
INTRODUCTION
Treatments targeting aberrant androgen receptor (AR) signaling initially show impressive efficacy [1-5]. Unfortunately, therapy response usually only lasts for about 18 months, after which castration-resistant prostate cancer (CRPC) emerges. Several resistance mechanisms centering on the AR have been evidenced: rise in androgen supply by conversion of weak adrenal androgens into dihydrotestosterone (DHT) or by intratumoral de novo synthesis, increase of AR expression and gene copy number, mutations of AR leading to promiscuity and response to non-androgen ligands, and occurrence of splice variants with ligand-independent activity [6-9]. In addition, a number of genomic alterations may arise in the AR signaling pathway, which further underscores the essential role of the androgen axis in CRPC [10, 11].
Elevated androgen levels and AR overexpression can be addressed to some extent with AR antagonists possessing higher activity for the target, as exemplified by the recent approval of the second-generation AR antagonist enzalutamide [2, 4]. However, the development of antagonists addressing the most important AR mutants is compounded by the number of different variants identified, and the limited and sometimes conflicting information on their prevalence. Most AR mutations identified in CRPC are located in the ligand-binding domain (LBD) and alter the ligand-induced conformation of this region so that coactivator recruitment is still possible in the presence of antagonists, non-androgen steroids or weak adrenal androgens [12, 13]. In addition, different sets of downstream genes are controlled by AR mutants, implying that ligand- and mutation-selective conformations may take place [10, 14].
Conversion of antagonism to agonism in the presence of different AR mutants has been observed for approved AR antagonists. Cyproterone acetate, hydroxyflutamide and nilutamide stimulate AR T877A, the first AR mutation identified in prostate cancer [15]. Hydroxyflutamide and bicalutamide activate the AR V715M mutant [16]. Bicalutamide, but not hydroxyflutamide, becomes an agonist for the AR W741L and W741C mutants, due to the activation of an androgenic-like programme [10], further confirming that ligands with distinct chemical scaffolds have different allosteric effects on receptor conformation [17]. The E709Y mutant is strongly stimulated by bicalutamide, but less so by hydroxyflutamide or nilutamide [18]. TThe AR mutation F876L, which leads to activation by the recently approved enzalutamide and the related ARN-509 compound, has been identified by an in vitro selection procedure and an in vivo model selected for growth in the presence of the antagonist [19-21]. This mutation has already been detected in patients developing resistance to ARN-509 or enzalutamide [22, 23]. The AR H874Y mutant is stimulated by anti-androgens, adrenal androgens and non-androgen steroids, leading to enhanced coactivator recruitment [24, 25]. Several AR mutants not stimulated by anti-androgens but activated by various physiological steroids have also been found. For example, AR L701H is stimulated by glucocorticoids, whose novel interactions were revealed in modeling experiments [26]. Since this mutant shows little response to AR antagonists, the broad activation by non-androgen steroids is probably responsible for the tumor growth observed in prostate models bearing this mutation [27]. The mutations L701H, H874Y and T877A were also reported in patients with resistance to the C17,20 lyase inhibitor abiraterone. This may be due to previous treatment with AR antagonists or to co-medication with glucocorticoids, which activate AR mutants [20, 28].
In view of the persistent crucial role of the AR in most CRPC patients, there is a high need for novel antagonists addressing the adaptive mutations that emerge following anti-hormone therapy. Here we describe BAY 1024767, a novel and potent competitive antagonist of wild-type and mutated AR forms, and with potent in vivo efficacy. The prevalence of selected AR mutations was assessed in CRPC patients using the newly described BEAMing (Beads, Emulsions, Amplification, and Magnetics) technology to analyze circulating tumor DNA (ctDNA), and found to be at least 12%.
RESULTS
Identification of BAY 1024767
The synthesis of BAY 1024767 is described in patent WO 2011/029537 (A1) as example 10. The compound was discovered during a lead optimization project aiming at identifying highly potent AR antagonists with strong activity against wild-type and mutated AR forms. The crystal structure of AR mutant W741L bound to bicalutamide shows the influence of ligand shape on helix 12 conformation [29]. We developed novel antagonists that extend beyond the space occupied by the fluorophenyl ring of bicalutamide in order to displace helix 12 into an antagonist conformation, even in AR forms with an expanded ligand-binding pocket due to mutation. BAY 1024767 is a representative of the thiohydantoin type of anti-androgens substantiating this hypothesis (Figure 1).
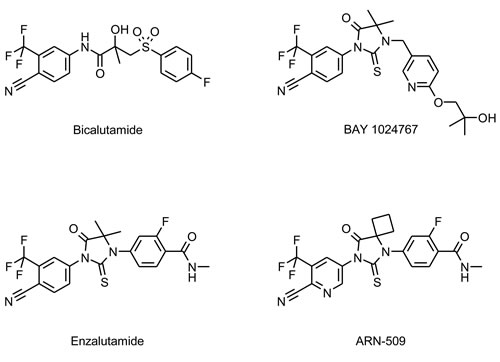
Figure 1: Chemical structures of the anti-androgens investigated.
BAY 1024767 is a strong antagonist for wild-type and mutated AR
Binding of BAY 1024767 to human wild-type AR was determined in a competitive assay and found to be 450 nM. Target engagement was verified in VCaP (AR wild-type) and LNCaP (AR T877A mutant) cells treated with R1881 (Figure 2A and 2B). Comparable dose-dependent down-regulations of PSA and FKBP5 were observed following BAY 1024767 application. In light of AR alterations previously described in CRPC patients, the following nine mutations were chosen for testing: L701H, E709Y, V715M, W741C, H874Y, F876L, T877A, M895T and M895V. PC-3 cells were transfected with plasmids harboring each mutation and an MMTV-controlled luciferase reporter. The levels of transfected AR mutants were comparable and in the same range as endogenous AR in LNCaP cells, when measured by ELISA (Supplementary Figure S1). The synthetic androgen R1881 stimulated AR wild-type and most mutants similarly, with however weaker responses of the L701H, W741C, M895T and M895V forms (Figure 3A). No dose-dependent agonistic activity was observed with BAY 1024767 for any of the mutants tested (Figure 3A). Enzalutamide did not stimulate the mutants tested, with the remarkable exception of F876L, reaching nearly 40% of the R1881 activity at the highest concentration used (Figure 3A), as previously described [19, 21, 23]. In sharp contrast, bicalutamide did not activate F876L, but did stimulate several of the other mutants already at low concentrations, reaching almost the activity seen with 1 nM R1881 at elevated levels in the case of W741C and M895T. Less but still significant stimulation was observed for E709Y and M895V. No stimulatory effect of bicalutamide was detected for the 5 other mutants tested (Figure 3A). Next, the antagonistic activity of the compounds was compared using different R1881 concentrations for stimulation (Figure 3B). BAY 1024767 was a strong antagonist for all mutants tested and superior to the other anti-androgens examined, with the exception of the T877A mutant (Figure 3B). Enzalutamide was a strong antagonist at low R1881 concentration, however lost most of its activity for both wild-type and mutated AR when increasing the treatment to 10 nM R1881 (Figure 3B). Bicalutamide displayed a comparatively weak antagonism for AR wild-type in presence of 0.1 nM R1881, which was rapidly lost when increasing the stimulation to 1 or 10 nM R1881. Reduced or no antagonism was observed for all mutants tested especially at low R1881 concentrations. The novel clinical AR antagonist ARN-509 was weaker than enzalutamide but slightly stronger than bicalutamide. When comparing the overall resistance of the AR mutants tested, it is clear that T877A is the most challenging to address. Altogether BAY 1024767 was the only anti-androgen tested which displayed antagonism for all AR mutants and retained activity at elevated androgen stimulation.
