
INTRODUCTION
Genomic DNA is constantly exposed to endogenous and exogenous insults, which cause DNA damage. If not repaired, this DNA damage may result in genetic mutations, leading to genome instability and cancer initiation [1, 2]. Removal of DNA damage and maintenance of genomic integrity depend on robust cellular DNA repair systems [2]. Base excision repair (BER), which removes DNA base damage caused by endogenous and exogenous agents, is a major repair pathway in eukaryotic cells [3–6]. It is estimated that BER repairs about 104 damaged/modified bases per cell per day [7–9]. BER is initiated with the excision of the damaged base by a specific DNA glycosylase, resulting in an apurinic/apyrimidinic site (AP site). The AP site is then cleaved by AP endonuclease 1 (APE1), leaving 3’ hydroxyl and 5’ deoxyribosephosphate termini (5’-dRp) [10, 11]. This intermediate structure can be processed through either the short patch BER (SP-BER) or the long patch BER (LP-BER) pathway [12, 13]. In SP-BER, Pol β adds only one nucleotide to the 3’-end of the nicked AP site, and then the dRP lyase activity of Pol β catalyzes β-elimination of the 5’-sugar phosphate residue, resulting in a ligatable nick that can then be sealed by XRCC1/Ligase IIIα [14, 15]. In LP-BER, Pol β performs strand displacement synthesis, generating a 2-10 nt short DNA flap, which is removed by flap endonuclease 1 (FEN1) [16–20]. DNA ligase I then seals the nick [12].
DNA polymerase β (Pol β) is a key player in both the SP-BER and LP-BER pathways [19, 21, 22]. Pol β, a 39 kDa protein, contains two domains, a dRP lyase domain (8 kDa) and a polymerase domain (31 kDa). These two domains correspond to the dRP lyase and polymerase activities, which are responsible for the removal of the sugar phosphate group and the incorporation of new deoxyribonucleotides, respectively [23]. The Pol β polymerase domain can be divided into the fingers, palm and thumb subdomains, based on crystallographic structures. These subdomains are responsible for dsDNA binding, nucleotidyl transferase and dNTP selection, respectively [23–25]. In addition, Pol β also interacts with many other proteins including APE1, PCNA and FEN1 [26, 27]. These interactions can recruit downstream factors to the DNA repair site, reciprocally stimulate enzyme activities, and coordinate the highly ordered chemical reactions of BER. Pol β deficiency impairs BER efficiency and promotes hypersensitivity to alkylating or oxidative agents [18, 28]. Knockout of Pol β in mice abolishes BER and induces hypersensitivity to DNA damaging reagents, including methyl methanesulfonate (MMS) and H2O2, leading to early embryonic lethality.
Most types of human cancers contain Pol β mutations (Figure 1A, [29, 30]). These findings suggest that Pol β mutation may promote carcinogenesis [31–35], but a clear link between Pol β mutation and cancer has not been established. In the current study, we identified Pol β R152C as a candidate cancer-causing mutation. We expressed and purified Pol β WT and R152C and found that R152C significantly reduces Pol β polymerase activity and BER efficiency. Furthermore, cells harboring Pol β R152C accumulated more genomic DNA damage in response to DNA damaging agents, which induced aneuploidy and a higher cellular transformation efficiency. These results suggest that the R152C mutation reduces Pol β biochemical activity, resulting in defective BER, which might contribute to genome instability and cancer development.
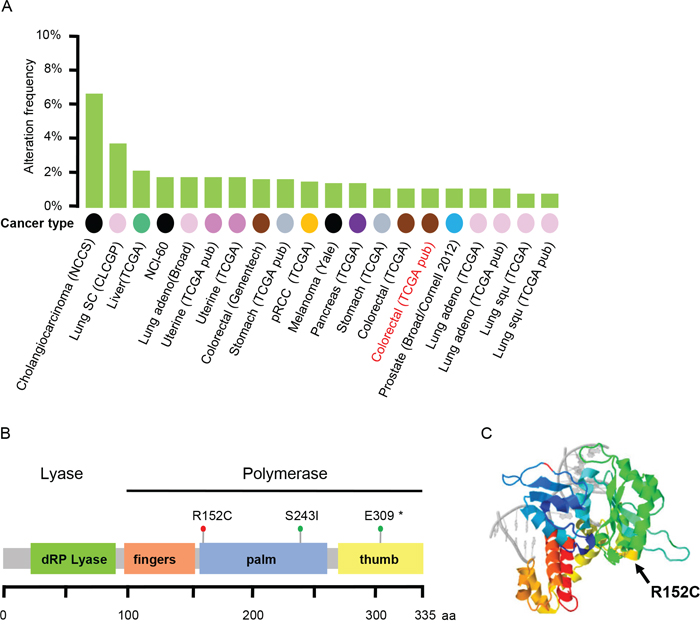
Figure 1: The R152C mutation of Pol β in colorectal cancer. A. Pol β mutation frequency across each cancer type in the Cancer Genome Atlas (TCGA) dataset. B. Three colorectal cancer-associated Pol β mutations from the TCGA dataset. C. The position of R152C in the 3D structure of Pol β.
RESULTS
The R152C mutation of Pol β is associated with colorectal cancer
By searching the Cancer Genome Atlas (TCGA) Data, we found that the Pol β mutation exists in all human tumors (Figure 1A). In colorectal cancer samples, we found 3 Pol β mutations: R152C, S243I, and E309* (*stop codon) (Figure 1B). Among the three mutations, R152C, an amino acid substitution of arginine by cysteine, was interesting because of its location on the protein, its effect on the net charge of the protein, and the sequence conservation of R152 among different species (Figure 1C) [36]. Furthermore, R152 has also been reported to be a methylation site of DNA Pol β [37]. Methylation on R152 could enhance the Pol β DNA binding activity and stimulate its DNA polymerase activity. The R152C mutation is methylation defective and, therefore, we speculated that this mutation could disrupt cellular functions.
