
Introduction
Hepatocellular carcinoma (HCC) is the fifth most common cancer type around the world. The incidence of HCC remains particularly high in East Asia and sub-Saharan Africa [1], and has been rising in North American and Western Europe [2, 3]. Its epidemics correlates with the prevalence of hepatitis B and C virus in Asia and U.S./Europe, respectively [4, 5]. In fact, chronic viral infections account for more than 70% cases of HCC globally.
The prognosis for patient with advanced HCC remains poor. Although sorafenib has been approved for patients with advanced HCC in 2007 [6], the benefits with sorafenib are modest. In the past years, there have been renewed and continued interests in identifying novel molecular targets in HCC.
Mechanistically, alterations of multiple signaling pathways were detected in HCC patients, including WNT, FGFR, VEGFR, and PDGFR. Among them, deregulation of WNT/beta-catenin signaling was found in 40-70% HCC patients, which is responsible for cell proliferation, migration, and self-renewal. Currently, pharmacological inhibition of WNT pathway is being tested at early stage of clinical trials. Further, activation of FGFR, VEGFR, and PDGFR signaling pathways is closely related to angiogenesis, a key process during HCC carcinogenesis. Therefore, several multi-kinase inhibitors have been evaluated for HCC patients, including sunitinib and dovitinib.
Another important signaling pathway for HCC is JAK-STAT pathway, which is critical to transferring extracellular signal into nucleus for transcriptional regulation. JAK-STAT pathway resides at downstream of various transmembrane receptors, including cytokine receptors such as interferon (IFN) and interleukin 6 (IL-6), as well as aforementioned growth-factor receptors, FGFR, VEGFR, and PDGFR. Once activated, Janus Kinase (JAK) family, JAK1, JAK2, JAK3, and TYK2, could phosphorylate the cytoplasmic portion of cytokine receptors, which in turn recruit signal transducer and activator of transcription (STAT) via its SH2 domain. Dimerization of STATs then occurs when they are associated with tyrosine kinase receptors, leading to their translocation into nucleus and increased transcription of downstream genes, such as c-MYC, CCND1, and VEGF. Therefore, functional JAK-STAT pathway is required for proliferation and survival of normal cells. [7-9]
During the carcinogenesis, several cytokine and growth factor receptor kinases are constitutively activated by different mechanisms. As a result, JAK-STAT pathway is essential for the uncontrolled growth of cancer cells, angiogenesis and metastasis. Several JAK1 mutations were found in different cancer types, such as leukemia, breast cancer, lung cancer, and HCC. JAK1V658F was found in leukemia patients, leading to constitutional activation of JAK1 [10, 11]. Further, seven distinct protein-altering JAK1 mutations were previously identified in tumors from HCC patients by whole-genome sequencing (WGS). Moreover, both of JAK1S729C and JAK1S703I mutations were recurrent and proved to be activating mutations in vitro [12]. On the other hand, a point mutation of JAK2V617F, was found in polycythemia vera (PV), essential thrombocythemia (ET), and primary myelofibrosis (PMF) patients [13-15]. This mutation causes the constitutive activation of JAK2 kinase via disrupting its auto-inhibition.
In the wake of strong correlation between JAK2V617F mutation and myeloproliferative neoplasms (MPN), the search for JAK inhibitors has been accelerated. Multiple molecules targeting different members of JAK kinase family have been synthesized and characterized. Among them, ruxolitinib was approved by FDA for patients with MPN. According to the results of two phase III clinical trials for myelofibrosis (COMFORT-I and COMFORT-II), ruxolitinib could alleviate the splenomegaly and other symptoms for 30-40% of patients [16, 17]. Mechanistically, ruxolitinib targets both JAK1 and JAK2 with similar IC50 by competitive inhibition of these two kinases [18]. The IC50 of ruxolitinib for JAK1 and JAK2 in vitro were 3.3 nM and 2.8 nM, respectively [19]. In the preclinical study, ruxolitinib could effectively inhibit the proliferation of JAK2V617F-positive cell line and alleviate MPN symptoms in JAK2V617F transgenic mice model [19]. However, the effect of ruxolitinib has not been extensively studied in solid tumors.
In the present study, we aimed to identify novel therapeutic targets in HCC and discovered four JAK1 mutations in HCC PDX models through WES. Their identities were confirmed by targeted sequencing, and they were then characterized for activation of JAK-STAT pathway and oncogenic potential via Western blot analysis and in vitro proliferation assay, respectively. Moreover, in vivo efficacy studies of ruxolitinib were conducted in JAK1-mutant PDX models. Taken together, our data suggest that activating JAK1 mutations may be molecular targets for the treatment of HCC.
