
Introduction
Male breast cancer is a rare disease that is 100-fold less common than female breast cancer and accounts for less than 1% of all cancers in men [1, 2]. In the United States, approximately 210,000 women and 2000 men will be diagnosed annually with the disease [3, 4]. Globally, male breast cancer has an annual average incidence of <1 case per 100,000 men [5]. Recent epidemiological studies show a slow but steady increase in the annual occurrence of this rare cancer [2, 6, 7].
Due to the low incidence of male breast cancer, most clinical and laboratory research has focused on female breast cancers [8]. However, studies from female breast cancers may not be entirely applicable to men since the etiology of male breast cancer remains unclear. Clinically, breast cancers are traditionally classified according to their receptor status, namely estrogen receptor (ER), progesterone receptor (PR), and HER2 (Human Epidermal Growth Factor Receptor 2). Similar to female breast cancer, the majority of male breast cancers are ER positive and/or PR positive [9]. A notable difference in male breast cancers compared to their female counterparts is the relatively lower percentage of ER, PR, HER2 negative (triple negative) and HER2 positive breast cancers [10-12]. Recent studies also highlight global genomic, transcriptomic, and proteomic differences between female and male breast cancers, with global gene expression profiling showing differences in at least 1,000 genes between female and male breast cancers [13]. In addition, using a computer framework dubbed COpy Number and EXpression In Cancer (CONEXIC), cancer driver genes were shown to be distinct between male and female breast cancers [14].
Although it is unquestionable that exposure to estrogens is a major risk factor for breast cancer development in women, the fact that men can and do develop breast cancer, albeit at much lower incidence, speaks to the fact that other factors likely contribute to breast carcinogenesis. In this study, we hypothesized that the human Y chromosome may be involved in the etiology of male breast cancers. The Y chromosome is one of the smallest human chromosomes, and consists of a minute pseudoautosomal region (PAR) that is homologous to the X chromosome, and a larger male-specific Y region (MSY) [15]. Approximately 450 transcribed genes have been annotated on the Y chromosome, of which 90 are protein-coding [16]. Early studies of male breast cancer genetics have provided some evidence that the Y chromosome may harbor a male specific tumor suppressor since partial or whole Y loss has been reported [17-19]. However, these reports involved small numbers of patients with low resolution genetic techniques. In addition, none of these studies examined Y chromosome in situ loss within tumor tissue, hindering the evaluation of clonal Y loss. To our knowledge, there are no reports evaluating in situ Y chromosome status in male breast cancers.
In this study, we addressed whether loss of the Y chromosome contributes to male breast carcinogenesis. Using fluorescent in situ hybridization (FISH) and droplet digital PCR (ddPCR), our results show clonal Y chromosome loss at a frequency of ~16% (5/31) in two independent cohorts of male breast cancer patients. Furthermore, we observed that Y chromosome loss can occur in ductal carcinoma in situ (DCIS) lesions. In order to identify a possible tumor suppressor within the Y chromosome, we used sequence-tagged-site PCR (STS-PCR) in male breast cancer specimens without Y chromosome loss, and show somatic deletion of the TMSB4Y gene in a male breast cancer patient, confirming prior reports showing loss of this region. We then created tetracycline-inducible clones of TMSB4Y in the human non-tumorigenic female breast epithelial cell line MCF-10A. Our results show that induced expression of TMSB4Y led to aberrant morphological changes, persistent reduction in cell proliferation, and a corresponding reduction in the fraction of metaphase cells. Using proximity ligation assays (PLA) and immunoprecipitation with western blotting, we show that TMSB4Y interacts directly with β-actin, a main component of the actin cytoskeleton and a modulator of cell cycle progression. Taken together, our results show that in situ clonal loss of the human Y chromosome may play an important role in male breast cancer tumorigenesis, and suggest that TMSB4Y has tumor suppressive properties.
Results
Clonal loss of Y chromosome in male breast cancer is a frequent event
To address the hypothesis that Y chromosome loss may have a role in breast carcinogenesis, we first evaluated its loss in male breast cancers. We obtained FFPE tissue blocks of male breast cancers from 15 patients (cohort 1, Table 1) and used these to synthesize a tissue microarray (TMA). This TMA was then analyzed for Y chromosomal loss by FISH, along with an X chromosome FISH probe as a control (Figure 1). In order to survey the entire Y chromosome, we used various combinations of FISH probes specific for the short arm, centromere, and long arm of the Y chromosome (Figure S1). By these criteria, we observed clonal loss of the whole Y chromosome in 2 out of 15 (~13.33%) male breast cancer patients.
Table 1: Clinical characteristics of patients. The five Y loss cases are highlighted. nd, not done; na, not available.
Cohort 1 Patient |
ER |
PR |
HER2 |
Stage |
01 |
+ |
+ |
- |
IA |
02 |
+ |
+ |
nd |
IA |
03 |
+ |
+ |
nd |
IA |
04 |
+ |
+ |
nd |
IA |
05 |
+ |
+ |
- |
IA |
06 |
+ |
+ |
nd |
IA |
07 |
+ |
Focally + |
Weakly + |
IIIC |
08 |
+ |
+ |
nd |
IIA |
09 |
+ |
+ |
nd |
IIA |
10 |
+ |
+ |
Equivocal |
IIB |
11 |
+ |
- |
- |
IIIA |
12 |
nd |
nd |
nd |
IA |
13 |
+ |
- |
- |
IIA |
14 |
+ |
- |
- |
IB |
15 |
na |
na |
na |
na |
Cohort 2 Patient |
||||
01 |
+ |
+ |
na |
IIIB |
02 |
+ |
nd |
na |
1 |
03 |
+ |
+ |
na |
na |
04 |
+ |
+ |
na |
IIIB |
05 |
nd |
nd |
na |
DCIS |
06 |
nd |
nd |
na |
DCIS |
07 |
+ |
+ |
+ |
IIIB |
08 |
+ |
+ |
na |
IIA |
09 |
+ |
+ |
na |
IIA |
10 |
+ |
+ |
na |
IA |
11 |
+ |
+ |
na |
IIB |
12 |
na |
na |
na |
IV |
13 |
+ |
+ |
na |
IIA |
14 |
+ |
+ |
na |
IA |
15 |
+ |
+ |
na |
IIA |
16 |
+ |
- |
na |
IA |
17 |
+ |
- |
nd |
IV |
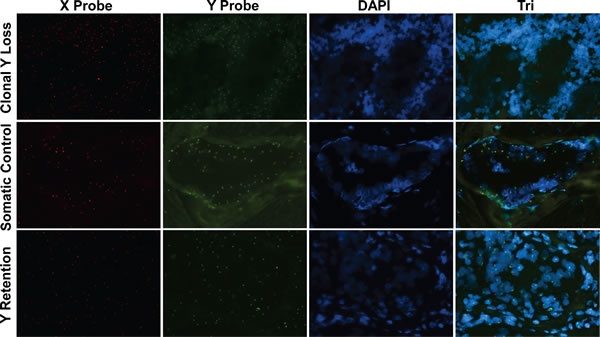
Figure 1: Clonal loss of the Y chromosome in male breast cancer. FISH was performed to evaluate Y and X chromosomes on a male breast cancer tissue sample from a patient with clonal Y loss (top panels), normal breast tissue from the same patient as a somatic control (middle panels) and a male breast cancer sample without Y loss (bottom panels). Red, X chromosome FISH Probe; Green, Y chromosome FISH Probe; Blue, DAPI nuclear labeling. Original magnification: 20X.