The R152C mutation is defective in polymerase activity
We first determined the effects of R152C mutation on Pol β function. WT and R152C Pol β were purified from E. coli to homogeneity (Figure 2A). Circular dichroism analysis showed that WT and R152C Pol β had the same overall structure (Figure 2B). We then assayed primer extension, gap filling, dRP lyase, and substrate-binding activity in vitro, using synthetic DNA substrates (Table 1). We found that R152C mutation dramatically reduced primer extension (Figure 2C) and gap filling (Figure 2D) activity compared to the WT enzyme. However, no differences were observed for DNA-binding activity (Figure 2E) or dRP lyase activity (data not shown). These results were consistent with the fact that R152C is located in the 31 kD polymerase catalytic domain, whereas the 8 kD domain is responsible for dRP lyase and DNA-binding activity.
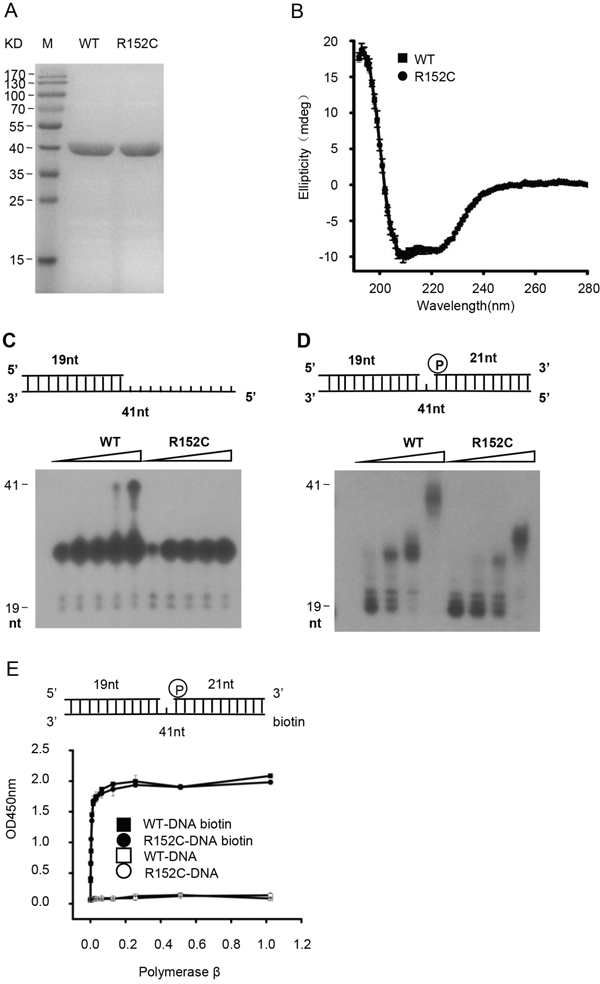
Figure 2: The Pol β R152C mutant is defective in polymerase activity. A. SDS-PAGE of the Pol β WT and R152C recombinant protein. Proteins were expressed in E. coli and purified by a His-tag column. B. Circular dichroism spectroscopy analyses of WT and R152C Pol β. C. Pol β primer extension activity assay. D. Pol β gap-filling activity assay. In (C) and (D), the top part of each panel shows the schematic structure of the corresponding DNA substrates. The bottom shows PAGE-separated products. E. ELISA-based isotherm adsorption assay of the DNA-binding affinity of WT and R152C Pol β.
Table 1: Prime and DNA substrates
Name |
Oligonucleotide sequence |
Application |
|---|---|---|
Pol β R152C F |
5'-gggactttgaaaaaagaattccttgtgaagagatgttacaaatg-3' |
Mutagenesis |
Pol β R152C R |
5'-ttgcatttgtaacatctcttcacaaggaattcttttttcaaagtccc-3' |
|
Pol β -2oli |
5'-ct tacacgttgact accgt 5'-ggatccactcttgcctcaaaagacggtagtcaacg tgtaag-3' |
Polymerase activity |
Pol-β -3oli |
5'-ct tacacgttgact accgt ttttgaggcaagagt ggat cc-3 5'-ggatccactcttgcctcaaaagacggtagtcaacgtgtaa g-3' |
Polymerase activity & DNA binding assay |
Pol β -U |
5'-c ttacacgttgac taccttUtttgaggcaagagtgga t cc-3' 5'-ggatc cactc ttgcc tcaaa gaagg tagtcaacgt gtaag-3' |
SP-BER assay |
Pol β -F |
5'-c ttacacgttgac tacctt Ftttgaggcaagagtgga tcc-3' 5'-ggatccactcttgcctcaaagaaggtagtcaacgtgtaag-3' |
LP-BER assay |
The R152C mutation has lower BER efficiency
Because Pol β is a key enzyme for BER, we speculate that R152C mutation of Pol β, which impairs its polymerase activity, may also impair its BER function. To test this, BER proteins were purified (Figure 3A). SP- and LP-BER were assayed using G/U mismatched oligonucleotides to mimic the SP-BER substrate and G/F (tetrahydrofuran, THF or F) mismatched oligonucleotides to mimic the LP-BER substrate (Pol β-F) (Table 1). As shown in Figure 3B and 3C, R152C has significantly lower SP- and LP-BER efficacy.
