
INTRODUCTION
Multiple myeloma (MM) is a plasma cell cancer characterized by clonal proliferation of plasma cells in the bone marrow (BM). A great improvement in the survival of MM patients was achieved the past decade due to the development of novel treatment options including novel agents and autologous stem cell transplantation (ASCT) for younger patients. These novel agents include the proteasome inhibitor bortezomib and the immunomodulatory drug lenalidomide. Nevertheless, these agents are not curative for the majority of patients and therefore investigation of novel targets to enhance therapy is necessary [1, 2].
The ubiquitin-proteasome system (UPS) is highly regulated and controls different protein functions and processes such as proliferation, metabolism and apoptosis by targeting cellular proteins for degradation. The ubiquitination process is dependent on ATP and requires the E1, E2 and E3 enzymes. First theubiquitin-activating enzyme E1 activates ubiquitin which is then delivered to the E2 ubiquitin-conjugating enzyme. The E2 subsequently forms a complex with the ubiquitin-ligase E3 and the target protein. The E3 transfers ubiquitin to the substrate to form a polyubiquitin chain, targeting the protein for degradation by the proteasome [3]. The success of the proteasome inhibitor bortezomib in the treatment of MM highlights the importance of the UPS in this disease. Selective targeting of more disease-specific components of the UPS might result in more effective treatment [4, 5]. There is an increasing interest in ubiquitin ligases as targets since they play a key role in the UPS by determining the protein to be ubiquitylated and thereby controlling cell function in a very specific way.
The anaphase promoting complex/cyclosome (APC/C) is a 13-subunit ubiquitin ligase protein complex that controls the cell cycle. The regulation of the APC/C is dependent on 2 co-activators, Cdc20 during the metaphase-anaphase transition and Cdh1 during mitotic exit and early G1-phase. Binding of the co-activator to the complex leads to the recruitment of substrates which will be targeted for proteasomal degradation. Key targets of APC/CCdh1 during mitotic exit and G1 maintenance are Skp2 and Ets2 leading to an increase in the levels of p27Kip1, p21Cip1 and cyclin D1 that are necessary to maintain the G1 phase [6]. Targets of the APC/CCdc20 complex are the cell cycle proteins cyclin B1 and securin. Their proteasomal degradation leads to the onset of the anaphase and mitotic exit. A major regulator of the APC/CCdc20 complex is the spindle assembly checkpoint (SAC). The SAC delays chromosome segregation until all kinetochores become properly attached to the mitotic spindle, then the APC/CCdc20 is activated [7].
Several studies suggest that the APC/C and its co-activator Cdc20 could be potential new therapeutic targets in cancer. Cdc20 overexpression has been correlated with poor prognosis in several solid cancers [8–14]. Knockdown of Cdc20 in various cancer cell lines caused mitotic arrest, cell death and increased sensitivity to chemotherapeutics and radiation [15–17]. Small molecule inhibitors of the APC/C called proTAME and apcin, have recently been discovered. ProTAME is a cell permeable prodrug that is converted to TAME (Tosyl-L-Arginine Methyl Ester) by intracellular esterases. TAME structurally mimics the IR-tail of the co-activators and therefore binds to APC/C, blocking the interaction of Cdc20 or Cdh1 with the APC/C [18]. Apcin is a small molecule that prevents substrate recognition by binding to Cdc20 [19]. In MM little is known about the APC/C and its co-activators. So far it has been shown that Cdc20 and the different components of the SAC are expressed in human MM cell lines (HMCLs) [20]. Our aim is to elucidate the importance and therapeutic potential of APC/C and its co-activator Cdc20 in MM.
RESULTS
Gene expression and prognostic value of Cdc20 and Cdh1 in MM patients
We assessed the distribution of Cdc20 expression in primary MM cells using three gene expression-based high-risk scores namely the RS, GPI and UAMS HRS scores [21–23]. In two independent MM patient cohorts we observed a significant higher Cdc20 expression in all high-risk groups (Figure 1A–1B). In addition, high Cdc20 expression correlated with a significant inferior overall survival in MM patients in both cohorts (Figure 1C). However there was no association between Cdh1 expression and gene expression-based risk scores in MM patients (Supplementary Figure 1A–1B). Low Cdh1 expression correlated with poor prognosis in MM in the TT-2 cohort but had no prognostic value in the HM-cohort (Supplementary Figure 1C). Next we evaluated the microarray expression level of Cdc20 and Cdh1 in HMCLs and observed a rather constant and low expression of Cdh1 while Cdc20 expression was higher and more variable (Supplementary Figure 1D). Moreover the expression of Cdc20 in primary MM cells correlated with proliferation measured as the plasma cell proliferation index with PI staining (n = 101, rs = 0,416, P < 0.001) (Supplementary Figure 2).
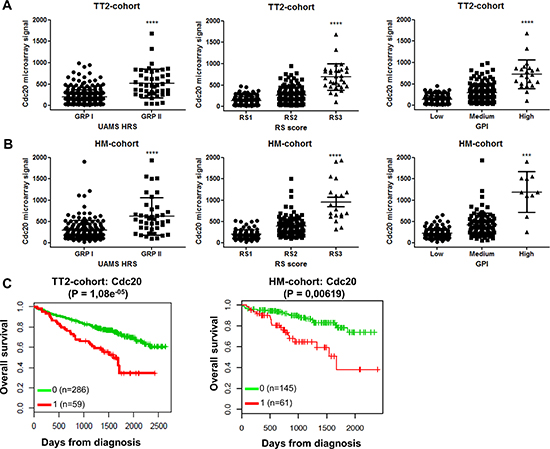
Figure 1: Cdc20 expression levels and prognostic value in MM patients. (A–B) Association of Cdc20 expression levels in MM patients with gene expression-based high-risk scores in TT2-cohort and HM-cohort. The high-risk groups are compared to the overall mean expression in all groups. ***indicates p-value is < 0.001 and ****p-value is < 0.0001 (student t-test). (C) The overall survival in the TT2-cohort and HM-cohort for low (green) or high (red) Cdc20 expression in MM patients.
Gene set enrichment analysis was performed comparing gene expression profiles of MM cells of patients with high or low Cdc20 or Cdh1 expression. A list of the top 10 GSEA pathways enriched in Cdc20 high, Cdc20 low, Cdh1 high and Cdh1 low MM patients can be found in Tables 1 and 2. MM cells of patients with a high Cdc20 expression showed a significant enrichment in genes associated with proliferation, while MM cells of patients with a low Cdc20 expression had a significant enrichment in genes under-expressed in the proliferation subgroup of the MM molecular classification (Supplementary Figure 3 and 4 and Supplementary Tables 1–4). Patients with high Cdh1 expression were characterized by a significant enrichment of genes related to mature bone marrow plasma cells and JAK/STAT signaling. Interestingly, patients with low Cdh1 expression showed a significant enrichment of MYC target genes (Supplementary Figure 5 and 6 and Supplementary Tables 5–7).