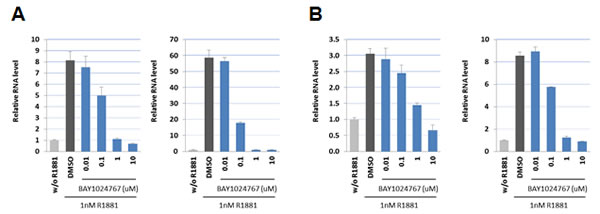
Figure 2: Down-regulation of androgen target genes in prostate cancer cells following BAY 1024767 treatment. A. PSA (left) and FKBP5 (right) expression in VCaP cells. B. PSA (left) and FKBP5 (right) expression in LNCaP cells.
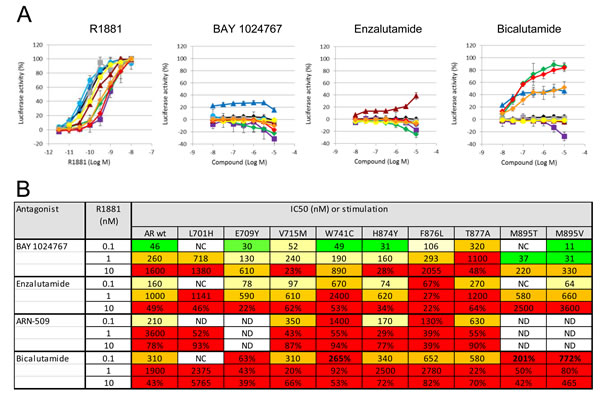
Figure 3: In vitro comparison of the activity of anti-androgens on AR wild-type and mutant forms. A. Determination of agonistic effects. Transfected PC-3 cells were treated with the indicated concentrations of compounds, and luciferase activity of the reporter gene was measured 20 h later. Standard deviation from 2-4 independent experiments is shown. The curves show responses of different AR variants: wild-type (black), L701H (magenta), E709Y (dark blue), V715M (turquoise), W741C (green), H874Y (grey), F876L (burgundy red), T877A (yellow), M895T (red), M895V (orange). B. Table summarizing the antagonistic activities in cell-based transactivation assays. The indicated compounds were used to treat AR mutants in presence of different androgen concentrations. The calculated IC50 values are shown or, in case more than 20% activity was observed at the highest concentration tested, the residual activity remaining. Color code: green: ≤50 nM; yellow: 50-250 nM; orange: 250-1000 nM; red: ≥1000 nM or residual activity >20%. NC: not calculated due to insufficient activity of the mutated AR form. ND: not determined.
BAY 1024767 promotes AR nuclear localization
Immunofluorescence studies using a FITC-labeled AR antibody were performed in transfected COS-7 cells treated with compounds for 4 h. AR wild-type and the mutants E709Y, V715M, W741C and H874Y were distributed in the cytoplasm in the absence of ligand but concentrated in the nucleus following R1881 treatment (Figure 4A and 4B). BAY 1024767 promoted nuclear localization of AR wild-type and mutants in the absence of R1881 with little residual cytoplasmic signal remaining upon antagonistic treatment alone, and did not prevent nuclear translocation in the presence of R1881. The results were similar following treatment with enzalutamide, however the translocation effects were slightly less pronounced, possibly due to weaker target engagement. Similar results were observed with endogenous AR in VCaP cells (Figure 4C).
BAY 1024767 releases AR from the PSA gene enhancer region
Given that BAY 1024767 antagonized R1881-stimulated gene transcription but was not capable of preventing the translocation of AR into the nucleus, its mode of action was investigated at the chromatin-binding level in VCaP cells. AR binding to PSA gene regulatory elements is well characterized [30]. Chromatin immunoprecipitation (ChIP) showed AR binding after agonist treatment to be mostly at the PSA enhancer region, but not at the middle region or at the promoter. The strong AR binding to the PSA enhancer was entirely prevented by BAY 1024767, but not by enzalutamide (Figure 4D). No significant changes were observed at the middle or promoter region of the PSA gene, however a slight increase of bound AR was observed after enzalutamide treatment, compared to the control or to BAY 1024767-treated samples.
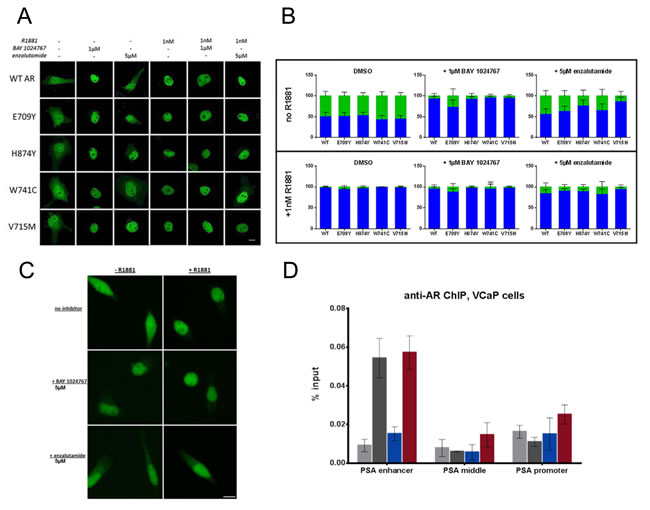
Figure 4: Effects of BAY 1024767 and enzalutamide on AR sub-cellular localization and chromatin binding. A. Transfected COS-7 cells were treated with the indicated compounds for 4 h and then analyzed by laser scanning microscopy, using a FITC-labeled AR antibody. The scale bar indicates 10 µm. B. AR nuclear and cytoplasmic distribution of 50 cells per treatment were quantitated using ImageJ. Results are presented as percentage of fluorescence signal overlapping with the DAPI nuclear region (blue bars) or in the cytoplasm (green bars). Error bars indicate the standard deviation. C. Cellular localization of endogenous AR in VCaP cells was analyzed using the antibody in part A. Representative images for each treatment are shown. The scale bar indicates 10 µm. D. ChIP was performed on VCaP cells following compound treatment for 6 h with 1 nM R1881 with DMSO (dark grey), 1 nM R1881 with 5 µM BAY 1024767 (blue), or 1 nM R1881 with 5 µM enzalutamide (red). No treatment controls (ethanol with DMSO) are shown in light grey. RT-PCR was performed with primers specific for the PSA enhancer or middle region, or the promoter. Error bars indicate the standard deviation from triplicate experiments.
