
Introduction
Compelling evidence demonstrates that altered chromatin remodeling plays an important role in tumorigenesis. Histone methylation is known to be regulated by histone methyltransferases and demethylases. LSD1/KDM1 was the first histone demethylase to be discovered; it can demethylate mono- and dimethylated lysine residues through a FAD-dependent amine oxidase reaction, and it has been considered an important contributor to some tumors [1, 2].
Recently, some members of jumonji C (JmjC) domain-containing protein family were identified as a second type of histone demethylase that can remove all methylation modifications on the lysine residues of histones [3, 4, 5]. All family members have a JmjC domain, which shares high similarity with the cupin metalloenzyme domain [6]. However, in addition to the subset of members with histone demethylase activity, many other members, including JMJD5, JMJD6 and TYW5, also possess protein or RNA hydroxylase activity [7, 8, 9]. Dysregulation of family members such as JMJD5, JMJD6, JARID1B, JARID2, PHF8 and JMJD2A causes abnormal embryonic development or promotes cancer cell proliferation and migration [5, 10, 11].
It is unknown if JmjC domain-containing proteins are involved in the pathogenesis of hepatocellular carcinoma (HCC). To define which JmjC domain-containing proteins might contribute to HCC, we mined the gene expression profiles of all family members in HCC by analyzing public databases. Very interestingly, nineteen of the twenty-nine members of this family were misregulated in HCC specimens. Among them, the most significantly down-regulated gene is JmjC domain-containing protein 5 (JMJD5); nearly 80% of HCC specimens showed at least two-fold downregulation of this gene. Our evidence showed that reduced JMJD5 promoted HCC cell proliferation and cell cycle progression by directly suppressing CDKN1A transcription. The present results provided novel insight into JMJD5’s function during HCC progression.
Results
The expression patterns of JmjC family members show JMJD5 downregulation in HCC specimens
To explore if JmjC family members are involved in HCC pathogenesis, we first used public GEO datasets to evaluate the gene expression patterns of known JmjC family members in human HCC. Two databases (GSE25097 and GSE14520) were chosen because they contain comprehensive gene expression profiles for more than 100 paired HCC samples. Interestingly, a few JmjC family members exhibited similar expression patterns in at least 30% of the HCC samples in both datasets (Figure 1A and Supplemental Figure 1A). KDM5B was significantly upregulated at least two-fold in 40% of the HCC specimens, whereas JMJD5 was significantly downregulated at least two-fold in 82% of the samples. KDM5B, also named JARID1B or PLU-1, has been considered an oncogene; it encodes a histone H3 lysine 4 (H3K4) demethylase and is localized on chromosome 1q32.1 [12, 13]. However, the function and mechanisms of action of JMJD5, also named KDM8, in tumorigenesis were relatively unclear. Although retroviral insertional mutagenesis originally identified JMJD5 as a tumor suppressor gene [14], it has recently been reported that JMJD5 overexpression promoted breast cancer cell proliferation [15]; this result suggests that JMJD5 may suppress tumors or promote cancer in a cell context-dependent manner. The JMJD5 expression pattern in HCC implied that JMJD5 may function as a tumor suppressor in this cancer. To address this possibility, in the present work, we focused on the role and mechanism of action of JMJD5 in HCC pathogenesis.
Using the public databases, GDS3834 and THE HUMAN PROTEIN ATLAS [16], we analyzed the JMJD5 expression pattern in human tissue. Very interestingly, JMJD5 was most highly expressed in the liver (Figure 1B), suggesting that JMJD5 could play an important role in hepatic functions and physiology. By analyzing TCGA data we surprisingly found that the lower expression of JMJD5 was significantly correlated with age (≤ 60 or > 60 year old, P = 0.0082), tumor stages (P < 0.05), overall survival (≤ 24 or > 24 months, P = 0.0222) and overall survival status (dead or alive, P = 0.034) (Table 1). But it was not associated with gender, metastasis stage and recurrence (P > 0.05) (Table 1). A Kaplan-Meier survival analysis exhibited that HCC patients with low JMJD5 expression had shorter survival time compared with those with high JMJD5 expression (P < 0.01) (Figure 1C), and the median survival of HCC patients with low JMJD5 expression and high JMJD5 expression was 987 and 2141 days respectively. Furthermore, we confirmed downregulation of JMJD5 in HCC samples and cell lines. JMJD5 was significantly downregulated in 29/46 (63%) samples, as shown by real-time RT-PCR (Figure 1D); in 43/63 (68.3%) different HCC samples, as shown by semi-quantitative PCR (Supplemental Figure 1B); as well as in five paired HCC samples, as shown by western blotting (Figure 1E). Moreover, compared with the normal adult liver, JMJD5 was markedly downregulated in all examined HCC cell lines, as shown by real-time RT-PCR and semi-quantitative RT-PCR (Figure 1F and Supplemental Figure 1C). The data collectively indicated that JMJD5 was significantly downregulated in most HCC samples.
Table 1: The correlations of JMJD5 expression with clinical characteristics of HCC
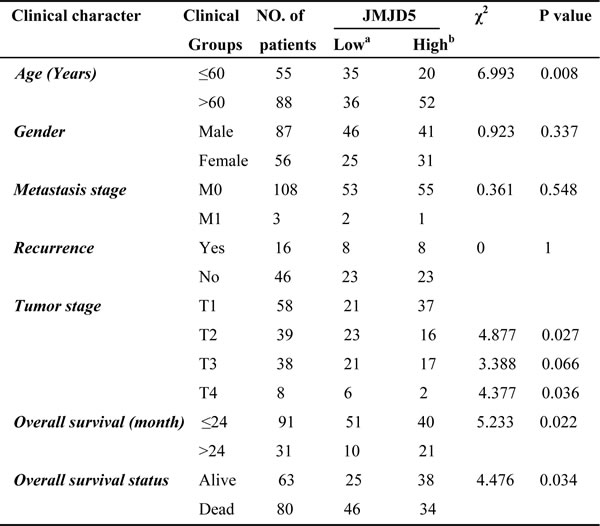
a. HCC patients were ranked according to JMJD5 expression from high to low, the bottom 50% were served as JMJD5 low group.
b. The top 50% were defined as JMJD5 high group.
All data were collected from TCGA. Chi-square test was utilized to estimate the significant difference. P< 0.05 was considered to be significant.
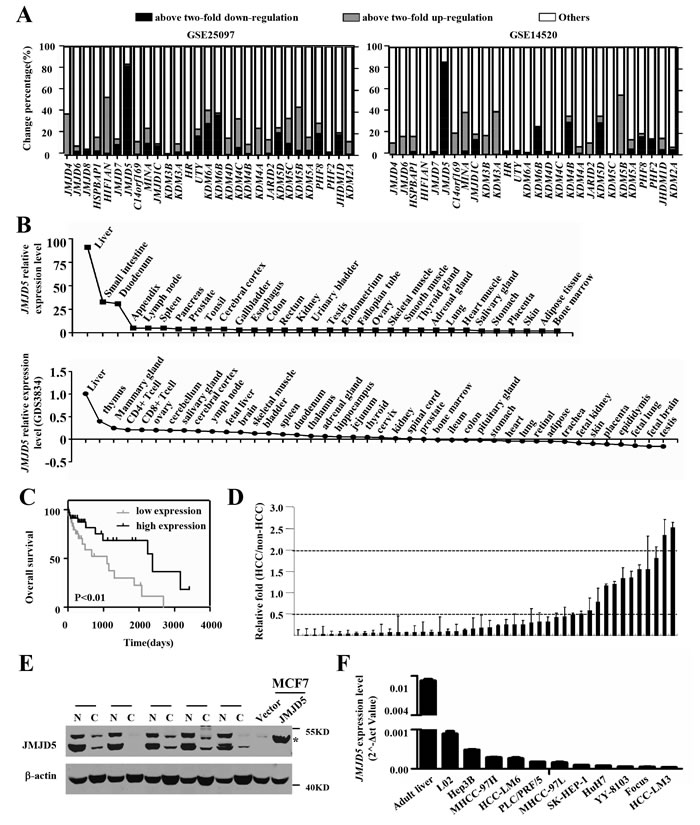
Figure 1: Tissue expression patterns and JMJD5 expression in HCC specimens and cell lines. A. The expression patterns of JmjC family members in HCC tissue were analyzed based on two public datasets (GSE25097 and GSE14520). Each column represents one gene, and the y-axis indicates the percent change in gene expression. B. The relative JMJD5 expression level in various human tissues and cells was evaluated based on THE HUMNA PROTEIN ATLAS and a public database (GDS3834). C. The result of Kaplan-Meier survival analysis exhibited that patients with low expression of JMJD5 (n = 47) had shorter overall survival time than those with high JMJD5 expression (n = 47). D. JMJD5 expression was measured in 46 paired HCC and adjacent, non-cancerous livers using real-time RT-PCR, each column represented one paired sample, and y axis indicated the fold change. 0.5 and 2 meant that JMJD5 was down-regulated or up-regulated two fold in HCC. E. Five paired HCC and corresponding adjacent, non-cancerous liver samples were chosen to evaluate the expression of JMJD5 protein by Western blot assay, where MCF7 cells expressing ectopic JMJD5 were served as positive control, asterisk indicated the JMJD5 band. N, adjacent, non-cancerous livers; C, HCC samples. F. mRNA expression of JMJD5 was detected in HCC cell lines and adult liver using real-time RT-PCR.
