
Introduction
The discovery of the transforming echinoderm microtubule-associated protein-like 4-anaplastic lymphoma kinase (EML4-ALK) fusion gene in lung cancer in 2007 revealed that ALK is not only a driver gene for very rare types of hematological cancer but can also serve as a therapeutic target for non-small cell lung cancers (NSCLC) [1-4]. ALK is present in approximately 3% to 5% of NSCLC patients, turns approximately 8,000 patients each year in the US alone [5]. A couple of months after the report of the ALK rearrangement in NSCLC, scientists identified rearrangement of the ROS1 oncogene in NSCLC [6]. Crizotinib (PF02341066) is a multi-targeted tyrosine kinase inhibitor with high clinical efficiency in NSCLC patients harboring ALK and ROS1 rearrangements [7, 8]. In light of its high efficiency and safety in phase I and phase II clinical trials, crizotinib has been granted accelerated approval by the Food and Drug Administration (FDA) as a front-line treatment for advanced ALK-rearranged NSCLC.
Despite rapid and dramatic effects, a common concern of targeted therapy is the development of resistance. In the case of ALK- and ROS1-rearranged NSCLC, crizotinib resistance inevitably occurs within years of treatment [9]. Analogous to resistance to epidermal growth factor receptor (EGFR) inhibitors, crizotinib resistance often occurs as a result of resistant mutations within the tyrosine kinase domains of ALK and ROS1 [10]. Preclinical studies using patient-derived cell lines have indicated that activation of bypass tyrosine kinase signaling contributes to crizotinib resistance [11]. Here, we review the discovery and biology of oncogenic ALK and ROS1 rearrangement and summarize several pivotal clinical trials that validated crizotinib as a therapeutic agent for NSCLC. We also focus on the established and ongoing research on crizotinib resistance. Finally, we will discuss several promising strategies to monitor and manage NSCLC patients in the clinic after crizotinib treatment failure.
Discovery of oncogenic ALK and ROS1 fusion genes in NSCLC
ALK is a highly conserved receptor tyrosine kinase (RTK) and it belongs to the insulin receptor superfamily. Like other RTKs, ALK is located in the cell membrane and is comprised of an extracellular ligand-binding domain, transmembrane domain and intracellular tyrosine kinase domain. Upon binding to ligands (such as pleiotrophin and midkine), ALK forms a homodimer, which enables ALK phosphorylation and protein kinase activation [12, 13]. ALK signaling has been implicated in central nervous system development, and its expression is significantly diminished after birth, except in rare scattered neural cells, endothelial cells and pericytes in the brain, suggesting a nervous system-specific distribution pattern of ALK under physiological conditions [14]. However, a variety of alterations in the ALK gene such as mutations, overexpression, amplification, translocations, or other structural rearrangements have been implicated in human cancer tumorigenesis [15]. Neuroblastoma is a pediatric cancer that possesses ALK abnormalities that arise from gene amplification and activating mutations [16, 17]. Chen and colleagues reported that ALK amplification together with MYCN amplification contributes to the tumorigenesis of neuroblastoma. DNA sequencing further revealed eight ALK missense mutations in 13 of 215 excised tumors and 8 of 24 neuroblastoma-derived cell lines. These mutations generate seven different amino acid substitutions at five different positions [18]. Mutant ALK undergoes autophosphorylation and has increased kinase activity (this is known as a kinase activating mutation) compared to wild-type ALK. Therefore, targeting kinase activating mutant ALK using a molecular approach is expected to improve the clinical outcomes of advanced ALK mutant neuroblastoma [19]. In addition to neuroblastoma, ALK amplification and mutation are reported in other types of cancers, including esophageal cancer, inflammatory breast cancer, colorectal cancer and rhabdomyosarcoma [20-23].
However, the most dominant ALK genomic alterations in cancer are caused by chromosomal rearrangement when different fusion partners and their associated promoter regions fuse the upstream of the ALK kinase domain leading to constitutive activation of ALK and the downstream PI3K/Akt and MAPK/Erk pathways. The first ALK rearrangement was described in 1994 in anaplastic large cell lymphoma (ALCL) as a t(2;5)(p23;q35) rearrangement that fuses ALK from chromosome 2 to nucleophosmin (NPM) from chromosome 5. The NPM-ALK fusion gene consists of the first four exons of NPM and the exons encoding the whole entire tyrosine kinase domain of ALK, and it is present in more than 50% of ALCL patients [1]. Other ALK fusion patterns, such as tropomyosin 3 (TPM3), TRK-fused gene (TFG) and tropomyosin 4 (TPM4), have also been detected. However, ALK rearrangement in NSCLC was not discovered until 2007 by Hiroyuki Mano’s research group in Japan [4]. Mano and colleagues created a cDNA library from a patient with lung adenocarcinoma and identified a fusion gene consisting of the intracellular tyrosine kinase domain of ALK and EML4, this fusion gene was shown to have oncogenic activity in mouse 3T3 cells and nude mice. Furthermore, a EML4-ALK K589M mutant with no kinase activity was incapable of tumorigenesis. Treatment with the ALK tyrosine kinase inhibitor WHI-P154 markedly inhibited the growth of EML4-ALK-transformed Ba/F3 cells [4]. The oncogenic potential of EML4-ALK in vivo was further highlighted by the Mano group. In contrast to transgenetic mouse models driven by other oncogenes that usually develop tumors around 3 to 6 months after birth, the lung-specific EML4-ALK transgenetic mice have develop multiple lung cancer nodules in both lungs without latency after birth, and inhibition of ALK leads to dramatic tumor regression in vivo. [24]. A recent study using CRISPR/Cas9-mediated in vivo EML4-ALK engineering in mice also showed that the mice expressing EML4-ALK were born with lung cancer, indicating that EML4-ALK is definitely a strong cancer promoter and a good therapeutic target [25].
In addition to EML4-ALK, other fusion patterns that have been identified include KIF5B-ALK, KLC1-ALK and TFG-ALK, and the most common fusion pattern is EML4-ALK (Table 1). Depending on the proportion of the EML4 gene that is fused to ALK, more than nine EML4-ALK variants have been identified, and all of those ALK variants show a remarkable response to ALK tyrosine kinase inhibitors in vitro and in vivo [26, 27] (Figure 1A). Therefore, the kinase function of ALK is critical for cell transformation, and ALK fusion proteins are therapeutic targets for NSCLC (discussed below).
Another insulin receptor superfamily, RTK ROS1 (chromosome 6q22), defines a unique molecular subset of NSCLC. Unfortunately, very little is known about the function of wild-type ROS1. Mice lacking ROS1 seem to be healthy, although males display abnormalities in the epididymis that prevent normal reproduction [28]. Ligands for ROS1 have not yet been identified [29]. ROS1 somatic mutations and focal amplification were reported at a very low frequency in cancer, and the significance of these genetic alterations is largely unknown. The first ROS1 fusion protein was discovered in the glioblastoma cell line U118MG. The fusion gene was formed by a deletion in chromosome 6 that resulted in fusion of the 3’ region of ROS1 to the 5’ region of FIG (Table 1). Expression of the FIG-ROS1 fusion protein results in anchorage-independent growth, foci formation, and tumorigenicity [30, 31]. In addition, FIG-ROS1, SLC34A2-ROS1, CD74-ROS1, EZR-ROS1, TPM3-ROS1 and SDC4-ROS1 and related variants are recognized as new cancer driver genes and are associated with sensitivity to ROS1 inhibition [32-34] (Table 1). It is estimated that approximately 1% of NSCLC harbor ROS1 rearrangement [35]. Interestingly, a recent biochemical study revealed that ROS1 proteins shares 77% amino acid sequence homology with ALK in the ATP binding sites of the tyrosine kinase domain; this leads to the observation that the ALK inhibitors crizotinib and TAE684 profoundly inhibit ROS1 kinase activity and lead to tumor regression [36]. The EGFR inhibitor gefitinib was shown to sensitize ROS1 inhibition in HCC78 cells, which harbor SLC34A2-ROS1, suggesting that ROS1 might cooperate with EGFR to promote proliferation in this cell line [37]. In contrast, ROS1 rearrangement was found to be mutually exclusive to ALK rearrangement in NSCLC [38]. Although no ROS1 specific tyrosine kinase inhibitors are currently in clinical trials, crizotinib and other ALK inhibitors may also have activity against ROS1. Recently, Pfizer has announced that crizotinib has received Breakthrough Therapy designation by the FDA for the potential treatment of ROS1-rearranged NSCLC.