Next, we obtained 19 additional male breast cancer FFPE samples, however, only 17 had adequate tissue for analysis (cohort 2, Table 1). Because blocks were unavailable for these samples, we could not create a separate TMA. Therefore, we analyzed each specimen by FISH (Table 2), and demonstrated clonal somatic Y loss in 3 additional patients. Due to DNA degradation and limited material, 3 of 17 samples were inadequate for FISH yielding an overall Y loss frequency of 17% (5 of 29 patients combining both cohorts). In order to analyze the remaining 3 samples that were unevaluable by FISH, we performed ddPCR on FFPE DNA using Taqman probes and primers specific to the X and Y chromosomes to assay for Y chromosome loss. As we have previously reported, fragmented DNA is optimal for ddPCR and minute amounts of DNA can be used for accurate quantification of alleles using FFPE derived material [20]. As seen in Table 2, the ratio of Y versus X positive signals (ratio Y/X) as measured by ddPCR was first validated using a commercial source of female and male control gDNA, with ratios of 0 and 0.966, respectively. We also included as a control a patient from cohort 1 with Y loss that showed a Y/X ratio of 0.193, supporting that ddPCR could be used to assess Y loss. As seen in Table 2, due to variability in tumor cellularity and non-tumor normal tissue contamination, tumors with Y loss generally had a Y/X ratio of less than 0.200, though the exception was patient 5 who had a Y/X ratio of 0.410, likely due to a higher than observed amount of normal tissue contamination. Notably, we observed two cases, patients 3 and 4, with Y/X ratios of 0.354 and 0.383 suggestive of Y loss. However, for these two patients, FISH analysis showed Y retention with somatic duplication of the X chromosome, yielding an artificially lower Y/X ratio by ddPCR. These results highlight some of the potential pitfalls and caveats of allelic enumeration and ratios, and that in situ analysis is often needed for definitive conclusions. Although one of the three patients with unevaluable samples for FISH yielded an equivocal result (patient 6), two patients clearly had Y/X ratios demonstrating retention of Y, though X loss could not be excluded in these patients (patients 14 and 16). Thus, combining results from the two independent cohorts of male breast cancer patients using both FISH and ddPCR, we observed a Y loss frequency of ~17% (5 of 29 male breast cancer patients), though if patients 14 and 16 are included via the ddPCR results, the frequency is marginally decreased to ~16% (5 of 31 patients).
For one of the patient samples with Y loss in cohort 1, a corresponding DCIS sample was available (Figure S2A), presenting an opportunity to ascertain whether Y loss occurred prior to invasive disease. As shown in Figure S2B, the DCIS from this patient was confirmed as non-invasive using an anti-smooth muscle actin stain. We further showed via FISH that this lesion had somatic Y loss, with the adjacent stromal tissue retaining the Y chromosome as an internal control (Figure S2C).
Table 2: Y chromosome loss in male breast cancer patients.
Patient Number |
Tumor Cellularity (%) |
Positive ddPCR Droplets |
Ratio Y/X |
ddPCR Interpretation |
FISH |
|
Y Chr Taqman |
X Chr Taqman |
|||||
01 |
70 |
7 |
524 |
0.013 |
Y Loss |
Y Loss |
02 |
60 |
6 |
114 |
0.053 |
Y Loss |
Y Loss |
03 |
80 |
357 |
1009 |
0.354 |
Equivocal |
Y Retention * |
04 |
80 |
217 |
566 |
0.383 |
Equivocal |
Y Retention * |
05 |
60 |
84 |
205 |
0.410 |
Equivocal |
Y Loss |
06 |
60 |
54 |
116 |
0.466 |
Equivocal |
Unevaluable |
07 |
70 |
47 |
73 |
0.644 |
Y Retention |
Y Retention |
08 |
70 |
17 |
24 |
0.708 |
Y Retention |
Y Retention |
09 |
70 |
158 |
196 |
0.806 |
Y Retention |
Y Retention |
10 |
80 |
57 |
52 |
1.096 |
Y Retention |
Y Retention |
11 |
70 |
134 |
117 |
1.145 |
Y Retention |
Y Retention |
12 |
70 |
334 |
214 |
1.561 |
Y Retention |
Y Retention |
13 |
70 |
482 |
293 |
1.645 |
Y Retention |
Y Retention |
14 |
60 |
156 |
91 |
1.714 |
Y Retention |
Unevaluable |
15 |
80 |
2067 |
1188 |
1.740 |
Y Retention |
Y Retention |
16 |
50 |
53 |
23 |
2.304 |
Y Retention |
Unevaluable |
17 |
70 |
252 |
86 |
2.930 |
Y Retention |
Y Retention |
Cohort 1 patient with Y loss |
80 |
101 |
523 |
0.193 |
Y Loss |
Y Loss |
Male gDNA |
NA |
2628 |
2720 |
0.966 |
Y Retention |
Male Control |
Female gDNA |
NA |
0 |
3451 |
0.000 |
No Y |
Female Control |
Tumor gDNA was extracted from FFPE tissue slides from male breast cancer patients and analyzed using ddPCR Taqman probes specific for X and Y chromosomes. Fourteen of 17 ddPCR-analyzable samples were validated by FISH; three patients had FFPE tissue that could not be validated with FISH.
* Denotes male breast cancer patients who have duplication of X chromosome.
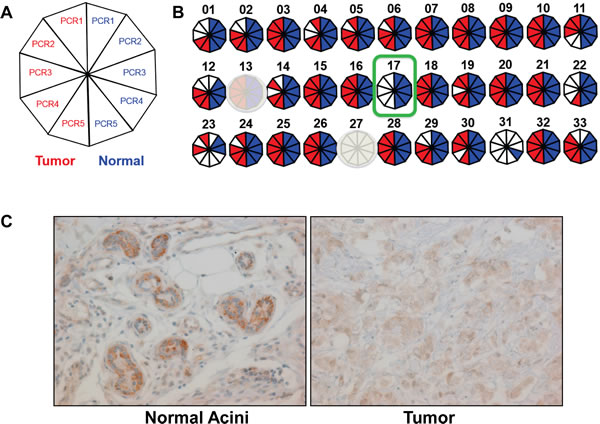
Figure 2: TMSB4Y is lost in male breast cancer and expressed in normal male breast tissue. A. Regional Y loss was assessed by STS-PCR that was repeated 5 times using 5 serial slides from matched tumor and normal FFPE gDNA from three patients as described in the text. A summary of each STS-PCR is depicted as a decagon, where a positive reaction is red for tumor, and the corresponding normal somatic control is blue. B. Results summary for a patient who retained the Y chromosome in his breast cancer by FISH, but demonstrated consistent loss of the STS-PCR marker S17, a region containing the TMSB4Y gene, in tumor but not in adjacent normal tissue (outlined in green). Primer sets S13 and S27 were uninformative (shaded out) due to non-specific amplicons and lack of amplicons (amplicon size too large as described in text), respectively. C. Immunohistochemistry labeling for TMSB4Y was performed on normal breast tissue (left) and corresponding breast cancer (right) from a male breast cancer patient with known Y chromosome loss by FISH. Original magnification: 20X.
