
INTRODUCTION
Thanks to an effective integration of surgery, chemotherapy and, in selected cases, radiotherapy, the overall survival rate for patients affected with Wilms tumor (WT), the most frequent pediatric renal tumor, now exceeds 90%. However, only approximately half of children who suffer from tumor relapse reach second durable remission. Aiming at tailoring therapeutic intensification at relapse, an international consensus has recognized three post-relapse risk groups according to the initial treatment received (which in turn is largely dictated by tumor stage and histology) [1]. However, the clinical behavior within the different risk subgroups has yet to be established, since the probability of response to conventional therapies at recurrence is extremely variable [2]. Therefore, the understanding of molecular features underlying recurrent tumors could help to develop effective treatments. Noteworthy, the next generation of early-phase clinical trials, initially conducted in the context of resistant/recurrent tumors, will probably rely on molecular-targeted drugs.
Different studies, aimed at characterizing the molecular genetics of WTs that are likely to relapse, identified some anomalies possibly associated with an adverse outcome, mainly loss of heterozygosity (LOH) at chromosomes 1p and/or 16q [3–5], copy number (CN) gain at chromosome 1q [6–9], and allelic imbalances at chromosome arms 1q, 3p, 3q, and 14q [8].
WT is genetically heterogeneous, and different genes are associated with its development, among which WT1 at 11p13, the WT2 locus at 11p15.5, WTX on chromosome Xq11.2 and CTNNB1 on 3p22.1 (reviewed in Huff [10]). Mutations of TP53, mapped to 17p13.1, are associated with anaplastic histology [11].
Recently, mutations affecting the SIX1 and SIX2 genes and miRNA processor genes (miRNAPGs), including DICER1, DROSHA and DGCR8, have been reported in WTs [12, 13]. SIX1/2 and DROSHA/DGCR8 mutations underlie high-risk blastemal-type WTs in patients pre-operatively treated according to the protocols of the Sociètè International d’Oncologie Pediatrique (SIOP). In addition, the co-occurrence of SIX1/2 and DROSHA/DGCR8 mutations resulted in worse outcome in favorable histology WTs in patients receiving primary nephrectomy according to the protocols of the Children’s Oncology Group (COG) [12, 13]. Hence, anomalies affecting these novel WT genes seem to be potentially associated with poor prognosis in WT.
The extreme difficulty in recruiting paired primary/recurrent WT samples of the same patient has so far precluded comprehensive studies comparing their genetic profiles. Only one study previously focused on the genomic evolution that leads to tumor recurrence [14]. From a comparison of 28 primary relapsing WTs and 10 unpaired recurrences, CN gains at a series of non contiguous clones spanning more than 80 Mb from region 1q23.3 to 1q44, and CN losses at 17p were statistically more frequent in recurrent tumors. Comparison of 10 paired tumors at diagnosis and relapse demonstrated CN gains at 5p, 8p12, 15q, 16p and 20q and CN losses of 11q and 17p as events acquired de novo in two recurrent tumors [14].
To identify genetic and genomic anomalies acquired and/or shared by recurrences, we performed genomic profiling by whole-genome single nucleotide polymorphism (SNP) arrays with an average resolution of 8 Kb, and characterized alterations affecting WT-associated genes, in nine primary tumors from eight patients (including a bilateral case) and in their respective eight recurrences. Through this approach, we identified common features possibly underlying the evolution that leads a primary disease to recur.
RESULTS
Chromosomal regions involved in CN anomalies (CNAs) and allelic ratio (AR) anomalies
Whole-genome chromosomal anomalies were examined by SNP array analysis in a set of paired primary tumors and corresponding recurrences (Figure 1). Out of four patients with pre-treated primary tumors, two (36I and 39I) showed multiple gross anomalies involving entire chromosomes/chromosome arms, including 1q CN gain and allelic imbalance and 16q CN loss and LOH, and three tumors from the two remaining cases (74I and 262I, both right and left side) only small focal anomalies (Figure 1). Out of four patients treated with initial nephrectomy at diagnosis, three (30I, 51I and 111I) displayed multiple gross chromosomal anomalies, including 1q CN gain and allelic imbalance (30I), 1p and 16q CN loss and LOH (51I), and one (113I) had only small focal anomalies. Recurrent tumor specimens had aberrations involving entire chromosomes/chromosome arms in all the patients.