In vitro anti-proliferative activity of BAY 1024767
The efficacy of BAY 1024767 was determined in different prostate cancer cell lines representative of clinically relevant resistance mechanisms (Figure 5A). In LAPC-4 cells which express AR wild-type, BAY 1024767 was superior to enzalutamide and to bicalutamide. In VCaP cells which contain 4- to 5-fold more AR protein than other prostate cancer cell lines (Figure 5B), BAY 1024767 was more effective at inhibiting cell proliferation than bicalutamide and enzalutamide. In LNCaP and 22Rv1 cells, containing mutated and/or splice variants of the AR, BAY 1024767 and enzalutamide had a comparable anti-proliferative effect, much stronger than that of bicalutamide. No anti-proliferative activity was observed when treating the AR-negative PC-3 cell line (Supplementary Figure S2).
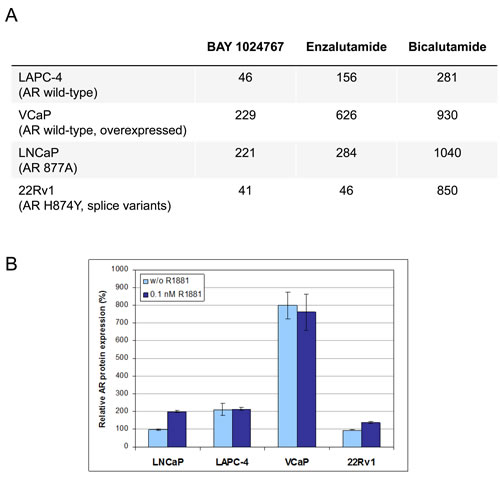
Figure 5: Anti-proliferative activity of anti-androgens in prostate cancer cell lines. A. GI50 values are indicated in nM. B. Determination of AR protein levels by ELISA. The amount of AR measured in untreated LNCaP cells (light blue bars) was set to 100%, and levels after 0.1nM R1881 treatment (dark blue bars) are shown for each cell line.
Pharmacokinetic properties of BAY 1024767
The pharmacokinetic properties of BAY 1024767 are summarized in Supplementary Table S1. Low hepatic clearance was measured in liver microsomes from different species including humans. The cell permeability was high and no efflux was observed. In vivo pharmacokinetic studies in male NMRI mice revealed a low clearance and a high oral bioavailability, thereby supporting oral administration as the application route for in vivo studies. After multiple oral doses of 50 mg/kg every second day to male SCID mice, unbound plasma concentrations of BAY 1024767 remained over the unbound anti-proliferative GI50 values determined in VCaP cells for the entire dosing interval. This was also true for bicalutamide dosed at 108 mg/kg daily and enzalutamide dosed at 160 mg/kg daily (Supplementary Figure S3).
In vivo efficacy of BAY 1024767
The KuCaP-1 model harbors the AR W741C mutation [31]. Mice with subcutaneous implantation of KuCAP-1 fragments were treated orally once every other day with BAY 1024767 or daily with bicalutamide, once the tumor reached an approximate mean of 100 mm3. BAY 1024767 applied at 50 mg/kg/administration (maximal tolerated dose) showed strong efficacy, and tumor volume remained almost constant, near the levels of the group treated by castration (Figure 6A). In sharp contrast, bicalutamide (60 mg/kg/day) did not affect tumor growth. Tumor weight measured at the end of the study confirmed the efficacy of BAY 1024767 (36% T/C on day 54 post-tumor implantation, significant difference (p < 0.05) versus control group), whereas bicalutamide tended to stimulate tumor growth (147% T/C) (Figure 6B), as previously described [31]. Treatments were well tolerated with mean body weight losses similar (bicalutamide) or inferior (BAY 1024767) to that of the control group, and mostly related to the tumor burden (Supplementary Figure S4). BAY 1024767 strongly reduced serum PSA levels (1.3 ng/ml, compared to 7.1 ng/ml in the control group (Figure 6C)). In contrast, bicalutamide treatment led to a 3-fold increase of serum PSA (21.3 ng/ml) when compared to the non-treated group. In a different study using the same tumor model, enzalutamide was compared to bicalutamide (Supplementary Figure S5). Enzalutamide, applied orally daily at 100 mg/kg was inactive (with 100% T/C calculated on tumor weights on day 52 post-tumor implantation). As previously, bicalutamide (100 mg/kg, orally, daily) was inactive with 112% T/C.
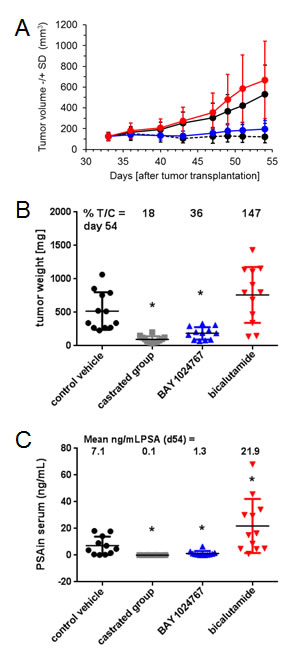
Figure 6: Comparison of the anti-tumor efficacy of BAY 1024767 and bicalutamide in the patient-derived KuCaP-1 model which contains a mutated AR form. A. Tumor volume -/+SD changes during the course of the experiment. Control (black, solid line), control castrated (black, dashed line), BAY 1024767 (50 mg/kg, q2dx11; blue) and bicalutamide (60 mg/kg, qdx21; red) are shown with standard deviation at each time point measured. B. Tumor weight at the end of the experiment. *p < 0.05 vs. control vehicle group, one-way ANOVA on ranks, Dunn´s method. C. Determination of serum PSA levels. *p < 0.05 vs. control vehicle group using Mann-Whitney rank sum test.
We additionally determined the in vivo efficacy of BAY 1024767 in the syngeneic Dunning rat R3327-G prostate cancer model [32]. Rats with subcutaneous implantation of tumor pieces were treated daily with oral doses of 40 mg/kg BAY 1024767, once the tumor reached 50 mm3. Comparison of tumor volumes showed a significant anti-tumor efficacy of BAY 1024767 (Figure 7A). This was also reflected at the level of tumor weights measured at the end of the study (11% T/C at day 113 post-tumor implantation, significant difference (p < 0.05) versus vehicle group (Figure 7B)). In addition, strong inhibition of seminal vesicle weight was observed following treatment with BAY 1024767, comparable to that seen in the castrated group (p < 0.05 versus vehicle group, data not shown). No significant body weight loss was observed, showing that treatment with BAY 1024767 at an efficacious dose was well tolerated in rats as well.
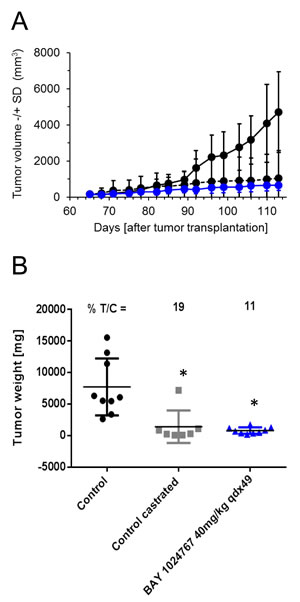
Figure 7: Anti-tumor efficacy of BAY 1024767 in the syngeneic Dunning rat R3327-G model. A. Tumor volume -/+ SD measured during the course of the experiment for control (black, solid line), control castrated (black, dashed line) and BAY 1024767 (40 mg/kg, qdx49; blue) are shown with the standard deviation of each time point measured. B. Tumor weight at the end of the experiment. *p < 0.05 vs. control vehicle group, one-way ANOVA on ranks, Dunn´s method.