The Y chromosome gene TMSB4Y is lost in male breast cancer
Although total Y loss was present in ~16% of our patient samples, we reasoned that if there was a tumor suppressor on the Y chromosome, deletion of this candidate tumor suppressor could occur with retention of the remainder of the Y chromosome. Microdeletions within the Y chromosome have been reported in males with varying degrees of frequency [21-23]. To further investigate this, we extracted gDNA from five male breast cancer patients who retained the Y chromosome, and amplified 33 Sequence Tagged Sites (STSs) within the MSY using standard PCR (Table S1, Figure S3). Primers were designed using published data for these genomic loci [24]. Paired tumor vs normal gDNA (identified by the study pathologist) were extracted from FFPE slides, but 2 of 5 samples had inadequate gDNA for further analysis. The STS primer pair S27 was not successfully amplified in any tumor or normal samples, likely due to fact that this amplicon size is ~1kb, and could not be amplified from fragmented FFPE gDNA.
From the remaining 32 STS primer pairs, S17 did not amplify for the tumor gDNA for one of the three analyzable male breast cancer patients, but was present in the germline paired normal gDNA (Figure 2B). This primer pair amplifies a region including exon 1 and intron 1 of TMSB4Y, a gene encoding the actin sequestering protein, Thymosin Beta 4, Y-linked. Prior literature also demonstrated that this region was lost in male breast patients with a frequency of 40% [25]. Furthermore, a query of the cBioPortal database using the parameter “TMSB4Y: HOMDEL” yielded a data set that shows TMSB4Y deletion in 16% (10/59) of metastatic prostate adenocarcinomas (Figure S4) [26]. To investigate whether TMSB4Y expression is consistent with a tumor suppressor, we sought to determine TMSB4Y expression in normal male breast tissue. We performed immunohistochemistry (IHC) to assess TMSB4Y protein expression in normal male breast tissue, using matched breast tumor tissue with Y loss as a negative control. Because TMSB4Y has a homolog located on the X chromosome, TMSB4X, we initially tested the specificity of the antibody. We transiently transfected TMSB4X and TMXB4Y cDNAs separately into HEK293 cells and harvested lysates for western blot. Unfortunately, none of our TMSB4X antibodies could detect TMSB4X protein, and therefore we obtained a FLAG-tagged TMSB4X cDNA and repeated the experiment. As shown in Figure S5, the TMSB4Y antibody specifically detected the TMSB4Y protein by western blot analysis but did not detect TMSB4X.
We then performed IHC on our tissue sample. As shown in Figure 2C, normal breast epithelial cells labeled with strong intensity compared to the adjacent tumor tissue, which demonstrated no labeling above a low level of nonspecific background. These results show that TMSB4Y is expressed in normal male breast tissue and its expression is lost in a patient with known Y chromosome loss. Unfortunately, the TMA and tissue slides of MBC patients (both cohort 1 and 2) were exhausted and we could not perform IHC to evaluate TMSB4Y expression in our other MBC patients.
TMSB4Y expression alters breast epithelial cell morphology
To investigate the effects of TMSB4Y in breast cells, we generated stable Dox-inducible clones in the MCF-10A cell line [27]. MCF-10A is a genetically stable non-tumorigenic breast epithelial cell line derived from reduction mammoplasty tissue, and a derivative cell line, TetHyg2.5, was created by stable transfection of the Tet repressor protein. MCF-10A cells are ideal for these studies, since they are non-cancerous, and therefore have less concern regarding pre-existing genetic alterations that could mask any given phenotype. In addition, they are derived from a female, so concerns regarding TMSB4Y expression are not applicable. TMSB4Y was cloned into the bidirectional pBI-EGFP vector, which contains the Tet response element, and expresses green fluorescent protein (GFP) simultaneously with TMSB4Y. We generated TmY1 and TmY2, two Dox-inducible cell lines that express both GFP and TMSB4Y when induced with Dox. A control cell line was also created called EV, which contains the pBI-EGFP empty vector and expresses only GFP when induced with Dox. Expression of TMSB4Y after Dox-induction was verified via western blotting (Figure 3A) and immunohistochemistry (IHC) on tissue cellblocks made from these cell lines (Figure 3B). We noted that TmY1 showed stronger induction of TMSB4Y compared to TmY2.
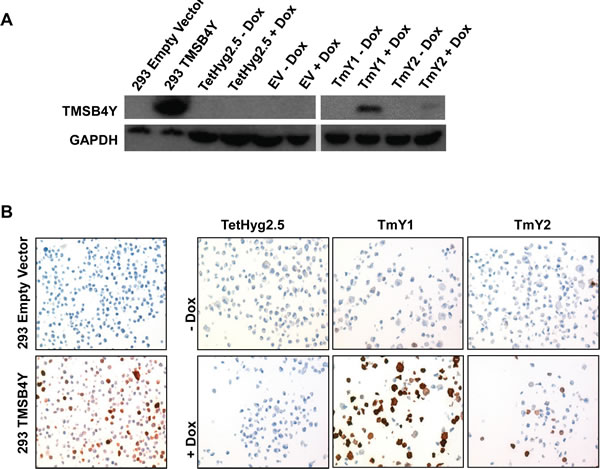
Figure 3: TMSB4Y expression in Dox-inducible clones. The TMSB4Y cDNA was cloned into the pBI-EGFP vector and stably transfected into the Dox-inducible MCF-10A cell line, TetHyg2.5, to the create TMSB4Y Dox-inducible clones TmY1 and TmY2. HEK293 cells were transiently transfected with empty vector (293 Empty Vector) or a TMSB4Y cDNA (293 TMSB4Y) to serve as negative and positive antibody controls, respectively. Parental TetHyg2.5, a stably transfected empty vector (EV) control cell line and the two inducible clones, TmY1 and TmY2, were placed in un-induced (-Dox) and induced (+Dox) media conditions for 48 hours prior to harvesting for lysates and fixation for FFPE blocks. TMSB4Y protein expression was verified via A.) western blotting using an anti-TMSB4Y antibody and GAPDH antibody as loading control, and B.) immunohistochemistry labeling of FFPE cell pellets using an anti-TMSB4Y antibody and counterstaining. Original magnification: 20X.