Figure 1: Whole-genome copy number and allelic ratio anomalies in the primary diseases and in their respective paired recurrences. Along the genome, data for CN aberrations as well as AR events are displayed for each sample individually on separate rows. CN aberrations are displayed above the dotted line: CN gains are represented by green marks, CN losses by red marks. Allelic events are displayed below the dotted line: allelic imbalances by purple marks and LOH by dark yellow marks. Regions involved in homozygous copy loss are indicated by thicker red marks. The position of constitutional CNVs is indicated in pink. I: primary tumor; II: recurrent tumor.
Several genetic events, both CNAs and AR anomalies, were observed in regions where constitutional CN variations (CNVs) are located (http://dgv.tcag.ca/dgv/app/home). Among the focal aberrations detected, case no. 30 displayed a focal MYCN amplification (chr2:15,495,567-16,243,938) both in the primary tumor (previously reported [8]) and in the recurrence (Figure 1).
Chromosomal regions involved in CNAs and AR anomalies acquired de novo in recurrences
We considered an anomaly as acquired de novo in recurrences when in the same chromosomal region the primary tumor had no aberrations or an aberration of different nature. Supplementary Table S1 reports the list of CNAs and AR anomalies that were acquired de novo in two or more recurrences. Allelic imbalance of chromosome 1q21.1-q44 was the only anomaly acquired by three recurrences. CN losses of chromosomes 7p22.1, 10p15.3-p14, 11q14.1, 11q14.3, 11q22.1-q25, 16q11.2-q22.1, 16q22.2-q23.3, allelic imbalances of chromosomes 3p26.3-p11.2, 3q11.2-q25.2, 6p22.1-p21.33, 6q24.3, 8p11.21-p11.1, 8q11.1-q11.21, 8q22.3, 16q11.2-q24.3, 17p13.3-p11.2, and LOH of chromosome 10p15.3-p14 were acquired by two recurrent tumors. Some of these anomalies clustered at specific chromosomal arms and encompassed large segments, while others were focal, scattered along the genome and often overlapped with constitutional CNVs (Supplementary Table S1), and therefore might represent spurious observations.
Chromosomal regions involved in CNAs and AR anomalies shared among recurrences
Supplementary Table S2 reports CNAs and AR anomalies that were present in at least three recurrent tumors. CN gains of chromosome 1q21.2-q32.1 and CN losses of chromosomes 16q11.2-q22.1 and 16q22.2-q23.3 were CNAs commonly present in recurrences. AR abnormalities included allelic imbalances involving chromosomes 1q21.1-q44, 3p26.3-p11.2, 3q11.2-q25.2, 8p11.21-p11.1, 8q11.1-q11.21, 8q22.3, 17p12-p11.2, and LOH of chromosomes 8q11.21-q11.22, 11q22.1, 13q31.2. Again, we could distinguish between anomalies involving large chromosomal segments, and focal, scattered events. Anomalies overlapping with constitutional CNVs are reported in Supplementary Table S2.
Comparison of chromosomal regions involved in CNAs and AR anomalies acquired de novo in recurrences with those shared among recurrences
We compared chromosomal regions involved in anomalies possibly acquired de novo in recurrences with those shared among them, and found that CN losses of chromosomes 16q11.2-q22.1, 16q22.2-q23.3, allelic imbalances of chromosomes 1q21.1-q44, 3p26.3-p11.2, 3q11.2-q25.2, 17p12-p11.2, together with a number of focal scattered anomalies were both acquired de novo in more than one recurrence and shared by at least three recurrences (Supplementary Table S3).
