
Introduction
This review is an updated and expanded version of a previous review on this topic [1]. This current review now discusses some of the types and classes of mutations which occurs in these pathways and their biochemical importance in terms of therapy. We will focus on the recent advancements in elucidating the roles of the Ras/Raf/MEK/ERK and Ras/PI3K/Akt/mTOR pathways and the types and classes of mutations which occur in these pathways. Since the discovery of the RAS, RAF, MEK, PIK3CA, and AKT oncogenes and NF1, DUSP5, PP2A, PTEN, TSC1 and TSC2 tumor suppressor genes, the Ras/Raf/MEK/ERK and Ras/PI3K/PTEN/Akt/mTOR signaling cascades have been extensively investigated with the ultimate goal of determining how these genes become activated/inactivated and whether it is possible to suppress their activity in cancer and other growth-related diseases [1-6]. Furthermore these pathways are also frequently implicated in the resistance and sometimes sensitivity to therapy [1-6]. Research has also resulted in the development of inhibitors that target critical components of these pathways with the ultimate goal to increase patient survival or in some cases to prevent or impede the development of other diseases (e.g., obesity, diabetes and premature aging) [7-9].
Before we discuss the Ras/Raf/MEK/ERK and Ras/PI3K/PTEN/Akt/mTOR signaling cascades, it is important to define some genetic terms as they are critical to understanding the importance of these pathways and the classes of genes and mutations that occur in components of these cascades. We briefly discuss certain classes of genes which play key roles in the development of cancer. Caretaker genes are involved in genomic stability and normally function to suppress the mutation rate [10]. Caretaker mutations occur mainly in tumor suppressor genes, such as TP53 (p53) and PTEN. TP53 and PTEN are caretaker genes. Caretaker genes help maintain the integrity of the genome.
Gatekeeper genes directly regulate cell growth and their loss can lead to tumorigenesis. They encode critical proteins which can regulate growth or the induction of apoptosis. Many genes fall into this class including: MAPK3/MAPK1 (ERK1/ERK2), TP53, PTEN, NF1, TSC1 TSC2, MTOR (mTOR), EIF4E (eIF4E). Obviously some genes can fall into multiple classifications [11].
The concept of a driver mutation is very important in cancer. If the driver mutation can be successfully targeted that may lead to elimination of the cancer. This is a mutation that is statistically enriched in a particular cancer and usually thought to be one of the first events in the malignant transformation of those particular cells to cancer cells [12]. Examples of driver mutations in these two pathways include RAS, NF1, BRAF, MEK1, PIK3CA (PI3K), and PTEN.
A second class of mutations is passenger mutations [6]. Passenger mutations may occur by different mechanisms. Passenger mutations may occur upon genomic deletion of the region of the chromosome which contains the driver mutation [6]. This has been observed in glioblastoma which have the glycolytic gene enolase 1 (ENO1) gene deleted as it is in the neighborhood of the 1p36 tumour-suppressor locus [6]. ENO1 is a member of a gene family and there are two other ENO genes. Normally, the cell can survive in the presence of ENO1 deletion, however, if ENO2 is silenced, the cancer cell with the ENO1 deletion dies. This provides a selective approach to kill cancer cells, illustrating the significance of identifying passenger mutations.
Next we discuss types of mutations which can result in therapeutic resistance. Gatekeeper mutations often occur in genes (often protein kinases) in either the inhibitor binding site or in the ATP binding site of the protein. They are detected in BRAF, ERK, BCRABL, and epidermal growth factor receptor (EGFR) and can mediate resistance to small molecule inhibitors as that is often where they bind and inhibit activity. They have also been detected in PIK3CA but not necessarily in the hot spot locations [13,14]. Hot spot locations are regions of the gene where mutations are most frequently detected and they can confer a biochemical advantage to the cells which allows abnormal growth.