JMJD5 downregulation in HCC is caused by an epigenetic mechanism
To investigate why JMJD5 was silenced in HCC, we used DAC, a DNA methyltransferase (DNMT) inhibitor, and TSA, a histone deacetylase (HDAC) inhibitor, to treat HCC cell lines. Interestingly, treatment with TSA, but not with DAC, could trigger JMJD5 expression in SK-HEP-1, HuH-7, YY-8103, MHCC-97H and HCC-LM6 cells (Figure 2A and Supplemental Figure 2A), implying that, although the JMJD5 promoter contains one CpG islands, it is histone modifications, not DNA methylation, that could be the major mechanism for regulating JMJD5 transcription. And the bisulfite sequencing of the CpG islands in the JMJD5 promoter also showed that there were no difference between the HCC specimens and the adjacent, non-cancerous liver tissue (data not shown). Next, we analyzed the histone modifications on the JMJD5 locus using the public chromatin immunoprecipitation (ChIP)-seq datasets from HepG2 hepatic cells. The data showed that acetylation of lysine 9 and 27 (H3K9ac and H3K27ac) as well as dimethylation and trimethylation of lysine 4 (H3K4me3 and H3K4me2) in histone H3, which are associated with active gene expression [17, 18, 19, 20, 21], was enriched in the JMJD5 promoter, whereas two known repressive histone modifications, H3K9 and H3K27 trimethylation (m3), were reduced in the same region (Figure 2B). To determine if these histone modifications in the region were altered during HCC tumorigenesis, we performed ChIP assays with anti-H3K9ac, anti-H3K27ac, anti-H3K4me2, anti-H3K4me3, anti-H3K9m3 and anti-H3K27m3 antibodies, and data showed that H3K9ac, H3K27ac, H3K4me2 and H3K4me3 were significantly decreased in HCC specimens; by contrast, both H3K9me3 and H3K27me3 were significantly enriched in HCC compared with the adjacent, non-cancerous liver tissue (Figure 2C and Supplemental Figure 2B). These collective data indicated that downregulation of JMJD5 in HCC may be primarily attributed to epigenetic regulation.
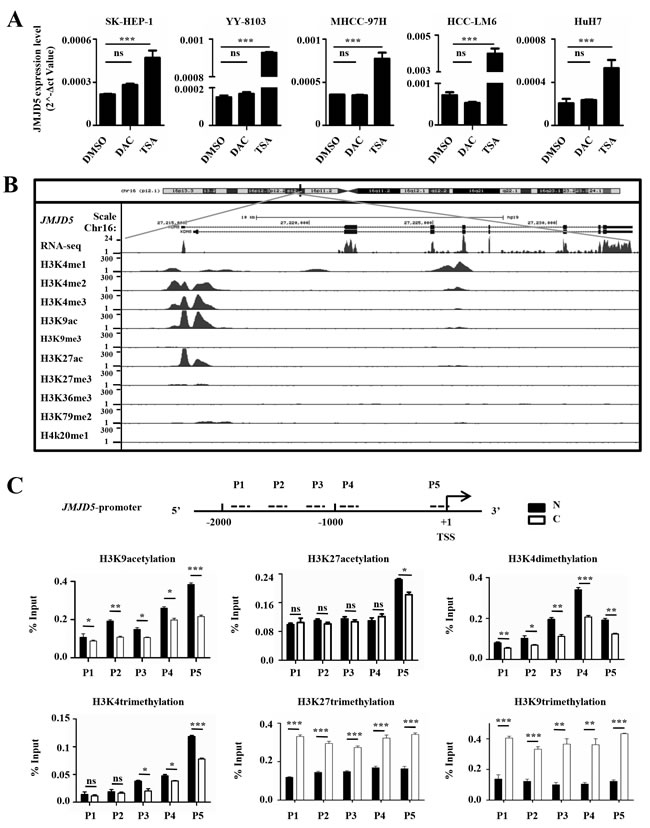
Figure 2: Epigenetic silencing of JMJD5 in HCC specimens and cells. A. The JMJD5 mRNA expression level was evaluated by real-time RT-PCR in five HCC cell lines after treatment with TSA or DAC. DMSO treatment served as a control. B. The histone modification patterns at the JMJD5 locus were analyzed using UCSC ChIP-seq data from HepG2 cells. C. ChIP assays followed by real-time RT-PCR, were performed to evaluate the enrichment of histone modifications on the JMJD5 promoter in HCC samples. The dotted lines indicate PCR primer (P1, P2, P3, P4 and P5) positions, and the numbers indicate the distance from transcription start site (TSS). *, P < 0.05; **, P < 0.01; ***, P < 0.001; ns, no significant difference.
JMJD5 knockdown promotes HCC cell growth and tumorigenicity
Abnormal proliferation is one property of malignant tumors [22]. To evaluate the effect of JMJD5 knockdown on HCC cell proliferation, we transiently transfected chemically synthesized small interfering RNAs (siRNAs) against JMJD5 into HCC cell lines, including MHCC-97H, SK-HEP-1 and YY-8103, and found that siRNA-mediated JMJD5 silencing significantly promoted the growth of these cells (Figure 3A). Subsequently we constructed a recombinant pSUPER plasmid encoding a short hairpin RNA (shRNA) against JMJD5 and then transfected it into MHCC-97H, SK-HEP-1, YY-8103, L02 and Hep3B cells. Interestingly, JMJD5 knockdown also significantly promoted colony formation in these cells (Figure 3B and Supplemental Figure 3A). Furthermore, SK-HEP-1 and YY-8103 cells containing a stable shRNA against endogenous JMJD5 were subcutaneously inoculated into the flanks of nude mice; as a control, equal quantities of cells containing the negative control shRNA were injected into the opposite flank of the same mice. Significantly, shRNA-mediated JMJD5 knockdown promoted the in vivo tumorigenicity of the HCC cells, as shown by their increased xenograft tumor size and weight compared with that of the cells with the empty vector (Figure 3C, 3D and Supplemental Figure 3B-3D). We also performed soft agar colony formation assays to investigate the anchorage-independent growth. The results showed that knockdown of endogenous JMJD5 clearly promoted the anchorage-independent growth of MHCC-97H, SK-HEP-1 and YY-8103 cells (Figure 3E). These data suggested that reduced JMJD5 expression may facilitate the tumorigenesis and progression of HCC.