Results
Identification of JAK1 mutations in HCC PDX models
More than 160 HCC PDX models were established at WuXi AppTec in the past three years, of which over 60 models were characterized by WES. Among them, four models (LI-03-0012, LI-03-0155, LI-03-0191, and LI-03-0257) were identified with non-synonymous mutations in JAK1 gene. These JAK1 mutations, including N451S in LI-03-0155, E483D in LI-03-0257, S703I in LI-03-0191, and A1086S in LI-03-0012 models, were then verified by Sanger sequencing with targeted primers (Data not shown).
Specifically, S703I mutation was found in the pseudo-kinase domain of JAK1 protein, and could potentially cause the disruption of auto-inhibition of JAK1 kinase. Notably, S703I was previously identified in tumors of two HCC patients, and proved to be an activating mutation of JAK1 gene [12]. For the other three JAK1 mutations, A1086S is located in catalytic kinase domain, whereas N451S and E483D are in the SH2 domain of JAK1 protein. (Figure 1A)
All four PDX models have been passaged in BALB/c nude mice for up to 3 generations. And their growth kinetics did not change much during this process (Data not shown). The growth rate of LI-03-0012 is noticeably faster than rest models. We suspected that other oncogenic mutations may contributed to this. In addition, H&E staining confirmed that pathological features were maintained in each PDX model, comparing to the patient’s tumor (PA) (Figure 1B, 1C, and 1D).
Taken together, mutations of JAK1 gene were identified in four HCC PDX models via WES, and validated by Sanger sequencing. These JAK1-mutant PDX models were established and ready for pharmacological studies.
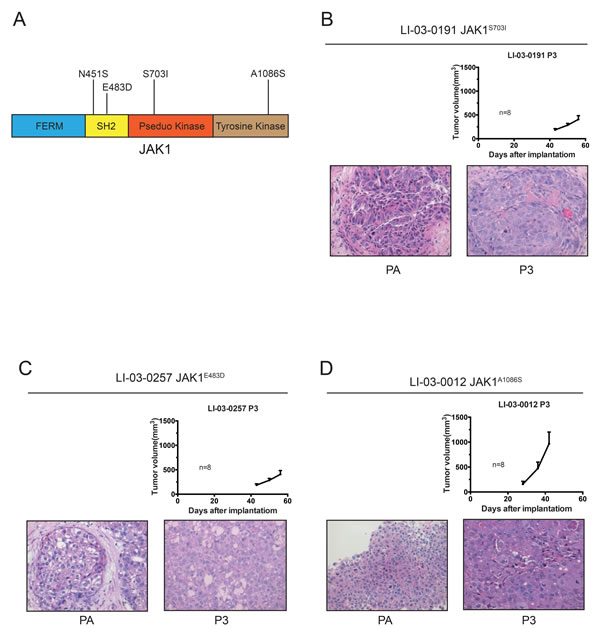
Figure 1: Growth curves and H&E staining of three JAK1-mutated PDX models. A. Diagram of JAK1 protein, illustrating non-synonymous mutations discovered in HCC PDX models. B.-D. Tumor growth potentials were measured in three JAK1-mutant PDX models (P3). Data were shown as mean ± SEM. The H&E staining sections of patient tumor (PA) and PDX tissues at P3 were shown (magnification, 40X).
Characterization of four JAK1 mutations
To explore the biological functions of JAK1 mutations in JAK-STAT signaling pathway, we introduced these mutations into pLVX-IRES-Neo-JAK1 plasmid. Plasmids containing EGFP, wild-type JAK1, JAK1N451S, JAK1E483D, JAK1S703I, JAK1A1086S, and JAK1S729C were constructed. The latter is a known and recurrent activating mutation of JAK1 [12]. Then, plasmids containing distinct JAK1 genes were transiently transfected into HEK293FT cells. The expression levels of phosphorylated JAK1, total JAK1, phosphorylated STAT3, total STAT3, phosphorylated STAT5, and total STAT5 were analyzed by Western blot, while β-actin served as a loading control. Notably, similar to the positive control JAK1S729C [12], ectopic expression of JAK1S703I in HEK293FT cells resulted in elevated expression levels of p-JAK1, p-STAT3, and p-STAT5 (Figure 2A).