A synthetic lethal mutation refers to a mutation that occurs in a second gene and results in the death of the cell. This terminology was coined by yeast geneticists. Synthetic lethal screening has resulted in the elucidation of how certain gene products interact with other gene products forming biochemical pathways [15,16]. For example, when there is an activated oncogene or inactivated tumor suppressor gene present in a cell which frequently leads to the abnormal proliferation of the cells, a synthetic lethal mutation may occur at a second gene which results in the death of the transformed cell [17-19]. In essence, there is the loss of a biochemical interaction between the mutant oncogene or tumor suppressor gene and the second gene and the cell dies. Hence the second mutation is referred to as synthetic lethal. In terms of the Ras/Raf/MEK/ERK pathway, which proliferates in response to mutant KRAS, silencing of genes such as voltage-dependent anion channel (VDAC1), serine/threonine kinase 33 (STK33), TANK- binding kinase 1 (TBK) or polo-like kinase-1 (PLK1) results in synthetic lethal interactions [15]. Synthetic lethal interactions are frequently identified by screening siRNA or shRNA libraries. In the PI3K/PTEN/Akt/mTOR pathway, a synthetic lethal interaction is observed in renal cell carcinoma (RCC) cells which lack the von Hippel–Lindau tumor suppressor protein (VHL) as treatment of the cells with rapamycin, an inhibitor of mTORC1 which the tumor cells are dependent on, results in death [15,16].
Lineage-specific mutations occur in genes which are abnormally expressed in certain types of cancers. In certain cell types, the cells become addicted to a lineage-specific gene as well as the mutant oncogene(s). An example is observed in melanoma cells which have mutant BRAF. These cells often have increased expression of the microphthalmia-associated transcription factor (MITF) which is believed to allow the survival of cells of the melanocyte lineage. MITF is sometimes amplified in certain subsets of melanoma cells and cooperates with mutant BRAF to regulate melanoma proliferation. In normal melanocytes, MITF induces cell cycle arrest, whereas in melanoma cells, mutant B-Raf may stimulate MITF transcription while this stimulation of transcription does not occur in normal melanocytes [20].
Oncogene-addiction is a widely-used term to describe the transformed cells addiction to a particular gene or pathway [20-27]. The transformed cells frequently contain a mutation at a particular oncogene, or correspondingly, inactivation of a tumor suppressor gene. The cells become addicted to the consequences of that mutation and grow under conditions where a normal cell would not persist [22]. Many malignant melanoma cells become addicted to mutant BRAF for proliferation [20]. Likewise either mutation of PIK3CA or silencing of PTEN and subsequent activation of Akt is a frequent form of oncogene addiction in many tumor types [1,2,20-27].
Oncogene bypass occurs when a cell bypasses the signal transduction component it normally depended upon for survival [4]. This has been observed in certain cells which were normally dependent upon EGFR for survival, however, when upon exposure to an EGFR inhibitor, cells emerged which displayed amplification of another oncogene, the MET oncogene (MNNG-HOS transforming gene) which allowed the growth of the cells in the presence of the EGFR inhibitor [22].
Kinase switching is a similar event. An example is when cells with the BRAF V600E mutation were cultured in the presence of the B-Raf inhibitor SB-590885, inhibitor-resistant cells arose which utilized the related Raf-1 and A-Raf isoforms [28]. The genetic mechanisms for oncogene bypass and kinase switching as well as many of the changes in inhibitor-resistant cells are complicated and may result from the outgrowth of a minority of the cells present in the original tumor or cell line.
Oncogenic shock is a term that is used to describe the biochemical consequences of inhibiting the oncogene. Interestingly, it has been observed that upon inactivation of the oncogene responsible for survival, the pro-survival and pro-apoptotic signals decay at different rates. In absence of the oncogene responsible for the oncogene addiction phenotype, the pro-survival signals decay more rapidly than the pro-apoptotic signals. This has led to the concept of oncogenic shock and provides the basics for the success of certain inhibitors in suppressing the growth of oncogene-transformed cells [25]. Oncogenic shock may be connected with the translation of “weak mRNAs” which are regulated by the mTOR complex 1(mTORC1) (see below). Both the Ras/Raf/MEK/ERK and PI3K/PTEN/Akt/mTOR pathways interact to regulate the activity of the mTORC1 complex. The half-lifes of proteins such as Akt and ERK are very short (within minutes), while the half-lifes of pro-apoptotic signals are much longer (hours). The decreased activity of Akt and ERK proteins will have a direct effect on the translation of weak mRNAs which often encode growth factors and other important proteins regulating cell growth (e.g., c-Myc). This is one reason why targeting the Ras/Raf/MEK/ERK and PI3K/PTEN/Akt/mTOR pathways has such profound effects on cell growth.