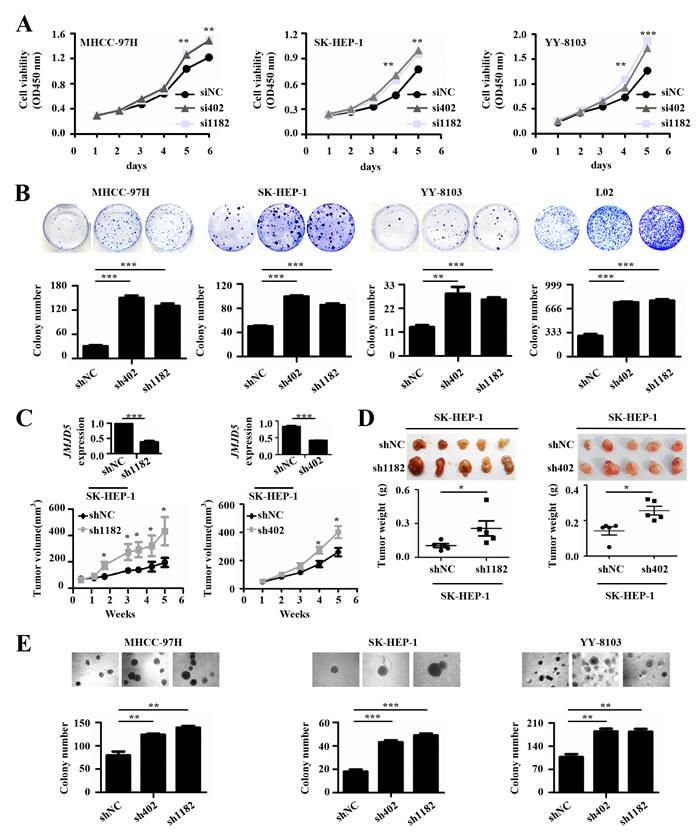
Figure 3: The effects of JMJD5 knockdown on the proliferation of HCC cells. A. Three HCC cell lines were transfected with siRNAs against JMJD5 (si402 and si1182), and cell viability was measured every day for 5 or 6 days to determine the cell growth curve. siNC served as a negative control. B. Four cell lines were transfected with plasmids containing shRNAs against JMJD5 (sh402 and sh1182), and colonies were selected with G418. Plate colony formation is shown with representative plates (upper), and colony numbers were statistically analyzed (lower). C.-D. In vivo tumorigenicity of SK-hep1 cells with JMJD5 stably knocked down. C. Tumor volumes were measured at the indicated time points. The JMJD5 expression level in these cells is indicated at the top. Each group contained five mice. D. Tumors were removed (upper), weighed and statistically analyzed (lower). E. Three HCC cell lines were transfected with shRNAs against JMJD5, and colony formation on soft agar medium was analyzed. Soft agar colony formation is shown with representative dishes (top), and the number of colonies was statistically analyzed (bottom). All experiments were repeated at least three times. *, P < 0.05; **, P < 0.01; ***, P < 0.001.
JMJD5 overexpression inhibits HCC cell growth and tumorigenicity
To further explore the effect of JMJD5 on HCC cell proliferation, we transiently transfected the recombinant plasmid pcDNA3.1B, which encodes JMJD5, into MHCC-97H, HuH-7, HCC-LM6 and L02; as a negative control, a plasmid containing the reverse JMJD5 sequence was used. As expected, compared with the controls, ectopic JMJD5 expression significantly inhibited the growth and colony formation of these cells (Figure 4A, 4B). Furthermore, ectopic JMJD5 expression significantly suppressed the in vivo tumorigenicity of HCC-LM6 and MHCC-97H cells after they were subcutaneously injected into the flanks of nude mice, as shown by the reduced size and weight of xenograft tumors (Figure 4C, 4D and Supplemental Figure 4A). JMJD5 overexpression also reduced the anchorage-independent colony formation of MHCC-97H, HuH-7, HCC-LM6 and HCC-LM3 cells (Figure 4E and Supplemental Figure 4B). The above results collectively implied that JMJD5 may function as a tumor suppressor in HCC cells by suppressing cell proliferation.
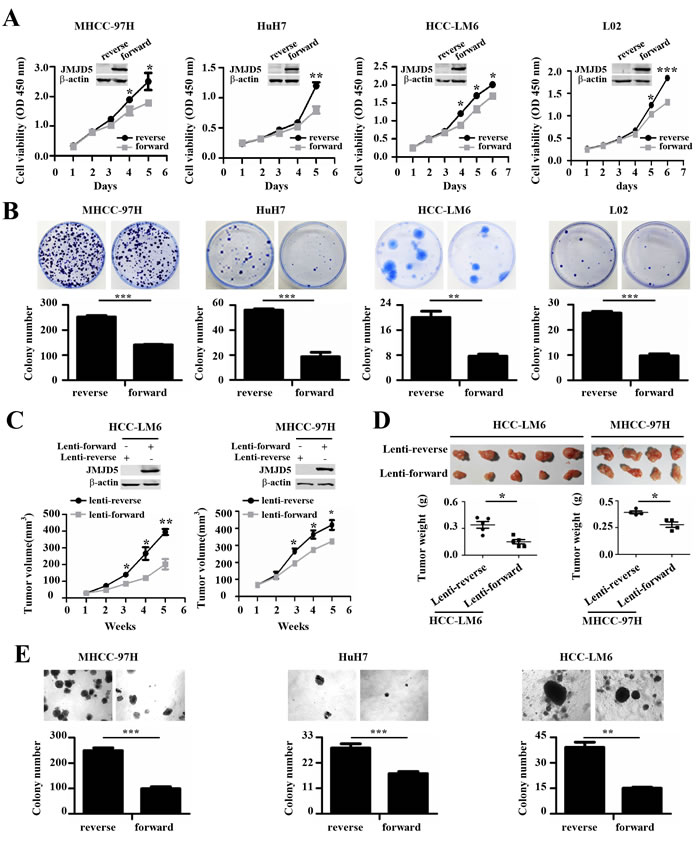
Figure 4: The anti-proliferative role of JMJD5 in HCC cells. A.-B. The proliferation of four hepatic cell lines transfected with plasmids expressing JMJD5 (forward) was evaluated. A. Cell viability was calculated every day for 5 or 6 days to determine the cell growth curve. A plasmid containing the reverse JMJD5 sequence was used as a control, and JMJD5 protein expression, as indicated by western blot, is shown at the top. B. Plate colony formation is shown with representative dishes (top) and the number of colonies was statistically analyzed (bottom). C.-D. In vivo tumorigenicity of MHCC-97H and HCC-LM6 HCC cells stably expressing JMJD5. C. Tumor volumes were measured at the indicated time points. The JMJD5 expression level in these cells is indicated at the top. Each group contained four or five mice. D. Tumors were removed (upper), weighed, and then statistically analyzed (lower). E. Three HCC cell lines were transfected with JMJD5, and colony formation in soft agar medium was analyzed. All experiments were repeated at least three times. *, P < 0.05; **, P < 0.01; ***, P < 0.001.
JMJD5 regulates the G1-S transition of the cell cycle
To explore the mechanism by which JMJD5 regulates cell proliferation, we carried out Gene Set Enrichment Analysis (GSEA) using public datasets (GSE14520), the results showed that lower JMJD5 expression was positively associated with Wnt/β-catenin pathway which is well-known to be activated during HCC development [23, 24], and cell cycle progress (Figure 5A and Supplemental Figure 5A). In addition, we also found that lower JMJD5 expression was negatively associated with metabolism processes such as lipid and amino acid catabolism (Figure 5A). Subsequently, we used FACS to assess the effect of JMJD5 knockdown or overexpression on cell cycle progression. Interestingly, siRNA-mediated JMJD5 knockdown significantly promoted the G1-S transition of MHCC-97H, SK-HEP-1, YY-8103 and L02 cells, whereas ectopic JMJD5 expression delayed the G1-S transition in MHCC-97H and HCC-LM6 cells (Figure 5B, 5C and Supplemental Figure 5B, 5C). This suggested that JMJD5 may regulate cell cycle progression in HCC cells. To further test this hypothesis, we performed 5-bromo-2’-deoxyuridine (BrdU) incorporation assays to assess the effect of JMJD5 knockdown or overexpression on newly synthesized DNA during the cell cycle. The result showed that siRNA-mediated JMJD5 knockdown indeed led to an increased proportion of cells in S phase, as indicated by BrdU incorporation in MHCC-97H, SK-HEP1, YY-8103 and L02 cells; by contrast, ectopic JMJD5 expression reduced BrdU incorporation in HCC-LM6 cells (Figure 5D, 5E and Supplemental Figure 5D). Collectively, these observations suggested that JMJD5 may influence the G1-S transition in HCC cells.