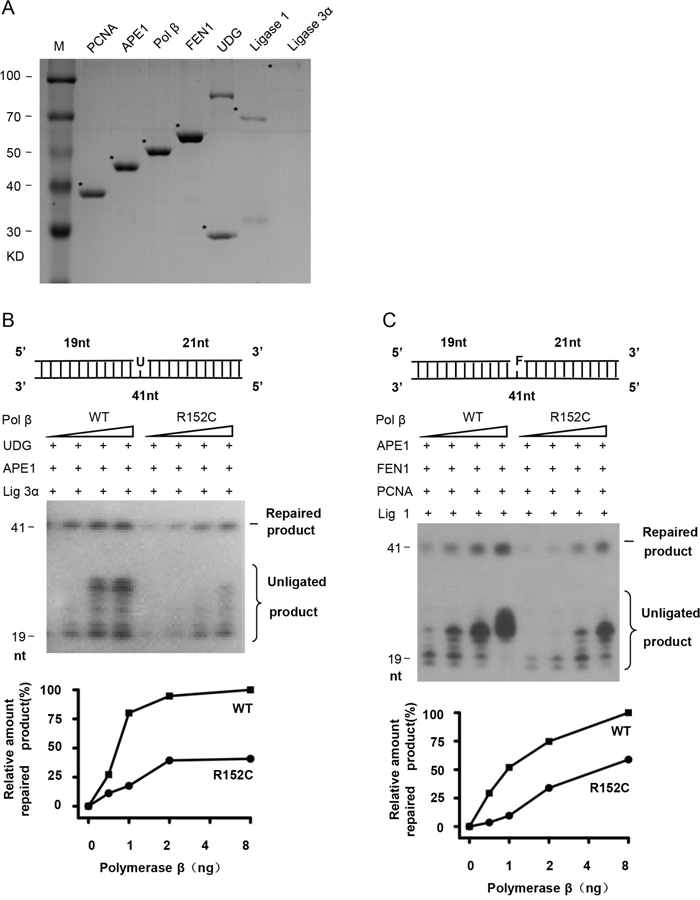
Figure 3: R152C mutation impairs BER efficiency in vitro. A. SDS-PAGE of purified BER proteins. SP-BER B. or LP-BER C. reconstitution with purified BER proteins. The top part of each panel shows the schematic structures of the corresponding DNA substrates. The middle shows PAGE-separated products and the bottom the relative percentage of repaired product obtained with the indicated amounts of Pol β. Values represent the mean ±SD of three independent assays.
To validate that Pol β R152C reduces cellular BER efficiency, we expressed human WT and R152C Pol β in 293 cells. Cells expressing similar levels of Pol β were selected (Figure 4A). Nuclear extracts (NE) from these cell lines were prepared and their BER efficiencies were assayed. We found that WT NE efficiently repaired the uracil (U) or THF (F) lesion, resulting in a 40 nt band, whereas the repair efficiency by R152C NE was only approximately 10% and 5% for SP-BER and LP-BER, respectively (Figure 4B and 4C).
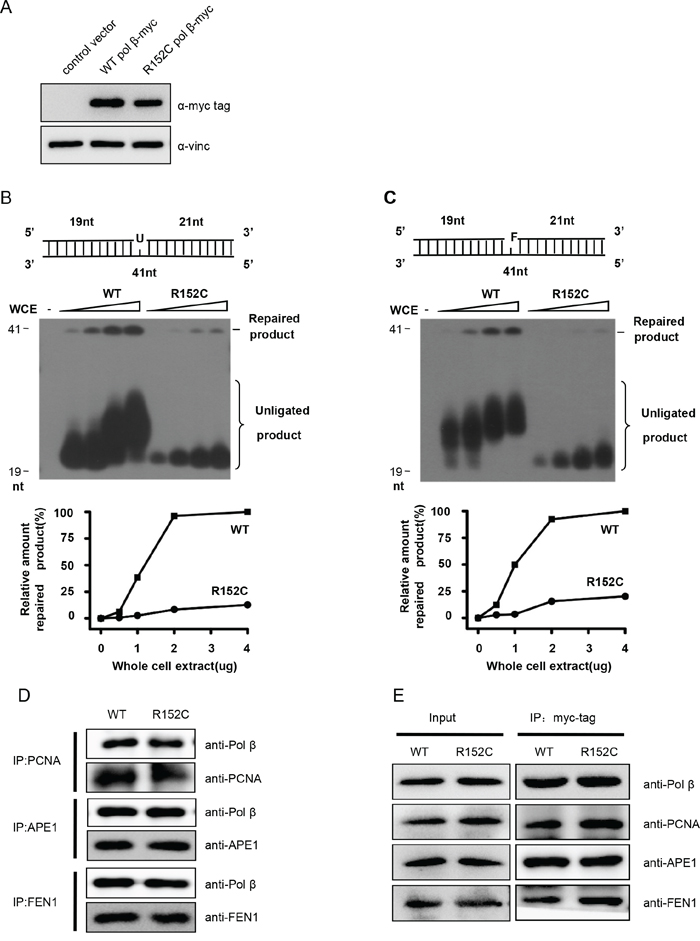
Figure 4: R152C mutation impairs BER efficiency in vivo. A. Western blotting shows expression of exogenous Pol β in 293 cells. SP-BER B. or LP-BER C. reconstitution with whole cell extracts (WCEs). The top part of each panel shows the schematic structures of the corresponding DNA substrates. The middle shows PAGE-separated products and the bottom the relative percentage of repaired product obtained with the indicated amounts of Pol β. The values represent the mean ±SD of three independent assays. D. Pull-down assay. Purified PCNA, APE1 and FEN1 were mixed in Tris buffer. The interaction between two proteins was determined by pull-down assay and western blotting. E. Co-immunoprecipitation assay. C-myc-tagged WT or R152C Pol β was expressed in 293 cells. C-myc-tagged proteins were precipitated using an anti-c-myc antibody, followed by western blot analysis using PCNA, APE1 and FEN1 antibodies.
Pol β has been reported to interact with many other proteins, including APE1 [38–42], PCNA [43], and FEN1 [27, 44]. These interactions play important roles in recruiting downstream factors to the DNA repair site, reciprocally stimulating enzyme activities and coordinating the highly ordered chemical reactions of BER. Disruption of these interactions would impede BER efficiency [27, 45]. To test if R152C disrupts the interaction of DNA Pol β with these proteins, we performed interaction assays in vitro (Figure 4D) and in vivo (Figure 4E). We found that R152C mutation has no influence on the interaction of Pol β with other major BER proteins, including APE1, FEN1 and PCNA (Figure 4D and 4E), so it is unlikely that defective protein interaction causes the decreased BER efficiency.
R152C Pol β-expressing cells are hypersensitive to DNA damage
Our data suggested R152C Pol β is defective in BER activity; we therefore suspected that R152C Pol β would sensitize cells to DNA damaging agents such as MMS and H2O2. This hypersensitivity could lead to accumulation of incompletely repaired DNA intermediates and chromatid breakage. Indeed, R152C Pol β-expressing cells were more sensitive to 1 hour of MMS or H2O2 treatment, compared with cells harboring WT Pol β (Figure 5A and 5B). To determine whether the R152C Pol β mutation increased the number of double strand breaks (DSBs) in cells, we counted the numbers of γH2AX foci in cells treated with 1 mM MMS. As shown in Figure 5C, more γH2AX foci were found in R152C-expressing cells, which means there were more DSBs. Consistently, western blotting data showed that the cells expressing R152C Pol β mutation increased γH2AX levels, with or without MMS treatment. (Figure 5D).