To further evaluate the transformation ability of these JAK1 mutations, Ba/F3 cells were stably infected with lentivirus expressing EGFP, wild-type JAK1, JAK1N451S, JAK1E483D, JAK1S703I, JAK1A1086S, and JAK1S729C, respectively. In the absence of IL-3, only the cells expressing JAK1S703I or positive control JAK1S729C were capable of continual proliferation, whereas the cells expressing the EGFP, wild-type and the other mutations of JAK1 gene ceased to grow in vitro (Figure 2B). In addition, no other obvious driver mutations for HCC, such as CTNNB1, have been found in genome of LI-03-0191 PDX model. These data suggest that, among all of four JAK1 mutations identified in the PDX models, only JAK1S703I mutation is able to activate JAK-STAT signaling pathway and exhibits a transforming potential.
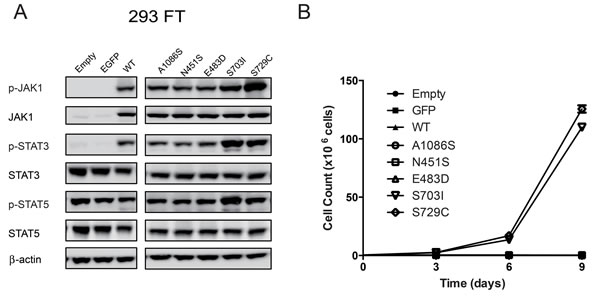
Figure 2: Characterization of four JAK1 mutations. A. 293FT cells were transiently transfected with pLVX-IRES-Neo plasmids containing open reading frames (ORFs) of wild-type and five mutant forms of JAK1 (JAK1N451S, JAK1E483D, JAK1S703I, JAK1A1086S, and JAK1S729C), EGFP, and empty vector control. The expression levels of phosphorylated and total JAK1, STAT3, and STAT5 were evaluated by western blot analyses. Beta-actin served as a loading control. The experiment was performed twice. The representative results were shown. B. Ba/F3 cells stably infected with lentivirus expressing wild-type JAK1, the five mutants, EGFP, and empty vector control were cultured in the absence of IL3. The cell number of each line were counted every three days and plotted against time. The experiment was performed twice with triplicates. The representative results were shown.
Anti-tumor activity of ruxolitinib in JAK1S703I-mutant PDX model
In vivo efficacy studies of JAK1/2 inhibitor, ruxolitinib, were conducted in these four JAK1-mutant models and a JAK1-WT PDX model as a control (Figure 3A). The results showed that, only in LI-03-0191 model bearing JAK1S703I mutation, treatment with ruxolitinib resulted in a tumor growth inhibition (TGI, 48%) in comparison with the vehicle control group. No significant anti-tumor activity of ruxolitinib was observed in rest three JAK1-mutant models, as well as JAK1-WT model. These results indicate that tumor growth in LI-03-0191 PDX model is sensitive to inhibition of JAK kinase, suggesting that JAK1S703I may play a critical role for tumorigenesis in this HCC model.
Pharmacodynamics analysis of JAK-STAT signaling pathway in tumor tissues
To further investigate the molecular mechanisms behind ruxolitinib-induced growth inhibition, tumor tissues were harvested from LI-03-0191 model in the above efficacy study. Constitutively activated STAT3 per se has been shown to be able to function as an oncogene, and STAT3 has been widely accepted as a cancer therapeutic target [20-22]. Thus, the expression levels of phosphorylated and total STAT3 in these samples were analyzed by Western blot, in comparison with the tumor samples obtained from two HCC patients.
Firstly, the level of total STAT3 in two HCC patient samples appeared to be higher than that in corresponding adjacent normal tissues. Interestingly, treatment of ruxolitinib led to a significant reduction (50%) of the phosphorylation level of STAT3 in tumors collected from all five animals in LI-03-0191 model, compared with that in vehicle group. In addition, a modest reduction of total STAT3 level was also observed in ruxolitinib-treated group in comparison with vehicle group (Figure 3B and 3C). These results suggest that ruxolitinib may cause the suppression of JAK-STAT signaling pathway in PDX models bearing JAK1S703I mutation.