Prevalence of AR mutations in CRPC patients
In order to determine the relevance of the AR mutations analyzed in this study in the clinical setting, we collected plasma samples from 100 patients who had received anti-hormone therapy and developed resistance. These samples were used for ctDNA purification and subsequent analysis with the very sensitive BEAMing technology [33]. Probes addressing six selected AR mutations were designed and in each case only one possible nucleotide change was selected, based on previously published information (Supplementary Table S2) [34]. The results (Table 1) reveal that 12 out of 100 CRPC patients had at least one out of the six investigated AR mutations (= 12%, 95% Jeffreys confidence interval: 6.73%, 19.43%). No correlation was observed with the Gleason score or the total PSA value. In two cases, two mutations were present in the same patient, indicating tumor heterogeneity. Interestingly, the T877A mutation was observed most frequently, accounting for five out of 14 cases in twelve different patients. Mutations at positions V715, W741 and H874 were each observed three times. The mutation frequency varied significantly among samples and was in several cases below 0.1%. In patient 5 however, a very high frequency of the T877A mutation was observed. These ratios should however be interpreted with caution since the amount of contaminating lymphocyte DNA may vary in each sample. Nonetheless the very high frequency and the fact that it was found in four additional patients underscore the importance of the T877A mutation in the acquisition of resistance to hormonal treatment in prostate cancer. It should also be noted that all the mutation frequencies determined may represent an underestimation since other DNA point mutations leading to the same amino acid exchange are possible but would not have been detected by the primers used in this study.
Table 1: Frequency of AR mutations in plasma DNA from CRPC patients as determined using the BEAMing technology
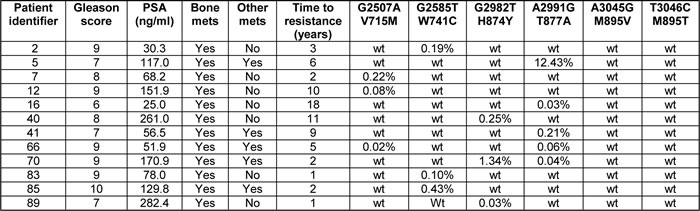
PSA: prostate-specific antigen; mets: metastases.
DISCUSSION
Resistance mechanisms described in CRPC mostly concern the AR pathway so that the identification of novel anti-androgens with improved properties remains an essential research focus in this field [3-5]. Indeed several new AR antagonists have moved to the clinical stage in recent years, raising the hope that new therapy options will be available soon [35-37].
BAY 1024767 is a novel competitive AR antagonist that exhibits strong activity against AR wild-type and mutated forms found in therapy-resistant patients, keeps antagonistic activity with increased androgen stimulation and in prostate cancer models with elevated AR protein levels. It also shows anti-proliferative activity in a model expressing splice variants. Its potent antagonism when androgen or AR levels are high is most probably due to strong target engagement whereas its activity towards mutants may be linked to its extended structure that presumably displaces helix 12 towards an antagonist position. The efficacy of BAY 1024767 was evidenced in various in vitro transactivation assays using mutated AR forms. In vivo, we observed very strong activity in the KuCaP-1 model which bears the comparatively frequent W741C AR mutation. Importantly, the compound also strongly impaired AR binding to the regulatory enhancer region of the PSA gene while reducing PSA serum levels in tumor-bearing mice, which is routinely used as a pharmacodynamic marker in the clinic. The efficacy of BAY 1024767 was further evidenced in the syngeneic Dunning R3327-G rat model with high AR expression where a significant anti-tumor effect was observed. In addition, a dramatic reduction of seminal vesicle weight, comparable to that seen in castrated animals, was evidenced.
AR mutations identified in CRPC patients usually do not merely lead to loss of interaction with antagonists but rather represent gain-of-function alterations. The mutations identified in ctDNA are likely to represent drivers of acquired resistance and among them T877A was the most frequent. This was unexpected since this mutation was originally identified in a patient treated with hydroxyflutamide [38], which is now rarely used for treatment. The recent finding that progesterone stimulates this mutant and is synthesized by the tumor may explain why this position is a mutation hotspot [39, 40]. Bicalutamide exhibits antagonism for the T877A mutant, which can be explained by the different positioning of AR LBD helix 12 [29]. Nonetheless, we found bicalutamide to be a weaker antagonist for this mutant in comparison to AR wild-type following stimulation with 0.1 nM androgen, and to lose most of its antagonistic activity at higher androgen levels. Indeed, loss of antagonism for the AR T877A mutant was seen for all compounds tested, but BAY 1024767 and enzalutamide maintained the best profile. This was mirrored in the proliferation assays with the LNCaP cell line, which harbors the T877A mutation, where BAY 1024767 and enzalutamide were also more efficacious than bicalutamide. The data underline that this position is key in the switch between an inactive and active AR conformation necessary for recruitment of co-activators and binding to target genes.
The occurrence of AR splice variants in CRPC patients has been reported [9, 41]. Many have lost their LBD and can therefore not be directly addressed by a competitive antagonist. However splice variants may form heterodimers with full-length AR wild-type, which are still blocked by anti-androgens [42, 43], and it will be interesting to find out whether this is also the case for heterodimers formed with AR LBD mutants.
The 12% frequency rate measured in our study for AR mutations is probably an underestimation. Firstly, despite the very high sensitivity of the BEAMing method, some of the interrogated mutations may have gone undetected, as the plasma preparations used in our study had not been optimized for enrichment of ctDNA. Secondly, for each selected position only one specific primer sequence was used to analyze the plasma DNA samples even though additional options existed in most cases. Thirdly, additional AR LBD mutations not addressed in this study have been identified recently, including F876L which was detected in 3 out of 27 patients treated with the novel AR antagonist ARN-509 [23]. In a very recent study on ctDNA from 62 patients progressing on enzalutamide or abiraterone, the AR mutation rate was found to be 18% by targeted next-generation sequencing of exon 8, which covers the C-terminal part of the LBD [22]. As in our study, the H874Y and T877A mutations were found several times. In addition, the M895V substitution was detected once, underlining the importance of having a compound such as BAY 1024767 for blockade of this AR mutant which was originally identified in a primary prostate tumor [44]. A sequencing approach was also used in another study focusing on circulating tumor cells (CTCs) from CRPC patients [45]. Here, 35 CTC samples were analyzed and 19 AR missense mutations identified in 15 patients. These mutations include several positions that we detected, and again M895V. Such unbiased sequencing approaches have the potential to identify all AR mutations arising in refractory prostate cancer patients provided the complete coding region is analyzed, but are unlikely to be used routinely in the near future. Also, they necessitate the isolation of CTCs, which is more cumbersome than purification of plasma DNA, and cannot be used in patients with low or no CTC counts.
Precise determination of the respective prevalence of individual AR mutations and their variation along treatment duration should now be more readily feasible using blood-based assays and the BEAMing technology described here, compared to analysis of CTCs which is not yet routinely established in the clinic, or of tumor biopsies which is challenging, especially in the case of metastases. Such approaches will deliver essential information for guiding the future treatment of CRPC, which is multifocal in nature and will necessitate bespoke drugs addressing both wild-type and mutated AR, while keeping efficacy in the presence of high androgen levels and at elevated AR expression.