Table 1: Summary of ALK and ROS1 fusion patterns in cancer
Cancer type |
ALK pattern |
Cancer type |
ROS1 pattern |
Hematological |
|||
ALCL |
NPM1-ALK, TFG-ALK, ATIC-ALK, CLTC-ALK, ALO17-ALK, MSN-ALK, TPM3/4-ALK, MYH9-ALK |
||
Diffuse large B cell lymphoma |
NPM-ALK, CLTC-ALK, SQSTM1-ALK, SEC31A-ALK, RANBP2-ALK |
||
Systemic histocytosis |
TPM3-ALK |
||
Mesenchymal |
Mesenchymal |
||
IMT |
TPM3/4-ALK, RANBP2-ALK, CARS-ALK, CLTC-ALK, SEC31A-ALK, PPFIBP1-ALK, DCTN1-ALK |
IMT |
YWHAE-ROS1, TFG-ROS1 |
Epithelioid inflammatory myofibroblastic sarcoma |
RANBP2-ALK |
Angiosarcoma |
CEP85L-ROS1 |
Ovarian sarcoma |
FN1-ALK |
||
Epithelial |
Epithelial |
||
NSCLC |
EML4-ALK, HIP1-ALK, KIF5B-ALK, KLC1-ALK DCTN1-ALK, SQSTM1-ALK, TRP-ALK,TFG-ALK, PTPN3-ALK, STRN-ALK |
NSCLC |
SDC4-ROS1, FIG-ROS1, SLC34A2-ROS1, CD74-ROS1, TPM3-ROS1, EZR-ROS1, LRIG3-ROS1, KDELR2-ROS1, CCDC6-ROS1 |
Renal cancer |
TPM3/4-ALK, VCL-ALK |
Gastric cancer |
SLC34A2-ROS1 |
Breast cancer |
EML4-ALK |
Colorectal adenocarcinoma |
SLC34A2-ROS1 |
Colorectal adenocarcinoma |
EML4-ALK |
Ovarian cancer |
FIG-ROS1 |
Esophageal squamous cell carcinoma |
TPM3/4-ALK |
Cholangiocarcinoma |
FIG-ROS1 |
Anaplastic thyroid cancer |
EML4-ALK |
Neural |
|
Neuroblastoma |
FIG-ROS1 |
||
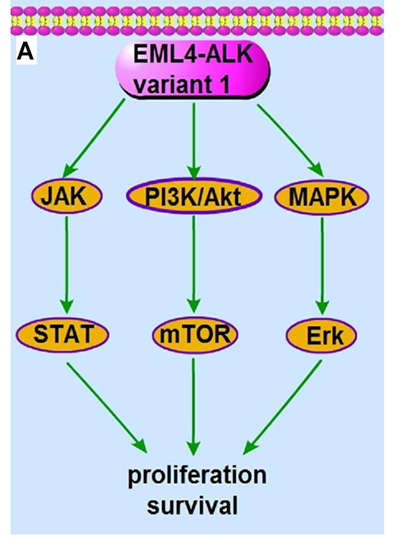
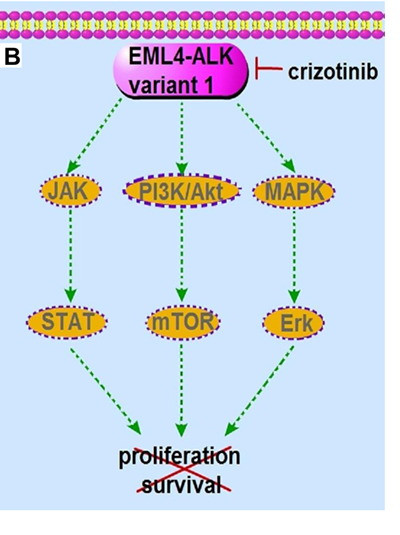
Figure 1: The signaling pathway and the molecular actions of crizotinib on EML4-ALK variant 1 fusion protein. A. The EML4-ALK variant 1 fusion protein constitutively activates PI3K/Akt, MAPK/Erk and Jak/Stat signaling, which promotes cell proliferation, survival and tumorigenesis. B. The ALK inhibitor crizotinib inhibits the kinase activity of EML4-ALK variant 1 and subsequently abrogates the downstream PI3K/Akt, MAPK/Erk and Jak/Stat signaling, leading to cancer cell apoptosis and death.
Crizotinib as a new treatment for ALK- and ROS1-rearranged cancer
ALK and ROS1 rearrangement are detected in approximately 5% and 1% of NSCLC patients, respectively, which constitutes a great number of patients worldwide. Fluorescence in situ hybridization (FISH) with break-apart probes is currently the most effective diagnostic technology for the detection of chromosomal rearrangement, and it has been approved for the detection of ALK rearrangement in clinical settings [39]. Reverse transcriptase PCR assay and highly sensitive immunohistochemistry are also feasible for pre-screening tests before FISH [5, 40, 41]. While a large proportion of biopsy samples are not suitable for the preparation of formalin-fixed, paraffin-embedded (FFPE) tissue for these detection assays, it is important to note the distinct clinicopathologic features of ALK- and ROS1-rearranged patients. Unlike EGFR, the prevalence of ALK- and ROS1-rearrangement is similar in Caucasians and Asians. These patients tend to be younger at the time of diagnosis. ALK and ROS1 alterations are also associated with never smoking or having a light smoking history, female gender, and adenocarcinoma with signet ring cell histology and seem to be mutually exclusive to other oncogenic driver genes [42-44]. However, recent studies have indicated that 8% of ALK-rearranged NSCLC are also positive for either an EGFR or k-RAS activating mutation [45-48].
Crizotinib, a potent ATP-competitive type-II aminopyridine tyrosine kinase inhibitor that inhibits ALK, has already been approved by the FDA for the treatment of ALK-rearranged NSCLC. Interestingly, crizotinib was initially designed to target the c-Met proto-oncoprotein [49]. A kinase inhibition screening assay showed that crizotinib inhibited the phosphorylation activities of up to 13 kinases (c-Met, ALK, RON, Axl, Tie-2, TrkA, TrkB, Abl, IRK, Lck, Sky, VEGFR2 and PDGFRβ) among 102 kinases tested. Preclinical models of Met-amplified NSCLC revealed showed that addition of crizotinib results in regression of xenograft tumors [49-51]. These findings have prompted clinical trials in patients with Met amplifications (NCT01121575). Crizotinib also inhibits proliferation of ALK-rearranged lymphoma cells with IC50 of approximately 50 nM. In a xenograft model with H3122 cells (a NSCLC cell line positive for EML4-ALK variant 1 rearrangement), crizotinib treatment resulted in a significant tumor regression [52] (Figure 1B). In addition to crizotinib, GSK1838705A, TAE684, CEP-14083, AP26113, ASP3026, NMS-E628 and other potent tyrosine kinase inhibitors that can act against oncogenic ALK are still under clinical development [53] (Table S1).
The potential clinical benefit of crizotinib in ALK-rearranged NSCLC was quickly appreciated after an open-label, multicenter dose-escalating phase I trial (PROFILE 1001, NCT00585195) [8]. Of note, patients were already being enrolled in the dose-escalation phase of study of crizotinib before the discovery of EML4-ALK in 2007 (Table 2). A total of 82 ALK-rearranged NSCLC patients were enrolled in the PROFILE 1001 trial and treated with crizotinib. Crizotinib was started at 50 mg once daily, and then the dose was escalated. The crizotinib dosage of 250 mg twice a day was reported to achieve an overall response rate (ORR) of 57% with mild manageable adverse effects, and it was defined as the recommended phase II dose [8]. The updated results of the PROFILE 1001 trial included a median progression free survival (PFS) of 9.7 months and estimated overall survival (OS) at 6 and 12 months of 87.9% and 74.8%, respectively [54]. The phase II crizotinib trial (PROFILE 1005, NCT00932451) demonstrated an ORR of 60% with a median PFS of 8.1 months [55]. Furthermore, a phase III crizotinib trial (PROFILE 1007, NCT00932893) compared crizotinib with pemetrexed or docetaxel as single agent chemotherapy regimens in NSCLC patients who had experienced disease progression after prior platinum-based chemotherapy and showed that crizotinib was superior to standard chemotherapy in ALK-rearranged NSCLC patients, with a higher ORR (65% vs 20%, P < 0.001), longer PFS (7.7 vs 3.0 months, HR = 0.49, P < 0.001) and significant improvement in quality of life [56]. Additionally, the very recently published PROFILE 1014 study (NCT01154140) demonstrated that crizotinib was superior to standard first-line pemetrexed-platinum chemotherapy in patients with previously untreated advanced ALK-rearranged NSCLC. The PFS was 10.9 months in the crizotinib group, which is significantly longer than the PFS of 7.0 months in the chemotherapy group (HR = 0.45, P < 0.001). The ORR was 74% in the crizotinib group compared to 45% in the chemotherapy group (P < 0.001), and crizotinib was associated with a greater reduction of lung cancer symptoms and greater improvement in quality of life [57]. The multi-center PROFILE 1029 study evaluating the safety and efficiency of crizotinib in ALK-rearranged East Asian NSCLC patients (NCT01639001) is currently ongoing (Table 2). Unfortunately, OS was similar between the crizotinib and chemotherapy groups in the PROFILE 1007 and PROFILE 1014 studies. The striking clinical efficiency of crizotinib has been tested in other ALK-rearranged cancers. Preliminary studies showed that ALK-rearranged advanced ALCL patients who relapsed after standard chemotherapy have high and durable responses to crizotinib. The ORR of crizotinib was 90.9% (10 out of 11), and 4 patients achieved a complete response. The OS and PFS rates at 2 years were 72.7% and 63.7%, respectively [58]. Shapiro and colleagues described crizotinib treatment of ALK-rearranged inflammatory myofibroblastic tumors (IMTs) [59]. One IMT patient who harbored an ALK rearrangement experienced a sustained partial response to crizotinib, while the ALK-negative IMT patient had no response. This suggests that the ALK-rearranged IMT was addicted to ALK-mediated signaling, making it a potential therapeutic target for this unique molecular subtype of soft tissue tumors (NCT00585195).
Fortuitously, crizotinib is also a potent ROS1 inhibitor. Preliminary results in 14 ROS1-rearranged NSCLC patients have been very impressive, with an ORR of 57% and a disease control rate of 79% at 8 weeks [43]. A recently published study also showed that crizotinib was highly effective in ROS1-rearranged NSCLC (NCT00585195). That study enrolled a total of 50 advanced ROS1-rearranged NSCLC patients in an expansion cohort of the PROFILE 1001 trial (Table 2). Most of the patients had received prior standard therapy, and they were treated with 250 mg of crizotinib twice a day during the trial. The following findings of that study are of particular interest: 3 patients achieved a complete response, 33 patients achieved a partial response, 9 patients had stable disease and the ORR was 72%. Crizotinib also had long-lasting clinical benefits, with an estimated median response duration of 17.6 months and a median PFS of 19.2 months [7]. The trial was still ongoing at the time of publication of this review, and the median OS had not yet been reached. Other kinase inhibitors, such as AP26113, have been reported to be potent ROS1 inhibitors. A clinical trial of AP26113 in ROS1-rearranged patients has been planned, and the results of that trial are eagerly awaited (NCT01449461).
ALK and ROS1 inhibition share several similarities, such as the time to first response (median 7.9 weeks for both) and ORR (61% and 72%, respectively), and the response of cancers to crizotinib were independent of the specific ALK and ROS1 gene rearrangements they possessed. One apparent difference between ALK and ROS1-rearranged NSCLC patients is their PFS with crizotinib treatment. The average PFS of patients treated with crizotinib in the PROFILE 1007 and PROFILE 1014 trials was approximately 9.3 months (7.7 and 10.9 months, respectively). In contrast, the estimated PFS in ROS1-rearranged patients was much longer at 19.2 months, and at the time of data cut-off, half of the ROS1-rearranged patients were still being followed to analyze disease progression. Therefore, the duration of disease control by crizotinib seems to be much longer and resistance to crizotinib seems to develop much later in ROS1- than ALK-rearranged NSCLC patients. While the FDA approval of crizotinib currently only covers ALK-rearranged NSCLC, further clinical trials comparing crizotinib to standard chemotherapy in ROS1-rearranged patients are warranted.