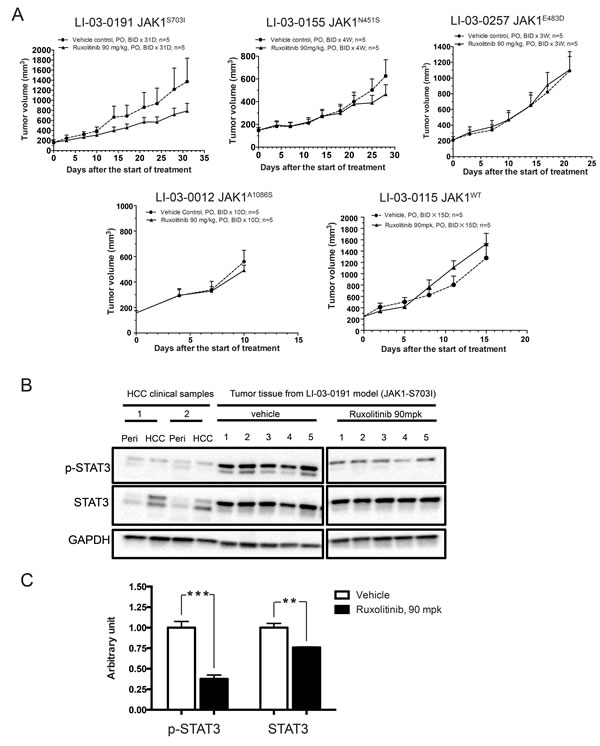
Figure 3: Anti-tumor efficacy of ruxolitinib in JAK1-mutant and JAK1-WT PDX models. A. BALB/c nude mice bearing JAK1-mutant or JAK1-WT PDX tumors were administered with either vehicle (●) or ruxolitinib (90 mpk, BID) (▲) for an indicated length of time. Tumor volumes were plotted against time. B. PDX tumor tissues of JAK1S703I-mutant model were harvested from animals of vehicle and ruxolitinib-treated group from the efficacy study. The expression levels of phosphorylated STAT3, total STAT3, and GAPDH in these PDX tumor tissues, as well as tumor and adjacent normal tissues from clinical HCC patients, were assessed by western blot analyses. The experiment was performed twice and the representative results were shown. C. Quantification of expression levels of phosphorylated STAT3 and total STAT3 in PDX tumor tissues from vehicle and ruxolitinib-treated group of JAK1S703I-mutant model (***, p < 0.001; **, P < 0.01).
Discussion
In this study, we have characterized four JAK1 mutations in over 60 HCC PDX models established at WuXi AppTec. These mutations were identified by WES analysis, confirmed by Sanger sequencing, and then evaluated for their transforming potential in vitro. Among them, JAK1S703I was able to activate JAK-STAT signaling pathway and drove in vitro cell proliferation independent of IL-3 stimulation in otherwise IL-3 dependent Ba/F3 cell line. Further, the tumor growth of HCC PDX model bearing JAK1S703I mutation was sensitive to treatment of a JAK1/2 inhibitor, ruxolitinib. These data indicated that JAK1S703I may be an activating mutation in HCC.
Currently, multi-kinase inhibitor sorafenib is used as first line therapy for unresectable advanced HCC patients, which only yields limited elongation in patients’ survival time [23]. Therefore, the search for new therapeutic targets in HCC is urgently needed.
Previous studies have demonstrated that pharmacological inhibition of STAT protein and upregulation of Src-homology protein tyrosine phosphatases (SHPs), a negative regulator of JAK-STAT pathway, could prevent growth of HCC cell line in vitro and in vivo [24-26]. In addition, an earlier study reported that targeting JAK-STAT pathway in HCC cell lines by ruxolitinib led to apoptosis in vitro [27]. These evidences strongly suggested that abnormal activation of JAK-STAT pathway is associated with HCC. However, since most studies were conducted in vitro or in cell line derived xenograft models, it is hard to directly translate these findings to clinical development of HCC therapies. In addition, though ruxolitinib was approved by FDA for myelofibrosis in 2007, it is not clear whether ruxolitinib is able to suppress in vivo growth of tumors with aberrant JAK-STAT signaling pathway.
Intriguingly, as shown in our study, inhibition of activated JAK-STAT signaling pathway caused by JAK1S703I mutation produced an obvious delay of tumor progression in HCC PDX models. PDX models prove to be biologically stable, which is superior to cancer-cell-derived xenograft model [28]. HCC PDX models can also maintain the tumor microenvironment and vascular structure of patient tumors. Given that JAK-STAT signaling pathway has been demonstrated to play an important role in tumor microenvironment [29], our results in PDX models will shed light on the clinical development of HCC therapies. Further, these results demonstrated that ruxolitinib is capable of shrinking solid tumors, such as HCC, with activating JAK1 mutation.
In addition, since JAK-STAT pathway is important for various physiological functions, only HCC patients with upregulated JAK-STAT signaling could be treated with JAK inhibitors, thus a therapeutic window may exist. And accurate detection of mutations in related cancer genes is also critical before treatment strategy can be finalized for each individual patient.