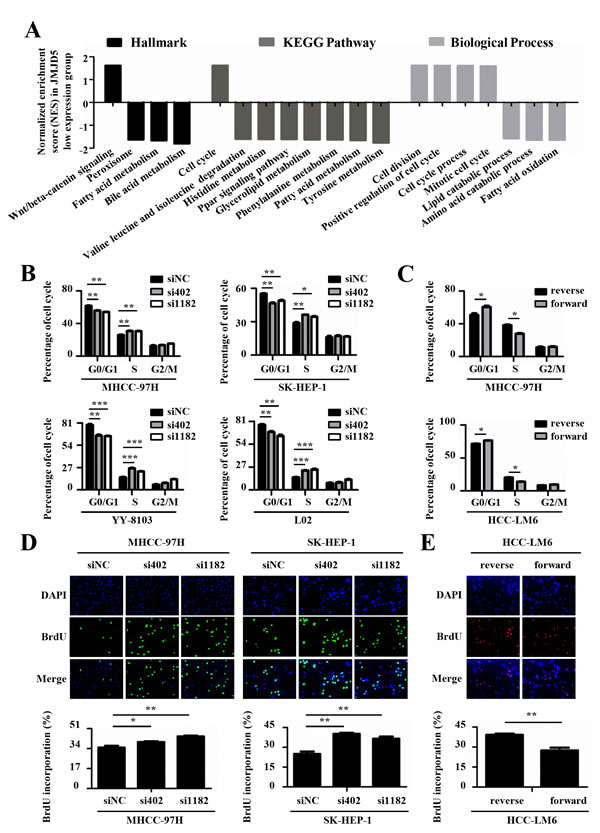
Figure 5: JMJD5 suppresses the cell cycle progression of HCC cells. A. Public dataset (GSE14520) was divided into two groups, higher JMJD5 expression group and lower JMJD5 expression group, and the normalized enrichment score (NES) of three gene set categories was calculated by Gene Set Enrichment Analysis (GSEA). Each column represented one statistically significant gene set (P < 0.05). The positive and negative NES represented these up-regulated and down-regulated gene sets in the lower JMJD5 expression group, respectively. B. Cell cycle analysis of HCC cells transfected with siRNAs against JMJD5, and C. cell cycle analysis of HCC cells ectopically expressing JMJD5. The percentages of the cell subpopulations at different stages of the cell cycle were statistically analyzed. D.-E. Immunofluorescence images (200x) showing HCC cells with BrdU incorporation after JMJD5 knockdown D. or overexpression E., where the percentages of cells with BrdU incorporation were statistically analyzed (bottom). All experiments were repeated at least three times. *, P < 0.05; **, P < 0.01; ***, P < 0.001.
JMJD5 affects HCC cell proliferation by regulating CDKN1A transcription
It is well-known that cell cycle progression is regulated by cyclins, cyclin-dependent kinases (CDKs), and cyclin-dependent kinase inhibitors [25, 26]. To clarify the molecular mechanism by which JMJD5 regulates the G1-S transition in HCC cells, we used real-time RT-PCR to analyze the mRNA levels of known molecules involved in the G1-S transition, including RB, CDKN1A, CDK2 and CCNE, in MHCC-97H and L02 cells that were silenced for JMJD5. Interestingly, only the mRNA level of CDKN1A, a well-known negative regulator of the G1-S transition, was significantly reduced at least two-fold in both cell lines after JMJD5 knockdown (Figure 6A). We then determined the expression levels of CDKN1A and its well-known upstream transcription factor, TP53, in L02, MHCC-97H, SK-HEP-1, YY-8103 and the TP53-null Hep3B cells after JMJD5 silencing. We found that siRNA-mediated JMJD5 knockdown dramatically reduced the mRNA and protein levels of CDKN1A, but not TP53 (Figure 6B, 6C). To further confirm the finding, we next determined the CDKN1A premature mRNA (pre-mRNA) levels in MHCC-97H, YY-8103, SK-HEP-1 and Hep3B cells after JMJD5 knockdown, and we found that they were also significantly reduced (Supplemental Figure 6A, 6B). Moreover, we observed that JMJD5 overexpression elevated CDKN1A mRNA levels in MHCC-97H, Hep3B and HCC-LM6 cells, and it even rescued the JMJD5 knockdown-mediated silencing of CDKN1A in MHCC-97H cells (Figure 6D and Supplemental Figure 6C, 6D). These data indicated that JMJD5 may directly activate CDKN1A transcription in a TP53-independent manner.
To further verify the notion that JMJD5 suppresses HCC cell proliferation mainly by activating CDKN1A, we transfected siRNA against CDKN1A into HCC cells with JMJD5 knockdown or overexpression. The results showed that CDKN1A knockdown could abrogate the effect of JMJD5 knockdown and overexpression on HCC cell proliferation (Figure 6E, 6F and Supplemental Figure 6E, 6F).
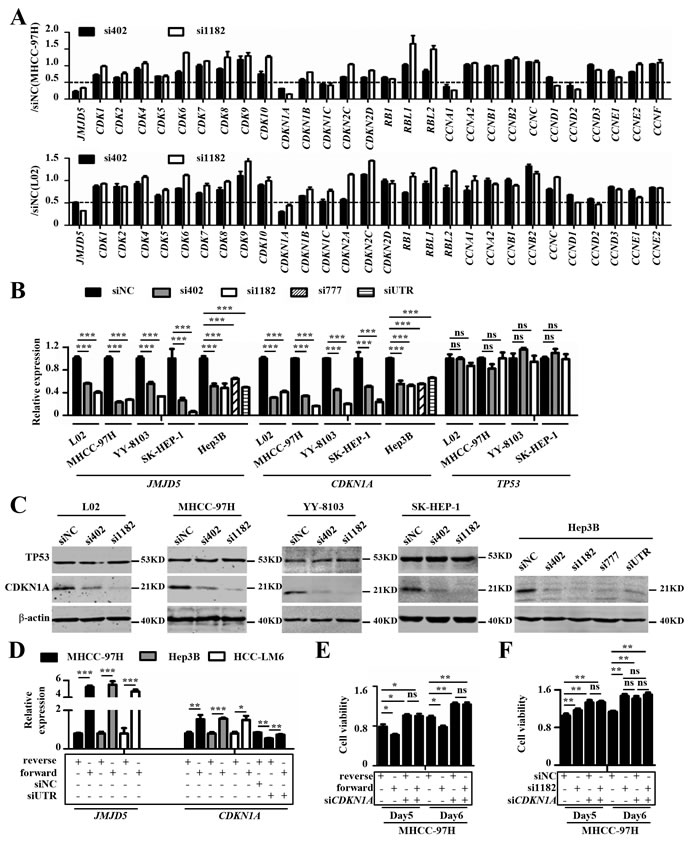
Figure 6: JMJD5 influences HCC cell proliferation primarily by regulating CDKN1A transcription. A. The expression of cell cycle-related regulators were performed using real-time RT-PCR in MHCC-97H and L02 cells after JMJD5 knockdown. The y-axis indicates the ratio of the mRNA levels of the given genes after siRNA-mediated JMJD5 knockdown to that of siNC. B. Real-time RT-PCR was used to measure CDKN1A and TP53 mRNA levels in HCC cell lines with siRNA-mediated JMJD5 knockdown. C. Five cell lines were transfected with siRNAs against JMJD5 and the levels of CDKN1A and TP53 proteins were tested by western blot. D. Ectopic JMJD5 expression increased CDKN1A mRNA expression and rescued the decrease in CDKN1A mRNA mediated by JMJD5 knockdown. E. CDKN1A knockdown abrogated the decreased cell viability mediated by JMJD5 overexpression. F. CDKN1A knockdown also abrogated the increased cell viability induced by JMJD5 knockdown. All experiments were repeated at least three times. *, P < 0.05; **, P < 0.01; ***, P < 0.001; ns, no significant difference.