Non-oncogene addiction is a more recently devised term to describe the addiction of a cell on another gene which is not an oncogene per se [26]. For example, rapamycin and modified rapamycins (rapalogs) target mTORC1 which is not normally considered an oncogene, but the cells are dependent upon the mTORC1 complex for their survival. RCC which lack the pVHL tumor suppressor protein exhibit non-oncogene addiction [27]. Now that we have discussed some general genetic terms, we can discuss in more detail the Ras/Raf/MEK/ERK and PI3K/PTEN/Akt/mTOR pathways.
The Ras/Raf/MEK/ERK Pathway
Usually signaling commences upon ligation of a growth factor/cytokine/interleukin/mitogen (ligand) to its cognate receptor at the cell surface. This event can result in the activation of many downstream signaling cascades including the Ras/Raf/MEK/ERK and Ras/PI3K/PTEN/Akt/mTOR pathways. These cascades can further transmit their signals to the nucleus to control gene expression, to the translational apparatus to enhance the translation of “weak” mRNAs, to the apoptotic machinery to regulate apoptosis or to other events involved in the regulation of cellular proliferation (for example, interactions with the p53 pathway to regulate cell cycle progression). Regulation of the Ras/Raf/MEK/ERK and Ras/PI3K/PTEN/Akt/mTOR pathways is mediated by a series of kinases, phosphatases, GTP:GDP exchange and scaffolding proteins. There are also many tumor suppressor proteins which interact with these cascades which frequently serve to fine tune or limit activity (e.g., PTEN, RKIP, PP2A, DUSP5, DUSP6, TSC1, TSC2). Mutations occur in many of the genes in these pathways leading to uncontrolled regulation and aberrant signaling [5,28-32]. Certain of these tumor suppressor genes can be regulated by oncogenic micro (mi) RNAs [33]. An overview of the effects of mutations and the activation of the Ras/Raf/MEK/ERK and Ras/PI3K/PTEN/Akt/mTOR signaling pathways and how they interact is presented in Figure 1. In this review, we will point out which genes are abnormally expressed in human cancer to illustrate the importance of these genes and pathways.
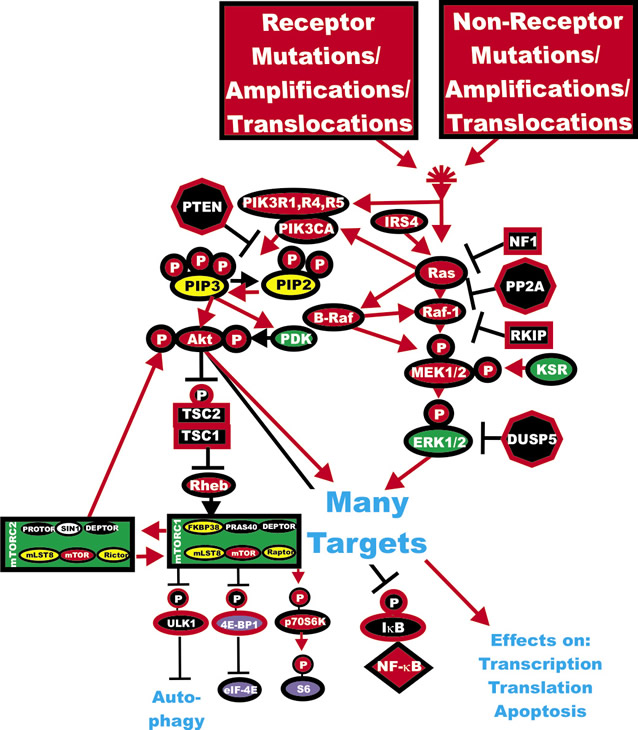
Figure 1: Activation of the Ras/Raf/MEK/ERK and Ras/PI3K/PTEN/Akt/mTOR Pathways by Genetic Mutations. Activation of the Ras/Raf/MEK/ERK and Ras/PI3K/PTEN/Akt/mTOR pathways can occur by upstream mutations in growth factor receptors or by mutations in upstream kinases and coupling molecules. In addition, intrinsic members of the two pathways are frequently mutated in human cancer. Genes in the Ras/Raf/MEK/ERK and Ras/PI3K/PTEN/Akt/mTOR pathways that have activating mutations detected in human cancer and proliferative diseases are indicated in red ovals. Four mutations have been detected in the RHEB gene in 903 samples present in the Sanger Institute COSMIC database. Four mutations have been observed in the p70S6K gene (RBS6KB1) out of 1047 samples examined (Sanger Institute COSMIC database. Also loss of tumor suppressor genes such as PTEN, PP2A, NF1 DUSP5, TSC1, TSC2 can contribute