Admittedly, further investigations are warranted to answer whether other activating mechanisms exist for JAK-STAT pathway in HCC. Nevertheless, the present study suggests that, in addition to standard-of-care therapy, a portion of HCC patients could benefit from targeting JAK-STAT signaling pathway. Combinational or sequential therapies based on sorafenib and JAK inhibitors would likely provide larger therapeutic margin to HCC patients with activated JAK-STAT pathway.
Materials and Methods
Establishment of HCC PDX Models
In compliance with the protocol approved by the Institutional Review Board of Eastern Hepatobiliary Surgery Hospital/Institute of Shanghai and with the subject’s informed consent, primary tumor tissues were collected for PDX establishment as described earlier [30]. In brief, surgically resected primary tumor tissues (designated as PA) were implanted subcutaneously in female BALB/c nude mice (Shanghai SLAC Laboratory Animal Co., Ltd., Shanghai, China). Once the first generation of xenograft (designed as P0) was established, serial implantations in BALB/c nude mice were performed to expand the xenograft tumors (i.e., P1, P2, P3, and beyond). Tumor volume was calculated as 0.5 × length × width2, and plotted against time to generate growth curves. All procedures and protocols were approved by the Institutional Animal Care and Use Committee of WuXi AppTec.
Histology
Pieces of patient samples (PA) or PDX tissues at each passage were collected and stained with hematoxylin and eosin [30]. Histopathology was reviewed by a board-certified pathologist (X.X.).
Tissue processing for genomic studies
Genomic DNA was isolated using a QIAamp DNA mini kit (Qiagen, Germantown, MD). The concentrations were quantified using NanoDrop ND-1000 spectrophotometer (NanoDrop, Wilmington, DE). DNA samples with A260/280 ratios between 1.8 and 2.0 and A260/230 ratios above 2.0, and proven to be high quality by gel electrophoresis were used for WES and SNP 6.0 array analyses.
Whole exome sequencing (WES)
One microgram of each DNA sample isolated from P3 xenograft tumors was used for library construction using a TruSeq DNA sample preparation kit (Illumina, San Diego, CA). Libraries were pooled (500 ng each) for exome capture and amplification using the TruSeq Exome Enrichment kit (Illumina). Sequencing was then performed with paired-end 2x100 base reads on the HiSeq 2000 platform (Illumina). Raw FASTQ files were first processed by a proprietary algorithm to filter out mouse sequence contaminations. We have shown that this filter step does not affect the human SNP detection [31, 32]. After mouse sequence removed, sequencing reads were aligned to human reference genome hg19/GRCh37 by BWA 0.6.1 and processed to variants calling by GATK 1.6.
Reverse transcription polymerase chain reaction and direct sequencing
Total RNA (1 μg) was reversely transcribed into single-stranded cDNAs using High Capacity cDNA Reverse Transcription Kits (Life Technologies, Carlsbad, CA) following the manufacturer’s instructions. One microliter of cDNA was used for a subsequent 25-μl polymerase chain reaction (PCR) amplification using a Premix Prime STAR HS PCR amplification kit (Takara Bio, Otsu, Shiga, Japan). To detect JAK1 mutations, we designed the forward and reverse primers targeting the sequences flanking the mutation points detected by whole-exome sequencing. PCR primers: LI-03-0257-Forward TCGGTATTTCCGCTACGCTC; LI-03-0257-Reverse ACACCTCATGGCTGTATGGC; LI-03-0191-Forward CCTTGTGACATTTAGCCGCC; LI-03-0191-Reverse TAAGCACCAGGCACACCTTT; LI-03-0012-Forward AGATACGTGCTACCGAGAGC; LI-03-0012-Reverse AACCAAGCAGAGGGATGGAC; LI-03-0257-Forward CGGTATTTCCGCTACGCTCA; LI-03-0257-Reverse CAGGCTACACTGGACACCTC.