In summary, we have identified BAY 1024767, a novel AR antagonist which addresses three essential mechanisms responsible for CRPC. This may offer new therapy options by overcoming and/or delaying resistance, either by treating patients who do not respond or who are stimulated by approved anti-hormonal agents, or by sequential use of AR antagonists with different profiles.
MATERIALS AND METHODS
Cell culture and materials
All anti-androgen compounds were synthesized in-house and are shown in Figure 1. The cell lines were obtained from ATCC (Manassas, VA, USA) or DSMZ (Braunschweig, Germany) and authenticated at the DSMZ by short tandem repeat DNA typing. The KuCaP-1 patient-derived model [46] was from Prof. O. Ogawa, University of Kyoto, Japan. The Dunning R3327-G rat prostatic tumor [47] and the AR expression plasmid [48] were described before. Site-directed mutagenesis was performed using the appropriate primer pairs and the QuickChange II XL Site-Directed Mutagenesis kit (Agilent Technologies, Santa Clara, CA, USA).
Binding and transient transfection assays
Competitive binding to recombinant human AR was determined in presence of 5 nM tritiated R1881. PC-3 transfection and determination of luciferase activity were performed as before [49]. The test compounds were added with 0.1% ethanol (agonistic mode) or the indicated R1881 concentrations (antagonistic mode). The average value of six wells treated in parallel was taken. The experiments were repeated at least twice independently. The NR Sandwich AR ELISA 49696 (Active Motif, Carlsbad, CA, USA) was used to determine endogenous AR levels and compare the levels of expressed AR protein in transfected PC-3 cells.
mRNA expression assays
VCaP or LNCaP cells were seeded into 6-well plates in RPMI 1640 medium without phenol red, supplemented with 10% charcoal-stripped fetal calf serum (cFCS) and 2 mM stable glutamine, and maintained for 2 days prior to treatment. R1881 (or ethanol control) was added to cells at 1 nM end concentration simultaneously with DMSO, or the AR antagonist BAY 1024767 at the indicated concentrations. After 24 h treatment, total RNA was extracted using the RNeasy Mini Kit (Qiagen, Hilden, Germany) and reverse-transcription was performed using the SuperScript™ III First-Strand Synthesis SuperMix for qRT-PCR (Invitrogen, Carlsbad, Germany). Transcript levels of AR target genes were determined by real-time PCR using the following assays (Applied Biosystems, Foster City, CA, USA): PSA Hs02576345_m1 and FKBP5 Hs00188025_m1. Human PPIA (Cyclophilin A) 4326316E was used as an internal control.
Immunofluorescence
COS-7 cells were grown to 60% confluence and then incubated in phenol red-free DMEM medium containing 10% cFCS. After two days, cells were transfected with AR expression plasmids (1.25 ng) using Lipofectamine 2000 (Invitrogen) for 20 h, then were treated for 4 h with 1 nM R1881 or ethanol along with DMSO, 1 µM BAY 1024767 or 5 µM enzalutamide.
VCaP cells were grown to 30% confluence and then incubated in phenol red-free DMEM medium containing carbon-filtered FCS (DMEM-cFCS) for 3 days. The cells were treated for 6 h with 1 nM R1881 or ethanol along with DMSO, 5 µM BAY 1024767 or 5 µM enzalutamide.
For staining, culture medium was removed and the cells were washed three times with PBS. The cells were then fixed in 4% paraformaldehyde in PBS for 15 min at room temperature, washed, permeabilized with PBS containing 0.5% Triton X-100 for 10 min, washed again, treated with blocking solution (1% BSA, 0.1% Tween-20 in PBS) for 30 min, and incubated overnight at 37°C in blocking solution in the presence of anti-AR-FITC antibody (sc-815-FITC; Santa Cruz Biotechnology, Dallas, TX, USA). The cells were then washed, stained with 300 nM DAPI in PBS for 5 min, and sealed with a coverslip. Cells were imaged on an LSM700 confocal laser scanning microscope (Zeiss, Göttingen, Germany), using a 25X objective. Quantification of COS-7 cells was performed on 25 cells in two replicates using ImageJ software.
Chromatin immunoprecipitation
VCaP cells were grown in 10 cm plates to 80% confluence. One plate of cells was used per immunoprecipitation. The cells were maintained in DMEM-cFCS for 3 days prior to treatment. R1881 (or ethanol control) was added to cells at 1 nM simultaneously with DMSO, or the AR antagonists BAY 1024767 or enzalutamide at 5 µM. After 6 h treatment, the cells were cross-linked in 1% formaldehyde in PBS for 8 min, quenched in 125 mM glycine, washed 3 times with PBS, and frozen. After cell lysis (5 mM PIPES pH 8, 85 mM KCl, 0.5 % Nonidet P40) and nuclei isolation using RIPA buffer, chromatin was sheared to 100-2000 base pair-long fragments using a S220 ultrasonicator (Covaris, Woburn, MA, USA).
Immunoprecipitations were performed using the IPstar device (Diagenode, Liège, Belgium) with 20 µL of magnetic Protein A bead suspension. 2 µg of antibodies anti-AR (sc-815-x; Santa Cruz) and rabbit IgG (Sigma-Aldrich, Taufkirchen, Germany) were used per immunoprecipitation. The buffers used were: bead wash (10 mM Tris-HCl pH 8, 1 mM EDTA, 0.02 % Tween-20), wash 1 (20 mM Tris-HCl pH 8, 2 mM EDTA, 150 mM NaCl, 1 % Triton X-100, 0.1 % SDS), wash 2 (20 mM Tris-HCl pH 8, 2 mM EDTA, 500 mM NaCl, 1 % Triton X-100, 0.1 % SDS), wash 3 (10 mM Tris-HCl pH 8, 1 mM EDTA, 250 mM LiCl, 1 % Nonidet P40, 1 % sodium deoxycholate) and elute (0.1 M NaHCO3, 1 % SDS).
Reverse cross-linking of immunoprecipitation elutions and inputs was done for 16 h at 65°C, with 160 µM NaCl final concentration. The samples were treated with RNaseA and Proteinase K, and were purified using the Qiagen PCR purification kit before analysis by qPCR.
The primers used were for the prostate specific antigen (PSA) gene [30, 50]: promoter forward CCT AGA TGA AGT CTC CAT GAG CTA CA; promoter reverse GGG AGG GAG AGC TAG CAC TTG; enhancer forward GCC TGG ATC TGA GAG AGA TAT CAT C; enhancer reverse ACA CCT TTT TTT TCT GGA TTG; middle forward CTG TGC TTG GAG TTT ACC TGA; middle reverse GCA GAG GTT GCA GTG AGC C.
Cell proliferation assays
All assays were performed in 96-well microtiter plates. Cells were grown in RPMI-1640 media without phenol red and supplemented with 10% cFCS and 2 mM L-glutamine. LAPC-4 cells were plated at 4,000 cells/well, VCaP cells at 16,000 cells/well, LNCaP cells at 2,000 cells/well, 22Rv1 cells at 4,000 cells/well. After 1 day, the cells were treated with R1881 (0.1 nM final concentration, except for LAPC-4 where the final concentration was 1 nM) and compound (day 0). Cell number was determined by Alamar Blue staining (2 h). Fluorescence was measured in a Victor3 device (PerkinElmer, Waltham, MA, USA) with the excitation filter set at 530 nm and the emission filter set at 590 nm. C0 was defined as the signal measured at day 7 for cells treated only with R1881 and CI was defined as the signal measured at day 7 for cells grown without R1881 or compound.