Table 2: Summary of crizotinib trials.
General information |
Patients baseline characteristics |
Diagnostic method |
Crizotinib treatment outcomes |
||||||||||||||
Profile |
NCT |
Subject |
n |
Age (year) |
Male (%) |
Ethnics |
Smoking status (%) |
ADC (%) |
ECOG |
FISH |
IHC |
NGS |
RT-PCR |
Dose |
ORR |
PFS (months) |
OS (months) |
10018 |
NCT00585195 |
ALK- rearranged advanced NSCLC |
82 |
25-78 |
43 (52) |
Caucasian 46 Asian 29 Other 7 |
Never 62 (76) ≤10 pack/year 15 (18) >10 pack/year 5 (6) |
79 (96) |
0-3 |
+ |
+ |
- |
+ |
50-300 mg |
57% |
72% at 6 months |
Not reached |
100154,a |
NCT00585195 |
ALK- rearranged stage III/IV NSCLC |
149 |
21-86 |
73 (49) |
Caucasian 95 Asian 41 Other 13 |
Never 106 (71) Former 42 (28) Present 1 (1) |
114 (97) |
0-2 |
+ |
- |
- |
- |
250 mg |
60.8% |
9.7 |
74.8% at 12 months |
100756,b |
NCT00932893 |
previously treated advanced ALK- rearranged NSCLC |
173/347 |
22-81 |
75 (43) |
Caucasian 90 Asian 79 Other 4 |
Never 108 (62) Former 59 (34) Present 5 (3) |
164 (95) |
0-2 |
+ |
- |
- |
- |
250 mg |
65% |
7.7 |
20.3 |
101457,c |
NCT01154140 |
previously untreated ALK- rearranged advanced nonsquamous NSCLC |
172/ 343 |
22-76 |
68 (40) |
Caucasian 91 Asian 77 Other 4 |
Never 106 (62) Former 56 (33) Present 10 (6) |
161 (94) |
0-2 |
+ |
- |
- |
- |
250 mg |
74% |
10.9 |
84% at 1 year |
1029d |
NCT01639001 |
ALK-rearranged East Asian NSCLC |
11 |
19-55 |
7 (64) |
- |
- |
- |
1-4 |
+ |
+ |
+ |
+ |
250 mg |
90.9% |
63.7% at 2 years |
72.7% at 2 years |
10017,e |
NCT00585195 |
ROS1- rearranged advanced NSCLC |
50 |
25-77 |
22 (44) |
Caucasian 27 Asian 21 Other 2 |
Never 39 (78) Former 11 (22) Present 0 (0) |
49 (98) |
0-2 |
+ |
- |
- |
+ |
250 mg |
72% |
19.2 |
85% at 12 months |
a The PROFILE 1001 trial is initially published in 2011 and it has been updated in 2012.
b This is the first phase 3, open-label trial comparing crizotinib with pemetrexed/docetaxel chemotherapy in 347 patients with locally advanced or metastatic ALK-rearranged lung cancer who had received one prior platinum-based regimen. This table only reflects information and therapeutic outcomes of patients in the crizotinib cohort.
c This is an open-label, phase 3 trial comparing crizotinib with chemotherapy in 343 patients with advanced ALK-rearranged nonsquamous NSCLC who had received no previous systemic treatment. This table only reflects information and therapeutic outcomes of patients in the crizotinib cohort.
d The PROFILE 1029 trial is still ongoing.
e The ROS1-rearranged NSCLC patients was included in phase 1 PROFILE 1001 trial.
ALK/ROS1-dominant crizotinib resistance
Although most ALK- and ROS1-rearranged NSCLC patients benefit substantially from crizotinib, resistance to targeted therapy remains to be a common clinical challenge. Resistance to EGFR inhibitors occurs with the emergence of the “gatekeeper” T790M mutation within the EGFR tyrosine kinase domain; analogously resistance to crizotinib partially arises from the emergence of resistance mutations. T790M is present in one-half of EGFR targeted resistant cases, and it is the most commonly observed EGFR resistance mutation [60, 61]. The first ALK-rearranged NSCLC patient who relapsed after crizotinib treatment was described in 2010. DNA deep sequencing of a pleural effusion specimen from that patient identified two additional mutations, L1196M and C1156Y. Ectopic expression of L1196M and C1156Y leads to resistance to crizotinib in EML4-ALK-transformed Ba/F3 cells, indicating that these tumor-derived L1196M and C1156Y mutations decrease the tumor’s sensitivity to ALK inhibitors [62]. The underlying mechanisms have been determined to a large extent. Similar to the T790M mutation in EGFR, the “gatekeeper” L1196M mutation adds new bulky residues to the ATP binding domain of ALK kinase, which hinders crizotinib binding (Figure 2A). Although C1156Y is not a “gatekeeper” mutation, it also causes the dislocation between crizotinib and ALK and indirectly leads to conformational changes in the binding cavity of ALK, which results in a marked decrease in crizotinib sensitivity [63]. However, one large difference between EGFR resistance and ALK resistance is that multiple resistance mutations have only been detected in ALK inhibitor resistant patients. In addition to L1196M and C1156Y, G1202R, S1206Y, F1174C, F1174L, D1203N, I1171T, 1151T-ins, L1152P and G1269A mutations also confer crizotinib resistance [9, 64, 65]. To date, resistance mutations have been identified in approximately 25% patients who failed on crizotinib treatment. In 2013, resistance to crizotinib caused by mutation was also reported in ROS1-rearranged patients. Repeated biopsy specimens revealed a G2032R mutation within ROS1, which was not detected in the pretreatment specimen. After the patient died, the autopsy results showed that all sites of disease had the G2032R mutation, suggesting that the expansion and spread of G2032R clones may have been responsible for crizotinib resistance in this patient. Unlike “gatekeeper” mutations in EGFR and ALK, the G2032R mutation was located in the solvent front of the ROS1 kinase domain and was analogous to the G1202R resistance mutation in ALK [10, 66].
The above-mentioned resistance mutations sometimes occur simultaneously with gene copy number gain (CNG). H3122 CR0.6 cells, which are partially resistant to crizotinib, only harbor the EML4-ALK CNG, while the fully resistant cell line H3122 CR harbors both the CNG and L1196M mutation, suggesting a stepwise evolution of crizotinib resistance involving gene amplification followed by mutation [67]. Fortunately, H3122 CR cells are still addicted to ALK signaling and are sensitive to ALK inhibition by molecular approaches, such as siRNA. Thus, resistance to crizotinib driven by CNG and additional mutations was defined as the ALK/ROS1-dominant resistance mechanism [67]. Several second generation ALK inhibitors have been developed and are entering clinical trials. Ceritinib, also called LDK378, is one such next generation ALK inhibitor. Unlike crizotinib, ceritinib selectively inhibits ALK without acting on c-Met. Ceritinib also inhibits ALK resistance mutations [68]. Preclinical studies showed remarkable activity of ceritinib against L1196M, G1269A, S1206Y and I1171T; however, it had less activity against C1156Y, G1202R, 1151T-ins, L1152P, and F1174C. In crizotinib resistant xenograft models and patient-derived cell lines, ceritinib was found to be 20-fold more efficient than crizotinib [69]. Computationally molecular dynamics and crystal structure analysis revealed that ceritinib showed increased potency against these resistant mutations [70]. A phase I dose-escalation trial showed that ceritinib has high efficiency in ALK-rearranged NSCLC patients regardless of whether they were previously treated with crizotinib. For 114 ALK-rearranged NSCLC patients who received at least 400 mg of ceritinib per day, the ORR was 58% and the median PFS was 7.0 months. Responses to ceritinib were also observed in patients with various ALK resistance mutations, suggesting that sequential treatment with ceritinib is a feasible method for overcoming crizotinib resistance (NCT01283516) [71]. Moreover, sequential crizotinib and ceritinib treatment has been shown to extend PFS (15.5-19.4 months) and OS (35.5-63.1 months) in ALK-rearranged NSCLC patients (n = 73) (NCT01283516) [72]. Another encouraging property for ceritinib is its intracranial efficacy [73]. An ORR of 54% and a median PFS of 6.9 months was achieved in 124 brain metastatic ALK-rearranged NSCLC patients treated with ceritinib at the recommended dosage of 750 mg per day. Among the 14 patients with measurable brain lesions at baseline (10 crizotinib-pretreated and 4 crizotinib-naïve), ceritinib treatment achieved an intracranial ORR of 50% in all patients, 40% in crizotinib-pretreated patients and 75% in crizotinib-naïve patients [74]. Therefore, ceritinib was granted accelerated approval by the US FDA in 2013 for the treatment of ALK-rearranged NSCLC patients and for NSCLC patients who failed on or are intolerant to crizotinib [75]. Other second generation ALK inhibitors, including alectinib/CH5424802, ASP3026 and AP26113, are under clinical development in ALK-rearranged patients [53].