After Dox-induction of TMSB4Y, we observed morphologic changes in TmY1 and TmY2 cells (Figure 4), with cells displaying a flattened appearance with abnormal borders. Although EV control cells did not show morphology changes, we used as an additional control transient overexpression of GFP in MCF-10A cells and found this was non-toxic (Figure S6). Additionally, we performed immunofluorescence with antibodies against F-actin to better visualize the cellular architecture. As seen in Figure S7, F-actin labeling after Dox-induction confirmed aberrant cell morphology seen in TMSB4Y expressing cells. Because male breast cancers are most often ER positive, and MCF-10A are ER negative cells, we wanted to evaluate expression of TMSB4Y in an ER positive breast cancer cell line. As such, we then transiently co-expressed the TMSB4Y cDNA with GFP in the human breast epithelial adenocarcinoma cell line MCF-7. As seen in Figure S8, similar aberrant morphological changes were observed after 48hrs in GFP positive cells.
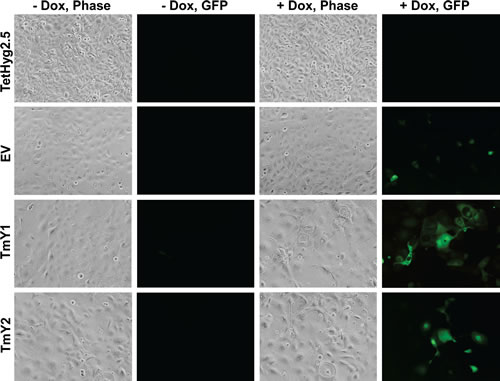
Figure 4: TMSB4Y expression leads to changes in cell morphology. Parental TetHyg2.5, a stably transfected empty vector (EV) control cell line and the two TMSB4Y Dox-inducible clones, TmY1 and TmY2, were placed in un-induced (-Dox) and induced (+Dox) media conditions for 48 hours and then imaged using phase contrast (Phase) and fluorescence (GFP) microscopy. Note the enlarged and aberrant cellular borders present in Dox-induced TMSB4Y clones, TmY1 and TmY2. Original magnification: 20X.
TMSB4Y expression reduces cell proliferation
Tumor suppressors often reduce the proliferation of cells when expressed. In order to assess this phenotype, we compared proliferation rates of our cell lines with and without TMSB4Y expression. Dox-induction of TMSB4Y significantly reduced the growth rates of TmY1 and TmY2 by approximately 30% when compared to TetHyg2.5 and EV (P < 0.05) in a 6-day growth assay (Figure 5A). This reduction in proliferation was persistent, as observed in a detailed growth assay where cell counts were taken on days 0, 3, 7, 13, and 18 (Figure 5B).
To further understand the mechanism of reduced proliferation, we then performed fluorescence activated cell sorting (FACS) analysis on our cells. FACS was used to sort out the GFP-positive TmY1 cells 48hrs after Dox-induction, and then these GFP-positive cells underwent cell cycle analysis based on their DNA content. TmY2 cells could not be used for these experiments due to the low level of GFP inducible expression. TMSB4Y expression in these GFP-positive cells sharply reduced the metaphase fraction (G2/M) in TmY1 when compared to EV (Figure 5C). This metaphase reduction correlates with the persistent reduction in cell proliferation observed, suggesting that TMSB4Y expression modulates cell cycle progression.
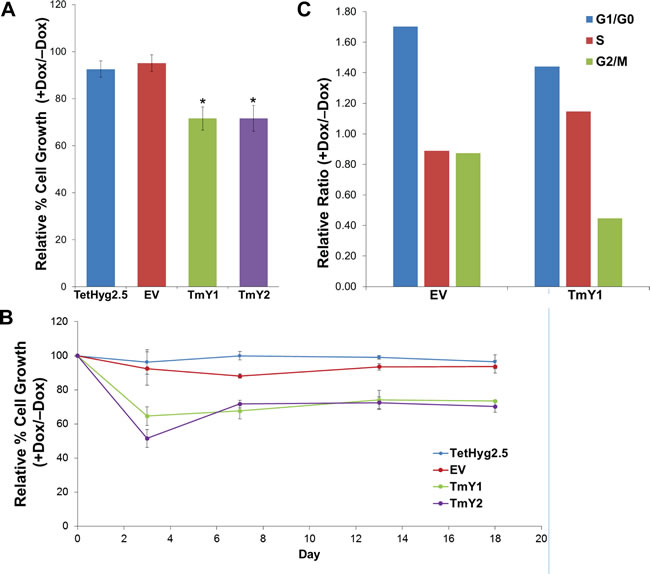
Figure 5: TMSB4Y expression reduces cell proliferation. Parental TetHyg2.5, a stably transfected empty vector (EV) control cell line and the two TMSB4Y Dox-inducible clones, TmY1 and TmY2, were placed in un-induced (-Dox) and induced (+Dox) media conditions and used for cell proliferation assays and FACS analysis. A. TmY1 and TmY2 exhibited ~30% of reduced proliferation upon Dox induction (*p < 0.05) relative to un-induced cells after 6 days of growth. B. Relative reduced cell proliferation after Dox-induction in both TmY1 and TmY2 was observed with prolonged culture as measured by cell counts at Days 3, 7, 13, and 18. C. TMSB4Y expression reduced the relative metaphase cell fraction in TMSB4Y expressing cells (TmY1) after Dox induction, but not in empty vector (EV) control cells after 48 hours and GFP-sorting for cell cycle analysis.
TMSB4Y interacts with β-actin
Thymosin beta proteins have been identified as actin monomer sequestering proteins in mammalian cells [28, 29]. However, to our knowledge, there are no studies demonstrating that TMSB4Y directly interacts with actin. We reasoned that TMSB4Y might modulate cell cycle progression through a possible interaction with actin, a main component of the cytoskeleton [30]. In support of this, the intracellular actin cytoskeletal state has been shown to regulate cell cycle progression via retrograde signaling [31, 32]. To demonstrate a TMSB4Y interaction with actin, we performed a proximity ligation assay (PLA) using antibodies specific to TMSB4Y and β-actin. PLA employs two specific antibodies of interest, each containing a linked nucleic acid sequence such that proteins within 0 to 40nm of one another can then be ligated via their nucleic acid sequences, with subsequent amplification and fluorescence detection. Using PLA, we observed that TMSB4Y and β-actin are in close proximity in situ only after Dox-induction, supporting a direct protein-protein interaction (Figure 6A). We further confirmed this direct interaction between TMSB4Y and β-actin by immunoprecipitating β-actin with an anti-TMSB4Y antibody in Dox-induced TmY1 lysates, however, this interaction was absent in Dox-induced EV lysates (Figure 6B). Together, these results suggest that TMSB4Y directly interacts with β-actin, and that this interaction may lead to the aberrant morphology and decreased proliferation seen upon TMSB4Y expression.