to activation of certain pathways. These are indicated in black octagons if they are phosphatases or black squares if they are coupling molecules. Other kinases not frequently mutated in human cancer are indicated in green symbols. PIP2 and PIP3 are indicated in yellow ovals. The 4E-BP1and eIF-4E proteins are indicated in purple ovals. mTORC1 phosphorylates the unc-51-like kinase 1 (ULK1) which results in the suppression of autophagy. ULK1 is indicated in a black oval. The mTORC1 inhibitor prevents phosphorylation of ULK1 and autophagy can occur. Red arrows indicate activating events in pathways. Black arrows indicate inactivating events in pathway. Activating phosphorylation events are depicted in red circles with Ps with a black outlined circle. Inactivating phosphorylation events are depicted in black circles with Ps with a red outlined circle.
Following stimulation of a growth factor receptor (GFR), a Src homology 2 domain containing protein (Shc) adaptor protein becomes associated with the C-terminus of the activated GFR, e.g., EGFR, insulin like growth factor-1 receptor (IGF-1R), vascular endothelial growth factor receptor (VEGFR) and many others [1-3, 34-38]. EGFR mutations can contribute to transformation of multiple cell lineages and these alterations are considered driver mutations
Shc recruits the growth factor receptor-bound protein 2 (Grb2) protein and the son of sevenless (SOS) homolog protein [a guanine nucleotide exchange factor (GEF)], resulting in the loading of the membrane-bound GDP:GTP exchange protein (GTPase) Ras with GTP [1,2]. RAS is frequently mutated in many diverse human cancers. RAS mutations are often driver mutations. GEFs promote Ras activation by displacing GDP from Ras which leads to GTP binding.
Ras activation is suppressed by the GTPase activating proteins (GAPs) that stimulate the GTPase activity of Ras. There are two prominent GAP proteins, p120GAP and NF1. NF1 is a tumor suppressor gene and has both driver and gatekeeper gene functions. Germline mutations at NF1 lead to neurofibromatosis [29].
Ras can also be activated by GFRs, such as insulin receptor (IR), via intermediates like insulin receptor substrate (IRS) proteins that bind Grb2 [34]. IRS4 has recently been documented to be mutated in melanoma [5]. Ras:GTP then recruits the serine/threonine (S/T) kinase Raf to the membrane where it becomes activated, likely via a Src-family tyrosine (Y) kinase [1,2]. Recently Ras-mediated Raf-1 activation has been shown to be dependent on calcium/calmodulin-dependent protein kinase II (CaMK-II) which phosphorylates Raf-1 at S338 in some experimental stimulation conditions [EGF, fetal bovine serum (FBS) treatment]. This dependency does not appear to occur with regards to B-Raf activation [39].
Both RAS and RAF are members of multi-gene families and there are three Ras members (KRAS, NRAS and HRAS) [1-4] and three RAF members [BRAF, RAF1 (a.k.a c-Raf) and ARAF) [1-3]. BRAF is frequently mutated in melanomas and certain other cancers and these mutations are frequently driver mutations [40].
