
Introduction
Antigen presentation through the binding of peptides to cellular HLA molecules modulates T cell response specificity, and thus determines cell antigenicity. CD8+ cytotoxic T lymphocytes (CTLs) have the potential to recognize tumor cells via specific peptide-HLA binding, leading to tumor regression following immunotherapy [1]. One class of antigens promoting this recognition is the tumor-associated self-antigens. However, neo-epitopes, that arise as a consequence of somatic mutations in the tumor, are another antigen subgroup that induces anti-tumor CTL responses [2, 3].
The main screening method currently used to identify neo-antigens relies on identification of tumor somatic mutations by whole-exome sequencing, followed by in silico analysis of candidate mutated peptides, to calculate their theoretical affinity for the patient’s HLA complex [4]. Synthetic peptides of the selected sequences are then pulsed on antigen presenting cells [2, 5], or loaded on tetramers [6], to evaluate their immunogenicity when introduced to autologous T- cells. The success of this method crucially depends on the accuracy of peptide-MHC binding prediction algorithms, which are often inadequate, and produce large numbers of candidate mutant peptides. Another method, that eliminates the need to predict peptide-MHC binding, is the use of tandem minigenes encoding each expressed cancer-specific mutation. These minigenes are expressed in the patients’ autologous antigen presenting cells and then tested for reactivity with the patients’ T cells [7, 8]. Subsequent validation of both the abovementioned neo-antigen screening methods is labor-intensive and time-consuming.
The collection of HLA class I (HLA-I) bound peptides expressed by a particular cell, otherwise known as the cell’s ‘HLA peptidome’, reflects the set of proteins found in that cell. The HLA peptidome serves as a unique immunological signature that can be selectively recognized by CTLs, potentially leading to cell lysis. Recent advances in mass spectrometry make it possible to analyze the HLA peptidome in great detail [9]. Here, we report the first analysis of the mutated HLA-I peptidome of human melanoma tumor cells, using a novel strategy, that combines whole-exome sequencing and mass spectrometry analysis. Using this method, we experimentally identified mutated peptides that are actually processed and presented by the tumor HLA molecules, and proceed to assess only those empirically identified candidates for testing immunogenicity.
Results
Identification of melanoma neo-antigens
To identify mutated antigens presented on human melanoma tumor cells, we re-sequenced the whole genomes of matched normal and metastatic tumor DNAs from a melanoma patient and identified their somatic mutations (Supplementary Table 1).
In parallel to our whole exome analysis, we performed immunoaffinity purification of the HLA molecules from the same patient’s melanoma cells, followed by capillary chromatography and tandem mass spectrometric analysis of HLA peptides. The mass spectrometry spectra were analyzed using MaxQuant [10] software tool and queried against the human proteome dataset (Uniprot) to which we manually added the amino acid changes corresponding to the mutations identified by our whole-genome sequencing.
Among the 4,958 peptides detected by mass spectrometry, we identified two patient-derived mutated peptides (Figure 1). The first peptide is a nonamer derived from the P677S alteration in Mediator of RNA polymerase II transcription subunit 15 (MED15) and the second peptide is an elevenmer derived from the S123L alteration in Tumor Protein D52-Like 2 (TPD52L2) protein. Peptide identification accuracy was validated by comparison of endogenous peptide spectra to synthetic peptide spectra (Supplementary Figure 1). Both wild-type and mutant transcripts corresponding to the peptides were expressed in the 12T tumor cell line (Supplementary Figure 2 and Supplementary Table 2).