Table 1: GSEA pathways significantly enriched in MM patients with high or low Cdc20 expression
|
Size |
Es |
Nes |
p-value |
|---|---|---|---|---|
GSEA pathways significantly enriched in MM patients with high Cdc20 expression |
||||
pujana_chek2_pcc_network |
414 |
–0.68 |
–2.66 |
< 0.0001 |
zheng_glioblastoma_plasticity_up |
207 |
–0.69 |
–2.65 |
< 0.0001 |
shedden_lung_cancer_poor_survival_a6 |
319 |
–0.78 |
–2.62 |
< 0.0001 |
pujana_brca2_pcc_network |
280 |
–0.75 |
–2.62 |
< 0.0001 |
tarte_plasma_cell_vs_plasmablast_dn |
194 |
–0.77 |
–2.61 |
< 0.0001 |
wong_embryonic_stem_cell_core |
190 |
–0.77 |
–2.60 |
< 0.0001 |
reactome_cell_cycle |
251 |
–0.72 |
–2.58 |
< 0.0001 |
vecchi_gastric_cancer_early_up |
313 |
–0.73 |
–2.58 |
< 0.0001 |
patil_liver_cancer |
473 |
–0.61 |
–2.58 |
< 0.0001 |
lindgren_bladder_cancer_cluster_3_up |
218 |
–0.75 |
–2.58 |
< 0.0001 |
GSEA pathways significantly enriched in MM patients with low Cdc20 expression |
||||
reactome_inhibition_of_voltage_gated_ca2_channels_via_gbeta_gamma_subunits |
21 |
0.63 |
1.85 |
< 0.0001 |
zhan_multiple_myeloma_pr_dn |
32 |
0.62 |
1.81 |
0.023 |
vilimas_notch1_targets_up |
47 |
0.49 |
1.75 |
0.004 |
reactome_inwardly_rectifying_k_channels |
26 |
0.58 |
1.73 |
0.004 |
biocarta_cardiacegf_pathway |
15 |
0.62 |
1.73 |
0.008 |
sumi_hnf4a_targets |
27 |
0.54 |
1.72 |
0.013 |
kondo_prostate_cancer_with_h3k27me3 |
129 |
0.45 |
1.70 |
0.002 |
meissner_brain_hcp_with_h3_unmethylated |
29 |
0.56 |
1.69 |
0.008 |
reactome_nuclear_receptor_transcription_pathway |
37 |
0.50 |
1.69 |
0.012 |
Table 2: GSEA pathways significantly enriched in MM patients with high or low Cdh1 expression
|
Size |
Es |
Nes |
p-value |
|---|---|---|---|---|
GSEA pathways significantly enriched in MM patients with high Cdh1 expression |
||||
reactome_growth_hormone_receptor_signaling |
22 |
–0.58 |
–1.88 |
0.004 |
mueller_common_targets_of_aml_fusions_dn |
23 |
–0.60 |
–1.80 |
0.004 |
kegg_jak_stat_signaling_pathway |
132 |
–0.41 |
–1.80 |
< 0.0001 |
mccabe_hoxc6_targets_cancer_up |
22 |
–0.58 |
–1.75 |
0.006 |
kuuselo_pancreatic_cancer_19q13_amplification |
15 |
–0.62 |
–1.74 |
0.012 |
moreaux_multiple_myeloma_by_taci_up |
288 |
–0.42 |
–1.72 |
< 0.0001 |
mueller_methylated_in_glioblastoma |
39 |
–0.52 |
–1.73 |
< 0.0001 |
reactome_effects_of_pip2_hydrolysis |
19 |
–0.58 |
–1.73 |
0.011 |
mcmurray_tp53_hras_cooperation_response_up |
21 |
–0.57 |
–1.72 |
0.010 |
pomeroy_medulloblastoma_desmoplasic_vs_classic_dn |
42 |
–0.48 |
–1.72 |
0.002 |
GSEA pathways significantly enriched in MM patients with low Cdh1 expression |
||||
reactome_pyruvate_metabolism_and_citric_acid_tca_cycle |
18 |
0.73 |
2.07 |
0.006 |
schuhmacher_myc_targets_up |
42 |
0.64 |
2.00 |
0.002 |
gazda_diamond_blackfan_anemia_progenitor_dn |
34 |
0.57 |
1.97 |
< 0.0001 |
chow_rassf1_targets_dn |
15 |
0.74 |
1.95 |
< 0.0001 |
zhan_multiple_myeloma_cd1_up |
39 |
0.81 |
1.79 |
0.020 |
krige_amino_acid_deprivation |
28 |
0.72 |
1.79 |
0.026 |
chiang_liver_cancer_subclass_unannotated_dn |
93 |
0.50 |
1.78 |
0.022 |
zhan_multiple_myeloma_cd1_vs_cd2_up |
57 |
0.73 |
1.77 |
0.026 |
schlosser_myc_targets_and_serum_response_up |
24 |
0.65 |
1.75 |
0.018 |
shaffer_irf4_targets_in_myeloma_vs_mature_b_lymphocyte |
70 |
0.50 |
1.73 |
0.013 |
Pharmacological inhibition of the APC/C with proTAME results in a metaphase arrest and reduced viability of MM cells
To assess if the APC/C could be a potential target in MM we used the APC/C inhibitor proTAME. We first examined protein levels of substrates APC/CCdc20 (cyclin B1) and APC/CCdh1 (Skp2) after 6, 18 and 24 hours proTAME treatment (Figure 2A). We observed an increase in cyclin B1 at early time points. Skp2 levels however were not affected by proTAME treatment. This suggests that proTAME may preferentially inhibit APC/CCdc20 activity in MM cells.
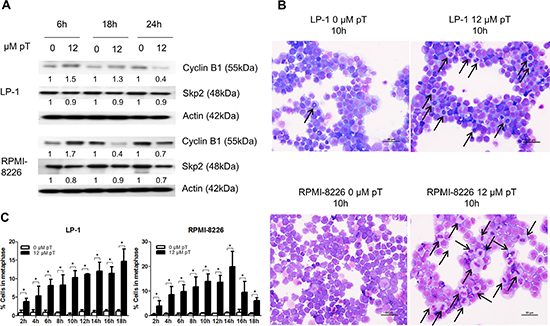
Figure 2: Pharmacological inhibition of the APC/C with proTAME results in a metaphase arrest. (A) LP-1 and RPMI-8226 cells were treated with 12 μM proTAME (pT) for 6, 18 and 24 hours. Western blot analysis was performed using cyclin B1, Skp2 and β-actin antibodies. This result is representative for 3 independent experiments. The pixel densities of proteins are normalized to β-actin and relative to control. (B) LP-1 and RPMI-8226 cells were treated with 12 μM proTAME and every 2 hours until 18 hours May-Grünwald Giemsa stained cytospins were made. Arrows point out the MM cells in metaphase. Scale bar measures 50 μm. (C) Quantification of the amount of cells in metaphase. Results shown in each graph are the mean of 3 independent experiments ± SD. *means the p-value is < 0.05 (Mann-Whitney U-test).
Since the APC/CCdc20 is involved in the metaphase-anaphase transition we investigated if inhibition of the APC/C could lead to an arrest of cells in the metaphase. May-Grünwald Giemsa stained cytospins of proTAME treated LP-1 and RPMI-8226 cells were analyzed using light microscopy (Figure 2B). Quantification of the percentage of cells in metaphase indicated a significant increase when the cell lines were treated with proTAME at each time point (Figure 2C).
As a mitotic arrest can lead to cell death we further investigated the effect of proTAME on the viability of MM cells. The HMCLs LP-1, RPMI-8226, JJN3, OPM-2, U266 and NCI-H929 were treated with proTAME and viability was measured 24 hours later (Figure 3A). We observed a dose-dependent decrease in the viability in all HMCLs. LP-1 was the least sensitive cell line with an IC50 of 12.1 μM and JJN3 was the most sensitive with an IC50 of 4.8 μM (Supplementary Table 8). In addition we tested proTAME on primary samples from 7 myeloma patients and observed again a dose-dependent reduction of the viability (Figure 3B). The IC50 varied among the different patients, ranging from 2.8 to 20.3 μM (Supplementary Table 8). Patient characteristics can be found in Supplementary Table 9. To investigate if proTAME influences the viability of cells from the BM microenvironment, we treated bone marrow stromal cells (BMSC) from different MM patients (Figure 3C) and observed that proTAME did not affect their viability. Moreover we treated peripheral blood mononuclear cells (PBMCs) of 3 healthy persons with proTAME (Figure 3D). ProTAME decreased the viability of PBMCs in a dose dependent way with an IC50 of 73,6 μM which is higher compared to the IC50 of MM cells (3,7–26,3x higher than MM patient cells and 6,1–15,3x higher as HMCLs).