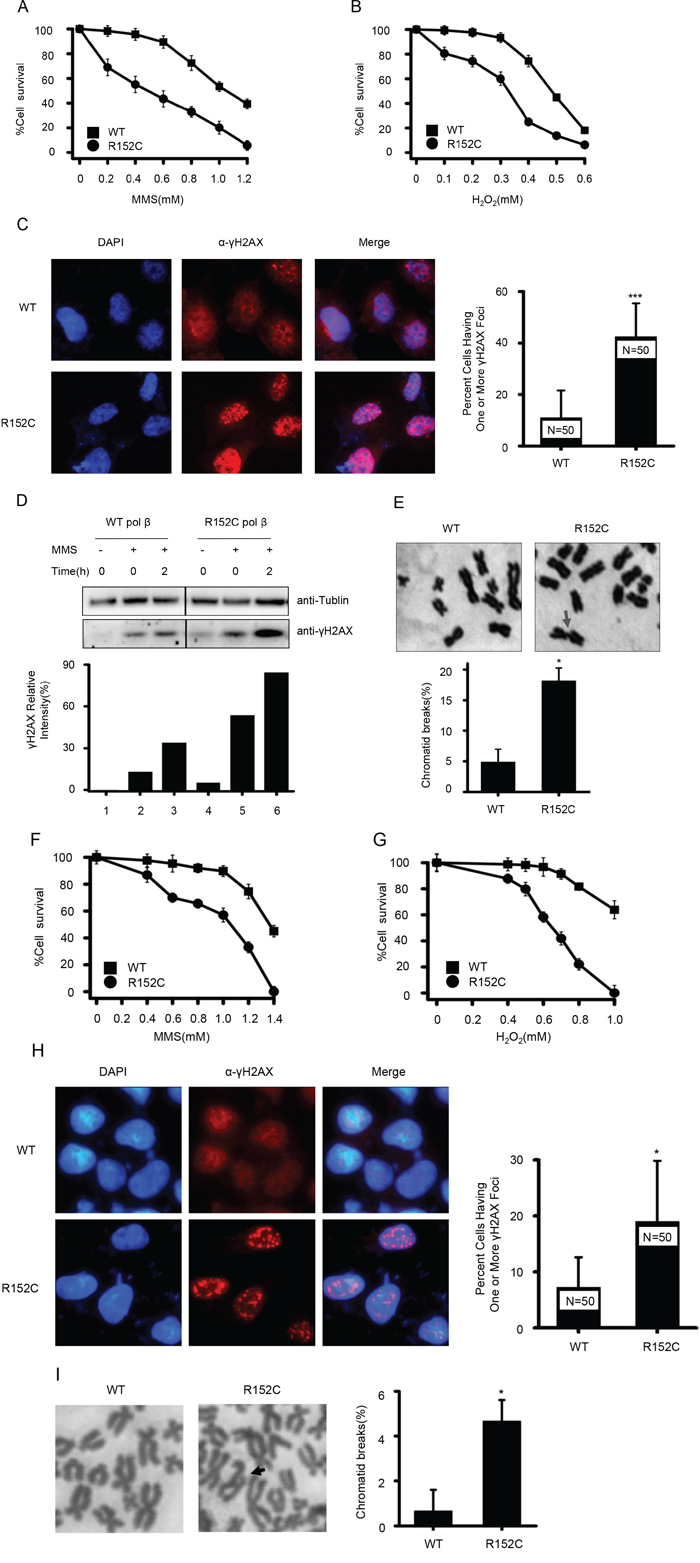
Figure 5: R152C cells are sensitive to the DNA damage agents, MMS and H2O2. MMS A. and H2O2 B. sensitivity assays. WT and R152C 293 cells were treated with MMS (A) and H2O2 (B) at the indicated concentrations. The number of cells was determined by the CellTiter 96 AQueous one-solution assay. C. Immunofluorescence staining for γH2AX (red) in 293 cells. The right panel shows the percentage of cells having one or more γH2AX focus. ***P<0.001, Student’s t-test. D. Western blotting of γH2AX levels before and after MMS treatment. E. Giemsa-stained WT and R152C 293 metaphase cells. Arrows indicate chromatid breaks. The bottom panel shows the quantification of mitotic cells with broken chromatids. *P=0.024, Student’s t-test. MMS F. and H2O2 G. sensitivity assays. WT and R152C SW480 cells were treated with MMS (F) and H2O2 (G) at the indicated concentrations. The number of viable cells was determined by the CellTiter 96 Aqueous one-solution assay. H. Immunofluorescence staining for γH2AX (red) in SW480 cells. The right panel shows the percentage of cells possessing one or more γH2AX focus. *P<0.05, Student’s t-test. I. Giemsa-stained WT and R152C SW480 metaphase cells. Arrows indicate chromatid breaks. The right panel shows quantification of mitotic cells with broken chromatids. *P<0.05, Student’s t-test.
Because the Pol β R152C mutation was identified from colon cancer cells, we also evaluated the cellular consequences of overexpressing WT or R152C Pol β in SW480 colon cancer cells. We found that SW480 cells harboring R152C Pol β displayed a reduced survival rate (Figure 5F, G) and accumulated more DSBs (Figure 5H) when challenged with DNA damaging agents.
Pol β R152C cells spontaneously accumulate chromosomal breaks and become aneuploid
Chromosomal instability and aneuploidy are associated with the accumulation of DNA breaks and cancer [46]. Therefore, we compared the number of spontaneous chromosomal breaks that occurred in WT and R152C cells. We found that the 293 cells carrying the R152C Pol β mutation had significantly more chromatid breaks than the WT cells (Figure 5E). This phenomenon was also confirmed in SW480 cells carrying the R152C mutation (Figure 5I).
The accumulation of chromosomal breaks is associated with aneuploidy. We found that the average percentage of aneuploidy in R152C cells was significantly higher than in WT cells (Figure 6A and 6B). Taken together, the data indicate that Pol β cells have higher levels of spontaneous chromatid breaks and aneuploidy than WT cells.