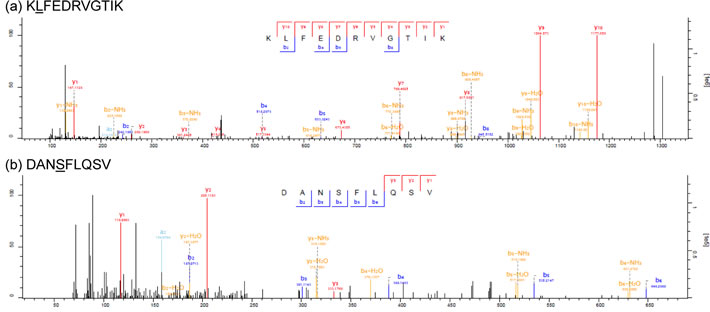
Figure 1: Tandem mass spectra of endogenous mutant peptides identified in 12T. a. KLFEDRVGTIK b. DANSFLQSV. Mutation sites are indicated with the amino acid underlined.
Immunogenicity evaluation of the identified neo-antigens
To evaluate the peptides’ ability to elicit an immune response we pulsed synthetic MED15 and TPD52L2 mutant peptides and their wild-type version, on Epstein bar virus (EBV)-transformed B cells established from the patients’ peripheral blood mononuclear cells (PBMCs) and co-cultured these cells with the autologous TILs. As seen in Figure 2, the mutated MED15 peptide stimulated interferon-γ (IFN-γ) release from the TILs, whereas the mutant TPD52L2 peptide did not show any response.
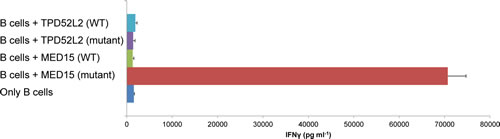
Figure 2: Response of the TIL to candidate neo-antigens identified from its autologous tumor. IFN-γ release measured after overnight co-culture of the TIL with autologous EBV transformed B cells that were pulsed with 1μM of the mutated or wild-type peptides. Error bars represent standard deviation of triplicates.
To determine the HLA allele to which the mutant MED15 peptide binds, we pulsed it on cells of the T2 line, which is B*51 positive [11], then cultured the cells with the patient’s TILs. Remarkably, the IFN-γ released following pulsing these cells with mutant MED15 peptide was comparable to that measured after TILs stimulation by the patient’s autologous tumor cells (17,000 pg ml−1 compared to 18,700 pg ml−1 with the autologous melanoma), indicating that the peptide binds the B*51 allele. Importantly, when cells of the T2 line were pulsed with the wild-type version of the MED15 peptide, a substantially weaker response was detected (300 pg ml−1 in the experiment detailed in Figure 3A).
Peptide titration assays further confirmed that the patient’s TILs were highly reactive with T2 cells pulsed with the mutated MED15 peptide, which was recognized at a minimum concentration of 10 nM (Figure 3B). Meanwhile, a response to the MED15 wild-type peptide was only observed when T2 cells were pulsed with peptide concentrations two orders of magnitude higher. Mutant MED15 peptide therefore appears to represent a naturally processed epitope recognized by the patient’s TILs (Figure 3B).
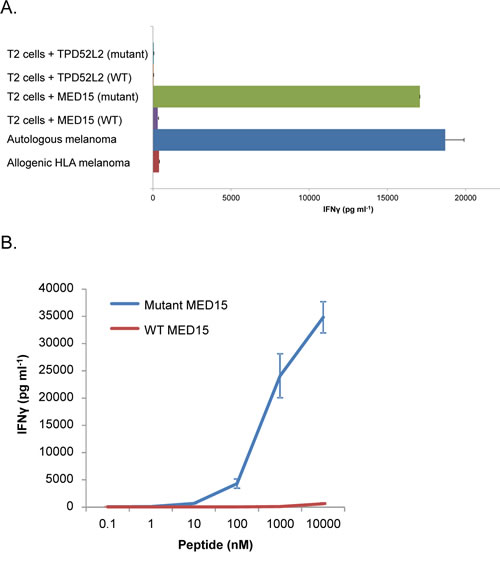
Figure 3: Response of the TILs to MED15 and TPD52L2 mutant and wild-type peptides in the context of HLA-B*51. A. IFN-γ release measured after overnight co-culture of the TILs with T2 cells that were pulsed with 1μM of the mutated or wild-type peptides or with the autologous and allogenic melanoma cells. B. IFN-γ release measured after overnight co-culture of 12T TILs with T2 cells that were pulsed with tittered concentrations of the MED15 mutated and wild-type peptides. Error bars represent standard deviation of triplicates.