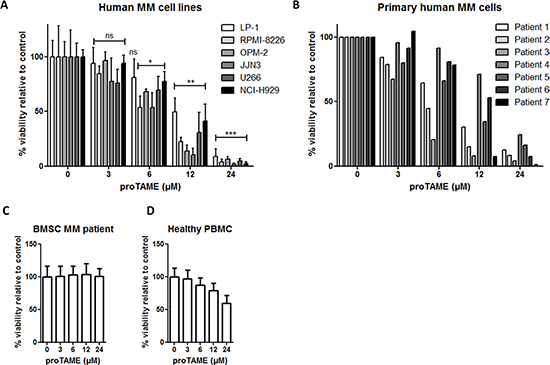
Figure 3: Pharmacological inhibition of the APC/C with proTAME results in a decreased viability of MM cells. (A) LP-1, RPMI-8226, JJN3, OPM-2, U266, NCI-H929, (B) primary human MM cells, (C) BMSC of MM patients and (D) healthy peripheral blood mononuclear cells were treated with 3, 6, 12 and 24 μM proTAME and after 24 hours viability was measured with CellTiter-Glo assay. Results shown are the mean of 4 independent experiments for HMCL, 2 for BMSC of MM patients and 3 for PBMC ± SD. *means the p-value is < 0.05, **means the p-value is < 0.001, ***means the p-value is < 0.0001 and represents a significant decrease compared to control (One-way ANOVA). For the primary human MM cells the result of each patient is shown individually.
proTAME is still active in the presence of the BM microenvironment
The BM microenvironment is an important source of MM survival and growth factors such as IL-6 and IGF-1. Here we tested if IL-6 and IGF-1 could abrogate the effect of proTAME on HMCLs by investigating the viability of LP-1, RPMI-8226 and NCI-H929 cells treated with proTAME with or without IL-6 or IGF-1. In all these MM cell lines IL-6 and IGF-1 induced an increased survival at both concentrations tested. Although in some conditions IL-6 and IGF-1 slightly diminished the effect of proTAME, at higher concentrations the effect of proTAME was not abrogated by these cytokines (Figure 4A). We also co-cultured the RPMI-8226 cells with healthy human bone marrow stromal cells (BMSCs) and treated them with proTAME. Although BMSCs reduced the percentage of apoptotic cells, proTAME was still able to significantly induce apoptosis in MM cells in the presence of BMSCs (Figure 4B).
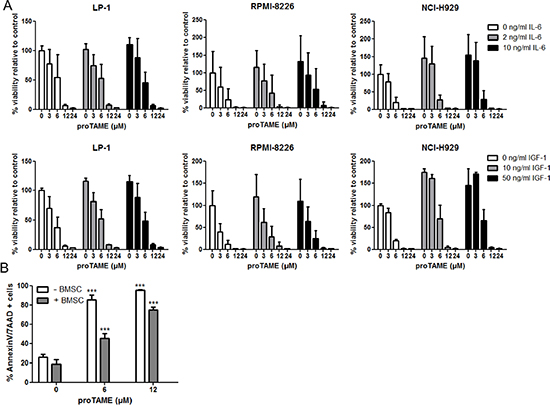
Figure 4: The effect of proTAME on MM cells in the presence of growth factors or BMSCs. (A) HMCL LP-1, RPMI-8226 and NCI-H929 were cultured in serum free conditions and treated with 0, 3, 6, 12 and 24 μM proTAME in combination with IL-6 or IGF-1. After 24 hours the viability was determined with the CellTiter-Glo assay. (B) RPMI-8226 cells were cultured in serum free conditions and treated with 0, 6, 12 μM proTAME in the presence or absence of human healthy BMSCs. After 24 hours apoptosis was examined with Annexin V/7AAD flow cytometry staining. Results shown in the graphs are the mean of 3 independent experiments ± SD. ***means the p-value is < 0.0001 (One-way ANOVA) and indicates a significant increase compared to control (0 μM proTAME).
Pharmacological inhibition of the APC/C with proTAME induces apoptosis in MM cells and is partially mediated by Bim and phosphorylation of Bcl-2 and Bcl-xL
Next we explored if APC/C inhibition could induce apoptosis in 4 cell lines. A significant increase in the number of apoptotic cells was found in each cell line after treatment with proTAME (Figure 5A). This was confirmed by an induction of apoptosis in primary samples from 3 myeloma patients (Figure 5B).
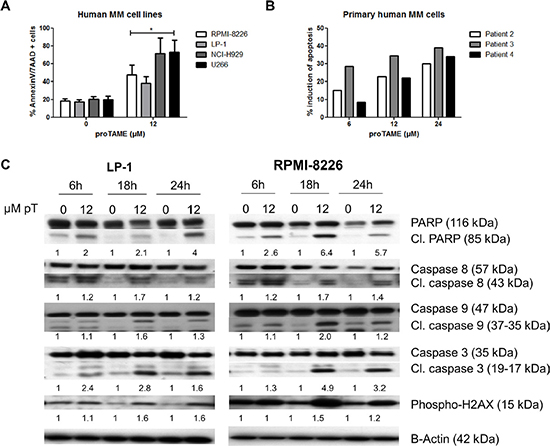
Figure 5: Pharmacological inhibition of the APC/C with proTAME induces apoptosis in MM cells. (A) LP-1, RPMI-8226, NCI-H929 and U266 cells weretreated with 12 μM proTAME and after 48 hours apoptosis was examined with Annexin V/7AAD flow cytometry staining. Results shown in the graph are the mean of 4 independent experiments ± SD,*means the p-value is < 0.05 and represents a significant increase compared to control (Mann-Whitney U-test). (B) Primary human MM cells were treated with indicated concentrations of proTAME and after 24 hours the induction of apoptosis was calculated with Annexin V/7AAD flow cytometry staining. The result of each patient is shown individually. (C) LP-1 and RPMI-8226 cells were treated with 12 μM proTAME for 6, 18 and 24 hours. Western blot was performed using caspase 3, 8, 9, PARP, phospho-H2AX and β-actin antibodies. The pixel densities of proteins were normalized to β-actin and relative to control. This result is representative for 3 independent experiments.
The apoptotic pathway was further investigated in LP-1 and RPMI-8226 cells by western blot. We observed cleavage of PARP, caspase 3, 8 and 9 beginning after 6 hours of proTAME treatment in both cell lines. Moreover, we observed a phosphorylation of H2AX after proTAME treatment (Figure 5C). Looking at Bcl-2 family proteins, no clear difference was observed in total Bcl-2, Bcl-xL and Mcl-1 however phosphorylation of Bcl-2 and Bcl-xL was clearly induced at earlier time points in HMCLs treated with proTAME (Figure 6A). In addition, Bim was increased when RPMI-8226 cells were incubated for 6 hours with proTAME (Figure 6B). To investigate the involvement of Bim in the induced apoptosis, Bim was silenced in RPMI-8226 cells with shRNA. Bim silencing was confirmed by western blot analysis of cells transduced with a shBim lentiviral vector compared to shScrambled transduced cells (Figure 6C). Reduction of Bim expression in RPMI-8226 cells significantly reduced the effect of proTAME, implying that Bim is at least partially responsible for the observed induction of apoptosis by proTAME (Figure 6D). LP-1 cells were not investigated for the involvement of Bim as Bim is not expressed in this cell line [24].