Raf-1 can be regulated by dephosphorylation by the protein serine/threonine phosphatase 2A (PP2A) and others [41,42]. PP2A has been reported to positively and negatively regulate Raf-1. PP2A is also considered a tumor suppressor gene and has gatekeeper gene functions [43].
Raf phosphorylates and activates the mitogen-activated protein kinase kinase-1 (MEK1) (a dual specificity kinase (T/Y) on S/T residues [1-4]). Other proteins such as kinase suppressor of Ras (KSR) have recently been shown to phosphorylate MEK1 [44-48]. KSR has scaffolding properties and interacts with Raf, MEK and ERK which regulate ERK activation [44-48]. KSR can form dimers with various Raf proteins which alter the effects of Raf inhibitors. KSR1 competes with Raf-1 for Raf inhibitor-induced binding to B-Raf which decreases the normal ERK activation observed after Raf-inhibitor treatment [47].
MEK1 phosphorylates extracellular signal regulated kinases 1/2 (ERK1 and 2) at specific T /Y residues [1-4]. MEK1 was originally not thought to be mutated frequently in human cancer. However, recent large scale mutation screening studies and studies aimed at determining mechanisms of resistance to small molecule inhibitors have observed that MEK1 is mutated in certain human cancers and also is mutated in certain inhibitor-resistant cells. MEK1 is also considered to be a driver oncogene in certain cancers [49]. Rac (Ras related gene) and p21-activating kinases (PAK) can also regulate MEK/ERK activation [50,51].
Activated ERK1 and ERK2 S/T kinases phosphorylate and activate a variety of substrates, including p90 Ribosomal six kinase-1 (p90Rsk1) and this pathway has been implicated in cancer progression [1-3]. ERK1/2 are considered by some as gatekeeper genes. ERK also phosphorylates MAPK signal integrating kinases (Mnk1/2) which can in turn phosphorylate (eukarytotic translation initiation factor 4E) eIF4E, a key protein involved in the translation of difficult mRNAs [1-3]. EIF4E is considered to be a gatekeeper gene.
p90Rsk1 can activate the cAMP response element binding protein (CREB) transcription factor as well as proteins involved in regulation of protein translation (e.g., Mnk-1, p70 ribosomal S6 kinase (p70S6K), eukaryotic translation initiation factor 4B, (eIF4B), and ribosomal protein S6 (rpS6) [52].
The number of ERK1/2 substrates/targets is easily in the hundreds. These substrate/targets include different types of molecules including: other kinases, phosphatases, growth factor receptors, cytokines, cell cycle regulator proteins, transcription factors, or proteins involved in mRNA translation or apoptosis. Suppression of MEK and ERK can have profound effects on cell growth, inflammation and aging. Activated ERK can also phosphorylate “upstream” Raf-1 and MEK1 which alter their activity. Depending upon the site phosphorylated on Raf-1, ERK phosphorylation can either enhance [53] or inhibit [54] Raf-1 activity. In contrast, some studies have indicated that when MEK1 is phosphorylated by ERK, its activity decreases [55]. Recent studies indicate that ERK does not negatively feedback inhibit B-Raf [56]. ERK also phosphorylates SOS at multiples sites leading to the dissociation of SOS from GRB2 and preventing Ras activation [4, 57]. ERK can also phosphorylate EGFR and suppress its activity [58]. The dual specificity phosphatases (DUSP) (aka MKPs) are transcriptionally induced by ERK phosphorylation of transcription factors (e.g., Ets) [59]. The DUSPs serve as negative feedback regulators to suppress ERK activity. Some of the events induced by ERK phosphorylation are rapid, such as post-trasnlational modification, while other events require gene transcription and translation (e.g., ERK phosphorylation of Ets which induces transcription of DUSPs). The DUSPs are potentially tumor suppressor genes and DUSP mutations have been detected in various cancers [60]. An overview of the regulatory loops in the Ras/Raf/MEK/ERK pathway is presented in Figure 2.