To further evaluate TILs reactivity with the mutant MED15 peptide, we preformed intracellular IFN-γ staining after co-culturing the TILs with the autologous melanoma as a reference for total reactivity or with T2 cells pulsed with the mutant or wild-type MED15 peptides. About 21% of the TILs were reactive against the melanoma cells and about 9% were reactive against the T2 cells pulsed with the mutant MED15 peptide. Therefore, we estimate that approximately 40% of the reactive TILs specifically react with the mutant peptide (Figure 4), suggesting that it is an immunodominant epitope.
Prediction of neo-antigens using NetMHCpan algorithm
We were interested to compare our method to the currently prevalent algorithmic prediction method. To that end, we used The NetMHCpan 2.8 algorithm, to predict all possible neo-antigens derived from 12T non-synonymous mutations, for all the cells’ HLA alleles and between 8-12 amino acids in length. In total, 5,404 different peptides were predicted (Supplementary Table 3).
Binding affinity predictions for wild-type and mutant peptides of MED15 and TPD52L2 are in Supplementary Table 4. The mutant and wild-type TPD52L2 peptides were predicted to bind the A*03 allele strongly and weakly, respectively and the MED15 mutant and wild-type peptide were predicted to weakly bind the B*51 allele.
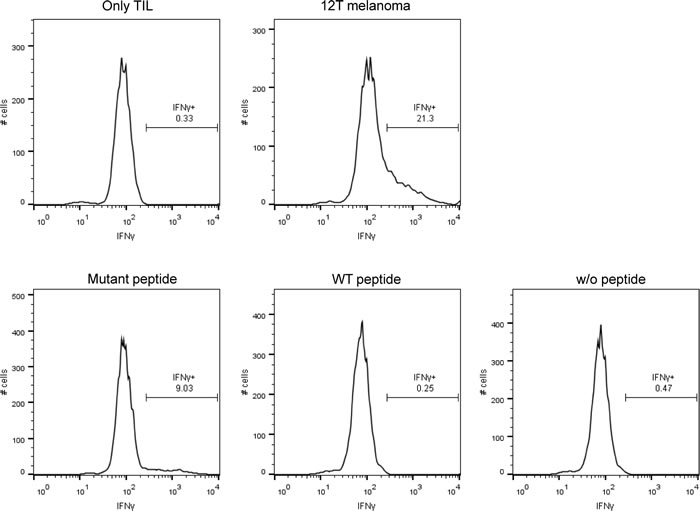
Figure 4: Intracellular staining of IFN-γ after stimulation with the peptides and melanoma. Intracellular IFN-γ was stained after 6 hours co-culture of the TILs with T2 cells that were pulsed with 1μM of the mutated or wild-type peptides or with the autologous melanoma.
Discussion
This report is the first to show that a combination of HLA peptidomics and autologous cancer exome data can be used to reveal cancer-specific antigens which expose neo-antigen, leading to a specific T cell response in humans. Currently, most neo-antigen identification studies use algorithms, such as NetMHCpan [4], to predict which mutated peptide sequences can bind the patients’ HLA alleles. As the HLA peptide processing steps are not fully known, and therefore cannot be taken into consideration within the prediction algorithm, these predictions yield a large number of false positive hits, that must then all be screened. Several criteria are usually used to reduce the number of screened peptides. These include: predicted binding affinity, length of the predicted peptides and their expression levels. Applying such limitations can exclude peptides such as the mutated MED15 peptide which was predicted to weakly bind the B*51 molecule. In contrast, isolation of HLA peptides directly from tumor cells ensures that only peptides that are actually processed in the cells, bind the HLA and are presented on the cells’ surface are tested for immunogenicity. Therefore more concerning than the added labor entailed by the multitude of candidate peptides, is the fact that the currently utilized filtering criteria also run the risk of producing false negative hits, thus missing peptides that might actually bind the HLA, as in the case demonstrated in this work.