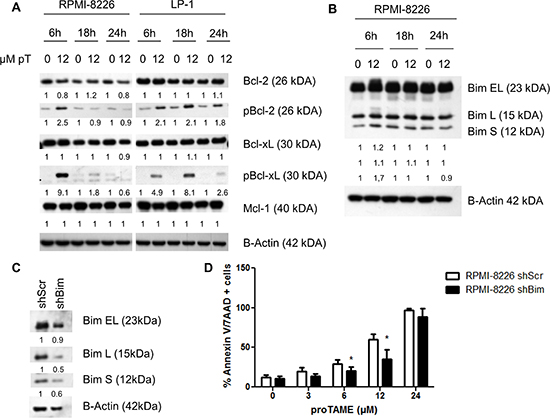
Figure 6: proTAME treatment induces Bim and phosphorylation of Bcl-2 and Bcl-xL. (A and B) LP-1 and RPMI-8226 cells were treated with 12 μM proTAME for 6, 18 and 24 hours. Western blot was performed using Bcl-2, pBcl-2, Bcl-xL, pBcl-xL, Mcl-1, Bim and β-actin antibodies. The pixel densities of proteins were normalized to β-actin and relative to control. This result is representative for 3 independent experiments. (C) Bim was knocked down in the RPMI-8226 using shRNA. These cells were treated with 0, 3, 6, 12 and 24 μM proTAME and after 48 hours apoptosis was measured with Annexin V/7AAD flow cytometry staining. Results shown in each graph are the mean of 4 independent experiments ± SD,*means the p-value is < 0.05 and indicates a significant difference compared to RPMI-8226shScr (Mann-Whitney U-test) (D).
Knockdown of Cdc20 results in a metaphase arrest and apoptosis in MM cells
To exclude that proTAME has non-specific effects we knocked down Cdc20 with siRNA in the HMCL RPMI-8226. Q-PCR and western blot analysis demonstrated a significant reduction of Cdc20 on respectively mRNA and protein levels. Knockdown of Cdc20 resulted in a significant induction of apoptosis after 48 and 72 h which was accompanied with cleavage of caspase 3, 8, 9 and PARP. Moreover, silencing Cdc20 resulted in phosphorylation of H2AX and accumulation of cells in metaphase (Figure 7A–7D).
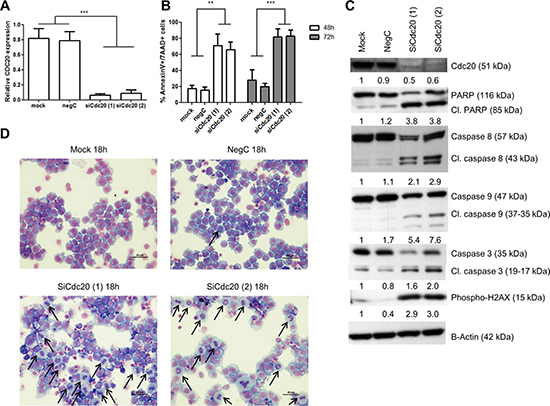
Figure 7: Knockdown of Cdc20 results in a metaphase arrest and cell death. RPMI-8226 cells were transfected using lipofectamine RNAiMAX reagent and two different siRNA duplexes against Cdc20. 48 hours after transfection (A) Q-PCR was performed. Results shown are the mean of 3 independent experiments ± SD. ***means the p-value is < 0.0001 (One-way ANOVA). (B) Apoptosis was examined with Annexin V/7AAD flow cytometry staining. Results shown in the graph are the mean of 3 independent experiments ± SD, **and ***mean the p-value is respectively < 0.001 and < 0.0001 (One-way ANOVA). (C) Western blot was performed using Cdc20, caspase 3, 8, 9, PARP, phospho-H2AX and β-actin antibodies. The pixel densities of proteins were normalized to β-actin and relative to mock. This result is representative for 3 independent experiments. (D) 18 hours after transfection May-Grünwald Giemsa stained cytospins were made. Arrows point out the MM cells in metaphase.
Inhibition of the APC/C with two chemical inhibitors significantly increases MM cell death
Recently it was described that combining proTAME with apcin leads to a synergistic inhibition of the APC/C [19]. We therefore incubated the HMCLs LP-1 and RPMI-8226 with a combination of proTAME and apcin. Although apcin alone had minor effects on MM cells, the combination of both inhibitors increased apoptosis significantly compared to single treatments (Figure 8A).
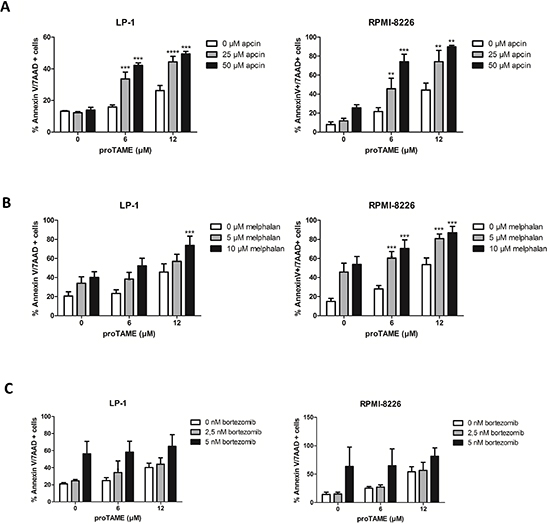
Figure 8: MM cell death is significantly increased when the APC/C is inhibited with two chemical inhibitors or in combination with melphalan. LP-1 and RPMI-8226 were treated with proTAME in combination with another APC/C inhibitor apcin (A), melphalan (B) or bortezomib (C). After 48 hours apoptosis was measured with Annexin V/7AAD flow cytometry staining. Results shown in each graph are the mean of at least 3 independent experiments ± SD. **means the p-value is < 0.001, ***means the p-value is < 0.0001 and represents a significant increase compared to single treatments (One-way ANOVA).
We also investigated the combination of proTAME with 2 important drugs used for the treatment of MM patients, namely melphalan and bortezomib. The combination of proTAME with melphalan increased apoptosis significantly compared to single treatments (Figure 8B). However, combining proTAME with bortezomib did not result in an increased apoptosis (Figure 8C) compared to single treatments.
DISCUSSION
Since the outcome of particularly high-risk patients has not yet improved, investigation of alternative treatment options of those patients is greatly needed [25]. High-risk MM patients are characterized by an overexpression of genes associated with cell cycle and a high labeling index [21, 26–28]. This gives a clear rationale to treat high-risk patients with (targeted) anti-mitotics. In this study we investigated if the APC/C, and its co-activator Cdc20 could be a potential target in MM. Based on three existing gene expression-based high-risk scores [21–23] we observed a higher Cdc20 expression in high-risk MM patient groups. Moreover we observed a correlation between Cdc20 expression in primary MM cells and proliferation. High Cdc20 was associated with poor overall survival in both MM patient cohorts. This is in agreement with other cancer types where an upregulated Cdc20 expression correlated with a poor prognosis [8–14]. Moreover a recent study shows that MM patients with a high expression of BUB1b, Cdc20 and CCNB1/2 had a poor survival outcome in the TT-2 cohort [29]. In addition we showed that MM cells of patients with a high Cdc20 expression showed a significant enrichment in genes associated with proliferation whereas MM cells of patients with low Cdc20 expression have a significant enrichment in genes underexpressed in the proliferation subgroup of the MM molecular classification. Cdh1 expression on the other hand was not associated with gene expression-based high-risk MM. Low Cdh1 expression was associated with poor prognosis in the TT-2 cohort but not in the HM-cohort. Cdh1 has been described as a potential tumor suppressor, since elevated Cdh1 levels inhibited breast tumor growth and depletion of Cdh1 induced proliferation of breast cancer cells [30–32]. We observed that patients with high Cdh1 expression had a significant enrichment of genes related to mature bone marrow plasma cells and JAK/STAT signaling whereas patients with low Cdh1 expression showed a significant enrichment in MYC target genes. From this we concluded that it would be interesting to target the APC/CCdc20 in MM.