WT1, CTNNB1, WTX, TP53 genetic and genomic anomalies
Sequence analysis disclosed no WT1 mutations in primary and recurrent tumors. However, SNP array analysis detected anomalies involving the 11p chromosomal region, where WT1 is mapped, in one case, no. 39 (I and II), that showed CN neutral LOH. No WTX gene mutations were identified in all tumor samples, but in a male patient, case no. 30 (I and II), a focal region of deletion on chromosome X, encompassing the WTX locus and thus deleting the only present allele, was found. Among female patients, case no. 111 (I and II) displayed a ca. 6.3 Mb region of CN neutral LOH affecting the WTX gene and the recurrent tumor 39II showed CN neutral LOH involving most of the X chromosome.
A heterozygous CTNNB1 c.133T>C, (p.S45P), mutation was found in one case, no. 36 (I and II). Chromosomal anomalies affecting the entire 3p region, where the CTNNB1 gene is mapped, were found in case no. 39 (I and II) and in the recurrence 113II, which showed allelic imbalance, and in recurrence 111II, which showed CN gain and allelic imbalance.
In one case, no. 111, both primary and recurrent tumors showed CN loss and LOH involving the 17p chromosomal region, where the TP53 gene is mapped. Although histologically this patient was described as showing diffuse anaplasia both in primary and recurrent disease, we identified a hemizygous TP53 c.817C>T (p.R273C) mutation only in the recurrence. This was possibly due to the absence of anaplastic cells in the primary tumor specimen that was used for DNA extraction.
Chromosomal anomalies affecting the 17p region were also identified in the recurrence 74II, which showed CN loss and LOH, in the primary tumor 51I, and in the recurrences 113II and 262II, which showed allelic imbalances.
SIX1, SIX2, and miRNAPGs genetic and genomic anomalies
The previously reported SIX1 hotspot c.530A>G (p.Q177R) mutation was found in heterozygosity in three recurrences: 51II, 74II and 262II. In these samples, cDNA sequencing demonstrated expression of both the mutated and wild-type alleles. While in two of the corresponding primary samples (51I and 262, both right and left side) we excluded the presence of the mutation, genomic sequencing of tumor no. 74I showed the subclonal presence of the mutated allele and cDNA sequencing showed the expression of both the wild-type and mutated alleles (Supplementary Figure S1). All SIX1 identified mutations were somatic. Chromosomal anomalies affecting the 14q region, where the SIX1 gene is mapped, were found in tumor no. 51I, which showed allelic imbalance, and in case no. 39II, which showed CN loss and LOH. No SIX2 mutations were found, and only one recurrence, 113II, showed allelic imbalance at 2p, where the SIX2 gene is mapped. DROSHA heterozygous mutations were found in case no. 51, where the c.3451G>C (p.D1151H) was present in both the primary tumor and in the recurrence, in recurrence 74II, bearing the c.3559C>A (p.Q1187K) mutation, and in recurrence 262II, showing the c.3452A>G (p.D1151G) mutation. All mutations, initially identified through cDNA sequencing, were eventually confirmed at genomic level. Primary tumors of cases no. 74I and 262I (both left and right side) showed no DROSHA mutation (Supplementary Figure S2). All DROSHA identified mutations were somatic. No chromosomal anomalies affecting the 5p13.3 region, where DROSHA is mapped, were found. No mutations affecting DICER1 and DGCR8 were found, but a rare germline variant, rs202208301 (allelic frequency in NHLBI Exome Sequencing Project (2) = 1/13006), was observed in case no. 36. Allelic imbalance for the 14q chromosomal region, in which DICER1 is mapped, was found in primary tumor no. 51I, and CN loss and LOH affecting the 22q DGCR8 chromosomal region in recurrence 111II.
The finding that SIX1 and DROSHA mutations were mainly identified in recurrences prompted us to expand the analysis on primary tumors using DNA from multiple blocks of formalin fixed paraffin embedded (FFPE) specimens and, when possible, from different histological components. While in cases no. 51I and no. 262I (both right and left side tumors) no SIX1 mutation was detectable in the DNA from any of the different blocks from the primary tumors investigated, case no. 74I showed SIX1 mutation in all the specimens. DROSHA mutations were identified in the DNA of one of six blocks of case no. 51I, in the DNA of two of three blocks of case no. 74I, and in the DNA of two of four specimens from the right side tumor of case no. 262I, but in none of the four specimens from the left side tumor of the same patient (Supplementary Figures S1 and S2).