Metabolic stability in vitro
The metabolic stability of BAY 1024767 was determined by incubating the compound with suspensions of liver microsomes from different species at a concentration of 1 µM. The intrinsic clearance was calculated from the half-life of the compound. Together with additional parameters like microsomal protein content, species-specific liver weight and liver blood flow, the hepatic in vivo blood clearance (CLH) was calculated.
Caco-2 permeation assay
Caco-2 cells were seeded on 24-well insert plates, 0.4 µm pore size, and grown for 15 days. The test compound was dissolved in DMSO and added either to the apical (A) or basolateral (B) compartment at a final concentration of 2 µM. Before and after 2 h incubation, samples were taken from both compartments and analyzed by LC/MS/MS. The apparent permeability (Papp) was determined for each direction and the efflux ratio was calculated as Papp B-A/Papp A-B.
In vivo pharmacokinetics in mice
BAY 1024767 was administered to 6-7 week-old male NMRI mice in the tail vein by a single dose of 0.5 mg/kg and intragastral by a single dose of 1 mg/kg formulated in PEG400/water (60/40). Three mice were sacrificed at different time points after dosing, and blood (for heparinized plasma) was sampled from the vena cava. Analysis of the samples was performed by LC/MS/MS and pharmacokinetic parameters were calculated by non-compartmental analysis.
In vivo efficacy studies
Animal experiments were approved by the relevant regulatory agency of the federal state of Berlin (Landesamt für Gesundheit und Soziales Berlin). Animals were housed according to institutional guidelines with access to food (pelleted diet) and water ad libitum. The KuCaP-1 patient-derived tumor model was maintained by serial passage in SCID/CB17 male mice. The presence of the W741C mutation was confirmed by DNA sequencing (not shown). For the efficacy studies, 11-12 week-old SCID/CB17 male mice (Charles River Laboratories, Erkrath, Germany) were inoculated subcutaneously with tumor fragments into the right flank. Tumors were allowed to grow and mice were randomly assigned to control or treatment groups with a mean tumor volume of 100 mm3 (n = 10-12 mice/group). Mice received vehicle, BAY 1024767 (50 mg/kg every 2 days [Q2D]), enzalutamide (100 mg/kg once daily [QD]) or bicalutamide (60 or 100 mg/kg once daily [QD]) orally for 18-21 days, depending on the study. The doses used were either the maximal tolerated dose or the dose reaching maximal exposure in mice. Compounds were solubilized in NMP/PEG300 1/9 (v/v). An additional group included mice castrated at the start of the study that received vehicle. Mouse body weight was determined at least twice a week. Tumors were measured twice a week using a caliper (tumor volume (mm3) = [length (mm) x width2 (mm2)]/2). Tumor/control ratio (T/C) % was calculated as [mean tumor weight in the treated group/mean tumor weight in the control vehicle group]×100). Efficacy was evaluated according to the National Cancer Institute standards (%T/C ≤ 42% = active). Statistical analysis was performed using a one-way analysis of variance (ANOVA) on ranks using Dunn’s method. The Quantikine® Human Kallikrein 3/PSA Immunoassay (R&D Systems, Minneapolis, MN, USA) was used to measure mouse serum PSA levels at the end of the study. Statistical analysis was performed using Mann-Whitney rank sum test.
The Dunning R3327-G tumor [51] was maintained by serial in vivo passages. For the efficacy experiment, tumor tissue pieces of 8 mm3 were inoculated subcutaneously (flank region) in 12-13 week-old male Copenhagen rats (COP/CrCrl strain, Charles River Laboratories). Treatment was started once the tumor reached 50 mm2 and rats were randomly assigned to control (n = 9), castrated (n = 10) or treatment group (n = 10). In the treatment group 40 mg/kg of BAY 1024767 was given orally once a day (p.o., QD). The vehicle was NMP/PEG 1/9 (v/v). Rat body weight was measured at least twice a week. Tumor volume and weight were determined as above.
AR mutation frequency
All patients received written information about the planned AR mutation analysis performed on serum samples and signed a document indicating their informed consent. Blood plasma was collected and prepared from CRPC patients under standardized clinical laboratory practices. A total of 100 patients of which 98 had received at least two different hormonal treatments and with a median time to CRPC status of 8 years (range 1-20 years) were analyzed. Seventy-seven of these patients had bone metastases, 59 had visceral metastases and 49 had both. Blood plasma (2 ml) obtained from each patient was collected in EDTA tubes, mixed gently and centrifuged at 4 °C for 10 min at 3400 g. It was then transferred by pipetting into 15 ml conical tubes which were centrifuged at 4 °C for 15 min at 1500 g. The cell-free plasma was then transferred into storage tubes and kept at -80 °C. The BEAMing experiments [52] were performed on cell-free plasma DNA by Sysmex Inostics (Hamburg, Germany). For the analysis, cell-free plasma DNA was first pre-amplified in a multiplex PCR reaction, amplified with nested primers and normalized. Emulsion PCR was then performed on the surface of magnetic beads in water-in-oil emulsions. Following destruction of the emulsion structure, the uncovered DNA fragments were hybridized to fluorescent probes specific to the mutations. These labeled beads were then quantified by flow cytometry. Statistical analysis was performed as described previously [53].
ACKNOWLEDGMENTS
We thank the numerous colleagues involved in prostate cancer and AR signaling research for many helpful discussions and support, and Dr. H.-F. Ulbrich for statistical analysis of the patient-derived data. The excellent technical help of H. Morack, A. Koehlke-Nork, F. Halwaß, H. Muckwar, F. Knoth, B. Hartmann, J. Laube, C. Maeßus, T. Jung, M. Klein, D. Trost, U. Reckling, F. Igogeit, R. Neumeister, K. Dörre, F. Fiske and J. Breusing is gratefully acknowledged. We are extremely thankful to Prof. O. Ogawa and Dr. T. Inoue (Kyoto University, Japan) for providing the KuCaP-1 xenograft model. The excellent support of Dr. P. Angenendt and Dr. F. Diehl at Sysmex Inostics GmbH (Hamburg, Germany) in the performance of the BEAMing analysis of plasma DNA samples is very gratefully acknowledged.
FUNDING
All studies were supported by Bayer Pharma AG.
CONFLICTS OF INTEREST
BAY 1024767 was identified at Bayer Pharma AG. S. Köhr, H. Faus, A. Cleve, U. Lücking and B. Haendler are inventors on the patent application disclosing BAY 1024767. T. Sugawara, P. Lejeune, S. Köhr, R. Neuhaus, H. Faus, K. A. Gelato, M. Brands, A. Cleve, U. Lücking, F. von Nussbaum, D. Mumberg and B. Haendler are employees of Bayer Pharma AG, which has a commercial interest in the development of AR antagonists.
