indeed, this scoring system was more powerful than single marker to separate patients into different prognostic groups. Further studies in independent cohorts are needed to validate the clinical implication of the proposed scoring system.
There was one potential flaw and limitation in this study. We did not analyze the mutations of all 21 spliceosome genes; the results we obtained might only reflect the clinical relevance of mutations in the three SF genes we analyzed. However, SF3B1, U2AF1 and SRSF2 mutations are the most frequent SF mutations in myeloid neoplasms and can be easily detected by Sanger’s sequencing.[2, 11] The finding that mutations in these three SF genes predict poor prognosis suggests routine test of these mutations may be helpful in the clinical management of AML patients.
In summary, this study demonstrated that SF-mutated patients had specific clinic-biologic features and cytogenetic changes. SF mutations were closely associated with RUNX1, ASXL1, IDH2 and TET2 mutations. Furthermore, the SF mutation was an independent poor-risk factor for OS and DFS among total cohort and patients with intermediate-risk cytogenetics. Incorporation of SF mutation with ten other prognostic factors into survival analyses can better stratify AML patients into different risk groups. Sequential study during the clinical course showed that SF mutations were quite stable during AML evolution. These mutations can be potential targets for novel therapies and biomarkers for disease monitoring.
Materials and Methods
Subjects
From March 1995 to Dec 2008, a total of 500 adult patients with newly diagnosed de novo AML according to the French-American-British (FAB) criteria at the National Taiwan University Hospital (NTUH) were enrolled as previously described.[14, 15] Patients with antecedent hematological diseases, history of cytopenia, family history of myeloid neoplasms or therapy-related AML were excluded. Among them, 363 (72.6%) patients received standard induction chemotherapy (Idarubicin 12 mg/m2 per day on days 1-3 and Cytarabine 100 mg/m2 per day on days 1-7) and then consolidation chemotherapy with 2-4 courses of high-dose Cytarabine (2000 mg/m2 q12h days 1-4, total 8 doses), with or without an anthracycline (Idarubicin or Novatrone), after achieving complete remission (CR).[14, 15] The patients with acute promyelocytic leukemia (M3 subtype) received concurrent all-trans retinoic acid and chemotherapy. The remaining 137 patients received palliative therapy due to underlying comorbidity or based on the decision of the patients. Forty-five patients received allogeneic hematopoietic stem cell transplantation (HSCT) in first CR and 52 in relapse/refractory status or second CR or beyond. This study was approved by the Institutional Review Board of the NTUH; and written informed consent was obtained from all participants in accordance with the Declaration of Helsinki.
Cytogenetics
Chromosomal analyses were performed as described previously.[26]
Immunophenotype analysis
A panel of monoclonal antibodies to myeloid associated antigens, including CD13, CD33, CD11b, CD15, CD14, and CD41a, as well as lymphoid-associated antigens, including CD2, CD5, CD7, CD19, CD10, and CD20, and lineage nonspecific antigens HLA-DR, CD34, and CD56 were used to characterize the phenotypes of the leukemia cells as previously described.[14]
Mutation analysis
Mutation analysis of SF genes, including SF3B1, SRSF2 and U2AF1, was performed by polymerase chain reaction (PCR) and direct sequencing.[17-19] Abnormal sequencing results were confirmed by at least two repeated analyses. Sequential analysis of SF mutations during the clinical course was performed in 489 samples from 163 patients. Mutation analyses of 18 other relevant molecular marker genes, including Class I mutations, such as FLT3/ITD and FLT3/TKD,[27] NRAS,[28] KRAS,[28] JAK2,[28] KIT[29] and PTPN11[29] mutations and Class II mutations, such as CEBPA[30] and RUNX1[13] mutations, as well as NPM1,[31] WT1,[32] TP53[33] and those genes related to epigenetic modification, such as MLL/PTD,[34] ASXL1,[35] IDH1,[36] IDH2,[37] TET2[38] and DNMT3A[14] mutations were performed as previously described. To detect SF mutations at diagnosis, we used DNA amplified in vitro from patients’ BM cells by IllustraTM GenomiPhi V2 DNA amplification kit as described by the manufacturer (GE Healthcare, Buckinghamshire, UK). All the mutations detected in such samples were verified in the original non-amplified samples.