The proposed method, depicted in Supplementary Figure 3, allows for identification of neo-antigens in significantly less time than currently used screening methods. While other methods require whole exome sequencing and identification of somatic mutations prior to wide-scale-screen with synthetic peptides, tetramers or construction of minigenes, mass spectrometry analysis of the peptides isolated from the tumor can be done in parallel, saving valuable time. Subsequent immunological assays for validation of the peptides identified by mass spectrometry only require synthesis of a few peptides compared to the hundreds of peptides that are needed for screening predicted sequences. Importantly, the number of reactive neo-antigens identified using this method is similar to the number of peptides identified by the other currently used methods (between 0-3 peptides per sample) [12, 13, 14, 6, 15].
As in all mass spectrometry analyses, identification is biased toward the most abundant peptides. Therefore, identifying a larger number of total HLA peptides will increase the chance of discovering neo-antigens. In this study HLA peptides were isolated from 2*108 cells, which is the estimated number of cells in clinically detectable solid human tumors [16], making this method applicable for neo-antigen identification directly from the isolated tumor, without the need to establish a tumor cell line.
Finally, using flow cytometry analysis of intracellular IFNγ, we demonstrated that about 40% of TILs reactivity against the melanoma cells is attributable to specific reactivity to the mutated MED15 peptide. This result corroborates previous observations showing that autologous T- cell response is driven mainly by mutated neo-antigens [17, 18].
Here we provide, to our knowledge, the first demonstration of an approach that combines two powerful tools, forming an alternative system for identifying immunogenic neo-epitopes in human cancer. This approach may be used clinically as a potential diagnostic strategy for personalized immunotherapy, not only for melanoma patients but for a variety of additional tumor types where neo-antigens play a critical role in anti-tumor reactivity.
Materials and Methods
Tumor tissues
All DNA samples used in this study were derived from metastases. Samples used for whole-exome capture were extracted from cell lines established directly from patient tumors as described previously [19]. We identified somatic mutations by comparing the tumor data to the matched normal tissue and identified 2,997 potential somatic mutations in 2,192 different genes. Of these alterations, 1019 caused amino acid changes consistent with prior data [19].
12T cells express the HLA-A*02, A*03, B*08, B*51, C*01 and C*07 class I alleles.
Production and purification of membrane HLA molecules
Cells lysate from two pellets of 12T cell lines were used for immunoaffinity purification of HLA molecules with their bound peptides, using the W6/32 mAb, bound to Amino-Link beads (Thermo Scientific, as in [20, 21]). The HLA peptides were recovered from HLA molecules with 1% TFA followed by separation of the peptides from the proteins contaminants by binding the eluted fraction to disposable reversed-phase C18 columns (Harvard Apparatus). Elution of the peptides was done with 30% acetonitrile and 1% TFA. The eluted peptides were cleaned also by C18 stage tip as in [22] .
Identification of the HLA peptides
The HLA peptides were dried by vacuum centrifugation, re-solubilized with 0.1% Formic acid and resolved on capillary reversed phase chromatography on 0.075x200 mm laser-pulled capillaries, self-packed with 3µ Reprosil-Aqua C18 [23]. Electrospray tandem mass spectrometry was performed with the Q-Exactive-Plus mass spectrometer (Thermo Scientific). The MS data was analyzed by MaxQuant [10] version 1.4.1.2, with 5% FDR and by Sequest using the Proteome Discoverer version 1.4.1.14 (Thermo Scientific). Peptide identifications were based on the human section of the Uniprot database (http://www.uniprot.org) from February 2014 combined with the wild type and mutant protein sequences.