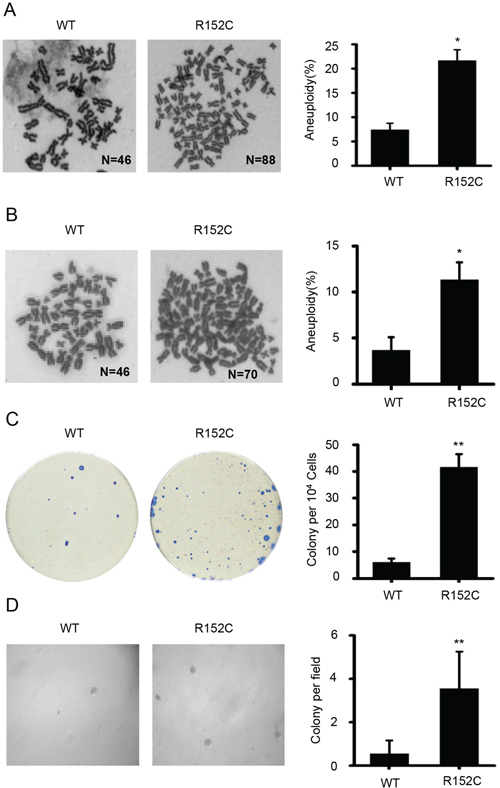
Figure 6: R152C cells have a high transformation potential. A. WT and R152C 293 metaphase cells were stained by Giemsa. The bottom panel shows the quantification of mitotic cells with aneuploidy, where the number of chromatids was >70. Here, approximately 180 mitotic cells per cell line were analyzed in three independent experiments. *P=0.017, Student’s t-test. B. WT and R152C SW480 metaphase cells were stained by Giemsa. C. Image of transformed cell colonies. WT and R152C SW480 cells were plated in a new dish for the colony-focus formation assays. The right panel shows the quantification of colonies, **P<0.01, Student’s t-test. D. Anchorage-independent growth. WT and R152C SW480 cells were plated in a new 6-well dish for anchorage-independent growth. The right panel shows the quantification of colonies, **P<0.01, Student’s t-test.
R152C cells have a high transformation potential
Our data indicate that R152C cells develop more chromatid breaks and are more likely to become aneuploid. We suspected that these cellular abnormalities would contribute to cellular transformation and lead to clonal expansion. To determine whether the R152C mutation promoted tumorigenesis, we performed cellular focus formation assays (Figure 6C). The number of colonies formed by R152C cells was six-fold higher than the number formed by WT cells, suggesting that R152C cells can develop aneuploidy-associated cancer. Anchorage-independent growth assays also showed that R152C Pol β-expressing cells possessed higher transformation activity than WT cells (Figure 6D).
DISSCUSSION
The Pol β R152C variant was recently identified in a colorectal carcinoma, but its role in cancer initiation was unknown. In this manuscript, we show that expression of the R152C colorectal carcinoma-associated Pol β variant in immortalized 293 cells results in cellular transformation. The mechanism of cellular transformation is likely to be genomic instability resulting from impaired BER efficiency by Pol β R152C mutation. This impaired BER activity, which increases the frequency of chromosomal aberrations, has been associated with several different types of cancers.
To further study the effects of the R152C Pol β variant in BER in vivo, we transfected human WT and R152C Pol β gene into 293 cells. Because R152C Pol β shows intact DNA binding activity (Figure 2E), we speculate that R152C Pol β competes with WT Pol β for DNA substrate binding, and therefore perturbs WT Pol β activity in cells. Indeed, R152C Pol β expression reduced both the SP-BER and LP-BER efficiency dramatically.
BER failure would lead to the accumulation of DNA damage, chromosome breakage, and aneuploidy [47]. In our study, R152C Pol β variants displayed defects in SP- and LP-BER. Furthermore, R152C Pol β expression, in both the 293 and SW480 cell lines, increased the sensitivity to MMS treatment, and increased the number of DSBs. In addition, karyotype analysis of R152C Pol β-expressing cells revealed more chromatid breaks and more aneuploid cells than WT controls. Cells usually have a precise surveillance system to detect DNA damage and chromatid aberrations, then they decide whether to die or not. However, a few cells with abnormal chromosomes could successfully bypass this surveillance system and transform into tumor cells. Our focus formation assay revealed that R152C cells formed clones, supporting a model where chromosome instability increases transformation and carcinogenesis [48, 49]. Altogether, our data demonstrate that the R152C mutation diminishes Pol β polymerase activity, impairs its capacity to conduct SP- and LP-BER, and promotes chromosome aberrations and genome instability, which contributes to carcinogenesis. Our results also indicate that R152 of Pol β could be a useful target for the diagnosis, prevention and treatment of cancer [11, 50, 51].
MATERIALS AND METHODS
Reagents and antibodies
All primers and DNA substrates used in this paper were synthesized by GenScript, Inc. using polyacrylamide gel electrophoresis (PAGE) purification. Four deoxynucleotide triphosphates (dNTPs) were purchased from New England Biolabs (N0446S). [γ-32P]-ATP (BLU002A, 250 uCi) and [α-32P]-dCTP (NEG513H, 250uCi) were purchased from PerkinElmer. The Pol β-specific antibody (ab26343) was purchased from Abcam. The anti-PCNA (SC-7907), APE1 (SC-55498), FEN1 (SC-56675), c-Myc (SC-40), Tubulin (sc-23950), goat anti-rabbit IgG-HRP (SC-2004), and goat anti-mouse IgG-HRP (sc-2005) antibodies were purchased from Santa Cruz Biotechnology.
Plasmid construction and preparation of recombinant proteins
Human WT Pol β cDNA (Genbank accession NM_002690) was from Dr. Binghui Shen’s laboratory in City of Hope National Medical Center, California, USA. The R152C mutant was generated using the QuikChange site-directed mutagenesis kit using the primers shown in Table 1. All recombinant proteins, including WT and R152C Pol β, FEN1, APE1, and Ligase IIIα, were purified from E.coli.