TA cloning analysis
For the patients with discrepancy of the mutation status of the SF genes in paired samples, Taq polymerase-amplified (TA) cloning was performed in the samples without detectable mutant by direct sequencing as previously described.[28]
Statistical analysis
The discrete variables of patients with and without SF mutation were compared using the chi-square tests, but if the expected values of contingency tables were smaller than 5, Fisher exact test was used. If the continuous data were not normally-distributed, Mann-Whitney U tests were used to compare continuous variables and medians of distributions. To evaluate the impact of SF mutation on clinical outcome, only the patients who received conventional standard chemotherapy, as mentioned above, were included in analysis.[14, 15] OS was measured from the date of first diagnosis to the date of last follow-up or death from any cause, whereas relapse was defined as a reappearance of at least 5% leukemic blasts in a BM aspirate or new extramedullary leukemia in patients with a previously documented CR.[39] Disease-free (DF) status indicated that the patient achieved CR and did not relapse by the end of this study. Cox regression survival estimation was used to plot survival curves and to test the difference between groups. Multivariate Cox proportional hazard regression analysis was used to investigate independent prognostic factors for OS and DFS. The proportional hazards assumption (constant hazards assumption) was examined by using Time-Dependent Covariate Cox regression before conducting multivariate Cox proportional hazard regression. The variables including age, WBC counts, karyotype, NPM1/FLT3-ITD, CEBPA, IDH2, WT1, RUNX1, ASXL1, DNMT3A and TP53 mutations were used as covariates. Those patients who received HSCT were censored at the time of HSCT in survival analysis to ameliorate the influence of the treatment.[14, 15] A P-value < 0.05 was considered statistically significant. All statistical analyses were performed with the SPSS 19 (SPSS Inc., Chicago, IL, USA) and Statsdirect (Cheshire, England, UK).
Acknowledgments/Grant Support
This work was partially sponsored by grants MOST 100-2628-B-002-003 -MY3,103-2628-B-002-008-MY3, 103-2923-B-002 -001 and 104-2314-B-002 -128-MY4 from the Ministry of Science and Technology (Taiwan), MOHW103-TD-B-111-04 from the Ministry of Health and Welfare (Taiwan) and NTUH 102P06 from the Department of Medical Research, National Taiwan University Hospital.
Authors’ contributions
Contribution: H.-A.H. was responsible for study design and plan, literature collection, data management and interpretation, statistical analysis and manuscript writing; C.-Y.L was responsible for statistical analysis and interpretation of the statistical findings; Y.-Y.K and L.-I.L. were responsible for mutation analysis and interpretation; W.-C.C., C.-H.T., C.-C.L., J.-L.T., M.Y., C.-C.,L, S.-Y.H., B.-S.K., S.-C.H., C.-Y.C., C.T.,L, -S.-J.W., and W.T. contributed patient samples and clinical data; M.-H.T., Y.-C.C., M.-C,L. and C.-W.L. performed the gene mutation and chromosomal studies and H.-F.T. planned, designed, wrote manuscript and coordinated the study over the entire period.
Conflicts of interest
The authors declare no competing financial interests.
Editorial note
This paper has been accepted based in part on peer-review conducted by another journal and the authors’ response and revisions as well as expedited peer-review in Oncotarget.
References
1. Chen M and Manley JL. Mechanisms of alternative splicing regulation: insights from molecular and genomics approaches. Nat Rev Mol Cell Biol. 2009; 10:741-754.
2. Yoshida K, Sanada M, Shiraishi Y, Nowak D, Nagata Y, Yamamoto R, Sato Y, Sato-Otsubo A, Kon A, Nagasaki M, Chalkidis G, Suzuki Y, Shiosaka M, Kawahata R, Yamaguchi T, Otsu M, et al. Frequent pathway mutations of splicing machinery in myelodysplasia. Nature. 2011; 478:64-69.
3. Papaemmanuil E, Cazzola M, Boultwood J, Malcovati L, Vyas P, Bowen D, Pellagatti A, Wainscoat JS, Hellstrom-Lindberg E, Gambacorti-Passerini C, Godfrey AL, Rapado I, Cvejic A, Rance R, McGee C, Ellis P, et al. Somatic SF3B1 mutation in myelodysplasia with ring sideroblasts. N Engl J Med. 2011; 365:1384-1395.
4. Thol F, Kade S, Schlarmann C, Loffeld P, Morgan M, Krauter J, Wlodarski MW, Kolking B, Wichmann M, Gorlich K, Gohring G, Bug G, Ottmann O, Niemeyer CM, Hofmann WK, Schlegelberger B, et al. Frequency and prognostic impact of mutations in SRSF2, U2AF1, and ZRSR2 in patients with myelodysplastic syndromes. Blood. 2012; 119:3578-3584.