Ceritinib, ASP3026 and AP26113 also exhibit activity against ROS1 kinase, but alectinib does not [53]. However, ceritinib, ASP3026 and AP26113 fail to inhibit crizotinib-resistant ROS1, and more potent ROS1 tyrosine kinase inhibitors are urgently needed. The multi-targeted tyrosine kinase inhibitors foretinib and cabozantinib exhibit more than 50-fold inhibition potency compared with crizotinib in ROS1-transformed Ba/F3 cells. Crizotinib-resistant G2032R mutants remain sensitive to foretinib and cabozantinib [76, 77]. More recently, PF-06463922 was reported to be the most potent ALK/ROS1 inhibitor with exquisite potency against ROS1 kinase. PF-06463922 is 10-fold more potent than crizotinib and foretinib and 100-fold more potent than ceritinib and alectinib in inhibition of both ROS1-addicted cell growth and ROS1 kinase activity. It inhibits cell proliferation and induces apoptosis in HCC78 cells harboring the SLC34A2-ROS1 rearrangement and in CD74-ROS1-transformed Ba/F3 cells at concentrations less than 1 nM. In Ba/F3 cells engineered to express the crizotinib L2026M “gatekeeper” and G2032R resistance mutations, PF-06463922 demonstrated nanomolar potency against both mutations in vitro and in vivo [78, 79] (Table S1). In addition, PF-06463922 is able to penetrate the blood-brain-barrier, and oral administration was found to be effective in a mouse model of ROS1-driven malignant glioma [80]. A phase I/II trial studying the safety and efficacy of PF-06463922 in ALK/ROS1-rearranged NSCLC was launched in January 2014; the trial will be complete in October 2017 (NCT01970865).
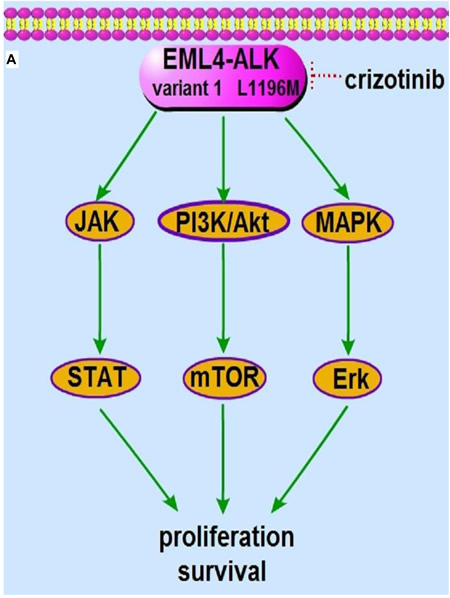
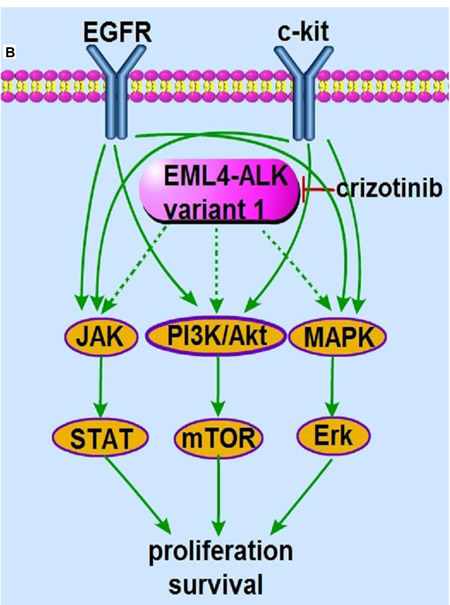
Figure 2: The molecular machinery responsible for crizotinib resistance. A. The presence of “gatekeeper” L1196M resistance mutation hinders crizotinib binding to the ATP binding domain of ALK kinase, which sustains the downstream PI3K/Akt, MAPK/Erk and Jak/Stat signaling in the presence of crizotinib. B. The activation of bypass EGFR and c-kit pathway rescues the PI3K/Akt, MAPK/Erk and Jak/Stat signaling, downstream of EML4-ALK variant 1 fusion protein.
ALK/ROS1 non-dominant crizotinib resistance
A growing number of studies suggest that resistance to targeted therapeutics is associated with reactivation of key downstream signaling pathways that are independent of the original oncogenic driver genes. This type of resistance model was initially described in EGFR mutant NSCLC, in which Met amplification reactivates PI3K/Akt and MAPK/Erk signals despite EGFR inhibition through the formation of Met-ErbB3 heterodimers. Treatment with a combination of EGFR and Met inhibitors efficiently diminished the constitutively activated PI3K/Akt and MAPK/Erk signals and overcame the resistance to gefitinib [81]. Clarification of such resistance mechanisms is expected to improve the prognosis of ALK- and ROS1- arranged patients. Sasaki and colleagues reported that H3122 TR3 cells, which are resistant to the ALK inhibitor TAE684, have a reduced ALK phosphorylation level in comparison with H3122 parental cells. They also found that H3122 TR3 cells secrete a greater amount of EGFR ligands, which leads to increased autophosphorylation of EGFR that cannot be inhibited by TAE684. Inhibition of EGFR signaling led to a significant decrease in proliferation and colony formation of H3122 TR3 cells, but not H3122 cells. Additionally, treatment with a combination of EGFR and ALK inhibitors led to complete inhibition of colony formation [82, 83]. More strikingly, increased EGFR phosphorylation was detected in 44% of ALK-rearranged crizotinib-resistant patients and in ROS1-rearranged crizotinib-resistant tumors, indicating that this mechanism may mediate resistance in some patients [11, 84]. Amplification of c-kit kinase also leads to crizotinib resistance. c-kit amplification has been detected in approximately 15% of crizotinib-resistant samples, and combining crizotinib with imatinib overcomes this resistance [11] (Figure 2B). More interestingly, RAS mutations have been shown to cause crizotinib resistance in both ALK- and ROS1-rearranged patients [85]. These results indicate that the cancer cells may have evolved to switch their dependence for survival from the original oncogeneic drivers ALK and ROS1 to other oncogenic drivers, including EGFR, kit and RAS. As the downstream PI3K/Akt and MAPK/Erk pathways can be reactivated despite ALK/ROS1 inhibition, a combinatorial treatment strategy that acts against these pathways may help to overcome resistance in ALK/ROS1 non-dominant settings.
One of the major concerns is to clearly identify the precious bypass oncogenic drivers that drive resistance in an individual patient. The preclinical resistant cell model has been extensively described. However, lack of cancer heterogeneity (high homogeneity of established parental cell lines) and defects in the microenvironment (absence of surrounding capillaries, immune cells, stromal cells, extracellular matrix, etc.) with this model are major concerns that limit the ability to translate laboratory findings directly into clinical applications. Therefore, there is an urgent need for more effective methods to identify specific bypass tracks responsible for promoting resistance in individual patients. In recent years, the patient-derived xenograft (PDX) model has emerged as a powerful tool for translational research, and it promises to enable a more personalized approach to patient care [86]. PDXs have significant similarities to their tumors of origin and are physiologically relevant to human cancers, making them ideal for evaluating the efficacy of therapeutic agents in vivo. Using the patient-derived model, the Engelman group recently described a combination strategy to predict resistance drivers and overcome resistance to targeted therapies [87]. For example, patient-derived MGH170-1BB cells, which are resistant to EGFR inhibitors, were solely responsive to a combination of EGFR and Met inhibitors among a panel of 76 targeted agents tested, suggesting that Met emerged as the resistance driver in this patient. Indeed, subsequent assessment of the patient biopsy specimen and MGH170-1BB cells has confirmed the presence of Met amplification. The MGH156-1A cells derived from another EGFR inhibitor-resistant patient were responsive to a combination of EGFR and fibroblast growth factor receptor (FGFR) inhibitors; genetic analysis of MGH156-1A cells and the corresponding specimen revealed a Y649C mutation in the FGFR3 tyrosine kinase domain. The combined use of a MAP kinase inhibitor and ALK inhibitor resulted in robust tumor regression in the ceritinib-resistant PDX. Next generation sequencing of the patient-derived MGH034-2A cells revealed a MAP2K1 K57N activating mutation, which underlies the remarkable response of the MGH034-2A cells and the PDX to simultaneous inhibition of ALK and MAP kinase [87]. Therefore, functional assessment of patient-derived models helps to identify resistance drivers. Using a combination screening strategy is a feasible method to predict resistance drivers and identify the most effective combination treatment for each patient.
Hsp90 inhibitors as a potent treatment strategy for ALK-positive NSCLC
Finally, another promising approach to treat ALK-rearranged NSCLC is the use of heat shock protein 90 (Hsp90) inhibitors. Hsp90 is a molecular chaperone that facilitates protein folding, maturation and stabilization. Inhibition of Hsp90 disrupts protein stability and leads to degradation of proteins, including the oncogenic ALK fusion protein [88, 89]. Hsp90 inhibitors have been shown to have single-agent activity in ALK-rearranged NSCLC xenografts. Phase 2 trials have shown that retaspimycin hydrochloride (IPI-504), ganetespib (STA-9090) and AUY922 have Hsp90 inhibitory activity in ALK-rearranged NSCLC (Table S1). The IPI-504 study recruited 76 patients who failed on EGFR inhibitor therapy, and 5 patients demonstrated a PR to IPI-504. Of note, 3 patients harbored an ALK rearrangement, 2 of whom had a PR and one of whom had stable disease. The estimated PFS for all the patients was 2.9 months, whereas the PFS of the 3 ALK-rearranged patients was 7 months [90, 91]. Similarly, the ganetespib study was not initially designed to evaluate the efficacy in ALK-rearranged patients, but the post-hoc analysis showed that a substantial proportion of ALK-rearranged patients had a PR, which was not observed in the patients with EGFR and RAS genotypes [92, 93]. Preliminary data from the phase 2 AUY922 trial showed an ORR of 29% (6 out of 21) in ALK-rearranged NSCLC. Of the 6 responders, 4 were crizotinib-naïve and 2 had previously been treated with crizotinib. The estimated median PFS rate was 42% at 18 weeks [94]. More strikingly, a preclinical study revealed the crizotinib-resistant L1196M and F1174L mutants were equally sensitive to Hsp90 inhibitors as cancers without those secondary mutations, and synergistic effects were observed when crizotinib was combined with Hsp90 inhibitors in ALK- and Met-driven cancers [67, 91]. Thus, the use of Hsp90 inhibitors is an optimal therapeutic strategy for ALK-rearranged NSCLC and a promising approach for overcoming crizotinib resistance. Multiple clinical studies of Hsp90 inhibitors as a single-agent or in combination with crizotinib or ceritinib in crizotinib-naïve ALK-rearranged, crizotinib-resistant cancers are underway (NCT01579994, NCT01712217, and NCT01772797).