Circular dichroism spectroscopy assay
CD spectroscopy analysis was performed as previously described [52]. Purified WT and R152C Pol β proteins were diluted to 1 μM with KH2PO4, and their CD spectra were detected at 20°C using a Chirascan CD spectrometer. Measurements were collected at intervals of 1 nm from 190 to 280 nm.
DNA polymerase activity assay
DNA substrates (Table 1) were incubated with 20 μl of reaction buffer A (50 mM Tris-HCl [pH 8.0], 10 mM MgCl2, 2 mM DTT, 20 mM NaCl, 10% glycerol), 50 μM of each dATP, dGTP, and dTTP (NEB), 8 μM [α-32P]-dCTP, and various amounts (0-20 ng) of WT or R152C recombinant Pol β for 30 mins at 37°C. The reaction was then stopped by adding equal volumes of gel loading buffer (90% formamide dye, 3 M EDTA, 0.02% bromophenol blue and 0.02% xylene cyanol), heated (5min, 95°C), separated by 15% PAGE containing 8 M urea, and visualized by autoradiography.
Base excision repair assay
The BER assay was performed as described previously [26, 39, 53, 54]. The reactions were carried out in 20 μl of reaction buffer B (40 mM HEPES-KOH [pH 7.8], 70 mM KCI, 7 mM MgCl2, 1 mM dithiothreitol, 0.5 mM EDTA, 2 mM ATP, 50 μM each of dATP, dTTP, and dGTP, and 8 μM 2 μCi [α-32P]-dCTP). For SP reconstitution with purified proteins, Uracil-DNA Glycosylase (UDG, 8 ng), APE1 (2 ng), Ligase IIIα (20 ng) and various amounts of Pol β (0-5 ng), were mixed and incubated with the SP-BER substrate Pol β-U (Table 1). For LP-BER, the Pol β-F substrate (Table 1) was incubated with a mixture of APE1 (2 ng), Pol β (0-5 ng), FEN1 (2 ng), and Ligase I (20 ng). For cell extract reconstitution, the SP- or LP-BER DNA substrate was incubated with the whole cell extract (WCE, 0-5 μg). Reactions (30 mins, 37°C) were then stopped by adding an equal volume of the gel loading buffer and visualized by autoradiography.
DNA-binding assay
ELISA-based affinity assays was used to assay WT or R152C Pol β and DNA binding. First, a 96-well ELISA plate was coated with streptavidin (1 μg/well) and incubated overnight (4°C). Then, biotin-labeled Pol-GAP DNA substrate (1 pmol/well) was immobilized onto a 96-well plate (overnight, 4°C) and washed 3 times with PBS, followed by the addition of 0-1 ug WT or R152C Pol β recombinant proteins. Binding of Pol β was detected using a rabbit anti-Pol β antibody (Abcam, ab26343) and goat anti-rabbit secondary antibody-conjugated HRP (SC-2004). The color was developed by adding tetramethylbenzidine (TMB) and stopped by addition of 1 M H2SO4. The OD450 value was read by a microplate reader.
Immunofluorescence
The cells were cultured in six-well plates containing acid-treated cover slides and incubated overnight. The cover slides were then washed with PBS, fixed with 4% formaldehyde in PBS for 30 min, and then washed with PBS. Triton X-100 (0.05%) was added for 5 min to permeabilize the cells. The slides were blocked with 2% BSA and then incubated with primary antibody. The slides were washed then incubated with secondary antibody conjugated with FITC, followed by washing with PBS and staining with DAPI. The mounted slides were viewed with a Zeiss Axioscope and the images were captured with a charge-coupled device camera.
MMS and H2O2 sensitivity assay
293 cells transfected with WT/R152C Pol β-N2 vector and N2-vector were seeded (1,500 cells/well) into 96-well plates, incubated (overnight, 37°C), treated (1 h, 37°C) with 0-1.5 mM MMS or 0-1 mM H2O2, washed with PBS, returned to fresh medium (DMEM containing 10% FBS), and incubated under normal growth conditions (37°C, 5% CO2, 72 h). The number of viable cells was determined by the CellTiter 96 AQueous one-solution cell proliferation assay (Promega). Each dilution of MMS or H2O2 included at least four replicates. The data are expressed as the percentage of growth relative to untreated controls.
Cell transformation assays
The focus formation assays were conducted according to a previously established protocol [55, 56]. Briefly, 1x104 cells were plated in 6 cm dishes and incubated for 30 days at 37°C. The cells were then washed with PBS and fixed with 4% formaldehyde in PBS for 30 mins. Giemsa was used to stain the cells overnight at room temperature. Stained plates were washed and dried prior to scoring the colonies. Anchorage-independent growth was assessed as previously described [56]. Approximately 8x103 cells were mixed with media containing 1% noble agar. This mixture was poured onto a layer of media containing 3% noble agar in a well of a 6-well dish and incubated for 30 days at 37°C. The number of colonies present in each of five microscope fields per well from a total of 6 wells per experiment were counted after 4 weeks of growth.
ACKNOWLEDGMENTS
The work is supported by Ministry of Science and Technology of China (2013CB911600), National Natural Science Foundation of China (31271449), Jiangsu Provincial Natural Science Foundation (BK20130044, BK20130061, BK2011783 ), The Research Fund for the Doctoral Program of Higher Education of China (RFDP) (20133207110005), The Program for New Century Excellent Talents in University of Ministry of Education of China (NCET-13-0868) and the Priority Academic Program Development Award for Jiangsu Higher Education Institutions. The work was also partially supported by an NIH grant RO1 CA085344 to B.H.S. and NCI designated cancer center support grant P30 CA033572. We also thank Dr. Nancy Linford for her professional editing of the manuscript.
CONFLICTS OF INTEREST
The authors declare no conflicts of interest.
Author contributions
PFY and ZT designed the study and wrote the paper. WXP and CHY provided TCGA data shown in Figure 1. HY and CY constructed vectors for expression of mutant proteins. ZXL performed pull down assay in Figure 4. SHF and ZJ performed drug sensitivity assay shown in Figure 5. HLF and HZG performed Giemsa-stained metaphase cells shown in Figure 5 and Figure 6. All authors analyzed the results and approved the final version of the manuscript.