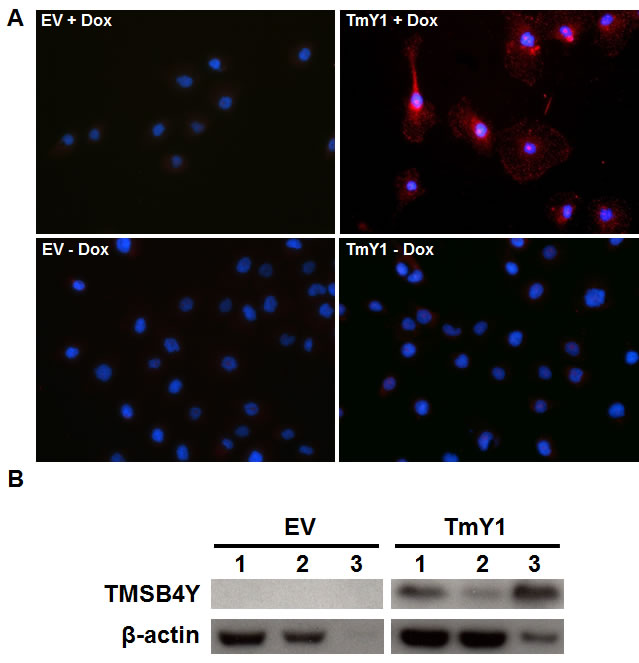
Figure 6: TMSB4Y interacts with β-actin. The empty vector control (EV) and TmY1 were grown in un-induced (-Dox) and induced (+Dox) conditions for 48 hours prior to assaying for TMSB4Y and β-actin protein interactions via A. a proximity ligation assay (PLA) using antibodies against TMSB4Y and β-actin as described in the text. The red signals (top right panel) demonstrate a TMSB4Y-β-actin interaction based on their in situ proximity of 0 to 40nm. Nuclei of cells are stained with DAPI (blue). Original magnification: 20X. B. Cell lysates were also used for immunoprecipitation (IP) experiments using a TMSB4Y antibody on Dox-induced lysates from EV and TmY1. The TMSB4Y antibody was used for IP, and then total lysate (1), supernatant after IP (2) or the IP fraction (3) was stained via western blot using anti-TMSB4Y and anti-β-actin antibodies.
Discussion
Cancer is a disease caused by a successive series of genetic alterations that lead to selective proliferative advantages in a single cell, which then expands into a tumorigenic clone [33]. Theoretically, all cells that originate from this clone will retain specific genetic “hits” acquired in this tumorigenic process. Therefore, our report of clonal in situ loss of the Y chromosome in ~16% of male breast cancers adds to the weight of evidence that Y loss contributes to male breast carcinogenesis. In accord with the clonal progression theory, we also observed clonal Y loss in a corresponding DCIS lesion from a male breast cancer patient, suggesting that Y loss can be an early event in male breast cancer, further supporting a role for early tumor formation.
We demonstrated that clonal Y loss was present in a percentage of male breast cancers via FISH. However, the capacity to perform FISH on FFPE tissues is variable due to the quality of DNA from this analyte. Therefore, we used ddPCR to determine Y loss in male breast cancer samples that were inadequate for FISH analysis. In theory, the loss of Y would skew the Y/X ratio towards zero. Previously, in an analysis of loss of heterozygosity of BRCA2 alleles, we showed that a basal level of contamination from surrounding normal stromal cells could be overcome using the highly sensitive and specific platform of ddPCR [20]. However, although normal cellular contamination is a common issue, X chromosomal duplication can also occur, as in 2 of our patients, making ddPCR less reliable to assess Y loss. However, our results do suggest that Y retention can be reliably concluded based upon the ratio of Y/X positive droplets using ddPCR. Thus, we included two of these patients in our overall assessment.
Our results showing Y loss support recent studies suggesting that the human Y chromosome has tumor suppressive functions. For example, mosaic loss of Y chromosome in peripheral blood is associated with a higher risk of non-hematologic cancers in men [34]. Furthermore, this loss of Y in peripheral blood is associated with smoking in a dose-dependent manner [35], suggesting a possible link with smoking and Y loss. Additionally, male microchimerism in women has been shown to reduce the risk of breast cancer by approximately one third [36]. In fact, male chimerism observed in the peripheral blood of women generally increases the survival of women affected by cancers and cardiovascular diseases [37]. Male microchimerism is a phenomenon that occurs in pregnant women with male fetuses, with subsequent exchange of fetal and maternal cells via the placenta and persistence of fetal cells [38]. In addition, a previous study demonstrated that transfer of the Y chromosome into a prostate cancer cell line with Y loss suppressed formation of xenografts in 59 out of 60 athymic nude mice [39]. Collectively, these previously described reports combined with our data strongly suggest that the human Y chromosome harbors tumor suppressive properties.
Upon observing Y loss in 5 out of 31 (~16%) of our male breast cancer patients, we hypothesized that male breast cancer without Y loss may still have deletion of a candidate tumor suppressor gene. Therefore, we utilized STS-PCR to investigate regional Y loss in male breast cancer patients with Y chromosome retention. Using STS-PCR, we showed somatic loss of an STS containing the TMSB4Y gene in one of three of our male breast cancer patients with Y retention (Figure 2A). A previous study using array comparative genomic hybridization (aCGH) demonstrated a 40% (10/25 patients) deletion of the Yq11.1-q11.221 locus in male breast cancer, a region that contains TMSB4Y [25]. Furthermore, an analysis of public databases via cBioPortal showed that TMSB4Y was deleted in 16% (10/59) prostate cancer samples (Figure S4) [40]. These combined data further support that TMSB4Y is a tumor suppressor whose loss contributes to male breast cancer.
Functional analysis of TMSB4Y also suggests this gene has tumor suppressor properties. When TMSB4Y is overexpressed in a Dox-inducible MCF-10A background, we observed altered cell morphology, reduced cell proliferation, and a reduced G2/M population. Mechanistically using PLA and immunoprecipitation, we showed that TMSB4Y directly interacts with β-actin. Since β-actin is a main component of the actin cytoskeleton, a major factor in cell morphology and a regulator of cell cycle progression [31, 32], the direct interaction of TMSB4Y and β-actin may modulate the actin cytoskeletal turnover in cells, and in turn could alter cell morphology with subsequent retardation of cell cycle progression.
In conclusion, we observed that ~16% of male breast cancers demonstrate in situ clonal loss of the Y chromosome. These data and those of others, support our hypothesis that the human Y chromosome may be a tumor suppressor in male breast cancer. Furthermore, the Y chromosome gene TMSB4Y may be a tumor suppressor gene that normally functions through its direct interaction with β-actin, which in turn regulates cell morphology and cell proliferation. These results lend new insights into the potential role of the Y chromosome as a tumor suppressor and implicate TMSB4Y in the etiology of male breast cancers.