Analysis of T cell responses
TILs were isolated and expanded from fresh tumor digests as previously described [24] and EBV-transformed B-cell lines were established from PBLs of the patient (EBV was a gift from Prof. Ephraim Gazit). The reactivity of the TILs were evaluated by incubating patient derived EBV transformed B cells or T2 cells (with antigen-processing defects that allow for the efficient loading of exogenous peptides [24]) with the candidate peptides at a concentration of 1 μM for 2 h at 37 °C. Following three washing steps, TILs were added at a ratio of 1:1 (1e5 cells), and the amounts of soluble IFN-γ secreted from the TILs cultured overnight with peptide-pulsed target cells or tumor cells were measured by ELISA assay (Biolegend).
Intracellular staining of IFN-γ
TILs were co-cultured with T2 cells pulsed with peptides or autologous melanoma cells for 6 hours. Then they were fixated using the Transcription Factor Buffer Set (BD Pharmingen), and stained anti IFN-γ, anti CD3 and anti CD8 (BD biosciences). Cells were analyzed using the FACSCalibur flow cytometer (BD biosciences). TILs were gated as positive for CD3 and CD8.
cDNA sequencing
RNA was purified from cultured cells using RNAeasy mini kit (Qiagen) and cDNA was prepared using iScript reverse transcription supermix for RT-qPCR (Bio-Rad). The regions containing the mutation sites were amplified by PCR using KAPA HiFi HotStart ReadyMix PCR Kit (KAPAbiosystems). The PCR primer pairs used were: MED15 (5′- GGTGTCCCCTGAAGACCTTG) and (5′- CTGGGCCCAGGTGTTGAG); TPD52L2 (5′- GCCGGCCAAGATATCAACCT) and (5′- GATCCGACAGGGGCTTGTC). PCR was performed using 100 ng cDNA and the following parameters: 1 cycle at 95°C for 3 min; 30 cycles at 98°C for 20 sec, annealing temperature of 65°C for 15 sec, 72°C for 60 sec; and 1 cycle at 72°C for 1 min. PCR products were cleaned with QIAquick PCR Purification Kit (Qiagen) and sequenced using a 3730 DNA Analyzer (ABI). Sequencing primers were the same as the PCR primers.
RNA sequencing
RNA was extracted from 12T cells using RNeasy mini kit (Qiagen). 1 μg of total RNA was processed using the TruSeq RNA Sample Preparation Kit v2 protocol (Illumina). Libraries were evaluated by Qubit and TapeStation. Sequencing libraries were constructed with barcodes to allow multiplexing of few samples on one lane of Illumina HiSeq 2500 V4 instrument. About 60 million single-end 60-bp reads were sequenced.
The TopHat (v2.0.10) was used to align the reads to the human genome (hg19), and counting reads on hg19 refseq genes (downloaded from igenomes) was done with HTSeq-count (version 0.6.1p1).
RPKM values were calculated using Total exonic length for each gene, which was calculated using the bioconductor GenomicFeatures package.
Prediction of neo-antigens using NetMHCpan 2.8 algorithm
The residues surrounding the amino acids resulting from nonsynonymous mutations were scanned to identify candidate 8-12 mer peptides that were predicted to bind with high affinity (strong binders, %Rank≤0.5 or IC50≤50) or low affinity (weak binders, 0.5≤%Rank≤2 or 50≤IC50≤500) to the cells’ HLA-I alleles using the NetMHCPan 2.8 algorithm [4].
Acknowledgments
We thank the Genomics and Bioinformatic units at the Israel National Center for Personalized Medicine (INCPM) for help with sample preparation, sequencing and analyzing the data, as well as Dr. Isadora Cohen and Dr. Alona Keren-Paz for critical reading and editing of the manuscript. This work was supported by the Intramural Research Programs of the National Cancer Institute. Y.S. is supported by the Israel Science Foundation grant numbers 1604/13 and 877/13, the ERC (StG-335377), by the Henry Chanoch Krenter Institute for Biomedical Imaging and Genomics, the estate of Alice Schwarz-Gardos, the estate of John Hunter, the Knell Family, the Peter and Patricia Gruber Award and the Hamburger Family. E.M., N.S., J.S. and M.J.B. are supported by Haya and Nehemia Lemelbaum. A.A. is supported by The Greta Koppel SCLC fund.