In this study we used the APC/C inhibitor proTAME. Previous research demonstrated that proTAME is able to inhibit the interaction of both co-activators with the APC/C in HeLa cells [18]. Inhibition by proTAME efficiently induced a metaphase arrest, which is in agreement with our study. Moreover we observed at early time points a clear increase in cyclin B1, a substrate of the APC/CCdc20, while the APC/CCdh1 substrate Skp2 was not affected. This could be due to the fact that cells arrest in metaphase first by the effect on APC/CCdc20 upon proTAME treatment making it difficult to see effects of proTAME on APC/CCdh1. The lack of cyclin B1 accumulation at later time points could be because of cells dying faster than they can accumulate cyclin B1. By western blot analysis we could indeed observe signs of cell death beginning as early as 6 hours following proTAME treatment. The observed metaphase arrest was followed by a reduction in the viability. The sensitivity of the different HMCLs is variable with an IC50 ranging from 4.8 to 12.1 μM. The variability in sensitivity can be attributed to the difference in proliferation rate between the different cell lines. Primary MM samples were sensitive to proTAME treatment with an IC50 ranging from 2.8 to 20.3 μM, comparable to the IC50 described in HeLA cells [18]. However non-malignant cells of MM patients and healthy PBMCs were not or only slightly (at the highest concentration) affected by proTAME. We furthermore investigated if BMSCs and soluble components of the BM microenvironment such as IL-6 and IGF-1, described as major survival and growth factors in MM, would affect the proTAME-induced cell death. Although there was some protection, proTAME was still able to significantly disrupt MM cell survival in the presence of BMSCs and soluble factors. These findings demonstrate that inhibition of the APC/Cdc20 would still be active in the BM microenvironment, making it a promising candidate for further preclinical investigation.
As a metaphase arrest is eventually able to trigger cell death, we investigated the activation of apoptotic pathways upon proTAME treatment [6]. ProTAME was able to significantly increase the number of apoptotic cells in different HMCLs. This was accompanied by caspase 3, 8, 9 and PARP cleavage, indicating the involvement of both intrinsic and extrinsic apoptotic pathways. Moreover H2AX was phosphorylated upon proTAME treatment indicating that proTAME treatment could be associated with DNA damage. This confirms previous findings that DNA damage occurs during mitotic arrest with microtubule inhibitors [33, 34]. On the other hand it is described that H2AX can be phosphorylated during cell cycle progression and cell cycle arrest in the absence of DNA damage [35]. Since proTAME is a chemical inhibitor we wanted to exclude non-specific effects of proTAME. Therefore Cdc20 was silenced with siRNA in the HMCL RPMI-8226 and this resulted in a metaphase arrest and induction of apoptosis similar as proTAME suggesting that the observed effect of proTAME is most likely mediated by inhibition of APC/CCdc20. These results are also in line with previous studies in other cancers where knockdown of Cdc20 resulted in a mitotic arrest and cell death [15–17].
A hallmark of cells arrested in mitosis by microtubule disrupting agents is the phosphorylation of Bcl-2 and Bcl-xL, two anti-apoptotic proteins. Phosphorylation of these proteins leads to inactivation of their anti-apoptotic function [36–38]. It is the upregulation of cyclin B1 which is responsible for the phosphorylation of Bcl-xL [39]. In this study, we observed a clear increase in cyclin B1 and phosphorylation of Bcl-2 and Bcl-xL followed by cell death upon proTAME treatment indicating a mitotic arrest. Mcl-1 is believed to be degraded during mitotic arrest and this process requires Cdc20 [38, 40]. Since Cdc20 is blocked with proTAME, this could explain the absence of Mcl-1 degradation in our experiments. Bim, a pro-apoptotic member of the Bcl-2 family was increased upon proTAME treatment. Bim was recently characterized as a substrate of the APC/CCdc20 complex [41]. The apoptotic consequences of depleting Cdc20 have been attributed to the accumulation of Bim. Reducing Bim expression in our cell line significantly reduces the anti-MM effect of proTAME. This might also explain why LP-1 cells which have no Bim expression are the least sensitive to proTAME treatment. Since p53 is a crucial cell cycle and apoptosis regulator we explored p53 protein expression after proTAME treatment. No changes were observed in p53 and phosphorylated p53 (data not shown). Also no link could be observed in the sensitivity of HMCL to proTAME treatment and their p53 status.
To further test whether the APC/CCdc20 complex is a valid target in MM, we combined proTAME with another APC/C inhibitor apcin. Sackton et al. demonstrated that simultaneously blocking the APC/C with proTAME and apcin is synergistic, suggesting that targeting several weak protein-protein interactions may be a good strategy to block large protein complexes such as the APC/C [19]. We confirmed this in our HMCLs as combination treatment with proTAME and apcin significantly induced more apoptosis compared to single treatments. As previously described, apcin alone did not significantly affect MM cell survival probably due to a less efficient inhibition of APC/C compared to proTAME alone. Moreover, we evaluated if proTAME could enhance the anti-MM activity of clinically relevant drugs such as bortezomib and melphalan. ProTAME significantly increased the effect of melphalan. However we did not observe a combination effect with bortezomib. This could be explained by the fact that proteasome inhibition generally induces an interphase arrest which would block the ability of proTAME to induce a metaphase arrest [18, 42].
In MM, vincristine in combination with doxorubicin and dexamethasone, has been used for relapsed patients and as induction therapy [43, 44]. However vincristine as a single agent has demonstrated low efficacy in MM [45]. Vincristine belongs to the microtubule-targeting drugs and disrupt microtubules leading to the activation of the SAC which subsequently blocks the APC/C [46]. However their use is associated with known side effects, such as peripheral neuropathy through disrupting the microtubules in the neurons. Since this mechanism is SAC dependent, the sensitivity to these microtubule-targeting drugs is variable due to variable SAC activity. Therefore, it is hypothesized that targeting mitotic exit downstream of the SAC, by for example blocking Cdc20 would be beneficial [15]. We indeed demonstrate in this study that the APC/CCdc20 could be an attractive and a new therapeutic target, especially in high-risk MM patients. This strategy can influence the proliferation and cell death of tumor cells without affecting the BM microenvironment. Moreover it can be combined with a clinically relevant drug such as melphalan, making it a promising candidate for further preclinical investigation. Since proTAME is not yet optimized for in vivo studies, new and more potent small molecules inhibiting the APC/CCdc20 should be developed and in vivo validated.
MATERIALS AND METHODS
Cell culture
The HMCLs LP-1, RPMI-8226, OPM-2 and NCI-H929 are obtained from the American Type Culture Collection and the U266 and JJN3 were kindly provided by Prof. Dr. I. Van Riet. All the HMCLs and the bone marrow stromal cells (BMSC) were cultured as previously described and regularly tested for mycoplasma contamination [47–50].
Reagents
The APC/C inhibitor proTAME, IL-6 and IGF-1 were obtained from R & D systems (Oxon, UK). Apcin was provided by Dr. R.W. King (Department of Cell Biology, Harvard Medical School). Bortezomib was obtained from Selleckchem (Munich, Germany) and melphalan was obtained from Sigma-Aldrich (St. Louis MO, USA).
Western blot analysis
Cells were harvested, lysed, and protein extracts were blotted as previously described [51]. Primary antibodies were used against cyclin B1 (#4138), Skp2 (#4313), caspase-3 (#9665), caspase-8 (#9746), caspase-9 (#9502), PARP (#9542), γH2AX (#5438), Bim (#2933), Mcl-1 (#5453), Bcl-xL (#2764), pBcl-2 (#2827), Cdc20 (#4823), horseradish peroxidase (HRP)-linked anti-mouse (#7076) and -rabbit (#7074) (Cell Signaling, Leiden, the Netherlands) and Bcl-2 (Sc-492), pBcl-xL (Sc-101644), HRP-linked anti-goat (Sc2020)(Santa Cruz, Heidelberg, Germany). β-actin (#4967) (Cell Signaling) was used as a loading control. The pixel densities of proteins were quantified by ImageJ (Wayne Rasband, NIH, USA).