JMJD5 regulates CDKN1A transcription by directly binding to the CDKN1A promoter in HCC cells
It has recently been reported that JMJD5 activates or inhibits transcription of downstream genes through binding to their promoters or gene body [15, 27, 28, 29]. Interestingly, the decreased JMJD5 expression could elevate Cdkn1a transcription through increasing H3K36me2 on the gene body in mouse embryonic fibroblast cells (MEF) [28], which appear to contrast to this study.
In order to clarify the conditions for the differential CDKN1A expression in HCC cells, we first detect whether JMJD5 could activate CDKN1A transcription by directly binding to CDKN1A gene locus in HCC cells, we transfected recombinant plasmids encoding the full length JMJD5 into MHCC-97H cells, and then we conducted ChIP-PCR assay with anti-JMJD5 antibody. The results showed that JMJD5 binds to the CDKN1A promoter, not gene body (Figure 7A). To explore the effect of JMJD5 knockdown and overexpression on CDKN1A promoter activity, we constructed a reporter gene system consisting of a luciferase reporter under the control of the CDKN1A promoter. We found that JMJD5 knockdown decreased luciferase activity; by contrast, JMJD5 overexpression increased luciferase activity, and the intensity of luciferase activity was positively associated with the length of the CDKN1A promoter (Figure 7B).
In addition, a previous study suggested that, in breast cancer, JMJD5 is a H3K36me2 demethylase [15] (Supplemental Figure 7A, 7B). However, western blots on whole-cell lysates from MHCC-97H, SK-HEP-1 and Hep3B cells showed that JMJD5 overexpression or knockdown had no obvious effect on H3K36me2 and other histone modifications (Supplemental Figure 7A, 7B), which is consistent with recent reports [7, 30, 31]. To further define whether JMJD5’s potential demethylase activity could influence CDKN1A transcription, we employed ChIP-PCR assay to assess the alteration of histone modifications on the CDKN1A locus, the results exhibited that the H3K36me2 modification had no significant difference on CDKN1A promoter and gene body whatever JMJD5 knockdown and overexpression (Figure 7C). Subsequently, we constructed a JMJD5 mutant with a H321A mutation in the ferrous iron binding site, which is responsible for its enzymatic activity [15]. Similarly, the JMJD5 mutant not only increased CDKN1A expression and luciferase reporter activity, but it also inhibited the colony formation of MHCC-97H and HCC-LM6 cells (Figure 7D-7F). Subsequently we further identified the necessary domain of JMJD5 responsible for CDKN1A activation by using the constructed JMJD5 deletion mutants, here we found that N-terminal amino acid residues (aa111-270), but not JmjC domain, of JMJD5 was required for CDKN1A activation (Supplemental Figure 8). Collectively our data suggested that JMJD5 could directly bind to the CDKN1A promoter, enhancing CDKN1A transcription in an enzyme activity-independent manner.
To further define the conditions for different Cdkn1a transcription in MEFs, we also analyzed the effect of Jmjd5 on Cdkn1a transcription. Our data exhibited that Jmjd5 knockdown increased Cdkn1a transcription and inhibited G1-S transition of the cell cycle (Supplemental Figure 9A, 9B), which were consistent with the published report [28]. However, in contrast to the data in HCC cells, Jmjd5 bond to Cdkn1a gene body, not its promoter (Supplemental Figure 9C). Interestingly, as Jmjd5 knockdown, the H3K36me2 modification was increased on Cdkn1a gene body but not promoter (Supplemental Figure 9D). These collective data suggested that the opposite effect of JMJD5 on CDKN1A transcription in HCC and MEF cells could be ascribed to the binding regions of JMJD5. In HCC cells, JMJD5 may function as a co-activator via binding to the CDKN1A promoter, whereas, in MEFs, Jmjd5, binding to Cdkn1a gene body, could exert the histone demethylase activity that modifies H3K36 methylation and alters the downstream gene transcription.
In addition, as comparison of HCC cells examined in this study, we also evaluated the effect of JMJD5 on breast and lung cancer cells. The results showed that JMJD5 overexpression promoted the proliferation of MCF7 breast cancer cells and A549 lung cancer cells, whilst JMJD5 knockdown inhibited their proliferation (Supplemental Figure 10). These data indicated that JMJD5 plays tumor-suppressive or cancer-promoting roles in a cell context-dependent manner.
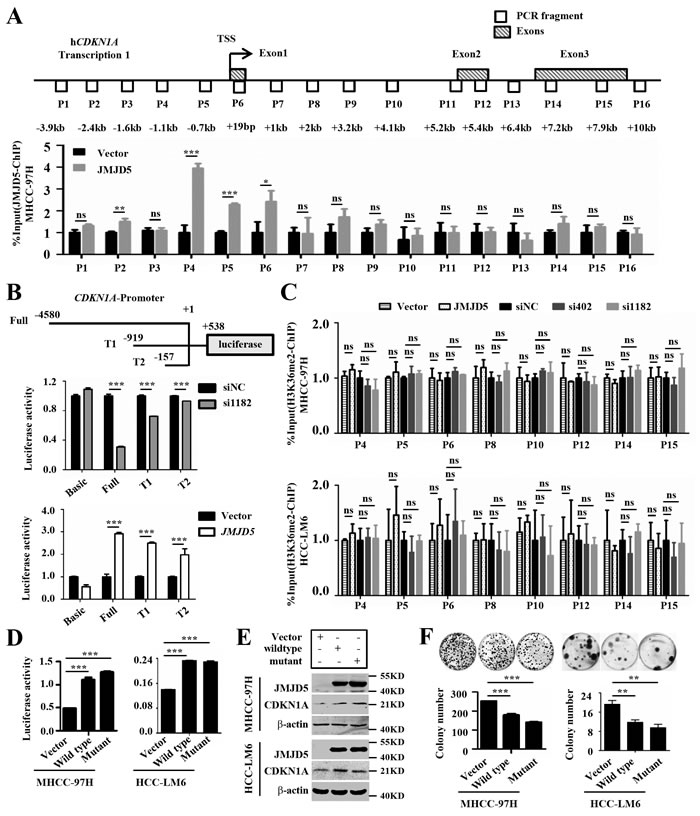
Figure 7: JMJD5 activates CDKN1A transcription by binding the CDKN1A promoter. A. MHCC-97H cells were transfected with plasmids encoding JMJD5, and then anti-JMJD5 antibody was utilized to conduct ChIP assay. JMJD5 enrichment on CDKN1A gene locus was measured by real-time quantitative PCR, the blank boxes represent the genomic positions of these primers for PCR, the shadowy boxes indicate the exons based on the transcript variant 1 (Transcription 1), and the numbers show the distance from the primers to transcription start site (TSS). B. Recombinant luciferase reporter plasmids (Full, T1 and T2) with three different upstream fragments of the CDKN1A regulatory element (upper) were employed to evaluate the effect of JMJD5 knockdown (middle) or overexpression (bottom) on CDKN1A transcription in MHCC-97H cells. A luciferase reporter vector without a promoter sequence was used to determine basal luciferase activity. C. Anti-H3K36me2 antibody was employed to carry out ChIP-qPCR assay, when JMJD5 knocked down and overexpressed in MHCC-97H and HCC-LM6 cells. D. A luciferase reporter system containing the upstream regulatory element of CDKN1A (Full) was used to evaluate the effect of the JMJD5H321A mutant on CDKN1A transcription in both MHCC-97H and HCC-LM6 cells. E. Both wild type JMJD5 and JMJD5H321A mutant increased CDKN1A protein expression in MHCC-97H and HCC-LM6 cells. F. JMJD5H321A mutant inhibited the colony formation of both MHCC-97H and HCC-LM6 cells. All experiments were repeated at least three times. *, P < 0.05; **, P < 0.01; ***, P < 0.001; ns, no significant difference.