5. Visconte V, Makishima H, Maciejewski JP and Tiu RV. Emerging roles of the spliceosomal machinery in myelodysplastic syndromes and other hematological disorders. Leukemia. 2012; 26:2447-2454.
6. Damm F, Kosmider O, Gelsi-Boyer V, Renneville A, Carbuccia N, Hidalgo-Curtis C, Della Valle V, Couronne L, Scourzic L, Chesnais V, Guerci-Bresler A, Slama B, Beyne-Rauzy O, Schmidt-Tanguy A, Stamatoullas-Bastard A, Dreyfus F, et al. Mutations affecting mRNA splicing define distinct clinical phenotypes and correlate with patient outcome in myelodysplastic syndromes. Blood. 2012; 119:3211-3218.
7. Ogawa S. Splicing factor mutations in myelodysplasia. Int J Hematol. 2012; 96:438-442.
8. Je EM, Yoo NJ, Kim YJ, Kim MS and Lee SH. Mutational analysis of splicing machinery genes SF3B1, U2AF1 and SRSF2 in myelodysplasia and other common tumors. Int J Cancer. 2013; 133:260-265.
9. Kihara R, Nagata Y, Kiyoi H, Kato T, Yamamoto E, Suzuki K, Chen F, Asou N, Ohtake S, Miyawaki S, Miyazaki Y, Sakura T, Ozawa Y, Usui N, Kanamori H, Kiguchi T, et al. Comprehensive analysis of genetic alterations and their prognostic impacts in adult acute myeloid leukemia patients. Leukemia. 2014; 28:1586-1595.
10. Taskesen E, Havermans M, van Lom K, Sanders MA, van Norden Y, Bindels E, Hoogenboezem R, Reinders MJ, Figueroa ME, Valk PJ, Lowenberg B, Melnick A and Delwel R. Two splice-factor mutant leukemia subgroups uncovered at the boundaries of MDS and AML using combined gene expression and DNA-methylation profiling. Blood. 2014; 123:3327-3335.
11. Cancer Genome Atlas Research N. Genomic and epigenomic landscapes of adult de novo acute myeloid leukemia. N Engl J Med. 2013; 368:2059-2074.
12. Gaidzik VI, Bullinger L, Schlenk RF, Zimmermann AS, Rock J, Paschka P, Corbacioglu A, Krauter J, Schlegelberger B, Ganser A, Spath D, Kundgen A, Schmidt-Wolf IG, Gotze K, Nachbaur D, Pfreundschuh M, et al. RUNX1 mutations in acute myeloid leukemia: results from a comprehensive genetic and clinical analysis from the AML study group. J Clin Oncol. 2011; 29:1364-1372.
13. Tang JL, Hou HA, Chen CY, Liu CY, Chou WC, Tseng MH, Huang CF, Lee FY, Liu MC, Yao M, Huang SY, Ko BS, Hsu SC, Wu SJ, Tsay W, Chen YC, et al. AML1/RUNX1 mutations in 470 adult patients with de novo acute myeloid leukemia: prognostic implication and interaction with other gene alterations. Blood. 2009; 114:5352-5361.
14. Hou HA, Kuo YY, Liu CY, Chou WC, Lee MC, Chen CY, Lin LI, Tseng MH, Huang CF, Chiang YC, Lee FY, Liu MC, Liu CW, Tang JL, Yao M, Huang SY, et al. DNMT3A mutations in acute myeloid leukemia: stability during disease evolution and clinical implications. Blood. 2012; 119:559-568.
15. Hou HA, Lin CC, Chou WC, Liu CY, Chen CY, Tang JL, Lai YJ, Tseng MH, Huang CF, Chiang YC, Lee FY, Kuo YY, Lee MC, Liu MC, Liu CW, Lin LI, et al. Integration of cytogenetic and molecular alterations in risk stratification of 318 patients with de novo non-M3 acute myeloid leukemia. Leukemia. 2014; 28:50-58.
16. Herold T, Metzeler KH, Vosberg S, Hartmann L, Rollig C, Stolzel F, Schneider S, Hubmann M, Zellmeier E, Ksienzyk B, Jurinovic V, Pasalic Z, Kakadia PM, Dufour A, Graf A, Krebs S, et al. Isolated trisomy 13 defines a homogeneous AML subgroup with high frequency of mutations in spliceosome genes and poor prognosis. Blood. 2014; 124:1304-1311.
17. Wu SJ, Kuo YY, Hou HA, Li LY, Tseng MH, Huang CF, Lee FY, Liu MC, Liu CW, Lin CT, Chen CY, Chou WC, Yao M, Huang SY, Ko BS, Tang JL, et al. The clinical implication of SRSF2 mutation in patients with myelodysplastic syndrome and its stability during disease evolution. Blood. 2012; 120:3106-3111.