Cell viability assay
The viability was measured with the CellTiter-Glo Luminiscent Viability assay (Promega, Leiden, The Netherlands) according to manufacturer’s instructions. The relative amount of viable cells was expressed as percentage of untreated cells.
Cell apoptosis assay
Apoptosis was measured with Annexin V-FITC and 7-AAD (BD Biosciences, Franklin Lakes, NJ, USA) followed by flow cytometric analysis (FACS Canto and Diva software, BD Biosciences) according to manufacturer’s instructions.
Microarray data of primary multiple myeloma cells and HMCLs
For the expression of Cdc20 and Cdh1 in HMCLs we used the Affymetrix data of 42 HMCLs from the University hospital of Heidelberg (Germany) and Montpellier (France). These data can be accessed through ArrayExpress database (E-TABM-1088, E-TABM-937 and E-MEXP-2360).
We used 2 independent cohorts of previously untreated MM patients for the association with gene expression-based high-risk [21–23] and survival analysis. The first cohort contains Affymetrix data of 345 MM patients from the University of Arkansas for Medical Science (UAMS, Little Rock, AR) and is termed the TT2-cohort [52]. These MM patients were treated with total therapy 2 [53]. These data can be accessed at the online Gene Expression Omnibus GSE4581. The second cohort consists of Affymetrix data of 206 MM patients from the University hospital of Heidelberg (Germany) and Montpellier (France) and is termed the HM-cohort. MM patients were treated with high dose therapy and autologous stem cell transplantation [21, 28, 54, 55]. These data can be accessed through ArrayExpress database (E-MTAB-372). Affymetrix probe 202870_at and a cut-off of 379.1 was used for Cdc20. For Cdh1 the 209416_at probe was used with a cut-off of 618.8 for the TT-2 cohort and 243 for the HM-cohort.
The plasma cell labeling index
Multiparameter flow cytometry immunophenotyping was performed as indicated [56]. The cell cycle was assessed using 4, 6-diamidino-2-phenylindole (DAPI) staining (Sigma Aldrich) and plasma cells in the S phase were quantified using incubation with bromodeoxyuridine (BrdU) for 1 hour, and labelling with an anti-BrdU antibody (APC BrdU flow kit, BD Pharmingen, Le Pont De Claix, France).
Assessment of metaphase arrest
MM cells were treated with proTAME and cytospins (1 × 105 cells/slide) were stained with May-Grünwald Giemsa (respectively VWR International, Leuven, Belgium and Merck, Darmstadt, Germany). Cells in metaphase were counted based on their morphology using a light microscope. For each cytospin, 3 different fields of at least 100 cells were counted.
Purification of primary human MM cells and peripheral blood mononuclear cells
BM samples were collected from MM patients for diagnosis and research purpose and blood was collected from healthy individuals after written informed consent in accordance with the Declaration of Helsinki. The research is approved by the Ethic Board of UZ Brussel (B.U.N. 143201316382) and the Tumourbank of Lille (CSTMT102). Mononuclear cells were obtained after Ficoll density gradient centrifugation (Nycomed, Lucron Bioproducts, Zurich, Switzerland) and purified as previously described [57]. Primary BMSCs were obtained from healthy subjects, and maintained in DMEM medium (Lonza, Basel, Switzerland) supplemented with 10% fetal calf serum (HyClone, USA), 10% horse serum (Invitrogen, USA), 100 U/mL penicillin/streptomycin (Lonza), and 2 mM L-glutamine (Lonza).
Lentiviral production and stable transduction
The RPMI-8226shBim and RPMI-shScr were produced as previously described [24].
Transfection of siRNA
For knockdown of Cdc20 Dicer-Substrate siRNA duplexes were used at a final concentration of 100 nM (HSC.RNAI.N001255.12.1 (duplex 1) and (HSC.RNAI.N001255.12.2 (duplex 2)). The DS NC1 was used as a control. All Dicer-Substrate siRNA duplexes were purchased at IDT (Leuven, Belgium) The RPMI-8226 were transfected using lipofectamine RNAiMax (Invitrogen, Gent, Belgium) according to the procedure provided by the manufacturer.
Statistical analysis
Gene expression was analyzed by the web tool GenomicScape [58]. Prognostic significance of Cdc20 and Cdh1 genes was calculated using Maxstat R function and Benjamini Hochberg multiple testing correction as previously described [59, 60]. Survival curves were plotted using the Kaplan-Meier method. All these analyses have been done with R.2.10.1 and bioconductor version 2.5. We compared the gene expression levels from patients with high Cdc20 or Cdh1 expression with patients with low Cdc20 or Cdh1 expression and picked up the genes which had significant different expression for Gene set enrichment analysis (GSEA). Gene set enrichment analysis was carried out by computing overlaps with canonical pathways and gene ontology gene sets obtained from the Broad Institute [61]. Graphical and statistical analysis were performed using Graph Prism 5.01 software. For statistical analysis, all experiments were performed at least 3 times as indicated in the Figure legends. The mean and SD of the results are shown in the graphs. The one-sided Mann-Whitney U-test, the student t-test and the one-way ANOVA with Bonferonni correction were used and p values ≤ 0.05 were considered statistically significant.
ACKNOWLEDGMENTS
The authors would like to thank Carine Seynaeve and Sofie Seghers for their technical assistance.
GRANT SUPPORT
This work was supported by European Commission’s Seventh Framework Programme (EU FP7) OVER-MyR, Universitaire Stichting van België and Vlaamse Liga tegen Kanker. S. Lub has a PhD fellowship from FWO-VI. E. De Bruyne and E. Van Valckenborgh are post-doctoral fellows of FWO-VI. E. Menu is an AXA research fund post-doctoral fellow. Work in the lab of R. W. King is supported by NIH grant GM66492. Work in the lab of J. Moreaux is supported by French INCA (Institut National du Cancer) Institute (2012–109/087437), University Hospital of Montpellier (CEP-IRB) and Fondation de France (201400047510).
Authors’ contributions
S.L., E.V.V, K.V. conceived and designed the experiments and wrote the manuscript, S.L., E.V.V., A.M., K.D.V., K.M., J.M., A.K. performed the experiments and analyzed the data, K.F. and X.L. provided BM samples of MM patients, R.K. provided reagent, S.L., A.M., K.M., K.D.V., E.D.B., E.M., K.F., A.K., J.M., D.H., X.L., R.K., K.V., E.V.V. provided crucial suggestions and revised the manuscript, K.V. and E.V.V. supervised the study.
CONFLICTS OF INTEREST
The authors declare no competing financial interest.
REFERENCES
1. Kumar SK, Rajkumar SV, Dispenzieri A, Lacy MQ, Hayman SR, Buadi FK, Zeldenrust SR, Dingli D, Russell SJ, Lust JA, Greipp PR, Kyle RA, Gertz MA. Improved survival in multiple myeloma and the impact of novel therapies. Blood. 2008; 111:2516–21.
2. Kumar SK, Lee JH, Lahuerta JJ, Morgan G, Richardson PG, Crowley J, Haessler J, Feather J, Hoering A, Moreau P, LeLeu X, Hulin C, Klein SK, et al. Risk of progression and survival in multiple myeloma relapsing after therapy with IMiDs and bortezomib: a multicenter international myeloma working group study. Leukemia. 2012; 26:149–57.
3. Hershko A. The ubiquitin system for protein degradation and some of its roles in the control of the cell division cycle. Cell Death Differ. 2005; 12:1191–7.
4. Cao B, Mao X. The ubiquitin-proteasomal system is critical for multiple myeloma: implications in drug discovery. Am J Blood Res. 2011; 1:46–56.
5. Nalepa G, Rolfe M, Harper JW. Drug discovery in the ubiquitin-proteasome system. Nat Rev Drug Discov. 2006; 5:596–613.
6. Penas C, Ramachandran V, Ayad NG. The APC/C Ubiquitin Ligase: From Cell Biology to Tumorigenesis. Front Oncol. 2012; 1:60.