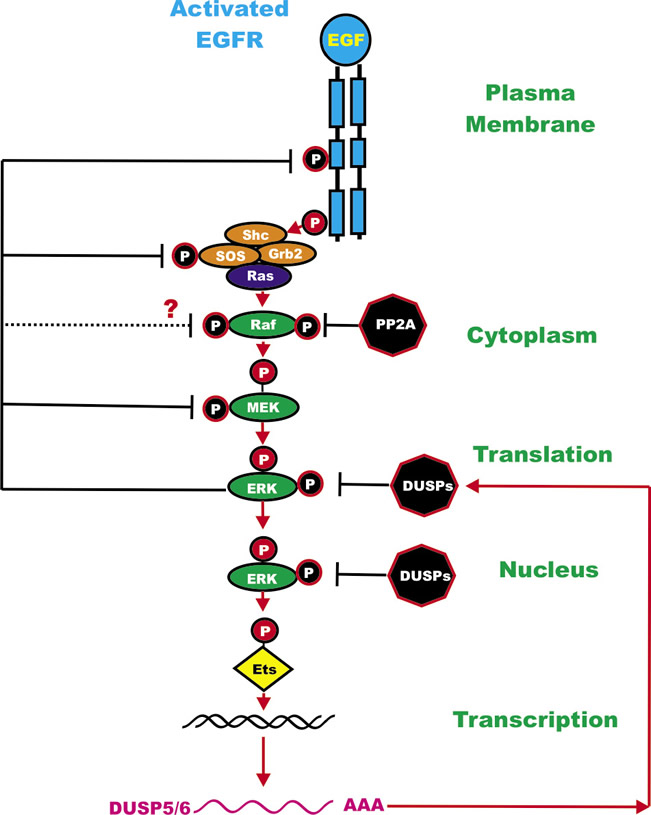
Figure 2: Regulatory Loops in the Ras/Raf/MEK/ERK Pathway. ERK can phosphorylate members of the Ras/Raf/MEK/ERK pathway and even upstream EGFR. Sometimes these phosphorylation events mediated by ERK can inhibit the activity of the phosphorylated molecule. ERK can also phosphorylate Ets which can lead to transcription of the DUSP genes which in turn can inactivate ERK by dephosphorylation. PP2A can also suppress Raf activity by dephosphorylation. The activated EGFR is depicted in blue. Kinases are indicated by green ovals. Phosphatases are indicated by black octagons. Coupling molecules are indicated by orange ovals. Ras is indicated by a purple oval. The transcription factors Ets is indicated by a yellow diamond. The activated EGFR is depicted in blue. Red arrows indicate activating events in pathways. Black arrows indicate inactivating events in pathway. Activating phosphorylation events are depicted in red circles with Ps with a black outlined circle. Inactivating phosphorylation/dephosphorylation events are depicted in black circles with Ps with a red outlined circle.
The Raf/MEK/ERK pathway exhibits properties of a negative feedback amplifier (NFA). In essence, NFA signaling is similar in biological design to those used in electronic circuits. NFAs in electronic circuits optimize robustness, stabilization of signal and linearization of non linear signal amplification. These properties of the Raf/MEK/ERK NFA are important in determining activation kinetics, response to drugs and various other downstream effects of activated ERK [56].
Phosphorylation events induced by ERK serve to alter the stability and/or activities of the proteins. These examples of feed-back loops become important in consideration of whether to just target MEK or to target both Raf and MEK in various cancers. It is important that the reader realize that certain phosphorylation events can either inhibit or repress the activity of the affected protein. This often depends on the particular residue on the protein phosphorylated which can confer a different configuration to the protein or target the protein to a different subcellular localization that may result in proteasomal degradation or association with certain scaffolding proteins.
There are numerous scaffolding/chaperonin proteins which interact with various components of the Raf/MEK/ERK cascade (e.g., 14-3-3) [61], MEK partner-1 (MP-1) [62], heat shock protein-90 (HSP-90) [63], KSR [64] Raf kinase inhibitory protein (RKIP) [65]. Heat shock proteins such as HSP-90 are considered caretakers as they normally serve to protect the activity of client proteins [66, 67]. Mutations at KRAS will confer sensitivity to HSP-90 inhibitors such as geldanamycin, documenting the importance of HSP-90 in regulation of this pathway [68,69].