Materials and Methods
Formalin fixed paraffin embedded (FFPE) tissue of male breast cancer patients
Thirty-two male breast cancer samples were collected from 2 independent cohorts. Cohort 1 consists of 15 male breast cancer patients from Johns Hopkins Hospital collected from 1990 to 2013. FFPE tissue blocks were available and samples were used to create a tissue microarray (TMA) for analysis under an IRB approved protocol. Cohort 2 consists of samples from 19 male breast cancer patients from the Veterans Affairs Medical Center, Washington DC collected between 1996 to 2012. The study was approved by the IRB, VAMC DC. We were able to extract genomic DNA from 17 of these 19 FFPE samples that was suitable for droplet digital PCR (ddPCR), though only 14 had suitable quality and quantity for both ddPCR and fluorescence in situ hybridization (FISH).
FISH
FFPE tissue slides were de-paraffinized and washed with xylene and EtOH. Slides were pretreated, hybridized with FISH probes, and then mounted for microscope observation. Pretreatment was performed using Pretreatment Kit I (02J02-032, Abbott Molecular). Probes were mixed with LSI/WCP Hybridization Buffer (06J67-001, Abbott Molecular). Slides were counterstained with DAPI, and mounted with ProLong® Gold (P36930, Invitrogen). All fluorescence microscopy photos were imaged with NIS-Elements BR2.30. All FISH probes used were from Abbott Molecular: Vysis CEP X (DXZ1) SO Probe (centromeric, 05J08-033), Vysis CEP X (DXZ1) SA Probe (centromeric, 05J09-033), Vysis CEP Y (DYZ1) SGn Probe (q arm, 05J10-034), Vysis CEP Y (DYZ3) SO Probe (centromeric, 05J08-035), and Vysis LSI SRY SO Probe (p arm, 05J27-089).
Droplet Digital PCR (ddPCR)
As previously described, ddPCR was performed using the QX100™ Droplet Digital PCR System according to the manufacturer’s recommendations (Bio-Rad) [41]. TaqMan® Copy Number Assays (Life Technologies) included Hs00314226_cn (FAM labeled) and Hs04125506_cn (VIC labeled) probes. Control genomic DNA used for these studies were commercially purchased male (Promega, G1471) and female (Promega, G1521) DNA. ddPCR was performed with Bio-Rad’s recommended two-step thermo-cycling protocol with a 58°C annealing/extension step. All data analysis was performed using QuantaSoft (Bio-Rad).
Genomic DNA (gDNA) extraction from FFPE tissue slides
Tumor and normal areas were identified and marked by the study pathologist (P.A.). Tissue slides were de-paraffinized and pinpoint solution (D3001-1, Zymo Research) was applied specifically onto the marked areas. DNA was then extracted using the QIAmp DNA FFPE tissue kit (56404, Qiagen) as per the manufacturer’s protocol.
STS-PCR
The male specific region of the Y chromosome Breakpoint mapper [24] was used to analyze the sequence-tagged sites (STS) status of gDNA extracted from patients. Thirty-three sets of STS primers across the MSY region (Table S1) were used. Each set of STS-PCR was repeated 5 times for reproducibility.
Cell culture
TetHyg2.5, a derivative of the non-tumorigenic human breast epithelial cell line MCF-10A [27] was grown in DMEM/F12 (1:1) supplemented with insulin at 10 µg/mL, hydrocortisone at 0.5 µg/mL, and cholera toxin at 0.1 µg/mL (hereafter denoted as “supplemented DMEM/F12”), 5% horse serum (Gibco), EGF at 20 ng/mL, and hygromycin B at 14.3 µg/mL. Dox-inducible TetHyg2.5 derivatives (EV, TmY1, TmY2, and UA3) were grown in supplemented DMEM/F12 with 5% Tetracycline (Tet)-free FBS (HyClone), EGF at 20 ng/mL, hygromycin B at 7.15 µg/mL, and G418 at 120 µg/mL. HEK293 and MCF-7 cells were grown in DMEM media with 5% FBS. All supplements were purchased from Sigma-Aldrich unless otherwise specified. MCF-10A, HEK293, and MCF-7 cells were purchased from American Type Culture Collection (ATCC).
Plasmids and transfections
The TMSB4Y cDNA was purchased as pCMV6-XL5-TMSB4Y (Origene) and was subcloned into the pBI-EGFP vector (Clontech). The TMSB4X cDNA was purchased as tagged and untagged plasmids as pCMV6-Entry-TMSB4X and pCMV6-XL5-TMSB4X, respectively (Origene) and used for transient transfection experiments in HEK293 cells. Plasmids were amplified using the Qiagen Hi-Speed Maxi kits (Qiagen) as per the manufacturer’s protocol. Transient transfections were carried with FUGENE6 (Promega) as per the manufacturer’s protocol.
Generation of Dox-inducible clones
TetHyg2.5 cells were seeded on day 0 and transfected with a cDNA of TMSB4Y cloned into the pBI-EGFP vector and a neomycin resistance vector on day 1. Selection media (hygromycin B at 7.15 µg/mL, and G418 at 120 µg/mL) was used from day 2 onwards. On day 5, cells were plated into 96-well plates and observed for clones expressing green fluorescent protein (GFP) upon 48-hr doxycycline (Dox)-induction at 2ug/ml. Two clones, TmY1 and TmY2, were identified as Dox-inducible cell lines, and an empty vector control (EV) was also generated.
Immunoblotting
Whole-cell protein lysates were prepared using Laemmli sample buffer and resolved by SDS-PAGE using NuPAGE gels (Invitrogen), transferred to a 0.2 µm pore size Invitrolon polyvinylidene difluoride (PVDF) membranes (Invitrogen), and probed with primary antibody followed by incubation with horseradish peroxidase-conjugated secondary antibodies. Antibodies used include anti-TMSB4Y (clone 6G4) mouse monoclonal antibody (SAB1403013, Sigma Aldrich), anti-FLAG M2 antibody (200472-21; Agilent), anti-rabbit IgG HRP-linked antibody (7074, Cell Signaling Technology), anti-mouse IgG HRP-linked antibody (7076, Cell Signaling Technology), anti-GAPDH (D16H11) XP rabbit monoclonal antibody (5174, Cell Signaling Technology), and anti-β-actin (clone 13E5) rabbit monoclonal antibody (4970, Cell Signaling Technology).
Cell line tissue block
Cells were trypsinized and fixed in 10% buffered formalin overnight. Cells were then centrifuged and resuspended with 1X PBS, then mixed via pipetting with 2% agarose solution. Agar plugs were processed into FFPE tissue blocks and slides.
Immunohistochemistry (IHC)
IHC was performed using the PowerVision + Poly-HRP anti-Mouse IHC Detection System (Immunovision). Briefly, slides were steamed in EDTA solution and incubated with anti-smooth muscle actin antibody mouse monoclonal (1:800 dilution, DAKO, m0851) or anti-TMSB4Y (clone 6G4) mouse monoclonal antibody (1:200 dilution, SAB1403013; Sigma Aldrich). Poly-HRP anti-mouse IgG antibody was applied and then visualized with 3,3’-diaminobenzidine (Sigma). Slides were counterstained with hematoxylin.