7. Peters J-M. The anaphase promoting complex/cyclosome: a machine designed to destroy. Nat Rev Mol Cell Biol. 2006; 7:644–56.
8. Kato T, Daigo Y, Aragaki M, Ishikawa K, Sato M, Kaji M. Overexpression of CDC20 predicts poor prognosis in primary non-small cell lung cancer patients. J Surg Oncol. 2012; 106:423–30.
9. Chang DZ, Ma Y, Ji B, Liu Y, Hwu P, Abbruzzese JL, Logsdon C, Wang H. Increased CDC20 expression is associated with pancreatic ductal adenocarcinoma differentiation and progression. J Hematol Oncol. 2012; 5:15.
10. Wu W, Hu K, Wang D, Zeng Z, Zhang D, Chen D, Bai L, Xu R. CDC20 overexpression predicts a poor prognosis for patients with colorectal cancer. J Transl Med. 2013; 11:142.
11. Choi J-W, Kim Y, Lee J-H, Kim Y-S. High expression of spindle assembly checkpoint proteins CDC20 and MAD2 is associated with poor prognosis in urothelial bladder cancer. Virchows Arch. 2013; 463:681–7.
12. Moura IMB, Delgado ML, Silva PM a, Lopes C a, do Amaral JB, Monteiro LS, Bousbaa H. High CDC20 expression is associated with poor prognosis in oral squamous cell carcinoma. J oral Pathol Med. 2014; 43:225–31.
13. Ding Z, Wu H, Zhang J, Huang G, Ji D. Expression characteristics of CDC20 in gastric cancer and its correlation with poor prognosis. Int J Clin Exp Pathol. 2014; 7:722–7.
14. Karra H, Repo H, Ahonen I, Löyttyniemi E, Pitkänen R, Lintunen M, Kuopio T, Söderström M, Kronqvist P. Cdc20 and securin overexpression predict short-term breast cancer survival. Br J Cancer. 2014; 110:2905–13.
15. Huang H-C, Shi J, Orth JD, Mitchison TJ. Evidence that mitotic exit is a better cancer therapeutic target than spindle assembly. Cancer Cell. 2009; 16:347–58.
16. Taniguchi K, Momiyama N, Ueda M, Matsuyama R, Mori R, Fujii Y, Ichikawa Y, Endo I, Shimada H. Targeting of CDC20 via Small Interfering RNA Causes Enhancement of the Cytotoxicity of Chemoradiation. Anticancer Res. 2008; 28:1559–63.
17. Kidokoro T, Tanikawa C, Furukawa Y, Katagiri T, Nakamura Y, Matsuda K. CDC20, a potential cancer therapeutic target, is negatively regulated by p53. Oncogene. 2008; 27:1562–71.
18. Zeng X, Sigoillot F, Gaur S, Choi S, Pfaff KL, Oh D, Hathaway N, Dimova N, Cuny GD, King W. Pharmacologic Inhibition of the Anaphase-Promoting Complex Induces A Spindle Checkpoint-Dependent Mitotic Arrest in the Absence of Spindle Damage. Cancer Cell. 2011; 18:382–95.
19. Sackton KL, Dimova N, Zeng X, Tian W, Zhang M, Sackton TB, Meaders J, Pfaff KL, Sigoillot F, Yu H, Luo X, King RW. Synergistic blockade of mitotic exit by two chemical inhibitors of the APC/C. Nature. 2014; 514:646–9.
20. Díaz-Rodríguez E, Álvarez-Fernández S, Chen X, Paiva B, López-Pérez R, García-Hernández JL, San Miguel JF, Pandiella A. Deficient spindle assembly checkpoint in multiple myeloma. PLoS One. 2011; 6:e27583.
21. Hose D, Rème T, Hielscher T, Moreaux J, Messner T, Seckinger A, Benner A, Shaughnessy JD, Barlogie B, Zhou Y, Hillengass J, Bertsch U, Neben K, et al. Proliferation is a central independent prognostic factor and target for personalized and risk-adapted treatment in multiple myeloma. Haematologica. 2011; 96:87–95.
22. Rème T, Hose D, Theillet C, Klein B. Gene expression Modeling risk stratification in human cancer. Bioinformatics. 2013; 29:1149–57.
23. Shaughnessy JD, Zhan F, Burington BE, Huang Y, Colla S, Hanamura I, Stewart JP, Kordsmeier B, Randolph C, Williams DR, Xiao Y, Xu H, Epstein J, et al. A validated gene expression model of high-risk multiple myeloma is defined by deregulated expression of genes mapping to chromosome 1. Blood. 2007; 109:2276–85.
24. De Bruyne E, Bos TJ, Schuit F, Van Valckenborgh E, Menu E, Thorrez L, Atadja P, Jernberg-wiklund H, Vanderkerken K. IGF-1 suppresses Bim expression in multiple myeloma via epigenetic and posttranslational mechanisms. Blood. 2010; 115:2430–41.
25. Barlogie B, Mitchell A, Rhee F Van, Epstein J, Morgan GJ, Crowley J. Perspectives Curing myeloma at last: defining criteria and providing the evidence. Blood. 2014; 124:3043–52.
26. Decaux O, Lodé L, Magrangeas F, Charbonnel C, Gouraud W, Jézéquel P, Attal M, Harousseau J-L, Moreau P, Bataille R, Campion L, Avet-Loiseau H, Minvielle S. Prediction of survival in multiple myeloma based on gene expression profiles reveals cell cycle and chromosomal instability signatures in high-risk patients and hyperdiploid signatures in low-risk patients: a study of the Intergroupe Francophone du Myélom. J Clin Oncol. 2008; 26:4798–805.
27. Stewart AK, Bergsagel PL, Greipp PR, Dispenzieri A, Gertz MA, Hayman SR, Kumar S, Lacy MQ, Lust JA, Russell SJ, Witzig TE, Zeldenrust SR, Dingli D, et al. A practical guide to defining high-risk myeloma for clinical trials, patient counseling and choice of therapy. Leukemia. 2007; 21:529–34.
28. Hose D, Rème T, Meissner T, Moreaux J, Seckinger A, Lewis J, Benes V, Benner A, Hundemer M, Hielscher T, Shaughnessy JD, Barlogie B, Neben K, et al. Inhibition of aurora kinases for tailored risk-adapted treatment of multiple myeloma. Blood. 2009; 113:4331–40.
29. Yang Y, Gu C, Luo C, Li F, Wang M. BUB1B promotes multiple myeloma cell proliferation through CDC20/CCNB axis. Med Oncol. 2015; 32:81.
30. Fujita T, Weijun L, Hiroyoshi D, Yong W. Regulation of Skp2-p27 Axis by the Cdh1/Anaphase-Promoting Complex Pathway in Colorectal Tumorigenesis. Am J Pathol. 2008; 173:217–28.
31. Fujita T, Liu W, Doihara H, Wan Y. An in vivo study of Cdh1/APC in breast cancer formation. Int J cancer. 2009; 125:826–36.
32. García-Higuera I, Manchado E, Dubus P, Cañamero M, Méndez J, Moreno S, Malumbres M. Genomic stability and tumour suppression by the APC/C cofactor Cdh1. Nat Cell Biol. 2008; 10:802–11.
33. Colin DJ, Hain KO, Allan LA, Clarke PR. Cellular responses to a prolonged delay in mitosis are determined by a DNA damage response controlled by Bcl-2 family proteins. Open Biol. 2015; 5:140156.
34. Dalton WB, Nandan MO, Moore RT, Yang VW. Human cancer cells commonly acquire DNA damage during mitotic arrest. Cancer Res. 2007; 67:11487–92.