18. Wu SJ, Tang JL, Lin CT, Kuo YY, Li LY, Tseng MH, Huang CF, Lai YJ, Lee FY, Liu MC, Liu CW, Hou HA, Chen CY, Chou WC, Yao M, Huang SY, et al. Clinical implications of U2AF1 mutation in patients with myelodysplastic syndrome and its stability during disease progression. Am J Hematol. 2013; 88:E277-282.
19. Lin CC, Hou HA, Chou WC, Kuo YY, Wu SJ, Liu CY, Chen CY, Tseng MH, Huang CF, Lee FY, Liu MC, Liu CW, Tang JL, Yao M, Huang SY, Hsu SC, et al. SF3B1 mutations in patients with myelodysplastic syndromes: the mutation is stable during disease evolution. Am J Hematol. 2014; 89:E109-115.
20. Renneville A, Roumier C, Biggio V, Nibourel O, Boissel N, Fenaux P and Preudhomme C. Cooperating gene mutations in acute myeloid leukemia: a review of the literature. Leukemia. 2008; 22:915-931.
21. Shih LY, Huang CF, Wu JH, Lin TL, Dunn P, Wang PN, Kuo MC, Lai CL and Hsu HC. Internal tandem duplication of FLT3 in relapsed acute myeloid leukemia: a comparative analysis of bone marrow samples from 108 adult patients at diagnosis and relapse. Blood. 2002; 100:2387-2392.
22. Shlush LI, Zandi S, Mitchell A, Chen WC, Brandwein JM, Gupta V, Kennedy JA, Schimmer AD, Schuh AC, Yee KW, McLeod JL, Doedens M, Medeiros JJ, Marke R, Kim HJ, Lee K, et al. Identification of pre-leukaemic haematopoietic stem cells in acute leukaemia. Nature. 2014; 506:328-333.
23. Malcovati L, Papaemmanuil E, Bowen DT, Boultwood J, Della Porta MG, Pascutto C, Travaglino E, Groves MJ, Godfrey AL, Ambaglio I, Galli A, Da Via MC, Conte S, Tauro S, Keenan N, Hyslop A, et al. Clinical significance of SF3B1 mutations in myelodysplastic syndromes and myelodysplastic/myeloproliferative neoplasms. Blood. 2011; 118:6239-6246.
24. Patnaik MM, Lasho TL, Hodnefield JM, Knudson RA, Ketterling RP, Garcia-Manero G, Steensma DP, Pardanani A, Hanson CA and Tefferi A. SF3B1 mutations are prevalent in myelodysplastic syndromes with ring sideroblasts but do not hold independent prognostic value. Blood. 2012; 119:569-572.
25. Lindsley RC, Mar BG, Mazzola E, Grauman PV, Shareef S, Allen SL, Pigneux A, Wetzler M, Stuart RK, Erba HP, Damon LE, Powell BL, Lindeman N, Steensma DP, Wadleigh M, DeAngelo DJ, et al. Acute myeloid leukemia ontogeny is defined by distinct somatic mutations. Blood. 2015; 125:1367-1376.
26. Tien HF, Wang CH, Lin MT, Lee FY, Liu MC, Chuang SM, Chen YC, Shen MC, Lin KH and Lin DT. Correlation of cytogenetic results with immunophenotype, genotype, clinical features, and ras mutation in acute myeloid leukemia. A study of 235 Chinese patients in Taiwan. Cancer Genet Cytogenet. 1995; 84:60-68.
27. Chou WC, Tang JL, Lin LI, Yao M, Tsay W, Chen CY, Wu SJ, Huang CF, Chiou RJ, Tseng MH, Lin DT, Lin KH, Chen YC and Tien HF. Nucleophosmin mutations in de novo acute myeloid leukemia: the age-dependent incidences and the stability during disease evolution. Cancer Res. 2006; 66:3310-3316.
28. Hou HA, Kuo YY, Tang JL, Chou WC, Yao M, Lai YJ, Lin CC, Chen CY, Liu CY, Tseng MH, Huang CF, Chiang YC, Lee FY, Liu MC, Liu CW, Huang SY, et al. Clinical implications of the SETBP1 mutation in patients with primary myelodysplastic syndrome and its stability during disease progression. Am J Hematol. 2014; 89:181-186.