Conclusions and perspectives
Extensive translational studies have demonstrated that lung cancer is no longer a single disease, and the “all-in-one-way” approach is no longer applicable. To date, more than ten cancer driver genes have been identified in lung adenocarcinoma. Therapeutic targets in lung squamous cell carcinoma, including fibroblast growth factor receptor (FGFR) and discoidin domain receptor 2 (DDR2), are currently being investigated [95-98]. However, the exact molecular drivers in nearly 50% of NSCLC patients have not been identified, and not all drivers can be targeted. For cases that the exact oncogenic drivers are not known, standard chemotherapy remains the foundation of treatment. However, The Cancer Genome Atlas (TCGA) analysis and next generation sequencing (NGS) may help to determine potential cancer drivers, and the PDX model can be applied to determine the best combination strategy for each patient.
In addition to the roles of ALK and ROS1 in NSCLC, their roles in other solid tumors have attracted increasing attention, indicating that one genomic alteration may be responsible for a wide range of cancers (known as pan-cancers). Furthermore, the similarities across different cancer types offers the possibility of repurposing targeted therapies directed by the molecular pathology of the cancer in addition to clinical classification (Table 1). The innovative ongoing Basket trial (NCT01121588) is evaluating the safety and activity of crizotinib in ALK-rearranged pan-cancers (also termed ALKoma). Reclassifying cancers based on genetic characters beyond tissue origination, such as EGFRoma, ALKoma and ROSoma, is promising, and these cancers could be treated with corresponding tyrosine kinase inhibitors, including gefitinib and crizotinib.
The origination of resistance mutations and bypass signaling pathways during the development of targeted therapy resistance are yet to be fully elucidated; however, several lines of evidence suggest that cancer heterogeneity is one of the driving forces. There is general consensusly agreed that the established parental cell lines are highly homologous; however, the derivative resistant cells are believed to be heterogeneous. For example, the lung adenocarcinoma PC-9 cells harboring the EGFR del E746_A750 activating mutation and are frequently used in studies of EGFR- targeted therapy. Zhang and colleagues generated PC-9 gefitinib- resistant (PC-9 GR) cells through chronic exposure to gefitinib and isolated a panel of resistant clones. Direct sequencing and amplification refractory mutation system (ARMS) assay showed that only half of the resistant clones had the T790M resistance mutation, indicating that some resistant clones may evade apoptosis through T790M-independent mechanisms [99]. In support of this notion, we generated PC-9 GR cells in our lab that did not have the T790M resistance mutation, and xenograft histology and immunohistochemistry showed that the cells underwent small cell lung cancer transformation. The PC-9 GR cells in our lab also underwent epithelial-mesenchymal transition (unpublished data), which reflects cancer heterogeneity during resistance to targeted therapeutics. Moreover, using more sensitive methodology, cells with T790M and Met amplification were found to preexist as minor clones in EGFR-mutant NSCLC [100]. It is highly likely that ALK/ROS1-dominant and non-dominant resistant cells preexist in crizotinib-naïve patients as a result of cancer heterogeneity, which would mean that the so-called acquired resistance is not actually “acquired” during treatment but is merely an outgrowth of preexisting clones under drug selection. If this is the case, it would have substantial clinical implications, as there would be feasible methods (such as repeat biopsy and non-invasive liquid biopsy) that could be implemented during the time course of the disease to choose the most appropriate targeted therapeutic agents.
Abbreviations
ALCL, anaplastic large cell lymphoma; ALK, anaplastic lymphoma kinase; CNG, copy number gain; DDR2, discoidin domain receptor 2; EGFR, epidermal growth factor receptor; EML4-ALK, echinoderm microtubule-associated protein-like 4-anaplastic lymphoma kinase; FDA, Food and Drug Administration; FFPE, formalin-fixed paraffin-embedded; FGFR, fibroblast growth factor receptor; FISH, fluorescence in situ hybridization; Hsp90, heat shock protein 90; IMT, inflammatory myofibroblastic tumor; NGS, next generation sequencing; NPM, nucleophosmin; NSCLC, non-small cell lung cancer; ORR, overall response rate; OS, overall survival; PDX, patient-derived xenograft; PFS, progression free survival; RTK, receptor tyrosine kinase; TCGA, The Cancer Genome Atlas; TFG, TRK-fused gene; TPM, tropomyosin.
conflicts of interest
This review was sponsored by grants from the National Natural Science Foundation of China (#81272518 and #81472192 to Jian Zhang). The authors have no other relevant affiliations or financial involvements with any organization or entity with a financial interest in or a financial conflict with the subject matter or materials discussed in the manuscript.
References
1. Morris SW, Kirstein MN, Valentine MB, Dittmer KG, Shapiro DN, Saltman DL, Look AT: Fusion of a kinase gene, ALK, to a nucleolar protein gene, NPM, in non-Hodgkin’s lymphoma. Science. 1994, 263:1281-4.
2. Elmberger PG, Lozano MD, Weisenburger DD, Sanger W, Chan WC: Transcripts of the npm-alk fusion gene in anaplastic large cell lymphoma, Hodgkin’s disease, and reactive lymphoid lesions. Blood. 1995, 86:3517-21.
3. Mathew P, Sanger WG, Weisenburger DD, Valentine M, Valentine V, Pickering D, Higgins C, Hess M, Cui X, Srivastava DK, Morris SW: Detection of the t(2;5)(p23;q35) and NPM-ALK fusion in non-Hodgkin’s lymphoma by two-color fluorescence in situ hybridization. Blood. 1997, 89:1678-85.
4. Soda M, Choi YL, Enomoto M, Takada S, Yamashita Y, Ishikawa S, Fujiwara S, Watanabe H, Kurashina K, Hatanaka H, Bando M, Ohno S, Ishikawa Y, et al.: Identification of the transforming EML4-ALK fusion gene in non-small-cell lung cancer. Nature. 2007, 448:561-6.
5. Wong DW, Leung EL, So KK, Tam IY, Sihoe AD, Cheng LC, Ho KK, Au JS, Chung LP, Pik Wong M, University of Hong Kong Lung Cancer Study G: The EML4-ALK fusion gene is involved in various histologic types of lung cancers from nonsmokers with wild-type EGFR and KRAS. Cancer. 2009, 115:1723-33.
6. Rikova K, Guo A, Zeng Q, Possemato A, Yu J, Haack H, Nardone J, Lee K, Reeves C, Li Y, Hu Y, Tan Z, Stokes M, et al.: Global survey of phosphotyrosine signaling identifies oncogenic kinases in lung cancer. Cell. 2007, 131:1190-203.
7. Shaw AT, Ou SH, Bang YJ, Camidge DR, Solomon BJ, Salgia R, Riely GJ, Varella-Garcia M, Shapiro GI, Costa DB, Doebele RC, Le LP, et al.: Crizotinib in ROS1-rearranged non-small-cell lung cancer. The New England journal of medicine. 2014, 371:1963-71.
8. Kwak EL, Bang YJ, Camidge DR, Shaw AT, Solomon B, Maki RG, Ou SH, Dezube BJ, Janne PA, Costa DB, Varella-Garcia M, Kim WH, Lynch TJ, et al.: Anaplastic lymphoma kinase inhibition in non-small-cell lung cancer. The New England journal of medicine. 2010, 363:1693-703.
9. Doebele RC, Pilling AB, Aisner DL, Kutateladze TG, Le AT, Weickhardt AJ, Kondo KL, Linderman DJ, Heasley LE, Franklin WA, Varella-Garcia M, Camidge DR: Mechanisms of resistance to crizotinib in patients with ALK gene rearranged non-small cell lung cancer. Clinical cancer research. 2012, 18:1472-82.
10. Awad MM, Katayama R, McTigue M, Liu W, Deng YL, Brooun A, Friboulet L, Huang D, Falk MD, Timofeevski S, Wilner KD, Lockerman EL, Khan TM, et al.: Acquired resistance to crizotinib from a mutation in CD74-ROS1. The New England journal of medicine. 2013, 368:2395-401.
11. Katayama R, Shaw AT, Khan TM, Mino-Kenudson M, Solomon BJ, Halmos B, Jessop NA, Wain JC, Yeo AT, Benes C, Drew L, Saeh JC, Crosby K, et al.: Mechanisms of acquired crizotinib resistance in ALK-rearranged lung Cancers. Science translational medicine. 2012, 4:120ra17.
12. Stoica GE, Kuo A, Aigner A, Sunitha I, Souttou B, Malerczyk C, Caughey DJ, Wen D, Karavanov A, Riegel AT, Wellstein A: Identification of anaplastic lymphoma kinase as a receptor for the growth factor pleiotrophin. The Journal of biological chemistry. 2001, 276:16772-9.
13. Stoica GE, Kuo A, Powers C, Bowden ET, Sale EB, Riegel AT, Wellstein A: Midkine binds to anaplastic lymphoma kinase (ALK) and acts as a growth factor for different cell types. The Journal of biological chemistry. 2002, 277:35990-8.
14. Iwahara T, Fujimoto J, Wen D, Cupples R, Bucay N, Arakawa T, Mori S, Ratzkin B, Yamamoto T: Molecular characterization of ALK, a receptor tyrosine kinase expressed specifically in the nervous system. Oncogene. 1997, 14:439-49.
15. Grande E, Bolos MV, Arriola E: Targeting oncogenic ALK: a promising strategy for cancer treatment. Molecular cancer therapeutics. 2011, 10:569-79.
16. Janoueix-Lerosey I, Lequin D, Brugieres L, Ribeiro A, de Pontual L, Combaret V, Raynal V, Puisieux A, Schleiermacher G, Pierron G, Valteau-Couanet D, Frebourg T, Michon J, et al.: Somatic and germline activating mutations of the ALK kinase receptor in neuroblastoma. Nature. 2008, 455:967-70.
17. Mosse YP, Laudenslager M, Longo L, Cole KA, Wood A, Attiyeh EF, Laquaglia MJ, Sennett R, Lynch JE, Perri P, Laureys G, Speleman F, Kim C, et al.: Identification of ALK as a major familial neuroblastoma predisposition gene. Nature. 2008, 455:930-5.
18. Chen Y, Takita J, Choi YL, Kato M, Ohira M, Sanada M, Wang L, Soda M, Kikuchi A, Igarashi T, Nakagawara A, Hayashi Y, Mano H, Ogawa S: Oncogenic mutations of ALK kinase in neuroblastoma. Nature. 2008, 455:971-4.