Figure 2 summarizes the clinical and molecular characteristics of the patients and of their tumors, including the occurrence of alterations in the investigated WT-associated genes, as well as the presence/absence of the foremost proposed molecular prognostic signatures.
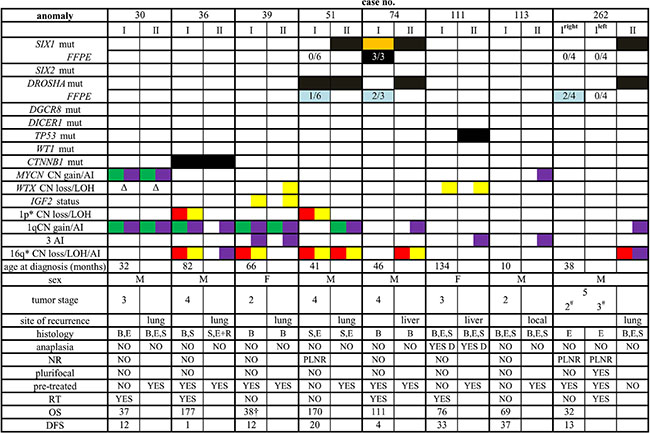
Figure 2: Clinical, pathological and molecular features of the patients investigated. I: primary tumor; II: recurrent tumor (“site of recurrence” indicates the site from which sample II was taken), mut: mutation, the presence of somatic mutation is indicated by a black box, by an orange box when subclonal, and by a light blue box when present only in a few of the investigated specimens (indicated by numbers). Chromosomal anomalies are reported as follows: green, CN gain; red, CN loss; yellow, LOH; purple, allelic imbalance (AI), *anomalies < 1 Mb have not been reported. Δ, deletion of the only present allele. All data but those in the line with “FFPE” (formalin fixed paraffin embedded) tag refer to results obtained on frozen material. M, male; F, female; #local stage; B, blastemal component; E, epithelial component; S, stromal component; R, rhabdomyoblastic elements; D, diffuse anaplasia; NR, nephrogenic rests; PLNR, perilobar nephrogenic rests; RT, radiotherapy; OS, overall survival; DFS, disease-free survival; †dead of disease.
DISCUSSION
In the present study we describe the findings observed in a cohort of eight pairs of primary/recurrent WTs analyzed by whole-genome SNP array, and we investigated the role in tumor progression of a group of genes, previously reported to be associated with poor outcome in this malignancy [12, 13]. We initially focused on genomic anomalies acquired de novo by at least two recurrences. Eventually, we selected aberrations present in at least three recurrent samples (shared events), irrespective of whether they were de novo or already present in the primary tumor. In fact, we argued that, while in some cases newly acquired anomalies, not present in the primary disease, are needed for the development of recurrences, in others the recurrence may originate from primary tumors that have already accumulated particularly aggressive molecular characteristics. Finally, in order to identify the possibly more relevant aberrations in recurrent disease, we selected anomalies in common between “de novo” and “shared” events.
Chromosomal events acquired de novo by more than one recurrence were CN losses at chromosomes 10p15.3-p14, 11q22.1-q25, 16q11.2-q23.3, allelic imbalances at chromosome 1q21.1-q44, at chromosome 3, 16q11.2-q24.3, 17p13.3-p11.2 and LOH at 10p15.3-p14, plus a number of small focal anomalies. Allelic imbalances at chromosomes 1q and 3 were previously identified with higher frequency in relapsing compared to non relapsing primary WTs [8]. The identified CN losses and allelic imbalances at 16q overlap with the small scattered regions that we found as associated with relapse and with prognostically significant chromosomal regions reported by others [3, 4, 8]. Although the different technical approach makes it difficult a comparison with previously published data on paired primary/recurrent samples, CN loss at chromosome 11q is the only anomaly identified as de novo event in at least two recurrences in both our and previous study [14]. CN loss and LOH at 10p15.3-p14 and allelic imbalance at 17p13.3-p11.2 represent newly reported de novo anomalies acquired by at least two recurrences. However, it must be noted that, since our SNP array-based analysis was performed on a single specimen from each tumor, our characterization of the repertoire of genetic aberrations present in the primary tumor mass might have been incomplete, because of tumor heterogeneity. This makes difficult to state with certainty that those events classified as acquired de novo were not already present in a different area of the primary tumor. Furthermore, in sample 36II, tumor cells were intermingled with normal lung cells, leading to a reduction in the representation of the genetic events of tumor cells.