Cell proliferation assay
Cells were seeded in supplemented DMEM/F12 with 1% Tet-free FBS (HyClone), EGF at a physiological concentration (0.2 ng/mL), hygromycin B at 7.15 µg/mL, and G418 at 120 µg/mL. Dox induction was performed by seeding cells in their respective media with 2µg/mL of Dox. 2 x 104 cells per well of a 6-well tissue culture dish were seeded on day 0. Media was changed every third day, and cells were harvested for cell counting on days 3, 6, 7, 13, and 18 using a Beckman Coulter® Vi-CELL™ XR Cell Viability Analyzer.
Proximity ligation assay (PLA)
Cells were seeded in chamber slides and Dox for induction x 48 hours. Slides were fixed in cold methanol for 15min in -20°C and treated with cold acetone for 1 minute at room temperature. Mouse monoclonal anti-TMSB4Y antibody (Clone 6G4, SAB1403013, Sigma Aldrich) at 1:200 and rabbit monoclonal anti-β-actin antibody (Clone 13E5, 4970, Cell Signaling Technology) at 1:200 were incubated overnight. PLA was performed as per instructions of the DuoLink® In Situ Red Starter Kit Mouse/Rabbit (DUO92101, Sigma Aldrich).
Immunoprecipitation
Cells were rinsed with ice-cold PBS, scraped, and lysed on ice in lysis buffer containing 1% Triton X-100, 10% glycerol, 100mM NaCl, 50mM Hepes (pH 7.2), 10mM NaF (all from Sigma Aldrich), 10mM Na3VO4 (New England Biosciences), and minitab cOmpleteTM protease inhibitor with EDTA (Roche). Lysates were quantified using the BCA protein assay reagent (Pierce). Immunoprecipitation was performed by incubating 1mg of lysate with 1µg of anti-TMSB4Y antibody (Clone 6G4, SAB1403013, Sigma Aldrich) overnight at 4ºC. Lysates were then incubated with Dynabeads Protein G (Life Technologies) for 4 hours at 4ºC. Dynabeads were then washed with cold lysis buffer and boiled at 100 ºC for 5 minutes in 2x Laemmli buffer before immunoblotting.
Cell-cycle analysis with flow cytometry
Cells were seeded at 50% confluency in T75 flasks and Dox-induced for 48hrs. GFP positive cells were isolated by fluorescence activated cell sorting using BD FACSAria II and fixed in PBS/3% formaldehyde/0.4% NP-40 containing 2 mg/mL Hoechst 33258 (Invitrogen). DNA content was measured with a BD LSR flow cytometer (BD Biosciences), and percentages of G1/G0, S, and G2/M phase cells were determined using Modfit LT software (Verity Software House).
Statistical analysis
All statistical analyses were conducted with GraphPad Prism software. For the t-tests conducted, p<0.05 was considered significant.
Acknowledgments
We thank our patients and veterans for their tissue samples used in this study. We thank the Tan Kah Kee Foundation for granting the Tan Kah Kee Postgraduate Scholarship to HYW. We also thank Ms. Ada Tam of the Flow Cytometry Core at the Sidney Kimmel Comprehensive Cancer Center at Johns Hopkins.
Conflicts of Interest
B.H.P. is a paid member of scientific advisory boards for Horizon Discovery, LTD and Loxo Oncology, and has research contracts with Genomic Health, Inc and Foundation Medicine. Under separate licensing agreements between Horizon Discovery, LTD and The Johns Hopkins University, B.H.P. is entitled to a share of royalties received by the University on sales of products. The terms of this arrangement are being managed by the Johns Hopkins University, in accordance with its conflict of interest policies. No potential conflicts of interest were disclosed by the other authors.
Grant Support
This work was supported by: The Avon Foundation (BHP, JL), NIH GM007309 (GMW, DJZ), CA168180 (RLC), CA167939 (SC) and the DOD PCRP PC130991 (PJH). We would also like to thank and acknowledge the support of NIH P30 CA006973, the Sandy Garcia Charitable Foundation, the Commonwealth Foundation, the Santa Fe Foundation, the Breast Cancer Research Foundation, the Health Network Foundation, the Marcie Ellen Foundation, The Helen Golde Trust, The Canney Foundation and The Robin Page/Lebor Foundation. None of the funding sources influenced the design, interpretation or submission of this manuscript.
References
1. Ottini L, Palli D, Rizzo S, Federico M, Bazan V and Russo A. Male breast cancer. Crit Rev Oncol Hematol. 2010; 73:141-155.
2. Rizzolo P, Silvestri V, Tommasi S, Pinto R, Danza K, Falchetti M, Gulino M, Frati P and Ottini L. Male breast cancer: genetics, epigenetics, and ethical aspects. Ann Oncol. 2013; 24 Suppl 8:viii75-viii82.
3. Jemal A, Siegel R, Ward E, Hao Y, Xu J and Thun MJ. Cancer statistics, 2009. CA Cancer J Clin. 2009; 59:225-249.
4. Jemal A, Siegel R, Xu J and Ward E. Cancer statistics, 2010. CA Cancer J Clin. 2010; 60:277-300.
5. Ly D, Forman D, Ferlay J, Brinton LA and Cook MB. An international comparison of male and female breast cancer incidence rates. Int J Cancer. 2013; 132:1918-1926.
6. Stang A and Thomssen C. Decline in breast cancer incidence in the United States: what about male breast cancer? Breast Cancer Res Treat. 2008; 112:595-596.
7. Speirs V and Shaaban AM. The rising incidence of male breast cancer. Breast Cancer Res Treat. 2009; 115:429-430.
8. Ruddy KJ and Winer EP. Male breast cancer: risk factors, biology, diagnosis, treatment, and survivorship. Ann Oncol. 2013; 24:1434-1443.
9. Cutuli B. Strategies in treating male breast cancer. Expert Opin Pharmacother. 2007; 8:193-202.
10. Nilsson C, Johansson I, Ahlin C, Thorstenson S, Amini RM, Holmqvist M, Bergkvist L, Hedenfalk I and Fjallskog ML. Molecular subtyping of male breast cancer using alternative definitions and its prognostic impact. Acta Oncol. 2013; 52:102-109.
11. Kornegoor R, Verschuur-Maes AH, Buerger H, Hogenes MC, de Bruin PC, Oudejans JJ, van der Groep P, Hinrichs B and van Diest PJ. Molecular subtyping of male breast cancer by immunohistochemistry. Mod Pathol. 2012; 25:398-404.
12. Ge Y, Sneige N, Eltorky MA, Wang Z, Lin E, Gong Y and Guo M. Immunohistochemical characterization of subtypes of male breast carcinoma. Breast Cancer Res. 2009; 11:R28.