Editorial note
This paper has been accepted based in part on peer-review conducted by another journal and the authors’ response and revisions as well as expedited peer-review in Oncotarget.
REFERENCES
1. Courtney KD and Taplin ME. The evolving paradigm of second-line hormonal therapy options for castration-resistant prostate cancer. Curr Opin Oncol. 2012; 24(3):272-277.
2. Ferraldeschi R, Welti J, Luo J, Attard G and de Bono JS. Targeting the androgen receptor pathway in castration-resistant prostate cancer: progresses and prospects. Oncogene. 2014.
3. Haendler B and Cleve A. Recent developments in antiandrogens and selective androgen receptor modulators. Mol Cell Endocrinol. 2012; 352:79-91.
4. Helsen C, Van den Broeck T, Voet A, Prekovic S, Van Poppel H, Joniau S and Claessens F. Androgen receptor antagonists for prostate cancer therapy. Endocr Relat Cancer. 2014; 21:T105-118.
5. McEwan IJ. Androgen receptor modulators: a marriage of chemistry and biology. Future Med Chem. 2013; 5:1109-1120.
6. Karantanos T, Evans CP, Tombal B, Thompson TC, Montironi R and Isaacs WB. Understanding the Mechanisms of Androgen Deprivation Resistance in Prostate Cancer at the Molecular Level. Eur Urol. 2014.
7. Lorente D and De Bono JS. Molecular alterations and emerging targets in castration resistant prostate cancer. Eur J Cancer. 2014; 50:753-764.
8. Perner S, Cronauer MV, Schrader AJ, Klocker H, Culig Z and Baniahmad A. Adaptive responses of androgen receptor signaling in castration-resistant prostate cancer. Oncotarget. 2015; 6:35542-55. doi: 10.18632/oncotarget.4689.
9. Steinestel J, Luedeke M, Arndt A, Schnoeller TJ, Lennerz JK, Wurm C, Maier C, Cronauer MV, Steinestel K and Schrader AJ. Detecting predictive androgen receptor modifications in circulating prostate cancer cells. Oncotarget. 2015. doi: 10.18632/oncotarget.3925.
10. O’Neill D, Jones D, Wade M, Grey J, Nakjang S, Guo W, Cork D, Davies BR, Wedge SR, Robson CN and Gaughan L. Development and exploitation of a novel mutant androgen receptor modelling strategy to identify new targets for advanced prostate cancer therapy. Oncotarget. 2015; 6:26029-26040. doi: 10.18632/oncotarget.4347.
11. Taylor BS, Schultz N, Hieronymus H, Gopalan A, Xiao Y, Carver BS, Arora VK, Kaushik P, Cerami E, Reva B, Antipin Y, Mitsiades N, Landers T, Dolgalev I, Major JE, Wilson M, et al. Integrative genomic profiling of human prostate cancer. Cancer Cell. 2010; 18:11-22.
12. Eisermann K, Wang D, Jing Y, Pascal LE and Wang Z. Androgen receptor gene mutation, rearrangement, polymorphism. Transl Androl Urol. 2013; 2:137-147.
13. Hay CW and McEwan IJ. The impact of point mutations in the human androgen receptor: classification of mutations on the basis of transcriptional activity. PLoS One. 2012; 7:e32514.
14. Brooke GN and Bevan CL. The role of androgen receptor mutations in prostate cancer progression. Curr Genomics. 2009; 10:18-25.
15. Veldscholte J, Berrevoets CA, Brinkmann AO, Grootegoed JA and Mulder E. Anti-androgens and the mutated androgen receptor of LNCaP cells: differential effects on binding affinity, heat-shock protein interaction, and transcription activation. Biochemistry. 1992; 31:2393-2399.
16. Fenton MA, Shuster TD, Fertig AM, Taplin ME, Kolvenbag G, Bubley GJ and Balk SP. Functional characterization of mutant androgen receptors from androgen-independent prostate cancer. Clin Cancer Res. 1997; 3:1383-1388.
17. Hara T, Miyazaki J, Araki H, Yamaoka M, Kanzaki N, Kusaka M and Miyamoto M. Novel mutations of androgen receptor: a possible mechanism of bicalutamide withdrawal syndrome. Cancer Res. 2003; 63:149-153.
18. Georget V, Bourguet W, Lumbroso S, Makni S, Sultan C and Nicolas JC. Glutamic acid 709 substitutions highlight the importance of the interaction between androgen receptor helices H3 and H12 for androgen and antiandrogen actions. Mol Endocrinol. 2006; 20:724-734.
19. Balbas MD, Evans MJ, Hosfield DJ, Wongvipat J, Arora VK, Watson PA, Chen Y, Greene GL, Shen Y and Sawyers CL. Overcoming mutation-based resistance to antiandrogens with rational drug design. Elife. 2013; 2:e00499.
20. Claessens F, Helsen C, Prekovic S, Van den Broeck T, Spans L, Van Poppel H and Joniau S. Emerging mechanisms of enzalutamide resistance in prostate cancer. Nat Rev Urol. 2014; 11:712-716.
21. Korpal M, Korn JM, Gao X, Rakiec DP, Ruddy DA, Doshi S, Yuan J, Kovats SG, Kim S, Cooke VG, Monahan JE, Stegmeier F, Roberts TM, Sellers WR, Zhou W and Zhu P. An F876L mutation in androgen receptor confers genetic and phenotypic resistance to MDV3100 (enzalutamide). Cancer Discov. 2013; 3:1030-1043.
22. Azad AA, Volik SV, Wyatt AW, Haegert A, Le Bihan S, Bell RH, Anderson S, McConeghy B, Shukin R, Bazov J, Youngren J, Paris PL, Thomas GV, Small EJ, Wang Y, Gleave ME, et al. Androgen receptor gene aberrations in circulating cell-free DNA: biomarkers of therapeutic resistance in castration-resistant prostate cancer. Clin Cancer Res. 2015.
23. Joseph JD, Lu N, Qian J, Sensintaffar J, Shao G, Brigham D, Moon M, Maneval EC, Chen I, Darimont B and Hager JH. A clinically relevant androgen receptor mutation confers resistance to second-generation antiandrogens enzalutamide and ARN-509. Cancer Discov. 2013; 3:1020-1029.
24. Duff J and McEwan IJ. Mutation of histidine 874 in the androgen receptor ligand-binding domain leads to promiscuous ligand activation and altered p160 coactivator interactions. Mol Endocrinol. 2005; 19:2943-2954.
25. Taplin ME, Bubley GJ, Shuster TD, Frantz ME, Spooner AE, Ogata GK, Keer HN and Balk SP. Mutation of the androgen-receptor gene in metastatic androgen-independent prostate cancer. N Engl J Med. 1995; 332:1393-1398.
26. van de Wijngaart DJ, Molier M, Lusher SJ, Hersmus R, Jenster G, Trapman J and Dubbink HJ. Systematic structure-function analysis of androgen receptor Leu701 mutants explains the properties of the prostate cancer mutant L701H. J Biol Chem. 2010; 285:5097-5105.
27. Zhao XY, Malloy PJ, Krishnan AV, Swami S, Navone NM, Peehl DM and Feldman D. Glucocorticoids can promote androgen-independent growth of prostate cancer cells through a mutated androgen receptor. Nat Med. 2000; 6:703-706.