Discussion
Some members of the JmjC family have recently been identified as a second type of histone demethylase, and dysregulation of a few family members, such as PLU-1, PHF8, JMJD3 and UTX, may contribute to various human diseases, including tumors, through epigenetic regulation [13, 32, 33]. However, the role of this family in HCC remains unclear.
JMJD5 participates in different physiological and pathological processes, including osteoclastogenesis, embryonic development, circadian clock regulation, stem cell differentiation and cancer progression [7, 15, 27, 28, 34, 35]. Suzuki et al used insertional mutagenesis in Blm-deficient mice to identify JMJD5 as a potential tumor suppressor that guaranteed genome integrity [14] and another group showed that JMJD5 was necessary for accurate chromosome segregation [36]. These data suggested that JMJD5 may function as a tumor suppressor in certain tumors. However, in lung cancer and breast cancer, JMJD5 functions as oncogene that promotes cancer cell proliferation [15, 37]. As well-known, many genes including some JmjC family members such as KDM2B, KDM6A and KDM6B, suppress or promote tumors in a cell context-dependent manner [38]. JMJD5 was reported to exert H3K36me2 demethylase activity or hydroxylase activity [7, 15]. In breast cancer, the H3K36me2 demethylase activity is essential for CCNA1 transcription and cell proliferation, by contrast, in HCC, our data revealed that the enzyme activity is seemingly unnecessary for CDKN1A activation and cell proliferation (Figure 7D-7F). This suggested that the transcription of different genes triggered by JMJD5 could depend on distinct mechanisms including its enzyme activity in diverse cancers.
With regard to molecular mechanism, it has recently been reported that JMJD5 can activate or suppress gene expression at transcriptional level and post-translational level. At transcriptional level, JMJD5 functions as a co-activator to promote HIF-1α target genes and miR302 transcription through binding to their promoters [27, 29], and also acts as the H3K36me2 demethylase to inhibit CDKN1A and CCNA1 transcription via binding to their gene bodies [15, 28]. At post-translational level, JMJD5 as hydroxylase may accelerate NFATc1 degradation. In the present work, our data showed that JMJD5, acting as a transcriptional co-activator, directly binds to the promoter of CDKN1A to enhance its transcription (Figure 7A, 7B), which is not to consistent with the published reports in MEF cells and human stem cells [27, 28]. In MEF cells, Jmjd5 binds to Cdkn1a gene body and negatively regulates Cdkn1a transcription, depending on the H3K36me2 demethylase activity [28]. In human stem cells, JMJD5 is inclined to suppress CDKN1A expression at post-transcriptional level through microRNA [27]. However, in HCC, JMJD5 mainly binds to CDKN1A promoter (Figure 7A) and functions as a transcriptional cofactor to positively promote CDKN1A transcription independent on the demethylase activity (Figure 7B-7E). These results indicated that the different binding regions of JMJD5 to CDKN1A gene locus, which could associate with the H3K36me2 demethylase activity, and the regulation fashion, may lead to the opposite outcome of CDKN1A transcription in different cell types.
Materials and Methods
Tissue specimens and cell lines
All specimens were harvested from patients who suffered from HCC and were informed consent. The liver tumor-derived cell lines SK-HEP-1, PLC/PRF/5, HuH-7, HepG2 and Hep3B were purchased from the Institute of Biochemistry and Cell Biology, Shanghai Institutes for Biological Sciences, Chinese Academy of Sciences. HCC cell lines MHCC-97H, MHCC-97L, HCC-LM3, HCC-LM6, YY-8103 and Focus were obtained from Zhong-Shan hospital, Fudan University. The immortalized human fetal liver cell line L02 was obtained from Shanghai Chang-Zheng Hospital, The Second Military Medical University. HCC cell lines including PLC/PRF/5, HuH-7, Hep3B, YY-8103, Focus and SK-HEP-1 were authenticated using short tandem repeats (STRs) sequencing and used within 6 months after authentication. All cells were permanently stocked at liquid nitrogen when obtained. The mouse embryonic fibroblast (MEF) cells were isolated from the 13.5 days of embryos.
Antibodies and reagents
The following antibodies were used in this research: JMJD5 (ab106391) from Abcam; β-actin (sc-47778), BrdU (sc-32323), P53 (sc-6243), CDKN1A (sc53870) and Flag (sc-166355) from Santa Cruz Biotechnology; and histone modification-specific antibodies were purchased from Abcam and Millipore. Propidium iodide (P4864), bromodeoxyuridine (B9258), DAC (A3656) and TSA (T1952) were obtained from Sigma.
Data mining of the gene expression profiles of JmjC family members
The expression patterns of JmjC family members in HCC specimens were analyzed based on public Gene Expression Omnibus (GEO) databases (GSE25097 and GSE14520). In brief, the ratios of expression in HCC versus in the adjacent normal liver specimens were calculated and log2 transformed. Genes with at least two-fold down-regulation or up-regulation were considered significant. The percentages of all samples with at least a two-fold change in gene expression were then calculated. The expression profile of JMJD5 in different human tissues and cells was analyzed in the GEO datasets (GDS: 3834) and THE HUMNA PROTEIN ATLAS (http://www.proteinatlas.org).
Overall survival analysis
TCGA data were obtained from cBioPortal (http://www.cbioportal.org/index.do). 94 HCC patients with RNA-seq data and clinical information including last communication time and survival status were ranked according to JMJD5 expression from high to low, the top 50% patients were defined as high expression group and the others were classified as low expression group. Kaplan-Meier survival analysis was utilized to draw the overall survival curve, and the statistical difference between two curve was calculated by log-rank test.
5-aza-2’-deoxycytidine (DAC) and trichostatin A (TSA) treatments
HCC cells were treated with 2 uM DAC for four days or with 300 ng/ml TSA for two days. The medium was refreshed every day, and RNA was isolated on the last day.
Public histone modifications ChIP-seq data mining
Public histone modifications ChIP-seq data were obtained from UCSC and the UCSC Accessions were listed as follows: wgEncodeEH000096, wgEncodeEH000083, wgEncodeEH001024, wgEncodeEH000095, wgEncodeEH000082, wgEncodeEH000081, wgEncodeEH001023, wgEncodeEH000094, wgEncodeEH003087, wgEncodeEH001749, wgEncodeEH000127. The distribution of these histone modifications on JMJD5 gene locus was visualized with UCSC browser according to the manual instruction.
Chromatin immunoprecipitation (ChIP) assay
ChIP assays were conducted using an EZ ChIP Kit (Millipore, Billerica, MA) according to the user manual.
Cell proliferation
Cells were cultured in 96-well plates with 2000-3000 cells/well. To measure cell viability, a Cell Counting Kit-8 (CCK-8, Dojindo Laboratories, Kumamoto, Japan) was used according to the manufacturer’s instructions.
Construction of recombinant plasmids
Two short hairpin RNAs (shRNAs) against JMJD5 were synthesized and inserted into the pSUPER vector (Oligoengine, Seattle, WA, USA), which was renamed sh402 and sh1182 based on their target sites. In addition, shRNA expressing irrelevant sequence served as a negative control and is referred to as shNC. To eliminate the effect of plasmid size on cell phenotype, the forward human JMJD5 open reading frame (ORF) (forward) and reverse sequence (reverse) were obtained by RT-PCR and inserted into pcDNA3.1/myc-His(-)B-3 X FLAG-IRES-hrGFP, which was derived from pcDNATM3.1/myc-His(-)B (Invitrogen, USA). The CDKN1A promoter sequence was cloned from normal human liver cDNA and inserted into pGL3 vector. For lentivirus construction, the shRNA was inserted into the PLKO-1 vector, and the JMJD5 forward and reverse sequences were inserted into the PCDH vector.