Chromosomal events shared by three or more recurrent tumors were CN gain at chromosome 1q21.2-q32.1, CN losses at 16q11.2-q23.3, allelic imbalances at chromosome 1q21.1-q44, chromosome 3 and chromosome 17p12-p11.2, together with a number of small focal anomalies. Anomalies in common between “de novo” and “shared” events included CN losses at chromosome 16q11.2-q23.3, allelic imbalances at chromosome 1q21.1-1q44, at chromosome 3 and chromosome 17p12-p11.2, and focal scattered anomalies, many of which representing CNVs.
The sequence analysis of a group of well established WT-related genes, including WT1, CTNNB1 and WTX, was not suggestive of any of these genes being involved in disease progression. TP53 mutation in recurrence 111II was related to the presence of anaplasia [11], which is considered to be associated with tumor resistance to therapy [15]. The SIX1 p.Q177R hotspot mutation was found in three out of the eight recurrences: in two cases it appeared to be acquired de novo, whereas in one it was already present in all the three different FFPE blocks investigated from the primary tumor and as a subclonal anomaly in the frozen primary tumor specimen. Mutations affecting the RNAse IIIB domain of DROSHA were identified in the same three recurrences: two of them derived from primary tumors displaying the presence of perilobar nephrogenic rests (PLNRs), in keeping with previous data [12]. In all the three cases, the mutation was already present in at least one of the DNA samples from multiple areas of the corresponding primary tumors, but never in all samples. Interestingly, primary tumor no. 74I, the only one in which some areas presented the co-occurrence of SIX1 and DROSHA mutations, was a pre-treated blastemal-type WT. Overall, our data suggest that in a fraction of relapsing WT patients SIX1 and DROSHA mutations might be (whether spatially or temporally) heterogeneous events within the primary tumor, which are positively selected in the process of tumor progression to recurrence. In addition, our findings provide clues on the molecular mechanism explaining the association of the above alterations with poor prognosis.
SIX1, SIX2 and miRNAPGs have been reported to be involved in regulating renal development [16–18]. This is consistent with the recent report of the occurrence of mutations of these genes in a fraction of WTs, a malignancy that develops as a consequence of a derangement of kidney embryogenesis [12, 13, 19]. In the latter studies, these mutations were observed at relatively high allelic fractions, suggesting that they represent early events in tumor development. Our data add to this scenario the possibility that alterations of the SIX1 and DROSHA genes occur also at later stage, driving tumor evolution toward chemotherapy resistance and recurrence.
In conclusion, we identified genomic and genetic events, previously associated with poor prognosis in primary relapsing WTs, that appear to be present in a significant fraction of recurrent tumors, possibly as de novo events in a subset of cases. We believe that these observations are noteworthy, because they confirm the importance of these anomalies in driving tumor progression and recurrence, and provide further evidence that primary tumors already bearing these defects may deserve particular clinical attention. In fact, the characterization of the full spectrum of molecular events involved in the evolution from primary to recurrent tumors might provide new insights into the design and planning of future clinical trials. A major limitation of this study is represented by the small number of cases analyzed, that might have biased our conclusions. As already anticipated, this is due to the rarity of WT recurrences. Therefore, only the exploitation of ongoing international cooperative initiatives will allow to verify, and possibly confirm on a greater number of paired cases, the observations here reported.