13. Callari M, Cappelletti V, De Cecco L, Musella V, Miodini P, Veneroni S, Gariboldi M, Pierotti MA and Daidone MG. Gene expression analysis reveals a different transcriptomic landscape in female and male breast cancer. Breast Cancer Res Treat. 2011; 127:601-610.
14. Johansson I, Ringner M and Hedenfalk I. The landscape of candidate driver genes differs between male and female breast cancer. PLoS One. 2013; 8:e78299.
15. Noordam MJ and Repping S. The human Y chromosome: a masculine chromosome. Curr Opin Genet Dev. 2006; 16:225-232.
16. Bachtrog D. Y-chromosome evolution: emerging insights into processes of Y-chromosome degeneration. Nat Rev Genet. 2013; 14:113-124.
17. Teixeira MR, Pandis N, Dietrich CU, Reed W, Andersen J, Qvist H and Heim S. Chromosome banding analysis of gynecomastias and breast carcinomas in men. Genes Chromosomes Cancer. 1998; 23:16-20.
18. Balazs M, Mayall BH and Waldman FM. Interphase cytogenetics of a male breast cancer. Cancer Genet Cytogenet. 1991; 55:243-247.
19. Mitchell EL. A cytogenetic study of male breast cancer. Cancer Genet Cytogenet. 1990; 47:107-112.
20. Cochran RL, Cravero K, Chu D, Erlanger B, Toro PV, Beaver JA, Zabransky DJ, Wong HY, Cidado J, Croessmann S, Parsons HA, Kim M, Wheelan SJ, Argani P and Park BH. Analysis of BRCA2 loss of heterozygosity in tumor tissue using droplet digital polymerase chain reaction. Hum Pathol. 2014; 45:1546-1550.
21. Zhu XB, Feng Y, Zhi EL, Fan WM and Zhang AJ. Y chromosome microdeletions: detection in 1,052 infertile men and analysis of 14 of their families [Article in Chinese]. Zhonghua Nan Ke Xue. 2014; 20:637-640.
22. Zhang YS, Dai RL, Wang RX, Zhang HG, Chen S and Liu RZ. Analysis of Y chromosome microdeletion in 1738 infertile men from northeastern China. Urology. 2013; 82:584-588.
23. Iijima M, Koh E, Izumi K, Taya M, Maeda Y, Kyono K, Yoshida A and Namiki M. New molecular diagnostic kit to assess Y-chromosome deletions in the Japanese population. Int J Urol. 2014; 21:910-916.
24. Lange J, Skaletsky H, Bell GW and Page DC. MSY Breakpoint Mapper, a database of sequence-tagged sites useful in defining naturally occurring deletions in the human Y chromosome. Nucleic Acids Res. 2008; 36(Database issue):D809-814.
25. Tommasi S, Mangia A, Iannelli G, Chiarappa P, Rossi E, Ottini L, Mottolese M, Zoli W, Zuffardi O and Paradiso A. Gene copy number variation in male breast cancer by aCGH. Cell Oncol (Dordr). 2011; 34:467-473.
26. Gao J, Aksoy BA, Dogrusoz U, Dresdner G, Gross B, Sumer SO, Sun Y, Jacobsen A, Sinha R, Larsson E, Cerami E, Sander C and Schultz N. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci Signal. 2013; 6:pl1.
27. Soule HD, Maloney TM, Wolman SR, Peterson WD, Jr., Brenz R, McGrath CM, Russo J, Pauley RJ, Jones RF and Brooks SC. Isolation and characterization of a spontaneously immortalized human breast epithelial cell line, MCF-10. Cancer Res. 1990; 50:6075-6086.
28. Yu FX, Lin SC, Morrison-Bogorad M, Atkinson MA and Yin HL. Thymosin beta 10 and thymosin beta 4 are both actin monomer sequestering proteins. J Biol Chem. 1993; 268:502-509.
29. Weber A, Nachmias VT, Pennise CR, Pring M and Safer D. Interaction of thymosin beta 4 with muscle and platelet actin: implications for actin sequestration in resting platelets. Biochemistry. 1992; 31:6179-6185.
30. Pollard TD and Cooper JA. Actin, a central player in cell shape and movement. Science. 2009; 326:1208-1212.
31. Thery M and Bornens M. Cell shape and cell division. Curr Opin Cell Biol. 2006; 18:648-657.
32. Heng YW and Koh CG. Actin cytoskeleton dynamics and the cell division cycle. Int J Biochem Cell Biol. 2010; 42:1622-1633.
33. Fearon ER and Vogelstein B. A genetic model for colorectal tumorigenesis. Cell. 1990; 61:759-767.
34. Forsberg LA, Rasi C, Malmqvist N, Davies H, Pasupulati S, Pakalapati G, Sandgren J, Diaz de Stahl T, Zaghlool A, Giedraitis V, Lannfelt L, Score J, Cross NC, Absher D, Janson ET, Lindgren CM, et al. Mosaic loss of chromosome Y in peripheral blood is associated with shorter survival and higher risk of cancer. Nat Genet. 2014; 46:624-628.
35. Dumanski JP, Rasi C, Lonn M, Davies H, Ingelsson M, Giedraitis V, Lannfelt L, Magnusson PK, Lindgren CM, Morris AP, Cesarini D, Johannesson M, Tiensuu Janson E, Lind L, Pedersen NL, Ingelsson E, et al. Smoking is associated with mosaic loss of chromosome Y. Science. 2015; 347:81-3.
36. Kamper-Jorgensen M. Microchimerism and survival after breast and colon cancer diagnosis. Chimerism. 2012; 3:72-73.
37. Kamper-Jorgensen M, Hjalgrim H, Andersen AM, Gadi VK and Tjonneland A. Male microchimerism and survival among women. Int J Epidemiol. 2014; 43:168-173.
38. Bianchi DW, Zickwolf GK, Weil GJ, Sylvester S and DeMaria MA. Male fetal progenitor cells persist in maternal blood for as long as 27 years postpartum. Proc Natl Acad Sci U S A. 1996; 93:705-708.
39. Vijayakumar S, Garcia D, Hensel CH, Banerjee M, Bracht T, Xiang R, Kagan J and Naylor SL. The human Y chromosome suppresses the tumorigenicity of PC-3, a human prostate cancer cell line, in athymic nude mice. Genes Chromosomes Cancer. 2005; 44:365-372.
40. Grasso CS, Wu YM, Robinson DR, Cao X, Dhanasekaran SM, Khan AP, Quist MJ, Jing X, Lonigro RJ, Brenner JC, Asangani IA, Ateeq B, Chun SY, Siddiqui J, Sam L, Anstett M, et al. The mutational landscape of lethal castration-resistant prostate cancer. Nature. 2012; 487:239-243.
41. Hindson CM, Chevillet JR, Briggs HA, Gallichotte EN, Ruf IK, Hindson BJ, Vessella RL and Tewari M. Absolute quantification by droplet digital PCR versus analog real-time PCR. Nat Methods. 10:1003-1005.