35. Tu W-Z, Li B, Huang B, Wang Y, Liu X-D, Guan H, Zhang S-M, Tang Y, Rang W-Q, Zhou P-K. γH2AX foci formation in the absence of DNA damage: mitotic H2AX phosphorylation is mediated by the DNA-PKcs/CHK2 pathway. FEBS Lett. 2013; 587:3437–43.
36. Fan M, Goodwin M, Vu T, Brantley-Finley C, Gaarde W a, Chambers TC. Vinblastine-induced phosphorylation of Bcl-2 and Bcl-XL is mediated by JNK and occurs in parallel with inactivation of the Raf-1/MEK/ERK cascade. J Biol Chem. 2000; 275:29980–5.
37. Du L, Lyle CS, Chambers TC. Characterization of vinblastine-induced Bcl-xL and Bcl-2 phosphorylation: evidence for a novel protein kinase and a coordinated phosphorylation/dephosphorylation cycle associated with apoptosis induction. Oncogene. 2005; 24:107–17.
38. Eichhorn JM, Sakurikar N, Alford SE, Chu R, Chambers TC. Critical role of anti-apoptotic Bcl-2 protein phosphorylation in mitotic death. Cell Death Dis. 2013; 4:e834.
39. Choi HJ, Zhu BT. Role of cyclin B1/Cdc2 in mediating Bcl-XL phosphorylation and apoptotic cell death following nocodazole-induced mitotic arrest. Mol Carcinog. 2014; 53:125–37.
40. Harley ME, Allan LA, Sanderson HS, Clarke PR. Phosphorylation of Mcl-1 by CDK1-cyclin B1 initiates its Cdc20-dependent destruction during mitotic arrest. EMBO J. 2010; 29:2407–20.
41. Wan L, Tan M, Yang J, Inuzuka H, Dai X, Wu T, Liu J, Shaik S, Chen G, Deng J, Malumbres M, Letai A, Kirschner MW, et al. Article APC Cdc20 Suppresses Apoptosis through Targeting Bim for Ubiquitination and Destruction. Dev Cell. 2014; 29:377–91.
42. Wójcik C, Schroeter D, Stoehr M, Wilk S, Paweletz N. An inhibitor of the chymotrypsin-like activity of the multicatalytic proteinase complex (20S proteasome) induces arrest in G2-phase and metaphase in HeLa cells. Eur J Cell Biol. 1996; 70:72–8.
43. Barlogie B, Smith L, Alexanian R. Effective Treatment of Advanced Multiple Myeloma Refractory to Alkylating Agents. N Engl J Med. 1984; 310:1353–6.
44. Barlogie B, Shaughnessy J, Tricot G, Jacobson J, Zangari M, Anaissie E, Walker R, Crowley J. Treatment of multiple myeloma. Blood. 2004; 103:20–33.
45. Jackson DV, Case LD, Pope EK, White DR, Spurr CL, Richards F, Stuart JJ, Muss HB, Cooper MR, Black WR. Single agent vincristine by infusion in refractory multiple myeloma. J Clin Oncol. 1985; 3:1508–12.
46. Chan K-S, Koh C-G, Li H-Y. Mitosis-targeted anti-cancer therapies: where they stand. Cell Death Dis. 2012; 3:e411.
47. Maes K, De Smedt E, Lemaire M, De Raeve H, Menu E, Van Valckenborgh E, Mcclue S, Vanderkerken K, De Bruyne E. The role of DNA damage and repair in decitabine-mediated apoptosis in multiple myeloma. Oncotarget. 2014; 5:3115–29. doi:10.18632/oncotarget.1821.
48. Wang J, Hendrix A, Hernot S, Lemaire M, De Bruyne E, Van Valckenborgh E, Lahoutte T, De Wever O, Vanderkerken K, Menu E. Bone marrow stromal cell – derived exosomes as communicators in drug resistance in multiple myeloma cells. Blood. 2014; 124:555–67.
49. De Veirman K, Van Ginderachter JA, Lub S, De Beule N, Thielemans K, Bautmans I, Oyajobi BO, De Bruyne E, Menu E, Lemaire M, Van Riet I, Vanderkerken K. Multiple myeloma induces Mcl-1 expression and survival of myeloid-derived suppressor cells. Oncotarget. 2015; 6:10532–47. doi:10.18632/oncotarget.3300.
50. Hu J, Dang N, Menu E, De Bryune E, Xu D, Van Camp B, Van Valckenborgh E, Vanderkerken K. Activation of ATF4 mediates unwanted Mcl-1 accumulation by proteasome inhibition. Blood. 2012; 119:826–38.
51. Menu E, Jernberg-Wiklund H, Stromberg T, De Raeve H, Girnita L, Larsson O, Axelson M, Asosingh K, Nilsson K, Van Camp B, Vanderkerken K. Inhibiting the IGF-1 receptor tyrosine kinase with the cyclolignan PPP: an in vitro and in vivo study in the 5T33MM mouse model. Blood. 2006; 107:655–60.
52. Zhan F, Huang Y, Colla S, Stewart JP, Hanamura I, Gupta S, Epstein J, Yaccoby S, Sawyer J, Burington B, Anaissie E, Hollmig K, Pineda-Roman M, et al. The molecular classification of multiple myeloma. Blood. 2006; 108:2020–8.
53. Barlogie B, Tricot G, Rasmussen E, Anaissie E, van Rhee F, Zangari M, Fassas A, Hollmig K, Pineda-Roman M, Shaughnessy J, Epstein J, Crowley J. Total therapy 2 without thalidomide in comparison with total therapy 1: role of intensified induction and posttransplantation consolidation therapies. Blood. 2006; 107:2633–8.
54. Seckinger A, Mei T, Ro A, Jauch A, Schnettler R, Ewerbeck V, Goldschmidt H, Klein B, Hose D. Clinical and prognostic role of annexin A2 in multiple myeloma. Blood. 2012; 120:1087–95.
55. Seckinger A, Meissner T, Goldschmidt H, Fuhler GM, Benner A. Bone morphogenic protein 6: a member of a novel class of prognostic factors expressed by normal and malignant plasma cells inhibiting proliferation and angiogenesis. Oncogene. 2009; 28:3866–79.
56. Caraux A, Vincent L, Bouhya S, Quittet P, Requirand G, Veyrune J, Olivier G, Rossi J, Klein B. Residual malignant and normal plasma cells shortly after high dose melphalan and stem cell transplantation. Highlight of a putative therapeutic window in Multiple Myeloma? Oncotarget. 2012; 3:1335–47. doi: 10.18632/oncotarget.650.
57. Bieghs L, Lub S, Fostier K, Maes K, Van Valckenborgh E, Menu E, Johnsen HE, Overgaard MT, Larsson O, Axelson M, Nyegaard M, Schots R, Jernberg Wiklund H. The IGF-1 receptor inhibitor picropodophyllin potentiates the anti-myeloma activity of a BH3-mimetic. Oncotarget. 2014; 5:11193–208. doi:10.18632/oncotarget.1933.
58. Kassambara A, Rème T, Jourdan M, Fest T, Hose D, Tarte K, Klein B. GenomicScape: an easy-to-use web tool for gene expression data analysis. Application to investigate the molecular events in the differentiation of B cells into plasma cells. PLoS Comput Biol. 2015; 11:e1004077.
59. Kassambara A, Hose D, Moreaux J, Walker BA, Protopopov A, Reme T, Pellestor F, Pantesco V, Jauch A, Morgan G, Goldschmidt H, Klein B. Genes with a spike expression are clustered in chromosome (sub)bands and spike (sub)bands have a powerful prognostic value in patients with multiple myeloma. Haematologica. 2012; 97:622–30.
60. Kassambara A, Hose D, Moreaux J, Rème T, Torrent J, Rossi JF, Goldschmidt H, Klein B. Identification of pluripotent and adult stem cell genes unrelated to cell cycle and associated with poor prognosis in multiple myeloma. PLoS One. 2012; 7:e42161.
61. Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci USA. 2005; 102:15545–50.