Cell culture
All cells were cultured in a humidified incubator with 5% CO2 at 37 °C. HEK293FT cells were maintained in DMEM medium (high glucose) supplemented with 10% FBS, 0.1mM nonessential amino acids (Invitrogen), 6 mM L-Glutamine and 1 mM Sodium Pyruvate. Ba/F3 cells were maintained in RPMI1640 medium containing 10% FBS and 2.5 ng/ml IL-3 (Invitrogen)
Plasmids construction, infection and transformation assay
Human JAK1 open reading frames (ORFs) for wild-type and mutant variants (A1086S, N451S, E483D, S703I and S729C) as well as an EGFP ORF were synthesized and constructed into the lentiviral vector pLVX-IRES-Neo (Clontech, Mountain View, CA). Lentivirus was packaged using HEK293T cells and concentrated using polyethylene glycol (Sigma, St. Louis, MO). Ba/F3 cells cultured in RPMI1640 medium containing 10% FBS and 2.5 ng/ml IL-3 were infected with viruses carrying JAK1 ORFs (wild-type or mutant) or EGFP as a control followed by G418 (Invitrogen) selection and subsequent expansion. For assaying transforming potential of JAK1 mutants, transduced Ba/F3 cells were cultured in the absence of IL-3 in triplicates. Cell counting was performed every 3 days for a total of 9 days.
Transfection and western blot analysis
HEK293FT cells were seeded the day before at 8x105 cells/well in a 6-well plate, and transfected with 2.5 ug the fore-mentioned lentiviral plasmid DNA (JAK1 wild-type, JAK1 mutants or EGFP) using Lipofectamine 2000 (Invitrogen). 48 h post-transfection, cells were serum starved for 4 hr and then lysed in RIPA buffer containing protease inhibitors (Roche, Basel, Swiss) and phosphatase inhibitors (Sigma). Protein lysates were then diluted by 4X LDS Sample Buffer and 10X Reducing Agent, denatured by boiling, and separated by SDS-PAGE. Western blotting was performed as described earlier [33]. Following antibodies from Cell Signaling Technology (Danvers, MA) were used: phosphorylated JAK1 (p-JAK1; Tyr1022/1023), JAK1, phosphorylated STAT3 (p-STAT3; Tyr705), STAT3α, phosphorylated STAT5 (p-STAT5; Tyr694), STAT5 and GAPDH. The antibody for β-actin was purchased from Sigma.
In vivo efficacy study
Ruxolitinib was synthesized and formulated in 5% DMSO, 0.5% methylcellulose and 0.1% Tween 80. Tumor tissues were cut into small fragments of approximately 30 mm3 under sterile conditions. BALB/c nude mice were implanted subcutaneously with a tumor fragment by using a trocar. When the average tumor size reached 150 to 200 mm3, tumor size-matched mice were randomly assigned to two groups with five mice in each group. The tumor-bearing mice were given ruxolitinib at 90 mg/kg twice per day orally [34], or vehicle alone for indicated time. Tumor volumes and body weights were measured using calipers twice a week.
Acknowledgments
The authors are grateful to Zhiqiang Cui, Yali Deng, Miaomiao Gu, Yuling Guo, Ling Hu, Xinchen Pan, Xue Tian, Bei Wang, Bing Wang, Liqing Wang, Yunbiao Yan, Guizhu Yang, and Tao Zhang of WuXi AppTec for technical support of this work.
Financial Support
This work was partially supported by the National Basic Research Program of China (973 Program) Grant No. 2012CB724500, and the program of Shanghai Municipal Commission of Health (XBR2013090) and the program of Shanghai subject chief scientist (14XD1400100).
Conflicts of Interest
The authors have declared no conflict of interest.
References
1. Venook AP, Papandreou C, Furuse J and de Guevara LL. The incidence and epidemiology of hepatocellular carcinoma: a global and regional perspective. The oncologist. 2010; 15 Suppl 4:5-13.
2. El-Serag HB and Mason AC. Rising incidence of hepatocellular carcinoma in the United States. N Engl J Med. 1999; 340:745-750.
3. El-Serag HB, Davila JA, Petersen NJ and McGlynn KA. The continuing increase in the incidence of hepatocellular carcinoma in the United States: an update. Ann Intern Med. 2003; 139:817-823.
4. Sherman M. Epidemiology of hepatocellular carcinoma. Oncology. 2010; 78 Suppl 1:7-10.
5. Bosetti C, Turati F and La Vecchia C. Hepatocellular carcinoma epidemiology. Best practice & research Clinical gastroenterology. 2014; 28:753-770.
6. Llovet JM, Ricci S, Mazzaferro V, Hilgard P, Gane E, Blanc JF, de Oliveira AC, Santoro A, Raoul JL, Forner A, Schwartz M, Porta C, Zeuzem S, Bolondi L, Greten TF, Galle PR, et al. Sorafenib in advanced hepatocellular carcinoma. N Engl J Med. 2008; 359:378-390.