19. George RE, Sanda T, Hanna M, Frohling S, Luther W, 2nd, Zhang J, Ahn Y, Zhou W, London WB, McGrady P, Xue L, Zozulya S, Gregor VE, Webb TR, Gray NS, Gilliland DG, Diller L, Greulich H, Morris SW, Meyerson M, Look AT: Activating mutations in ALK provide a therapeutic target in neuroblastoma. Nature. 2008, 455:975-8.
20. Schoppmann SF, Streubel B, Birner P: Amplification but not translocation of anaplastic lymphoma kinase is a frequent event in oesophageal cancer. European journal of cancer. 2013, 49:1876-81.
21. Tuma RS: ALK gene amplified in most inflammatory breast cancers. Journal of the National Cancer Institute. 2012, 104:87-8.
22. Bavi P, Jehan Z, Bu R, Prabhakaran S, Al-Sanea N, Al-Dayel F, Al-Assiri M, Al-Halouly T, Sairafi R, Uddin S, Al-Kuraya KS: ALK gene amplification is associated with poor prognosis in colorectal carcinoma. British journal of cancer. 2013, 109:2735-43.
23. Bonvini P, Zin A, Alaggio R, Pawel B, Bisogno G, Rosolen A: High ALK mRNA expression has a negative prognostic significance in rhabdomyosarcoma. British journal of cancer. 2013, 109:3084-91.
24. Soda M, Takada S, Takeuchi K, Choi YL, Enomoto M, Ueno T, Haruta H, Hamada T, Yamashita Y, Ishikawa Y, Sugiyama Y, Mano H: A mouse model for EML4-ALK-positive lung cancer. Proceedings of the National Academy of Sciences of the United States of America. 2008, 105:19893-7.
25. Maddalo D, Manchado E, Concepcion CP, Bonetti C, Vidigal JA, Han YC, Ogrodowski P, Crippa A, Rekhtman N, de Stanchina E, Lowe SW, Ventura A: In vivo engineering of oncogenic chromosomal rearrangements with the CRISPR/Cas9 system. Nature. 2014, 516:423-7.
26. Peters S, Taron M, Bubendorf L, Blackhall F, Stahel R: Treatment and detection of ALK-rearranged NSCLC. Lung cancer. 2013, 81:145-54.
27. Sasaki T, Rodig SJ, Chirieac LR, Janne PA: The biology and treatment of EML4-ALK non-small cell lung cancer. European journal of cancer. 2010, 46:1773-80.
28. Acquaviva J, Wong R, Charest A: The multifaceted roles of the receptor tyrosine kinase ROS in development and cancer. Biochimica et biophysica acta. 2009, 1795:37-52.
29. Sonnenberg-Riethmacher E, Walter B, Riethmacher D, Godecke S, Birchmeier C: The c-ros tyrosine kinase receptor controls regionalization and differentiation of epithelial cells in the epididymis. Genes & development. 1996, 10:1184-93.
30. Birchmeier C, Sharma S, Wigler M: Expression and rearrangement of the ROS1 gene in human glioblastoma cells. Proceedings of the National Academy of Sciences of the United States of America. 1987, 84:9270-4.
31. Birchmeier C, O’Neill K, Riggs M, Wigler M: Characterization of ROS1 cDNA from a human glioblastoma cell line. Proceedings of the National Academy of Sciences of the United States of America. 1990, 87:4799-803.
32. Charest A, Lane K, McMahon K, Park J, Preisinger E, Conroy H, Housman D: Fusion of FIG to the receptor tyrosine kinase ROS in a glioblastoma with an interstitial del(6)(q21q21). Genes, chromosomes & cancer. 2003, 37:58-71.
33. Gu TL, Deng X, Huang F, Tucker M, Crosby K, Rimkunas V, Wang Y, Deng G, Zhu L, Tan Z, Hu Y, Wu C, Nardone J, et al.: Survey of tyrosine kinase signaling reveals ROS kinase fusions in human cholangiocarcinoma. PloS one. 2011, 6:e15640.
34. El-Deeb IM, Yoo KH, Lee SH: ROS receptor tyrosine kinase: a new potential target for anticancer drugs. Medicinal research reviews. 2011, 31:794-818.
35. Pan Y, Zhang Y, Li Y, Hu H, Wang L, Li H, Wang R, Ye T, Luo X, Zhang Y, Li B, Cai D, Shen L, Sun Y, Chen H: ALK, ROS1 and RET fusions in 1139 lung adenocarcinomas: a comprehensive study of common and fusion pattern-specific clinicopathologic, histologic and cytologic features. Lung cancer. 2014, 84:121-6.
36. Ou SH, Tan J, Yen Y, Soo RA: ROS1 as a ‘druggable’ receptor tyrosine kinase: lessons learned from inhibiting the ALK pathway. Expert review of anticancer therapy. 2012, 12:447-56.
37. McDermott U, Iafrate AJ, Gray NS, Shioda T, Classon M, Maheswaran S, Zhou W, Choi HG, Smith SL, Dowell L, Ulkus LE, Kuhlmann G, Greninger P, Christensen JG, Haber DA, Settleman J: Genomic alterations of anaplastic lymphoma kinase may sensitize tumors to anaplastic lymphoma kinase inhibitors. Cancer research. 2008, 68:3389-95.
38. Puig de la Bellacasa R, Karachaliou N, Estrada-Tejedor R, Teixido J, Costa C, Borrell JI: ALK and ROS1 as a joint target for the treatment of lung cancer: a review. Translational lung cancer research. 2013, 2:72-86.
39. Salido M, Pijuan L, Martinez-Aviles L, Galvan AB, Canadas I, Rovira A, Zanui M, Martinez A, Longaron R, Sole F, Serrano S, Bellosillo B, Wynes MW, et al.: Increased ALK gene copy number and amplification are frequent in non-small cell lung cancer. Journal of thoracic oncology. 2011, 6:21-7.
40. Martelli MP, Sozzi G, Hernandez L, Pettirossi V, Navarro A, Conte D, Gasparini P, Perrone F, Modena P, Pastorino U, Carbone A, Fabbri A, Sidoni A, et al.: EML4-ALK rearrangement in non-small cell lung cancer and non-tumor lung tissues. The American journal of pathology. 2009, 174:661-70.
41. Shaw AT, Yeap BY, Mino-Kenudson M, Digumarthy SR, Costa DB, Heist RS, Solomon B, Stubbs H, Admane S, McDermott U, Settleman J, Kobayashi S, Mark EJ, Rodig SJ, Chirieac LR, Kwak EL, Lynch TJ, Iafrate AJ: Clinical features and outcome of patients with non-small-cell lung cancer who harbor EML4-ALK. Journal of clinical oncology. 2009, 27:4247-53.
42. Shaw AT, Engelman JA: ALK in lung cancer: past, present, and future. Journal of clinical oncology. 2013, 31:1105-11.
43. Bergethon K, Shaw AT, Ou SH, Katayama R, Lovly CM, McDonald NT, Massion PP, Siwak-Tapp C, Gonzalez A, Fang R, Mark EJ, Batten JM, Chen H, et al.: ROS1 rearrangements define a unique molecular class of lung cancers. Journal of clinical oncology. 2012, 30:863-70.
44. Awad MM, Shaw AT: ALK inhibitors in non-small cell lung cancer: crizotinib and beyond. Clinical advances in hematology & oncology. 2014, 12:429-39.
45. Won JK, Keam B, Koh J, Cho HJ, Jeon YK, Kim TM, Lee SH, Lee DS, Kim DW, Chung DH: Concomitant ALK translocation and EGFR mutation in lung cancer: a comparison of direct sequencing and sensitive assays and the impact on responsiveness to tyrosine kinase inhibitor. Annals of oncology. 2015, 26:348-54.
46. de Mello RA, Madureira P, Carvalho LS, Araujo A, O’Brien M, Popat S: EGFR and KRAS mutations, and ALK fusions: current developments and personalized therapies for patients with advanced non-small-cell lung cancer. Pharmacogenomics. 2013, 14:1765-77.
47. Baldi L, Mengoli MC, Bisagni A, Banzi MC, Boni C, Rossi G: Concomitant EGFR mutation and ALK rearrangement in lung adenocarcinoma is more frequent than expected: report of a case and review of the literature with demonstration of genes alteration into the same tumor cells. Lung cancer. 2014, 86:291-5.
48. Chiari R, Duranti S, Ludovini V, Bellezza G, Pireddu A, Minotti V, Bennati C, Crino L: Long-term response to gefitinib and crizotinib in lung adenocarcinoma harboring both epidermal growth factor receptor mutation and EML4-ALK fusion gene. Journal of clinical oncology. 2014, 32:e30-2.
49. Zou HY, Li Q, Lee JH, Arango ME, McDonnell SR, Yamazaki S, Koudriakova TB, Alton G, Cui JJ, Kung PP, Nambu MD, Los G, Bender SL, et al: An orally available small-molecule inhibitor of c-Met, PF-2341066, exhibits cytoreductive antitumor efficacy through antiproliferative and antiangiogenic mechanisms. Cancer research. 2007, 67:4408-17.
50. Chen X, Zhou JY, Zhao J, Chen JJ, Ma SN, Zhou JY: Crizotinib overcomes hepatocyte growth factor-mediated resistance to gefitinib in EGFR-mutant non-small-cell lung cancer cells. Anti-cancer drugs. 2013, 24:1039-46.
51. Gelsomino F, Facchinetti F, Haspinger ER, Garassino MC, Trusolino L, De Braud F, Tiseo M: Targeting the MET gene for the treatment of non-small-cell lung cancer. Critical reviews in oncology/hematology. 2014, 89:284-99.
52. Koivunen JP, Mermel C, Zejnullahu K, Murphy C, Lifshits E, Holmes AJ, Choi HG, Kim J, Chiang D, Thomas R, Lee J, Richards WG, Sugarbaker DJ, Ducko C, Lindeman N, Marcoux JP, Engelman JA, Gray NS, Lee C, Meyerson M, Janne PA: EML4-ALK fusion gene and efficacy of an ALK kinase inhibitor in lung cancer. Clinical cancer research. 2008, 14:4275-83.