REFERENCES
1. Bartkova J, Horejsi Z, Koed K, Kramer A, Tort F, Zieger K, Guldberg P, Sehested M, Nesland JM, Lukas C, Orntoft T, Lukas J, Bartek J. DNA damage response as a candidate anti-cancer barrier in early human tumorigenesis. Nature. 2005; 434:864-870.
2. Hoeijmakers JH. Genome maintenance mechanisms for preventing cancer. Nature. 2001; 411:366-374.
3. David SS, O’Shea VL, Kundu S. Base-excision repair of oxidative DNA damage. Nature. 2007; 447:941-950.
4. Zharkov D. Base excision DNA repair. Cell Mol Life Sci. 2008; 65:1544-1565.
5. Barnes DE, Lindahl T. Repair and genetic consequences of endogenous DNA base damage in mammalian cells. Annu Rev Genet. 2004; 38:445-476.
6. Seeberg E, Eide L, Bjoras M. The base excision repair pathway. Trends in biochemical sciences. 1995; 20:391-397.
7. Lindahl T. Instability and decay of the primary structure of DNA. Nature. 1993; 362:709-715.
8. Lindahl T, Wood RD. Quality control by DNA repair. Science. 1999; 286:1897-1905.
9. Nakamura J, Walker VE, Upton PB, Chiang SY, Kow YW, Swenberg JA. Highly sensitive apurinic/apyrimidinic site assay can detect spontaneous and chemically induced depurination under physiological conditions. Cancer Res. 1998; 58:222-225.
10. Demple B, Sung JS. Molecular and biological roles of Ape1 protein in mammalian base excision repair. DNA repair. 2005; 4:1442-1449.
11. Abbotts R, Jewell R, Nsengimana J, Maloney DJ, Simeonov A, Seedhouse C, Elliott F, Laye J, Walker C, Jadhav A. Targeting human apurinic/apyrimidinic endonuclease 1 (APE1) in phosphatase and tensin homolog (PTEN) deficient melanoma cells for personalized therapy. Oncotarget. 2014; 5:3273. doi: 10.18632/oncotarget.1926.
12. Pascucci B, Stucki M, Jonsson ZO, Dogliotti E, Hubscher U. Long patch base excision repair with purified human proteins. DNA ligase I as patch size mediator for DNA polymerases delta and epsilon. J Biol Chem. 1999; 274:33696-33702.
13. Frosina G, Fortini P, Rossi O, Carrozzino F, Raspaglio G, Cox LS, Lane DP, Abbondandolo A, Dogliotti E. Two pathways for base excision repair in mammalian cells. J Biol Chem. 1996; 271:9573-9578.
14. Srivastava DK, Berg BJ, Prasad R, Molina JT, Beard WA, Tomkinson AE, Wilson SH. Mammalian abasic site base excision repair. Identification of the reaction sequence and rate-determining steps. J Biol Chem. 1998; 273:21203-21209.
15. Matsumoto Y, Kim K. Excision of deoxyribose phosphate residues by DNA polymerase beta during DNA repair. Science. 1995; 269:699-702.
16. Dogliotti E, Fortini P, Pascucci B, Parlanti E. The mechanism of switching among multiple BER pathways. Prog Nucleic Acid Res Mol Biol. 2001; 68:3-27.
17. Klungland A, Lindahl T. Second pathway for completion of human DNA base excision-repair: reconstitution with purified proteins and requirement for DNase IV (FEN1). EMBO J. 1997; 16:3341-3348.
18. Sobol RW, Horton JK, Kuhn R, Gu H, Singhal RK, Prasad R, Rajewsky K, Wilson SH. Requirement of mammalian DNA polymerase-beta in base-excision repair. Nature. 1996; 379:183-186.
19. Nealon K, Nicholl ID, Kenny MK. Characterization of the DNA polymerase requirement of human base excision repair. Nucleic Acids Res. 1996; 24:3763-3770.
20. Horton JK, Prasad R, Hou E, Wilson SH. Protection against methylation-induced cytotoxicity by DNA polymerase beta-dependent long patch base excision repair. J Biol Chem. 2000; 275:2211-2218.
21. Allinson SL, Dianova, II, Dianov GL. DNA polymerase beta is the major dRP lyase involved in repair of oxidative base lesions in DNA by mammalian cell extracts. EMBO J. 2001; 20:6919-6926.
22. Beard WA, Prasad R, Wilson SH. Activities and mechanism of DNA polymerase beta. Methods Enzymol. 2006; 408:91-107.
23. Beard WA, Wilson SH. Structure and mechanism of DNA polymerase Beta. Chem Rev. 2006; 106:361-382.
24. Goellner EM, Svilar D, Almeida KH, Sobol RW. Targeting DNA polymerase β for therapeutic intervention. Mol Pharmacol. 2012; 5:68.
25. Idriss HT, Al-Assar O, Wilson SH. DNA polymerase β. Int J Biochem Cell. 2002; 34:321-324.
26. Guo Z, Zheng L, Dai H, Zhou M, Xu H, Shen B. Human DNA polymerase beta polymorphism, Arg137Gln, impairs its polymerase activity and interaction with PCNA and the cellular base excision repair capacity. Nucleic Acids Res. 2009; 37:3431-3441.
27. Prasad R, Dianov GL, Bohr VA, Wilson SH. FEN1 stimulation of DNA polymerase beta mediates an excision step in mammalian long patch base excision repair. J Biol Chem. 2000; 275:4460-4466.
28. Sobol RW, Wilson SH. Mammalian DNA beta-polymerase in base excision repair of alkylation damage. Prog Nucleic Acid Res Mol Biol. 2001; 68:57-74.
29. Starcevic D, Dalal S, Sweasy JB. Is there a link between DNA polymerase beta and cancer? Cell Cycle. 2004; 3:998-1001.
30. Rozacky J, Nemec A, Sweasy J, Kidane D. Gastric cancer associated variant of DNA polymerase beta (Leu22Pro) promotes DNA replication associated double strand breaks. Oncotarget. 2015; 6:24474-24487. doi: 10.18632/oncotarget.4426.
31. Lang T, Dalal S, Chikova A, DiMaio D, Sweasy JB. The E295K DNA polymerase beta gastric cancer-associated variant interferes with base excision repair and induces cellular transformation. Mol Cell Biol. 2007; 27:5587-5596.