7. Vainchenker W and Constantinescu SN. JAK/STAT signaling in hematological malignancies. Oncogene. 2013; 32:2601-2613.
8. Yu H and Jove R. The STATs of cancer—new molecular targets come of age. Nature reviews Cancer. 2004; 4:97-105.
9. Furqan M, Mukhi N, Lee B and Liu D. Dysregulation of JAK-STAT pathway in hematological malignancies and JAK inhibitors for clinical application. Biomarker research. 2013; 1:5.
10. Jeong EG, Kim MS, Nam HK, Min CK, Lee S, Chung YJ, Yoo NJ and Lee SH. Somatic mutations of JAK1 and JAK3 in acute leukemias and solid cancers. Clinical cancer research. 2008; 14:3716-3721.
11. Staerk J, Kallin A, Demoulin JB, Vainchenker W and Constantinescu SN. JAK1 and Tyk2 activation by the homologous polycythemia vera JAK2 V617F mutation: cross-talk with IGF1 receptor. J Biol Chem. 2005; 280:41893-41899.
12. Kan Z, Zheng H, Liu X, Li S, Barber TD, Gong Z, Gao H, Hao K, Willard MD, Xu J, Hauptschein R, Rejto PA, Fernandez J, Wang G, Zhang Q, Wang B, et al. Whole-genome sequencing identifies recurrent mutations in hepatocellular carcinoma. Genome research. 2013; 23:1422-1433.
13. Levine RL, Wadleigh M, Cools J, Ebert BL, Wernig G, Huntly BJ, Boggon TJ, Wlodarska I, Clark JJ, Moore S, Adelsperger J, Koo S, Lee JC, Gabriel S, Mercher T, D’Andrea A, et al. Activating mutation in the tyrosine kinase JAK2 in polycythemia vera, essential thrombocythemia, and myeloid metaplasia with myelofibrosis. Cancer Cell. 2005; 7:387-397.
14. Baxter EJ, Scott LM, Campbell PJ, East C, Fourouclas N, Swanton S, Vassiliou GS, Bench AJ, Boyd EM, Curtin N, Scott MA, Erber WN, Green AR and Cancer Genome P. Acquired mutation of the tyrosine kinase JAK2 in human myeloproliferative disorders. Lancet. 2005; 365:1054-1061.
15. Kralovics R, Passamonti F, Buser AS, Teo SS, Tiedt R, Passweg JR, Tichelli A, Cazzola M and Skoda RC. A gain-of-function mutation of JAK2 in myeloproliferative disorders. N Engl J Med. 2005; 352:1779-1790.
16. Harrison C, Kiladjian JJ, Al-Ali HK, Gisslinger H, Waltzman R, Stalbovskaya V, McQuitty M, Hunter DS, Levy R, Knoops L, Cervantes F, Vannucchi AM, Barbui T and Barosi G. JAK inhibition with ruxolitinib versus best available therapy for myelofibrosis. N Engl J Med. 2012; 366:787-798.
17. Verstovsek S, Mesa RA, Gotlib J, Levy RS, Gupta V, DiPersio JF, Catalano JV, Deininger M, Miller C, Silver RT, Talpaz M, Winton EF, Harvey JH, Jr., Arcasoy MO, Hexner E, Lyons RM, et al. A double-blind, placebo-controlled trial of ruxolitinib for myelofibrosis. N Engl J Med. 2012; 366:799-807.
18. Mascarenhas J and Hoffman R. Ruxolitinib: the first FDA approved therapy for the treatment of myelofibrosis. Clinical cancer research. 2012; 18:3008-3014.
19. Quintas-Cardama A, Vaddi K, Liu P, Manshouri T, Li J, Scherle PA, Caulder E, Wen X, Li Y, Waeltz P, Rupar M, Burn T, Lo Y, Kelley J, Covington M, Shepard S, et al. Preclinical characterization of the selective JAK1/2 inhibitor INCB018424: therapeutic implications for the treatment of myeloproliferative neoplasms. Blood. 2010; 115:3109-3117.
20. Shen Y, Devgan G, Darnell JE, Jr. and Bromberg JF. Constitutively activated Stat3 protects fibroblasts from serum withdrawal and UV-induced apoptosis and antagonizes the proapoptotic effects of activated Stat1. Proc Natl Acad Sci U S A. 2001; 98:1543-1548.
21. Furqan M, Akinleye A, Mukhi N, Mittal V, Chen Y and Liu D. STAT inhibitors for cancer therapy. Journal of hematology & oncology. 2013; 6:90.