53. Gridelli C, Peters S, Sgambato A, Casaluce F, Adjei AA, Ciardiello F: ALK inhibitors in the treatment of advanced NSCLC. Cancer treatment reviews. 2014, 40:300-6.
54. Camidge DR, Bang YJ, Kwak EL, Iafrate AJ, Varella-Garcia M, Fox SB, Riely GJ, Solomon B, Ou SH, Kim DW, Salgia R, Fidias P, Engelman JA, Gandhi L, Janne PA, Costa DB, Shapiro GI, Lorusso P, Ruffner K, Stephenson P, Tang Y, Wilner K, Clark JW, Shaw AT: Activity and safety of crizotinib in patients with ALK-positive non-small-cell lung cancer: updated results from a phase 1 study. The Lancet Oncology. 2012, 13:1011-9.
55. GJ Riely TE, R Salgia, SI Ou, SN Gettinger, GA Otterson, S Lanzalone, A Polli, AT Shaw: Results og a global phase II study og crizotinib in advanced ALK-positive non-small cell lung cancer. IASLC Chicago Multidisciplinary Symposium in Thoracic Oncology. 2012, 2012.
56. Shaw AT, Kim DW, Nakagawa K, Seto T, Crino L, Ahn MJ, De Pas T, Besse B, Solomon BJ, Blackhall F, Wu YL, Thomas M, O’Byrne KJ, Moro-Sibilot D, Camidge DR, Mok T, Hirsh V, Riely GJ, Iyer S, Tassell V, Polli A, Wilner KD, Janne PA: Crizotinib versus chemotherapy in advanced ALK-positive lung cancer. The New England journal of medicine. 2013, 368:2385-94.
57. Solomon BJ, Mok T, Kim DW, Wu YL, Nakagawa K, Mekhail T, Felip E, Cappuzzo F, Paolini J, Usari T, Iyer S, Reisman A, Wilner KD, Tursi J, Blackhall F, Investigators P: First-line crizotinib versus chemotherapy in ALK-positive lung cancer. The New England journal of medicine. 2014, 371:2167-77.
58. Gambacorti Passerini C, Farina F, Stasia A, Redaelli S, Ceccon M, Mologni L, Messa C, Guerra L, Giudici G, Sala E, Mussolin L, Deeren D, King MH, Steurer M, Ordemann R, Cohen AM, Grube M, Bernard L, Chiriano G, Antolini L, Piazza R: Crizotinib in advanced, chemoresistant anaplastic lymphoma kinase-positive lymphoma patients. Journal of the National Cancer Institute. 2014, 106:djt378.
59. Butrynski JE, D’Adamo DR, Hornick JL, Dal Cin P, Antonescu CR, Jhanwar SC, Ladanyi M, Capelletti M, Rodig SJ, Ramaiya N, Kwak EL, Clark JW, Wilner KD, Christensen JG, Janne PA, Maki RG, Demetri GD, Shapiro GI: Crizotinib in ALK-rearranged inflammatory myofibroblastic tumor. The New England journal of medicine. 2010, 363:1727-33.
60. Kobayashi S, Boggon TJ, Dayaram T, Janne PA, Kocher O, Meyerson M, Johnson BE, Eck MJ, Tenen DG, Halmos B: EGFR mutation and resistance of non-small-cell lung cancer to gefitinib. The New England journal of medicine. 2005, 352:786-92.
61. Pao W, Miller VA, Politi KA, Riely GJ, Somwar R, Zakowski MF, Kris MG, Varmus H: Acquired resistance of lung adenocarcinomas to gefitinib or erlotinib is associated with a second mutation in the EGFR kinase domain. PLoS medicine. 2005, 2:e73.
62. Choi YL, Soda M, Yamashita Y, Ueno T, Takashima J, Nakajima T, Yatabe Y, Takeuchi K, Hamada T, Haruta H, Ishikawa Y, Kimura H, Mitsudomi T, Tanio Y, Mano H, Group ALKLCS: EML4-ALK mutations in lung cancer that confer resistance to ALK inhibitors. The New England journal of medicine. 2010, 363:1734-9.
63. Sun HY, Ji FQ: A molecular dynamics investigation on the crizotinib resistance mechanism of C1156Y mutation in ALK. Biochemical and biophysical research communications. 2012, 423:319-24.
64. Tartarone A, Lazzari C, Lerose R, Conteduca V, Improta G, Zupa A, Bulotta A, Aieta M, Gregorc V: Mechanisms of resistance to EGFR tyrosine kinase inhibitors gefitinib/erlotinib and to ALK inhibitor crizotinib. Lung cancer. 2013, 81:328-36.
65. Qi X, Ma W, Li S, Zhou C: Overcoming crizotinib resistance in ALK-rearranged non-small cell lung cancer. Lung cancer. 2014, 85:335-6.
66. Sun H, Li Y, Tian S, Wang J, Hou T: P-loop conformation governed crizotinib resistance in G2032R-mutated ROS1 tyrosine kinase: clues from free energy landscape. PLoS computational biology. 2014, 10:e1003729.
67. Katayama R, Khan TM, Benes C, Lifshits E, Ebi H, Rivera VM, Shakespeare WC, Iafrate AJ, Engelman JA, Shaw AT: Therapeutic strategies to overcome crizotinib resistance in non-small cell lung cancers harboring the fusion oncogene EML4-ALK. Proceedings of the National Academy of Sciences of the United States of America. 2011, 108:7535-40.
68. Kanaan Z, Kloecker GH, Paintal A, Perez CA: Novel targeted therapies for resistant ALK-rearranged non-small-cell lung cancer: ceritinib and beyond. OncoTargets and therapy. 2015, 8:885-92.
69. Friboulet L, Li N, Katayama R, Lee CC, Gainor JF, Crystal AS, Michellys PY, Awad MM, Yanagitani N, Kim S, Pferdekamper AC, Li J, Kasibhatla S, Sun F, Sun X, Hua S, McNamara P, Mahmood S, Lockerman EL, Fujita N, Nishio M, Harris JL, Shaw AT, Engelman JA: The ALK inhibitor ceritinib overcomes crizotinib resistance in non-small cell lung cancer. Cancer discovery. 2014, 4:662-73.
70. Ni Z, Zhang TC: Computationally unraveling how ceritinib overcomes drug-resistance mutations in ALK-rearranged lung cancer. Journal of molecular modeling. 2015, 21:175.
71. Shaw AT, Kim DW, Mehra R, Tan DS, Felip E, Chow LQ, Camidge DR, Vansteenkiste J, Sharma S, De Pas T, Riely GJ, Solomon BJ, Wolf J, Thomas M, Schuler M, Liu G, Santoro A, Lau YY, Goldwasser M, Boral AL, Engelman JA: Ceritinib in ALK-rearranged non-small-cell lung cancer. The New England journal of medicine. 2014, 370:1189-97.
72. Gainor JF, Tan DS, De Pas T, Solomon BJ, Ahmad A, Lazzari C, de Marinis F, Spitaleri G, Schultz K, Friboulet L, Yeap BY, Engelman JA, Shaw AT: Progression-Free and Overall Survival in ALK-Positive NSCLC Patients Treated with Sequential Crizotinib and Ceritinib. Clinical cancer research. 2015, 21:2745-52.
73. Dong-Wam Kim RM, Daniel SW Tan, Enriqueta Felip, Laura QM Chow, D Ross Camidge, Johan Vansteenkiste, Sunil Sharma, Tommaso De Pas, Gregory J Riely, Benjamin J Solomon, Juergen Wolf, Michael Thomas, Martin Schuler, Geoffrey Liu, Armando Santoro, Margarida Geraldes, Anthony L Boral, Alejandro Yovine, Alice T Shaw.: Ceritinib in advanced anaplastic lymphoma kinase (ALK)-rearranged (ALK+) non-small cell lung cancer (NSCLC): Results of the ASCEND-1 trial. 2014 ASCO Annual Meeting, 2014.
74. Alice T Shaw RM, Daniel SW Tan, Enriqueta Felip, Laura QM Chow, D Ross Camidge, Johan Vansteenkiste, Sunil Sharma, Tommaso De Pas, Gregory J Riely, Benjamin J Solomon, Juergen Wolf, Michael Thomas, Martin Schuler, Geoffrey Liu, Armando Santoro, Paramita Sen, Anthony L Boral, Alejandro Yovine, Dong-Wan Kim: Evaluation of Ceritinib-treated Patients with Anaplastic Lymphoma Kinase Rearranged (ALK+) Non-small Cell Lung Cancer (NSCLC) and Brain Metastases in the ASCEND-1 Study. Annals of oncology. 2014, 25 (Supplement 4): iv426–iv470.
75. Khozin S, Blumenthal GM, Zhang L, Tang S, Brower M, Fox E, Helms W, Leong R, Song P, Pan Y, Liu Q, Zhao P, Zhao H, Lu D, Tang Z, Al Hakim A, Boyd K, Keegan P, Justice R, Pazdur R: FDA approval: ceritinib for the treatment of metastatic anaplastic lymphoma kinase-positive non-small cell lung cancer. Clinical cancer research. 2015, 21:2436-9.
76. Davare MA, Saborowski A, Eide CA, Tognon C, Smith RL, Elferich J, Agarwal A, Tyner JW, Shinde UP, Lowe SW, Druker BJ: Foretinib is a potent inhibitor of oncogenic ROS1 fusion proteins. Proceedings of the National Academy of Sciences of the United States of America. 2013, 110:19519-24.
77. Katayama R, Kobayashi Y, Friboulet L, Lockerman EL, Koike S, Shaw AT, Engelman JA, Fujita N: Cabozantinib overcomes crizotinib resistance in ROS1 fusion-positive cancer. Clinical cancer research. 2015, 21:166-74.
78. Zou HY, Li Q, Engstrom LD, West M, Appleman V, Wong KA, McTigue M, Deng YL, Liu W, Brooun A, Timofeevski S, McDonnell SR, Jiang P, Falk MD, Lappin PB, Affolter T, Nichols T, Hu W, Lam J, Johnson TW, Smeal T, Charest A, Fantin VR: PF-06463922 is a potent and selective next-generation ROS1/ALK inhibitor capable of blocking crizotinib-resistant ROS1 mutations. Proceedings of the National Academy of Sciences of the United States of America. 2015, 112:3493-8.