MATERIALS AND METHODS
Patients and material
The study included eight patients with WT enrolled into the Associazione Italiana Ematologia Oncologia Pediatrica (AIEOP) protocols TW-2003 (ongoing) and TW-1992 (closed). The treatment protocols had been approved by the participating institutions’ review boards, and specific informed consent to the use of biological samples for the aim of the study obtained from the parents or the guardians of all patients. Eligibility to the study was exclusively based on the availability of matched samples from diagnosis (primary tumor, I) and recurrence (recurrent tumor sample, II). Clinico-pathological characteristics and survival data are depicted in Figure 2. All patients had sporadic tumors, and without signs of any syndromic condition predisposing to WT development.
DNA and RNA were extracted from frozen tumor tissue fragments sampled by a pathologist, using conventional methods. A detailed histology of the frozen tumor specimen used in the study is not available. For selected cases, multiple blocks of FFPE material of the primary tumor mass/masses were checked and macro dissected (> 95% viable tumor cells) to investigate more exhaustively the primary neoplasm and its different histological components. In particular, six different blocks were investigated for tumor no. 51I, corresponding to the blastemal component (four blocks), stromal component with rhabdomyoblastic differentiation (one block), epithelial component with papillary features (one block). Three different blocks with blastemal component were investigated for tumor no. 74I. For the tumor no. 262I of the right side, three different blocks were investigated: one block with epithelial component with papillary features, one block with epithelial component with tubular structures, and one block from which two different areas were macro dissected, one with epithelial component with glomerular structures, and one with epithelial component with glomerular structures and stromal component with rhabdomyoblastic differentiation. For the tumor no. 262I of the left side, three different blocks were investigated: two blocks with epithelial component with papillary features and one block from which two different areas were macro dissected, one with epithelial and blastemal component, and one with epithelial component with glomerular structures. For this case, which was plurifocal, there were no further primary tumor masses not investigated. DNA was extracted using the GeneRead™ DNA FFPE Kit (Qiagen, Milan, Italy).
Genotyping and generation of copy number and allelic event calls
DNA from frozen tumor tissue was analyzed using Illumina 370CNV genotyping BeadChip arrays (370 K). The Infinium II Genotyping reaction steps were performed according to manufacturer’s specifications (Illumina, San Diego, CA, USA) at the CBM genotyping service, Trieste, Italy. Normalized bead intensity data obtained for each sample were analyzed with Illumina Genome Studio v1.0.2 software using the manufacturer’s default cluster settings, which generates SNP genotypes from fluorescent intensities, together with the normalized measure of the total signal intensity for the two alleles of a SNP, the Log R ratio (LRR), and the normalized measure of the allelic intensity ratio of the two alleles, the B allele frequencies (BAF). These values were used to detect CNAs and AR anomalies. The SNPRank Segmentation algorithm within Nexus Copy Number™ v5 (Biodiscovery Inc., El Segundo, CA, USA) was then used to process LRR and BAF and to perform the CN and allelic event calls. Low-level CN gain and loss were defined as LRR values varying from 0.25 to 0.6 or from −0.25 to −0.7, respectively, high level CN gain when LRR values were > 0.6 and homozygous copy loss (HCL) when LRR values were < −0.7. Three types of AR anomalies are automatically classified by Nexus on the basis of BAF and of the homozygous frequency threshold: LOH, allelic imbalance and total allelic loss (TAL). We set a homozygous frequency threshold of 95% and called an LOH with BAF values > 0.8 or < 0.2 and an allelic imbalance with BAF values varying from 0.2 to 0.4 or from 0.6 to 0.8. A TAL was called if the probes showed a noncanonical profile with bands corresponding to the different possible genotypes. During analyses, discontinuities < 1 Mb within aberrant regions have been smoothened.
All the annotations and map information were based on the hg18 release of the human genome, and retrieved from the different databases linked to Nexus (e.g. UCSC, Ensembl, miRNA database, Database of Genomic Variants, RepeatMasker). The methods here briefly summarized have been previously reported in detail [8]. Raw data are available from the authors on request.