Authors’ contributions
S.K., E.B., E.M.S, M.J.B, A.A and Y.S. designed the study. S.K., E.B., E.M.S. and N.S. performed the experiments. N.Q, J.K.T., J.S, S.A.R, M.J.B., and Y.S. collected and analyzed the melanoma samples. All authors contributed to the final version of the paper.
COnflicts of INTERESTS
The authors declare no competing financial interests.
References
1. Hinrichs CS and Rosenberg SA. Exploiting the curative potential of adoptive T-cell therapy for cancer. Immunological reviews. 2014; 257:56-71.
2. Robbins PF, Lu YC, El-Gamil M, Li YF, Gross C, Gartner J, Lin JC, Teer JK, Cliften P, Tycksen E, Samuels Y and Rosenberg SA. Mining exomic sequencing data to identify mutated antigens recognized by adoptively transferred tumor-reactive T cells. Nat Med. 2013.
3. Tran E, Turcotte S, Gros A, Robbins PF, Lu YC, Dudley ME, Wunderlich JR, Somerville RP, Hogan K, Hinrichs CS, Parkhurst MR, Yang JC and Rosenberg SA. Cancer immunotherapy based on mutation-specific CD4+ T cells in a patient with epithelial cancer. Science. 2014; 344:641-645.
4. Nielsen M, Lundegaard C, Blicher T, Lamberth K, Harndahl M, Justesen S, Roder G, Peters B, Sette A, Lund O and Buus S. NetMHCpan, a method for quantitative predictions of peptide binding to any HLA-A and -B locus protein of known sequence. PLoS ONE. 2007; 2:e796.
5. Pritchard AL, Burel JG, Neller MA, Hayward NK, Lopez JA, Fatho M, Lennerz V, Wolfel T and Schmidt CW. Exome sequencing to predict neoantigens in melanoma. Cancer immunology research. 2015; 3:992-8. doi: 10.1158/2326-6066.CIR-15-0088.
6. Cohen CJ, Gartner JJ, Horovitz-Fried M, Shamalov K, Trebska-McGowan K, Bliskovsky VV, Parkhurst MR, Ankri C, Prickett TD, Crystal JS, Li YF, El-Gamil M, Rosenberg SA and Robbins PF. Isolation of neoantigen-specific T cells from tumor and peripheral lymphocytes. The Journal of clinical investigation. 2015; 125:3981-3991.
7. Rosenberg SA and Restifo NP. Adoptive cell transfer as personalized immunotherapy for human cancer. Science. 2015; 348:62-68.
8. Tran E, Ahmadzadeh M, Lu YC, Gros A, Turcotte S, Robbins PF, Gartner JJ, Zheng Z, Li YF, Ray S, Wunderlich JR, Somerville RP and Rosenberg SA. Immunogenicity of somatic mutations in human gastrointestinal cancers. Science. 2015; 350:1387-90. doi: 10.1126/science.aad1253.
9. Pritchard AL, Hastie ML, Neller M, Gorman JJ, Schmidt CW and Hayward NK. Exploration of peptides bound to MHC class I molecules in melanoma. Pigment cell & melanoma research. 2015; 28:281-294.
10. Cox J and Mann M. MaxQuant enables high peptide identification rates, individualized p.p.b.-range mass accuracies and proteome-wide protein quantification. Nature biotechnology. 2008; 26:1367-1372.
11. Young NT, Mulder A, Cerundolo V, Claas FHJ and Welsh KI. Expression of HLA class I antigens in transporter associated with antigen processing (TAP)-deficient mutant cell lines. Tissue Antigens. 1998; 52:368-373.