29. Hou HA, Chou WC, Lin LI, Chen CY, Tang JL, Tseng MH, Huang CF, Chiou RJ, Lee FY, Liu MC and Tien HF. Characterization of acute myeloid leukemia with PTPN11 mutation: the mutation is closely associated with NPM1 mutation but inversely related to FLT3/ITD. Leukemia. 2008; 22:1075-1078.
30. Lin LI, Chen CY, Lin DT, Tsay W, Tang JL, Yeh YC, Shen HL, Su FH, Yao M, Huang SY and Tien HF. Characterization of CEBPA mutations in acute myeloid leukemia: most patients with CEBPA mutations have biallelic mutations and show a distinct immunophenotype of the leukemic cells. Clin Cancer Res. 2005; 11:1372-1379.
31. Falini B, Mecucci C, Tiacci E, Alcalay M, Rosati R, Pasqualucci L, La Starza R, Diverio D, Colombo E, Santucci A, Bigerna B, Pacini R, Pucciarini A, Liso A, Vignetti M, Fazi P, et al. Cytoplasmic nucleophosmin in acute myelogenous leukemia with a normal karyotype. N Engl J Med. 2005; 352:254-266.
32. Hou HA, Huang TC, Lin LI, Liu CY, Chen CY, Chou WC, Tang JL, Tseng MH, Huang CF, Chiang YC, Lee FY, Liu MC, Yao M, Huang SY, Ko BS, Hsu SC, et al. WT1 mutation in 470 adult patients with acute myeloid leukemia: stability during disease evolution and implication of its incorporation into a survival scoring system. Blood. 2010; 115:5222-5231.
33. Hou HA, Chou WC, Kuo YY, Liu CY, Lin LI, Tseng MH, Chiang YC, Liu MC, Liu CW, Tang JL, Yao M, Li CC, Huang SY, Ko BS, Hsu SC, Chen CY, et al. TP53 mutations in de novo acute myeloid leukemia patients: longitudinal follow-ups show the mutation is stable during disease evolution. Blood Cancer J. 2015; 5:e331.
34. Shiah HS, Kuo YY, Tang JL, Huang SY, Yao M, Tsay W, Chen YC, Wang CH, Shen MC, Lin DT, Lin KH and Tien HF. Clinical and biological implications of partial tandem duplication of the MLL gene in acute myeloid leukemia without chromosomal abnormalities at 11q23. Leukemia. 2002; 16:196-202.
35. Chen TC, Hou HA, Chou WC, Tang JL, Kuo YY, Chen CY, Tseng MH, Huang CF, Lai YJ, Chiang YC, Lee FY, Liu MC, Liu CW, Liu CY, Yao M, Huang SY, et al. Dynamics of ASXL1 mutation and other associated genetic alterations during disease progression in patients with primary myelodysplastic syndrome. Blood Cancer J. 2014; 4:e177.
36. Chou WC, Hou HA, Chen CY, Tang JL, Yao M, Tsay W, Ko BS, Wu SJ, Huang SY, Hsu SC, Chen YC, Huang YN, Chang YC, Lee FY, Liu MC, Liu CW, et al. Distinct clinical and biologic characteristics in adult acute myeloid leukemia bearing the isocitrate dehydrogenase 1 mutation. Blood. 2010; 115:2749-2754.
37. Lin CC, Hou HA, Chou WC, Kuo YY, Liu CY, Chen CY, Lai YJ, Tseng MH, Huang CF, Chiang YC, Lee FY, Liu MC, Liu CW, Tang JL, Yao M, Huang SY, et al. IDH mutations are closely associated with mutations of DNMT3A, ASXL1 and SRSF2 in patients with myelodysplastic syndromes and are stable during disease evolution. Am J Hematol. 2014; 89:137-144.
38. Chou WC, Chou SC, Liu CY, Chen CY, Hou HA, Kuo YY, Lee MC, Ko BS, Tang JL, Yao M, Tsay W, Wu SJ, Huang SY, Hsu SC, Chen YC, Chang YC, et al. TET2 mutation is an unfavorable prognostic factor in acute myeloid leukemia patients with intermediate-risk cytogenetics. Blood. 2011.
39. Cheson BD, Bennett JM, Kopecky KJ, Buchner T, Willman CL, Estey EH, Schiffer CA, Doehner H, Tallman MS, Lister TA, Lo-Coco F, Willemze R, Biondi A, Hiddemann W, Larson RA, Lowenberg B, et al. Revised recommendations of the International Working Group for Diagnosis, Standardization of Response Criteria, Treatment Outcomes, and Reporting Standards for Therapeutic Trials in Acute Myeloid Leukemia. J Clin Oncol. 2003; 21:4642-4649.