WT1, CTNNB1, WTX, TP53, SIX1, SIX2, and miRNAPGs sequence analysis
Sanger sequencing was performed on DNA/cDNA from frozen tumor material for the following analyses. The entire coding region of WT1 and WTX, the exons 3, 7, 8 of the CTNNB1 gene, and exons 4 to 9 of the TP53 gene were screened for mutations. SIX1 (CCDS9748) and SIX2 (CCDS1822) p.Q177R hotspot mutations were investigated on genomic DNA, and in mutated cases, the expression of the mutated and wild-type alleles was assessed by cDNA sequencing. DROSHA (CCDS47195) cDNA spanning aminoacids 936–1253, covering RNAse IIIa and RNAse IIIb domains mutations, was sequenced. Identified mutations were confirmed at genomic level sequencing the affected exon. For primary WT 262I of the left side, in which only genomic DNA was available, DROSHA exons 23, 24, 29 and 30 (ENST00000511367), covering previously identified mutations, were sequenced. DICER1 (CCDS9931) cDNA spanning aminoacids 1682–1829, covering all mutations identified in the RNAse IIIb domain, was sequenced. For primary WT 262I of the left side, corresponding region was investigated through DNA sequencing. DGCR8 (CCDS13773) p.E518K hotspot was screened by genomic DNA sequencing, whereas aminoacids 59–257 were screened through cDNA sequencing. For primary WT 262I of the left side, genomic DNA was amplified and screened (aminoacids 59–240). DNAs obtained from FFPE material from cases 51I, 74I, 262I (both left and right side) were screened for the specific SIX1 and DROSHA mutations identified. Primer sequences and conditions are described in Supplementary Table S4.
ACKNOWLEDGMENTS AND FUNDING
The Authors thank Associazione Bianca Garavaglia, Busto Arsizio, Varese, for financial support, Loris De Cecco and Edoardo Marchesi, Functional Genomic Unit, Fondazione IRCCS Istituto Nazionale dei Tumori, Milan, Italy, for technical support, Boschetti Luna, Pediatric Unit, Fondazione IRCCS Istituto Nazionale dei Tumori, Milan, Italy, for secretarial assistance, Antonio Fiorino Department of Preventive and Predictive Medicine, Fondazione IRCCS Istituto Nazionale dei Tumori, Milan, Italy, for helpful discussion.
CONFLICTS OF INTEREST
The authors indicate no potential conflicts of interest.
REFERENCES
1. Spreafico F, Pritchard Jones K, Malogolowkin MH, Bergeron C, Hale J, de Kraker J, Dallorso S, Acha T, de Camargo B, Dome JS, Graf N. Treatment of relapsed Wilms tumors: lessons learned. Expert Rev Anticancer Ther. 2009; 9:1807–15.
2. Ha TC, Spreafico F, Graf N, Dallorso S, Dome JS, Malogolowkin M, Furtwängler R, Hale JP, Moroz V, Machin D, Pritchard-Jones K. An international strategy to determine the role of high dose therapy in recurrent Wilms’ tumour. Eur J Cancer. 2013; 49:194–210.
3. Grundy PE, Breslow NE, Li S, Perlman E, Beckwith JB, Ritchey ML, Shamberger RC, Haase GM, D’Angio GJ, Donaldson M, Coppes MJ, Malogolowkin M, Shearer P, et al. Loss of heterozygosity for chromosomes 1p and 16q is an adverse prognostic factor in favorable-histology Wilms tumor: a report from the National Wilms Tumor Study Group. J Clin Oncol. 2005; 23:7312–21.
4. Messahel B, Williams R, Ridolfi A, A’hern R, Warren W, Tinworth L, Hobson R, Al-Saadi R, Whyman G, Brundler MA, Kelsey A, Sebire N, Jones C, et al. Allele loss at 16q defines poorer prognosis Wilms tumour irrespective of treatment approach in the UKW1-3 clinical trials: a Children’s Cancer and Leukaemia Group (CCLG) Study. Eur J Cancer. 2009; 45:819–26.