Screening for stable cell lines
SK-HEP-1 cells were transfected with shNC, sh402 and sh1182, then selected on 0.8 mg/ml G418 (Life Technologies Inc., Grand Island, NY, USA) for 3 weeks. Clones derived from single cells were collected and evaluated for their knockdown efficiency. YY-8103 cells were transfected with lentiviruses expressing shNC or sh1182, and MHCC-97H and HCC-LM6 cells were separately transfected with lentiviruses expressing JMJD5 forward or reverse; subsequently, 2 μg/ml puromycin was used to screen for positive clones.
Plate colony formation assays
Cells were seeded in triplicate in 35-mm dishes with 3000-5000 cells/dish. To kill non-transfected cells, 0.6-1 mg/ml G418 was applied three times a week. After 2-3 weeks, clones were stained with crystal violet and counted.
In vivo tumor xenografts
Stable HCC cells were subcutaneously injected near the scapulas of 6-week-old nude mice. Once a tumor formed, its volume was measured at different time points using the formula: Volume = 0.5 X width2 X length. After 5-6 weeks, the mice were euthanized, and the tumors were dissected and weighed.
Soft agar colony formation assays
Approximately 2000-3000 transfected cells were plated into 24-well plates containing 1% base agar and 0.5% top agar. After 2-3 weeks, the colonies were counted.
Gene set enrichment analysis (GSEA)
Gene set enrichment analysis was carried out using GSEA software according to the references [39, 40]. For the relationship analysis between JMJD5 expression level and three gene categories including Hallmark, KEGG Pathway and Biological Process, we firstly chose the top50 and bottom50 HCC tissues from the public database (GSE14520) according to JMJD5 expression. Subsequently the higher JMJD5 expression group, lower JMJD5 expression group and gene sets were conducted for GSEA analysis.
Cell cycle analysis
Cultured cells were collected using 1X trypsin and washed once in 1X PBS. The cells were resuspended in 1X PBS containing 0.2% Triton X-100, RNase A (100 µg/ml) and propidium iodide (50 µg/ml) and incubated for 30 min in the dark. The samples were subsequently measured using a FACSCalibur flow cytometer and CellQuest software (BD Biosciences, USA).
BrdU incorporation assay
Cells were incubated with 100 µM BrdU (Sigma-Aldrich, USA) for 2 hours and fixed with paraformaldehyde. After perforation with 0.5% Triton X-100 and DNA denaturation with 2 mol/L HCl, the cells were incubated with anti-BrdU antibody to perform immunofluorescence. A fluorescence microscope was used to analyze the cellular incorporation of BrdU.
Immunofluorescence assays
In brief, cells were fixed in paraformaldehyde at 4°C for 30 mins, then treated with 0.5% Triton X-100 at room temperature for 5 mins. Then, the cells were blocked with bovine serum albumin (BSA) and incubated with anti-BrdU antibody at room temperate for 2 hours, followed by incubation with AlexaFluor 488 (green)- or AlexaFluor 546 (red)-coupled secondary antibody for 1 hour.
Dual -luciferase reporter assay
MHCC-97H and HCC-LM6 cells were co-transfected with a combination of pRL-TK, encoding Renilla luciferase, and pGL3, encoding luciferase fused with the CDKN1A promoter, together with an siRNA against JMJD5 or a plasmid expressing JMJD5. Luciferase activity was measured 48 hours after transfection using the Dual-Luciferase Reporter Assay System Kit (Promega, Madison, WI, USA) according to the user manual.
Statistics
Log-rank test was utilized to compare the survival curve, Chi-square test was used to compare the correlations of JMJD5 expression with clinical characteristics of HCC, and Student’s t-test was used to evaluate statistically significant differences of quantitative variables. P < 0.05 was considered significant.
conflicts of interest
The authors disclose no potential conflicts of interest.
Grant Support
This work was supported by grants from the China National Key Projects for Infectious Disease (2012ZX10002012-008 and 2013ZX10002010-006), the National Natural Science Foundation of China (81472621, 81272271 and 81402329), the Chinese National Key Program on Basic Research (2010CB529200), the Doctoral Innovation Fund Projects from Shanghai Jiao Tong University School of Medicine (BXJ201316), and the Shanghai Pujiang Program (14PJ1405500).
REFERENCES
1. Niebel D, Kirfel J, Janzen V, Holler T, Majores M, Gutgemann I. Lysine-specific demethylase 1 (LSD1) in hematopoietic and lymphoid neoplasms. Blood. 2014; 124:151-152.
2. Shi YG, Tsukada Y. The discovery of histone demethylase. Cold Spring Harb Perspect Biol. 2013; 5(9).
3. Tsukada Y, Fang J, Erdjument-Bromage H, Warren ME, Borchers CH, Tempst P, Zhang Y. Histone demethylation by a family of JmjC domain-containing proteins. Nature. 2006; 439: 811-816.
4. Klose RJ, Kallin EM, Zhang Y. JmjC-domain-containing proteins and histone demethylation. Nat Rev Genet. 2006; 7: 715-727.
5. Kooistra SM, Helin K. Molecular mechanisms and potential functions of histone demethylases. Nat Rev Mol Cell Biol. 2012; 13: 297-311.
6. Clissold PM, Ponting CP. JmjC: cupin metalloenzyme-like domains in junonji, hairless and phospholipase A2beta. Trends Biochem Sci. 2001; 26: 7-9.
7. Youn MY, Yokoyama A, Fujiyama-Nakamura S, Ohtake F, Minehata K, Yasuda H, Suzuki T, Kato S, Imai Y. JMJD5, a Jumonji C(JmjC) domain-containing protein, negatively regulates osteoclastogenesis by facilitating NFATc1 protein degradation. J Biol Chem. 2012; 287: 12994-13004.
8. Wang F, He L, Huangyang P, Liang J, Si W, Yan R, Han X, Liu S, Gui B, Li W, Miao D, Jing C, Liu Z, Pei F, Sun L, Shang Y. JMJD6 promotes colon carcinogenesis through negative regulation of P53 by hydroxylation. PLoS Biol. 2014; 12: e1001819.
9. Noma A, Ishitani R, Kato M, Nagao A, Nureki O, Suzuki T. Expanding role of the jumonji C domain as an RNA hydroxylase. J Biol Chem. 2010; 285: 34503-34507.
10. Bjorkman M, Ostling P, Harma V, Virtanen J, Mpindi JP, Rantala J, Mirtti T,Vesterinen T, Lundin M, Sankila A, Rannikko A, Kaivanto E, Kohonen P, KallioniemiO, Nees M. Systematic knockdown of epigenetic enzymes identifies a novel histone demethylase PHF8 overexpressed in prostate cancer with an impact on cell proliferation, migration and invasion. Oncogene. 2012; 31: 3444-3456.
11. Li BX, Luo CL, Li H, Yang P, Zhang MC, Xu HM, Xu HF, Shen YW, Xue AM, Zhao ZQ. Effects of siRNA-mediated knockdown of jumonji domain containing 2A on proliferation, migration and invasion of the human breast cancer cell line MCF-7. Exp Ther Med. 2012; 4: 755-761.
12. Xiang Y, Zhu Z, Han G, Ye X, Xu B, Peng Z, Ma Y, Yu Y, Lin H, Chen AP, Chen CD. JARID1B is a histone H3 lysine 4 demethylase up-regulated in prostate cancer. Proc Natl Acad Sci U S A. 2007; 104: 19226-19231.