32. Dobashi Y, Kubota Y, Shuin T, Torigoe S, Yao M, Hosaka M. Polymorphisms in the human DNA polymerase beta gene. Hum Genet. 1995; 95:389-390.
33. Goode EL, Ulrich CM, Potter JD. Polymorphisms in DNA repair genes and associations with cancer risk. Cancer epidemiology, biomarkers & prevention. 2002; 11:1513-1530.
34. Dalal S, Chikova A, Jaeger J, Sweasy JB. The Leu22Pro tumor-associated variant of DNA polymerase beta is dRP lyase deficient. Nucleic Acids Res. 2008; 36:411-422.
35. Xi T, Jones IM, Mohrenweiser HW. Many amino acid substitution variants identified in DNA repair genes during human population screenings are predicted to impact protein function. Genomics. 2004; 83:970-979.
36. Pelletier H, Sawaya MR, Wolfle W, Wilson SH, Kraut J. Crystal structures of human DNA polymerase β complexed with DNA: implications for catalytic mechanism, processivity, and fidelity. Biochemistry. 1996; 35:12742-12761.
37. El-Andaloussi N, Valovka T, Toueille M, Steinacher R, Focke F, Gehrig P, Covic M, Hassa PO, Schar P, Hubscher U, Hottiger MO. Arginine methylation regulates DNA polymerase beta. Mol Cell. 2006; 22:51-62.
38. Marintchev A, Robertson A, Dimitriadis EK, Prasad R, Wilson SH, Mullen GP. Domain specific interaction in the XRCC1-DNA polymerase beta complex. Nucleic Acids Res. 2000; 28:2049-2059.
39. Kubota Y, Nash RA, Klungland A, Schar P, Barnes DE, Lindahl T. Reconstitution of DNA base excision-repair with purified human proteins: interaction between DNA polymerase beta and the XRCC1 protein. EMBO J. 1996; 15:6662-6670.
40. Gryk MR, Marintchev A, Maciejewski MW, Robertson A, Wilson SH, Mullen GP. Mapping of the interaction interface of DNA polymerase beta with XRCC1. Structure. 2002; 10:1709-1720.
41. Dianova, II, Sleeth KM, Allinson SL, Parsons JL, Breslin C, Caldecott KW, Dianov GL. XRCC1-DNA polymerase beta interaction is required for efficient base excision repair. Nucleic Acids Res. 2004; 32:2550-2555.
42. Wong HK, Wilson DM, 3rd. XRCC1 and DNA polymerase beta interaction contributes to cellular alkylating-agent resistance and single-strand break repair. J Cell Biochem. 2005; 95:794-804.
43. Kedar PS, Kim SJ, Robertson A, Hou E, Prasad R, Horton JK, Wilson SH. Direct interaction between mammalian DNA polymerase beta and proliferating cell nuclear antigen. J Biol Chem. 2002; 277:31115-31123.
44. Prasad R, Liu Y, Deterding LJ, Poltoratsky VP, Kedar PS, Horton JK, Kanno S, Asagoshi K, Hou EW, Khodyreva SN, Lavrik OI, Tomer KB, Yasui A, Wilson SH. HMGB1 is a cofactor in mammalian base excision repair. Mol Cell. 2007; 27:829-841.
45. Wong HK, Wilson DM. XRCC1 and DNA polymerase β interaction contributes to cellular alkylating-agent resistance and single-strand break repair. J Biol Chem. 2005; 95:794-804.
46. Ganem NJ, Storchova Z, Pellman D. Tetraploidy, aneuploidy and cancer. Curr Opin Genet Dev. 2007; 17:157-162.
47. Sancar A, Lindsey-Boltz LA, Ünsal-Kaçmaz K, Linn S. Molecular mechanisms of mammalian DNA repair and the DNA damage checkpoints. Annu Rev Biochem. 2004; 73:39-85.
48. Gorgoulis VG, Vassiliou L-VF, Karakaidos P, Zacharatos P, Kotsinas A, Liloglou T, Venere M, DiTullio RA, Kastrinakis NG, Levy B. Activation of the DNA damage checkpoint and genomic instability in human precancerous lesions. Nature. 2005; 434:907-913.
49. van Gent DC, Hoeijmakers JH, Kanaar R. Chromosomal stability and the DNA double-stranded break connection. Nat Rev Genet. 2001; 2:196-206.
50. Verschraegen C, Lounsbury K, Howe A, Greenblatt M. Therapeutic implications for ovarian cancer emerging from the Tumor Cancer Genome Atlas. Translational Cancer Research. 2015; 4:40-59.
51. Kassambara A, Gourzones-Dmitriev C, Sahota S, Rème T, Moreaux J, Goldschmidt H, Constantinou A, Pasero P, Hose D, Klein B. A DNA repair pathway score predicts survival in human multiple myeloma: the potential for therapeutic strategy. Oncotarget. 2014; 5:2487. doi: 10.18632/oncotarget.1740.
52. Nemec AA, Donigan KA, Murphy DL, Jaeger J, Sweasy JB. Colon cancer-associated DNA polymerase beta variant induces genomic instability and cellular transformation. J Biol Chem. 2012; 287:23840-23849.
53. Podlutsky AJ, Dianova, II, Podust VN, Bohr VA, Dianov GL. Human DNA polymerase beta initiates DNA synthesis during long-patch repair of reduced AP sites in DNA. EMBO J. 2001; 20:1477-1482.
54. Biade S, Sobol RW, Wilson SH, Matsumoto Y. Impairment of proliferating cell nuclear antigen-dependent apurinic/apyrimidinic site repair on linear DNA. J Biol Chem. 1998; 273:898-902.
55. Yamtich J, Nemec AA, Keh A, Sweasy JB. A germline polymorphism of DNA polymerase beta induces genomic instability and cellular transformation. PLoS Genet. 2012; 8:e1003052.
56. Sweasy JB, Lang T, Starcevic D, Sun K-W, Lai C-C, DiMaio D, Dalal S. Expression of DNA polymerase β cancer-associated variants in mouse cells results in cellular transformation. Proc Natl Acad Sci USA. 2005; 102:14350-14355.