79. Zou HY, Friboulet L, Kodack DP, Engstrom LD, Li Q, West M, Tang RW, Wang H, Tsaparikos K, Wang J, Timofeevski S, Katayama R, Dinh DM, Lam H, Lam JL, Yamazaki S, Hu W, Patel B, Bezwada D, Frias RL, Lifshits E, Mahmood S, Gainor JF, Affolter T, Lappin PB, Gukasyan H, Lee N, Deng S, Jain RK, Johnson TW, Shaw AT, Fantin VR, Smeal T: PF-06463922, an ALK/ROS1 Inhibitor, Overcomes Resistance to First and Second Generation ALK Inhibitors in Preclinical Models. Cancer cell. 2015, 28:70-81.
80. Johnson TW, Richardson PF, Bailey S, Brooun A, Burke BJ, Collins MR, Cui JJ, Deal JG, Deng YL, Dinh D, Engstrom LD, He M, Hoffman J, Hoffman RL, Huang Q, Kania RS, Kath JC, Lam H, Lam JL, Le PT, Lingardo L, Liu W, McTigue M, Palmer CL, Sach NW, Smeal T, Smith GL, Stewart AE, Timofeevski S, Zhu H, Zhu J, Zou HY, Edwards MP: Discovery of (10R)-7-amino-12-fluoro-2,10,16-trimethyl-15-oxo-10,15,16,17-tetrahydro-2H-8,4-(m etheno)pyrazolo [4,3-h.] [2,5,11.]-benzoxadiazacyclotetradecine-3-carbonitrile (PF-06463922), a macrocyclic inhibitor of anaplastic lymphoma kinase (ALK) and c-ros oncogene 1 (ROS1) with preclinical brain exposure and broad-spectrum potency against ALK-resistant mutations. Journal of medicinal chemistry. 2014, 57:4720-44.
81. Engelman JA, Zejnullahu K, Mitsudomi T, Song Y, Hyland C, Park JO, Lindeman N, Gale CM, Zhao X, Christensen J, Kosaka T, Holmes AJ, Rogers AM, Cappuzzo F, Mok T, Lee C, Johnson BE, Cantley LC, Janne PA: MET amplification leads to gefitinib resistance in lung cancer by activating ERBB3 signaling. Science. 2007, 316:1039-43.
82. Sasaki T, Koivunen J, Ogino A, Yanagita M, Nikiforow S, Zheng W, Lathan C, Marcoux JP, Du J, Okuda K, Capelletti M, Shimamura T, Ercan D, Stumpfova M, Xiao Y, Weremowicz S, Butaney M, Heon S, Wilner K, Christensen JG, Eck MJ, Wong KK, Lindeman N, Gray NS, Rodig SJ, Janne PA: A novel ALK secondary mutation and EGFR signaling cause resistance to ALK kinase inhibitors. Cancer research. 2011, 71:6051-60.
83. Yamada T, Takeuchi S, Nakade J, Kita K, Nakagawa T, Nanjo S, Nakamura T, Matsumoto K, Soda M, Mano H, Uenaka T, Yano S: Paracrine receptor activation by microenvironment triggers bypass survival signals and ALK inhibitor resistance in EML4-ALK lung cancer cells. Clinical cancer research. 2012, 18:3592-602.
84. Davies KD, Mahale S, Astling DP, Aisner DL, Le AT, Hinz TK, Vaishnavi A, Bunn PA, Jr., Heasley LE, Tan AC, Camidge DR, Varella-Garcia M, Doebele RC: Resistance to ROS1 inhibition mediated by EGFR pathway activation in non-small cell lung cancer. PloS one. 2013, 8:e82236.
85. Cargnelutti M, Corso S, Pergolizzi M, Mevellec L, Aisner DL, Dziadziuszko R, Varella-Garcia M, Comoglio PM, Doebele RC, Vialard J, Giordano S: Activation of RAS family members confers resistance to ROS1 targeting drugs. Oncotarget. 2015, 6:5182-94. doi: 10.18632/oncotarget.3311.
86. Choi SY, Lin D, Gout PW, Collins CC, Xu Y, Wang Y: Lessons from patient-derived xenografts for better in vitro modeling of human cancer. Advanced drug delivery reviews. 2014, 79-80:222-37.
87. Crystal AS, Shaw AT, Sequist LV, Friboulet L, Niederst MJ, Lockerman EL, Frias RL, Gainor JF, Amzallag A, Greninger P, Lee D, Kalsy A, Gomez-Caraballo M, Elamine L, Howe E, Hur W, Lifshits E, Robinson HE, Katayama R, Faber AC, Awad MM, Ramaswamy S, Mino-Kenudson M, Iafrate AJ, Benes CH, Engelman JA: Patient-derived models of acquired resistance can identify effective drug combinations for cancer. Science. 2014, 346:1480-6.
88. Proia DA, Sang J, He S, Smith DL, Sequeira M, Zhang C, Liu Y, Ye S, Zhou D, Blackman RK, Foley KP, Koya K, Wada Y: Synergistic activity of the Hsp90 inhibitor ganetespib with taxanes in non-small cell lung cancer models. Investigational new drugs. 2012, 30:2201-9.
89. Whitesell L, Lindquist SL: HSP90 and the chaperoning of cancer. Nature reviews Cancer. 2005, 5:761-72.
90. Sequist LV, Gettinger S, Senzer NN, Martins RG, Janne PA, Lilenbaum R, Gray JE, Iafrate AJ, Katayama R, Hafeez N, Sweeney J, Walker JR, Fritz C, Ross RW, Grayzel D, Engelman JA, Borger DR, Paez G, Natale R: Activity of IPI-504, a novel heat-shock protein 90 inhibitor, in patients with molecularly defined non-small-cell lung cancer. Journal of clinical oncology. 2010, 28:4953-60.
91. Sang J, Acquaviva J, Friedland JC, Smith DL, Sequeira M, Zhang C, Jiang Q, Xue L, Lovly CM, Jimenez JP, Shaw AT, Doebele RC, He S, Bates RC, Camidge DR, Morris SW, El-Hariry I, Proia DA: Targeted inhibition of the molecular chaperone Hsp90 overcomes ALK inhibitor resistance in non-small cell lung cancer. Cancer discovery. 2013, 3:430-43.
92. Miyajima N, Tsutsumi S, Sourbier C, Beebe K, Mollapour M, Rivas C, Yoshida S, Trepel JB, Huang Y, Tatokoro M, Shinohara N, Nonomura K, Neckers L: The HSP90 inhibitor ganetespib synergizes with the MET kinase inhibitor crizotinib in both crizotinib-sensitive and -resistant MET-driven tumor models. Cancer research. 2013, 73:7022-33.
93. Socinski MA, Goldman J, El-Hariry I, Koczywas M, Vukovic V, Horn L, Paschold E, Salgia R, West H, Sequist LV, Bonomi P, Brahmer J, Chen LC, Sandler A, Belani CP, Webb T, Harper H, Huberman M, Ramalingam S, Wong KK, Teofilovici F, Guo W, Shapiro GI: A multicenter phase II study of ganetespib monotherapy in patients with genotypically defined advanced non-small cell lung cancer. Clinical cancer research. 2013, 19:3068-77.
94. Edward B. Garon TM, Fabrice Barlesi, Leena Gandhi, Lecia V. Sequist, Sang-We Kim, Harry J.M. Groen, Benjamin Besse, Egbert F Smit, Dong-Wan Kim, Mikhail Akimov, Emin Avsar, Stuart Bailey, Enriqueta Felip.: Phase II study of the HSP90 inhibitor AUY922 in patients with previously treated, advanced non-small cell lung cancer (NSCLC). 2012 ASCO Annual Meeting: J Clin Oncol 30, 2012 (suppl; abstr 7543), 2012.
95. Kim HR, Kim DJ, Kang DR, Lee JG, Lim SM, Lee CY, Rha SY, Bae MK, Lee YJ, Kim SH, Ha SJ, Soo RA, Chung KY, et al.: Fibroblast growth factor receptor 1 gene amplification is associated with poor survival and cigarette smoking dosage in patients with resected squamous cell lung cancer. Journal of clinical oncology. 2013, 31:731-7.
96. Tang X, Xue Y, Su S, Harun N, Cai Y, Behrens C, Papadimitrakopoulou V, Blowers D, Moran C, Kalhor N, Rice DC, Beran G, Lee JJ, et al.: Fibroblast growth factor receptor 1 (FGFR1) gene copy number gain in adenocarcinoma and squamous cell carcinoma of the lung. 2013 ASCO Annual Meeting, 2013.
97. Ford CE, Lau SK, Zhu CQ, Andersson T, Tsao MS, Vogel WF: Expression and mutation analysis of the discoidin domain receptors 1 and 2 in non-small cell lung carcinoma. British journal of cancer. 2007, 96:808-14.
98. Hammerman PS, Sos ML, Ramos AH, Xu C, Dutt A, Zhou W, Brace LE, Woods BA, Lin W, Zhang J, Deng X, Lim SM, Heynck S, et al: Mutations in the DDR2 kinase gene identify a novel therapeutic target in squamous cell lung cancer. Cancer discovery. 2011, 1:78-89.
99. Zhang S, Zheng X, Huang H, Wu K, Wang B, Chen X, Ma S: Afatinib increases sensitivity to radiation in non-small cell lung cancer cells with acquired EGFR T790M mutation. Oncotarget. 2015, 6:5832-45. doi: 10.18632/oncotarget.3332.
100. Turke AB, Zejnullahu K, Wu YL, Song Y, Dias-Santagata D, Lifshits E, Toschi L, Rogers A, Mok T, Sequist L, Lindeman NI, Murphy C, Akhavanfard S, Yeap BY, Xiao Y, Capelletti M, Iafrate AJ, Lee C, Christensen JG, Engelman JA, Janne PA: Preexistence and clonal selection of MET amplification in EGFR mutant NSCLC. Cancer cell. 2010, 17:77-88.