22. Bromberg JF, Wrzeszczynska MH, Devgan G, Zhao Y, Pestell RG, Albanese C and Darnell JE. Stat3 as an Oncogene. Cell. 1999; 98:295-303.
23. Muntane J, De la Rosa AJ, Docobo F, Garcia-Carbonero R and Padillo FJ. Targeting tyrosine kinase receptors in hepatocellular carcinoma. Curr Cancer Drug Targets. 2013; 13:300-312.
24. Sethi G, Chatterjee S, Rajendran P, Li F, Shanmugam MK, Wong KF, Kumar AP, Senapati P, Behera AK, Hui KM, Basha J, Natesh N, Luk JM and Kundu TK. Inhibition of STAT3 dimerization and acetylation by garcinol suppresses the growth of human hepatocellular carcinoma in vitro and in vivo. Molecular cancer. 2014; 13:66.
25. Su JC, Chiang HC, Tseng PH, Tai WT, Hsu CY, Li YS, Huang JW, Ko CH, Lin MW, Chu PY, Liu CY, Chen KF and Shiau CW. RFX-1-dependent activation of SHP-1 inhibits STAT3 signaling in hepatocellular carcinoma cells. Carcinogenesis. 2014; 35:2807-2814.
26. Su JC, Tseng PH, Wu SH, Hsu CY, Tai WT, Li YS, Chen IT, Liu CY, Chen KF and Shiau CW. SC-2001 overcomes STAT3-mediated sorafenib resistance through RFX-1/SHP-1 activation in hepatocellular carcinoma. Neoplasia. 2014; 16:595-605.
27. Wilson GS, Tian A, Hebbard L, Duan W, George J, Li X and Qiao L. Tumoricidal effects of the JAK inhibitor Ruxolitinib (INC424) on hepatocellular carcinoma in vitro. Cancer letters. 2013; 341:224-230.
28. Tentler JJ, Tan AC, Weekes CD, Jimeno A, Leong S, Pitts TM, Arcaroli JJ, Messersmith WA and Eckhardt SG. Patient-derived tumour xenografts as models for oncology drug development. Nat Rev Clin Oncol. 2012; 9:338-350.
29. Eirini Bournazou JB. Targeting the tumor microenvironment. JAKSTAT. 2013; 2:e23828.
30. Fang DD, Zhang B, Gu Q, Lira M, Xu Q, Sun H, Qian M, Sheng W, Ozeck M, Wang Z, Zhang C, Chen X, Chen KX, Li J, Chen SH, Christensen J, et al. HIP1-ALK, a novel ALK fusion variant that responds to crizotinib. Journal of thoracic oncology. 2014; 9:285-294.
31. Fang DD, Gao YB, Xu Q, Sun HY, Zhang B, Chen ZL, et al. Identification of somatic mutations in esophageal squamous cell carcinoma and corresponding xenograft by next-generation sequencing [abstract]. In: Proceedings of the 103rd Annual Meeting of the American Association for Cancer Research; 2012 Mar 31-Apr 4; Chicago, Illinois. Abstract nr 2351.
32. Xu Q, Wang G, Fang DD, Gao YB, Chen YD, Sun LL, et al. Deep sequencing of Xenografts and case-matched blood and primary tumors reveals a 20 folds enrichment of loss of heterozygosity versus somatic mutations suggesting LOH plays an ever important role in tumorigenesis [abstract]. In: Proceedings of the 103rd Annual Meeting of the American Association for Cancer Research; 2012 Mar 31-Apr 4; Chicago, Illinois. Abstract nr LB-397.
33. Fang DD, Zhang CC, Gu Y, Jani JP, Cao J, Tsaparikos K, Yuan J, Thiel M, Jackson-Fisher A, Zong Q, Lappin PB, Hayashi T, Schwab RB, Wong A, John-Baptiste A, Bagrodia S, et al. Antitumor Efficacy of the Dual PI3K/mTOR Inhibitor PF-04691502 in a Human Xenograft Tumor Model Derived from Colorectal Cancer Stem Cells Harboring a Mutation. PLoS One. 2013; 8:e67258.
34. Evrot E, Ebel N, Romanet V, Roelli C, Andraos R, Qian Z, Dolemeyer A, Dammassa E, Sterker D, Cozens R, Hofmann F, Murakami M, Baffert F and Radimerski T. JAK1/2 and Pan-deacetylase inhibitor combination therapy yields improved efficacy in preclinical mouse models of JAK2V617F-driven disease. Clinical cancer research. 2013; 19:6230-6241.