12. Robbins PF, Lu YC, El-Gamil M, Li YF, Gross C, Gartner J, Lin JC, Teer JK, Cliften P, Tycksen E, Samuels Y and Rosenberg SA. Mining exomic sequencing data to identify mutated antigens recognized by adoptively transferred tumor-reactive T cells. Nature medicine. 2013; 19:747-52. doi: 10.1038/nm.3161.
13. Tran E, Turcotte S, Gros A, Robbins PF, Lu YC, Dudley ME, Wunderlich JR, Somerville RP, Hogan K, Hinrichs CS, Parkhurst MR, Yang JC and Rosenberg SA. Cancer Immunotherapy Based on Mutation-Specific CD4+T Cells in a Patient with Epithelial Cancer. Science. 2014; 344:641-645.
14. Tran E, Ahmadzadeh M, Lu YC, Gros A, Turcotte S, Robbins PF, Gartner JJ, Zheng Z, Li YF, Ray S, Wunderlich JR, Somerville RP and Rosenberg SA. Immunogenicity of somatic mutations in human gastrointestinal cancers. Science. 2015; 350:1387-1390.
15. Pritchard AL, Burel JG, Neller MA, Hayward NK, Lopez JA, Fatho M, Lennerz V, Wolfel T and Schmidt CW. Exome Sequencing to Predict Neoantigens in Melanoma. Cancer immunology research. 2015; 3:992-998.
16. Del Monte U. Does the cell number 10(9) still really fit one gram of tumor tissue? Cell Cycle. 2009; 8:505-506.
17. Lennerz V, Fatho M, Gentilini C, Frye RA, Lifke A, Ferel D, Wolfel C, Huber C and Wolfel T. The response of autologous T cells to a human melanoma is dominated by mutated neoantigens. Proc Natl Acad Sci USA. 2005; 102:16013-16018.
18. Lu YC, Yao X, Li YF, El-Gamil M, Dudley ME, Yang JC, Almeida JR, Douek DC, Samuels Y, Rosenberg SA and Robbins PF. Mutated PPP1R3B Is Recognized by T Cells Used To Treat a Melanoma Patient Who Experienced a Durable Complete Tumor Regression. Journal of immunology. 2013; 190:6034-6042.
19. Wei X, Walia V, Lin JC, Teer JK, Prickett TD, Gartner J, Davis S, Program NCS, Stemke-Hale K, Davies MA, Gershenwald JE, Robinson W, Robinson S, Rosenberg SA and Samuels Y. Exome sequencing identifies GRIN2A as frequently mutated in melanoma. Nature genetics. 2011; 43:442-446.
20. Bassani-Sternberg M, Barnea E, Beer I, Avivi I, Katz T and Admon A. Soluble plasma HLA peptidome as a potential source for cancer biomarkers. Proc Natl Acad Sci USA 2010; 107:18769–18776.
21. Milner E, Gutter-Kapon L, Bassani-Strenbrg M, Barnea E, Beer I and Admon A. The effect of proteasome inhibition on the generation of the human leukocyte antigen peptidome. Molecular & cellular proteomics. 2013; 12:1853-64. doi: 10.1074/mcp.M112.026013.
22. Rappsilber J, Ishihama Y and Mann M. Stop and go extraction tips for matrix-assisted laser desorption/ionization, nanoelectrospray, and LC/MS sample pretreatment in proteomics. Anal Chem. 2003; 75:663-670.
23. Ishihama Y, Rappsilber J, Andersen JS and Mann M. Microcolumns with self-assembled particle frits for proteomics. J Chromatogr A. 2002; 979:233-239.
24. Topalian SL, Muul LM, Solomon D and Rosenberg SA. Expansion of human tumor infiltrating lymphocytes for use in immunotherapy trials. Journal of immunological methods. 1987; 102:127-141.