28. Carreira S, Romanel A, Goodall J, Grist E, Ferraldeschi R, Miranda S, Prandi D, Lorente D, Frenel JS, Pezaro C, Omlin A, Rodrigues DN, Flohr P, Tunariu N, J SdB, Demichelis F, et al. Tumor clone dynamics in lethal prostate cancer. Sci Transl Med. 2014; 6:254ra125.
29. Bohl CE, Gao W, Miller DD, Bell CE and Dalton JT. Structural basis for antagonism and resistance of bicalutamide in prostate cancer. Proc Natl Acad Sci U S A. 2005; 102:6201-6206.
30. Kim J, Jia L, Tilley WD and Coetzee GA. Dynamic methylation of histone H3 at lysine 4 in transcriptional regulation by the androgen receptor. Nucleic Acids Res. 2003; 31:6741-6747.
31. Yoshida T, Kinoshita H, Segawa T, Nakamura E, Inoue T, Shimizu Y, Kamoto T and Ogawa O. Antiandrogen bicalutamide promotes tumor growth in a novel androgen-dependent prostate cancer xenograft model derived from a bicalutamide-treated patient. Cancer Res. 2005; 65:9611-9616.
32. Isaacs JT, Isaacs WB, Feitz WF and Scheres J. Establishment and characterization of seven Dunning rat prostatic cancer cell lines and their use in developing methods for predicting metastatic abilities of prostatic cancers. Prostate. 1986; 9:261-281.
33. Diehl F, Li M, Dressman D, He Y, Shen D, Szabo S, Diaz LA, Jr., Goodman SN, David KA, Juhl H, Kinzler KW and Vogelstein B. Detection and quantification of mutations in the plasma of patients with colorectal tumors. Proc Natl Acad Sci U S A. 2005; 102:16368-16373.
34. Gottlieb B, Beitel LK, Nadarajah A, Paliouras M and Trifiro M. The androgen receptor gene mutations database: 2012 update. Hum Mutat. 2012; 33:887-894.
35. Loddick SA, Ross SJ, Thomason AG, Robinson DM, Walker GE, Dunkley TP, Brave SR, Broadbent N, Stratton NC, Trueman D, Mouchet E, Shaheen FS, Jacobs VN, Cumberbatch M, Wilson J, Jones RD, et al. AZD3514: a small molecule that modulates androgen receptor signaling and function in vitro and in vivo. Mol Cancer Ther. 2013; 12:1715-1727.
36. Moilanen AM, Riikonen R, Oksala R, Ravanti L, Aho E, Wohlfahrt G, Nykanen PS, Tormakangas OP, Palvimo JJ and Kallio PJ. Discovery of ODM-201, a new-generation androgen receptor inhibitor targeting resistance mechanisms to androgen signaling-directed prostate cancer therapies. Sci Rep. 2015; 5:12007.
37. Rathkopf DE, Morris MJ, Fox JJ, Danila DC, Slovin SF, Hager JH, Rix PJ, Chow Maneval E, Chen I, Gonen M, Fleisher M, Larson SM, Sawyers CL and Scher HI. Phase I study of ARN-509, a novel antiandrogen, in the treatment of castration-resistant prostate cancer. J Clin Oncol. 2013; 31:3525-3530.
38. Berrevoets CA, Veldscholte J and Mulder E. Effects of antiandrogens on transformation and transcription activation of wild-type and mutated (LNCaP) androgen receptors. J Steroid Biochem Mol Biol. 1993; 46:731-736.
39. Cai C, Chen S, Ng P, Bubley GJ, Nelson PS, Mostaghel EA, Marck B, Matsumoto AM, Simon NI, Wang H and Balk SP. Intratumoral de novo steroid synthesis activates androgen receptor in castration-resistant prostate cancer and is upregulated by treatment with CYP17A1 inhibitors. Cancer Res. 2011; 71:6503-6513.
40. Chen EJ, Sowalsky AG, Gao S, Cai C, Voznesensky O, Schaefer R, Loda M, True LD, Ye H, Troncoso P, Lis RL, Kantoff PW, Montgomery RB, Nelson PS, Bubley GJ, Balk SP, et al. Abiraterone treatment in castration-resistant prostate cancer selects for progesterone responsive mutant androgen receptors. Clin Cancer Res. 2015; 21:1273-1280.
41. Lu J, Van der Steen T and Tindall DJ. Are androgen receptor variants a substitute for the full-length receptor? Nat Rev Urol. 2015; 12:137-144.
42. Watson PA, Chen YF, Balbas MD, Wongvipat J, Socci ND, Viale A, Kim K and Sawyers CL. Constitutively active androgen receptor splice variants expressed in castration-resistant prostate cancer require full-length androgen receptor. Proc Natl Acad Sci U S A. 2010; 107:16759-16765.
43. Xu D, Zhan Y, Qi Y, Cao B, Bai S, Xu W, Gambhir SS, Lee P, Sartor O, Flemington EK, Zhang H, Hu CD and Dong Y. Androgen receptor splice variants dimerize to transactivate target genes. Cancer Res. 2015.
44. Sanchez D, Rosell D, Honorato B, Lopez J, Arocena J and Sanz G. Androgen receptor mutations are associated with Gleason score in localized prostate cancer. BJU Int. 2006; 98:1320-1325.
45. Jiang Y, Palma JF, Agus DB, Wang Y and Gross ME. Detection of androgen receptor mutations in circulating tumor cells in castration-resistant prostate cancer. Clin Chem. 2010; 56:1492-1495.
46. Terada N, Shimizu Y, Yoshida T, Maeno A, Kamba T, Inoue T, Nakamura E, Kamoto T and Ogawa O. Antiandrogen withdrawal syndrome and alternative antiandrogen therapy associated with the W741C mutant androgen receptor in a novel prostate cancer xenograft. Prostate. 2010; 70:252-261.
47. Kager M, Spruss T, Schneider MR and von Angerer E. Dunning R3327-G prostate carcinoma of the rat: an appropriate model for drug evaluation. J Cancer Res Clin Oncol. 1992; 118:334-338.
48. Geserick C, Meyer HA, Barbulescu K and Haendler B. Differential modulation of androgen receptor action by deoxyribonucleic acid response elements. Mol Endocrinol. 2003; 17:1738-1750.
49. Faus H and Haendler B. Androgen receptor acetylation sites differentially regulate gene control. J Cell Biochem. 2008; 104:511-524.
50. Gaughan L, Logan IR, Neal DE and Robson CN. Regulation of androgen receptor and histone deacetylase 1 by Mdm2-mediated ubiquitylation. Nucleic Acids Res. 2005; 33:13-26.
51. Lubaroff DM, Canfield L and Reynolds CW. The Dunning tumors. Prog Clin Biol Res. 1980; 37:243-263.
52. Dressman D, Yan H, Traverso G, Kinzler KW and Vogelstein B. Transforming single DNA molecules into fluorescent magnetic particles for detection and enumeration of genetic variations. Proc Natl Acad Sci U S A. 2003; 100:8817-8822.
53. Brown LD, Cai TT and DasGupta A. Interval Estimation for a Binomial Proportion. Statist Sci. 2001; 16:101-133.