5. Spreafico F, Gamba B, Mariani L, Collini P, D’Angelo P, Pession A, Di Cataldo A, Indolfi P, Nantron M, Terenziani M, Morosi C, Radice P, Perotti D. AIEOP Wilms Tumor Working Group. Loss of heterozygosity analysis at different chromosome regions in Wilms tumor confirms 1p allelic loss as a marker of worse prognosis: a study from the Italian Association of Pediatric Hematology and Oncology. J Urol. 2013; 189:260–6. doi: 10.1016/j.juro.2012.09.009
6. Natrajan R, Williams RD, Hing SN, Mackay A, Reis-Filho JS, Fenwick K, Iravani M, Valgeirsson H, Grigoriadis A, Langford CF, Dovey O, Gregory SG, Weber BL, et al. Array CGH profiling of favourable histology Wilms tumours reveals novel gains and losses associated with relapse. J Pathol. 2006; 210:49–58.
7. Hing S, Lu YJ, Summersgill B, King-Underwood L, Nicholson J, Grundy P, Grundy R, Gessler M, Shipley J, Pritchard-Jones K. Gain of 1q is associated with adverse outcome in favorable histology Wilms’ tumors. Am J Pathol. 2001; 158:393–8.
8. Perotti D, Spreafico F, Torri F, Gamba B, D’Adamo P, Pizzamiglio S, Terenziani M, Catania S, Collini P, Nantron M, Pession A, Bianchi M, Indolfi P, et al. Genomic profiling by whole-genome single nucleotide polymorphism arrays in Wilms tumor and association with relapse. Genes Chromosomes Cancer. 2012; 51:644–53.
9. Gratias EJ, Jennings LJ, Anderson JR, Dome JS, Grundy P, Perlman EJ. Gain of 1q is associated with inferior event-free and overall survival in patients with favorable histology Wilms tumor: a report from the Children’s Oncology Group. Cancer. 2013; 119:3887–94.
10. Huff V. Wilms’ tumours: about tumour suppressor genes, an oncogene and a chameleon gene. Nat Rev Cancer. 2011; 11:111–21.
11. Bardeesy N, Beckwith JB, Pelletier J. Clonal expansion and attenuated apoptosis in Wilms’ tumors are associated with p53 gene mutations. Cancer Res. 1995; 55:215–9.
12. Walz AL, Ooms A, Gadd S, Gerhard DS, Smith MA, Guidry Auvil JM, Meerzaman D, Chen QR, Hsu CH, Yan C, Nguyen C, Hu Y, Bowlby R, et al. Recurrent DGCR8, DROSHA, and SIX homeodomain mutations in favorable histology Wilms tumors. Cancer Cell. 2015; 27:286–97.
13. Wegert J, Ishaque N, Vardapour R, Geörg C, Gu Z, Bieg M, Ziegler B, Bausenwein S, Nourkami N, Ludwig N, Keller A, Grimm C, Kneitz S, et al. Mutations in the SIX1/2 pathway and the DROSHA/DGCR8 miRNA microprocessor complex underlie high-risk blastemal type Wilms tumors. Cancer Cell. 2015; 27:298–311.
14. Natrajan R, Little SE, Sodha N, Reis-Filho JS, Mackay A, Fenwick K, Ashworth A, Perlman EJ, Dome JS, Grundy PE, Pritchard-Jones K, Jones C. Analysis by array CGH of genomic changes associated with the progression or relapse of Wilms’ tumour. J Pathol. 2007; 211:52–9.
15. Beckwith JB, Zuppan CE, Browning NG, Moksness J, Breslow NE. Histological analysis of aggressiveness and responsiveness in Wilms’ tumor. Med Pediatr Oncol. 1996; 27:422–8.
16. Christensen KL, Patrick AN, McCoy EL, Ford HL. The six family of homeobox genes in development and cancer. Adv Cancer Res. 2008; 101:93–126.
17. Dressler GR. The cellular basis of kidney development. Annu Rev Cell Dev Biol. 2006; 22:509–29.
18. Ho J, Kreidberg JA. MicroRNAs in renal development. Pediatr Nephrol. 2013; 28:219–25.
19. Rakheja D, Chen KS, Liu Y, Shukla AA, Schmid V, Chang TC, Khokhar S, Wickiser JE, Karandikar NJ, Malter JS, Mendell JT, Amatruda JF. Somatic mutations in DROSHA and DICER1 impair microRNA biogenesis through distinct mechanisms in Wilms tumours. Nat Commun. 2014; 2:4802.