13. Yamane K, Tateishi K, Klose RJ, Fang J, Fabrizio LA, Erdjument-Bromage H, Taylor-Papadimitriou J, Tempst P, Zhang Y. PLU-1 is an H3K4 demethylase involved in transcriptional repression and breast cancer cell proliferation. Mol Cell. 2007; 25: 801-812.
14. Suzuki T, Minehata K, Akaqi K, Jenkins NA, Copeland NG. Tumor suppressor gene identification using retroviral insertional mutagenesis in Blm-deficient mice. EMBO J. 2006; 25: 3422-3431.
15. Hsia DA, Tepper CG, Pochampalli MR, Hsia EY, Izumiya C, Huerta SB, Wright ME, Chen HW, Kung HJ, Izumiya Y. KDM8, a H3K36me2 histone demethylase that acts in the cyclin A1 coding region to regulate cancer cell proliferation. Proc Natl Acad Sci U S A. 2010; 107: 9671-9676.
16. Uhlen M, Fagerberg L, Hallstrom BM, Lindskog C, Oksvold P, Mardinoglu A,Sivertsson A, Kampf C, Sjostedt E, Asplund A, Olsson I, Edlund K, Lundberg E, Navani S, Szigyarto CA, Odeberg J, Djureinovic D, Takanen JO, Hober S, Alm T, Edqvist PH, Berling H, Tegel H, Mulder J, Rockberg J, Nilsson P, Schwenk JM, Hamsten M, von Ferlitzen K, Forsberg M, Persson L, Johansson F, Zwahlen M, von Heijine G, Nielsen J, Ponten F. Proteomics. Tissue-based map of the human proteome. Science 2015; 347: 1260419.
17. Fernandez-Sanchez A, Baragano Raneros A, Carvajal Palao R, Sanz AB, Ortiz A,Ortega F, Suarez-Alvarez B, Lopez-Larrea C. DNA demethylation and histone H3K9 acetylation determine the active transcription of the NKG2D gene in human CD8+ T and NK cells. Epigenetics. 2013; 8: 66-78.
18. Yu ZQ, Zhang BL, Ni HB, Liu ZH, Wang JC, Ren QX, Mo JB, Xiong Y, Yao RQ, Gao DS. Hyperacetylation of Histone H3K9 involved in the promotion of Abnormally High Transcription of the gdnf Gene in Glioma Cells. Mol Neurobiol. 2014; 50: 914-922.
19. Black JC, Van Rechem C, Whetstine JR. Histone Lysine Methylation Dynamics: Establishment, Regulation, and Biological Impact. Molecular cell. 2012; 48: 491-507.
20. Berger SL. The complex language of chromatin regulation during transcription. Nature. 2007; 447: 407-412.
21. Du Z, Li H, Wei Q, Zhao X, Wang C, Zhu Q, Yi X, Xu W, Liu XS, Jin W, Su Z. Genome-Wide Analysis of Histone Modifications: H3K4me2, H3K4me3, H3K9ac, and H3K27ac in Oryza sativa L. Japonica. Mol Plant. 2013; 6: 1463-1472.
22. Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011; 144: 646-674.
23. Laurent-Puig P, Zucman-Rossi J. Genetics of hepatocellular tumors. Oncogene. 2006; 25: 3778-3786.
24. Whittaker S, Marais R, Zhu AX. The role of signaling pathways in the development and treatment of hepatocellular carcinoma. Oncogene. 2010; 29: 4989-5005.
25. Viallard JF, Lacombe F, Belloc F, Pellegrin JL, Reiffers J. Molecular mechanisms controlling the cell cycle: fundamental aspects and implications for oncology. Cancer Radiother. 2001; 5: 109-129.
26. Johnson DG, Walker CL. Cyclins and cell cycle checkpoints. Annu Rev Pharmacol Toxicol. 1999; 39: 295-312.
27. Zhu H, Hu S, Baker J. JMJD5 regulates cell cycle and pluripotency in human embryonic stem cells. Stem Cells. 2014; 32: 2098-2110.
28. Ishimura A, Minehata K, Terashima M, Kondoh G, Hara T, Suzuki T. Jmjd5, an H3K36me2 histone demethylase, modulates embryonic cell proliferation through the regulation of Cdkn1a expression. Development. 2012; 139: 749-759.
29. Wang HJ, Hsieh YJ, Cheng WC, Lin CP, Lin YS, Yang SF, Chen CC, Izumiya Y, Yu JS, Kung HJ, Wang WC. JMJD5 regulates PKM2 nuclear translocation and reprograms HIF-1α-mediated glucose metabolism. Proc Natl Acad Sci U S A. 2014; 111: 279-284.
30. Del Rizzo PA, Krishnan S, Trievel RC. Crystal structure and functional analysis of JMJD5 indicate an alternate specificity and function. Mol Cell Biol. 2012; 32: 4044-4052.
31. Williams ST, Walport LJ, Hopkinson RJ, Madden SK, Chowdhury R, Schofield CJ, Kawamura A. Studies on the catalytic domains of multiple JmjC oxygenases using peptide substrates. Epigenetics. 2014; 9: 1596-1603.
32. Arteaga MF, Mikesch JH, Qiu J, Christensen J, Helin K, Kogan SC, Dong S, So CW. The histone demethylase PHF8 governs retinoic acid response in acute promyelocytic leukemia. Cancer Cell. 2013; 23: 376-389.
33. Ntziachristos P, Tsirigos A, Welstead GG, Trimarchi T, Bakoqianni S, Xu L, Loizou E, Holmfeldt L, Strikoudis A, King B, Mullenders J, Becksfort J, Nedjic J, Paietta E, Tallman MS, Rowe JM, Tonon G, Satoh T, Kruidenier L, Prinjha R, Akira S, Van Vlierberghe P, Ferrando AA, Jaenisch R, Mullighan CG, Aifantis I. Contrasting roles of histone 3 lysine 27 demethylases in acute lymphoblastic leukaemia. Nature. 2014; 514: 513-517.
34. Jones MA, Covington MF, DiTacchio L, Vollmers C, Panda S, Harmer SL. Jumonji domain protein JMJD5 functions in both the plant and human circadian systems. Proc Natl Acad Sci U S A. 2010; 107: 21623-21628.
35. Wang Z, Wang C, Huang X, Shen Y, Shen J, Ying K. Differential proteome profiling of pleural effusions from lung cancer and benign inflammatory disease patients. Biochim Biophys Acta. 2012; 1824: 692-700.
36. Marcon E, Ni Z, Pu S, Turinsky AL, Trimble SS, Olsen JB, Silverman-Gavrila R, Silverman-Gavrila L, Phanse S, Guo H, Zhong G, Guo X, Young P, Bailey S, Roudeva D, Zhao D, Hewel J, Li J, Graslund S, Paduch M, Kossiakoff AA, Lupien M, Emili A, Wodak SJ, Greenblatt J. Human-chromatin-related protein interactions identify a demethylase complex required for chromosome segregation. Cell Rep. 2014; 8: 297-310.
37. Huang X, Zhang S, Qi H, Wang Z, Chen HW, Shao J, Shen J. JMD5 interacts with P53 and negatively regulates P53 function in control of cell cycle and proliferation. Biochim Biophys Acta. 2015; 1853: 2286-2295.
38. Sarris M, Nikolaou K, Talianidis I. Context-specific regulation of cancer epigenomes by histone and transcription factor methylation. Oncogene. 2014; 33: 1207-1217.
39. Mootha VK, Lindgren CM, Eriksson KF, Subramanian A, Sihag S, Lehar J, Puigserver P, Carlsson E, Ridderstrale M, Laurila E, Houstis N, Daly MJ, Patterson N, Mesirov JP, Golub TR, Tamayo P, Spiegelman B, Lander ES, Hirschhorn JN, Altshuler D, Groop LC. PGC-1aplha-responsive genes involved in oxidative phosphorylation are coordinately downregulated in human diabetes. Nat Genet. 2003; 34: 267-273.
40. Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub TR, Lander ES, Mesirov JP. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci U S A. 2005; 102: 15545-15550.