RKIP is also considered a metastasis suppressor gene in certain cancers and has gatekeeper and caretaker effects. Raf-1 activation by Ras has been shown to be dependent on the prohibitin protein, a ubiquitously expressed protein which may also serve as a chaperonin protein [70].
The regulation of ERK1/2 activity in the nucleus and cytoplasm is complex as the p38MAPK-alpha splice isoform Max-interacting protein (Mxi-2) can bind ERK1/2 and ensure its translocation into the nucleus and prevent both MAPK phosphatase-1 (MKP1) and DUSP5 from dephosphorylating ERK1/2 in the nucleus and not the cytoplasm. Most phosphatases will probably eventually be shown to be tumor suppressor genes. Upon Mxi-2 binding ERK1/2, enhanced ERK1/2 activity is detected in the nucleus. Mxi-2 prevents the dephosphorylation of ERK1/2 by MKP1 and DUSP5. This allows activated ERK1,2 to phosphorylate the transcription factor c-Myc and other critical substrates [71-73].
In the nucleus ERK can phosphorylate transcription factors, such as: E twenty-six-like transcription factor 1 (Elk-1), estrogen receptor (ER), Fos, globin transcription factor 1 (Gata-1), c-Myc, signal transducer activation of transcription 1 & 3 (STAT1 & 3) and others [1-3,55,74-76]. These transcription factors bind the promoters of many genes, including growth factor and cytokine genes that are important in promoting growth and preventing apoptosis of multiple cell types.
ERK can also phosphorylate and modulate the activity of the Twist, Snail, Slug, and Zeb1 transcription factors either directly or indirectly which can regulate cellular proliferation, survival and some can modulate epithelial mesenchymal transition (EMT) [77-92]. Phosphorylation of the transcription factors by ERK1/2, or in some cases the related MAPK, p38MAPK, prevents their ubiquitination and results in their stabilization and increased activity in the nucleus and ability to promote EMT [83-92].
In the nucleus, ERK can also phosphorylate mitogen and stress-activated protein kinases (MSKs) [93,94] which in turn can phosphorylate transcription factors such as activator transcription factor-1 (ATF-1) that is important in the regulation of many immediate early genes controlled by activating protein-1 (AP-1) [95]. The ternary complex factors (TCF) such as Elk-1, Sap-1 and Net are also phosphorylated by ERK which results in their activation [96,97]. The TCFs form complexes with serum responsive factor (SRF) and activate many genes through their serum responsive elements (SRE) in their promoter regions [98,99].
MSKs also phosphorylate many proteins involved in modulating chromatin structure including: Histone H3, and (high-mobility-group protein-14) HMG14 which can result in the transcription of immediate early genes after mitogens/growth factor stimulation [100]. ERK1/2 can phosphorylate many proteins critical for cytoskeletal structure/reorganization including: calpain (Capn) [101], focal adhesion kinase (FAK) [102], myosin light polypeptide kinase (MLCK) and paxillin-6 (Pax6) [103]. Sometimes phosphorylation by ERK of FAK can result in FAK dephosphorylation [104].
Thus the Ras/Raf/MEK/ERK pathway is important in determining cellular shape and mobility/invasion. Under certain circumstances, aberrant regulation of this pathway can contribute to abnormal cellular growth, mobility and invasion which may lead to many abnormalities including; autocrine transformation, drug resistance, senescence, premature aging, or metastasis [1,2,105-119].
Thus the reader begins to understand how the Ras/Raf/MEK/ERK pathways can regulate the expression of many genes involved in the response to growth factors and mitogens. Furthermore many of the genes in this pathway, as well as other genes that regulate the activity of this pathway, have varying abilities to influence cancer development. They can sometimes be drivers of cancer development, gatekeeper or caretaker genes. An overview of the effects of the Ras/Raf/MEK/ERK and PI3K/PTEN/Akt/mTOR pathways on key regulatory pathways is presented in Figure 3.
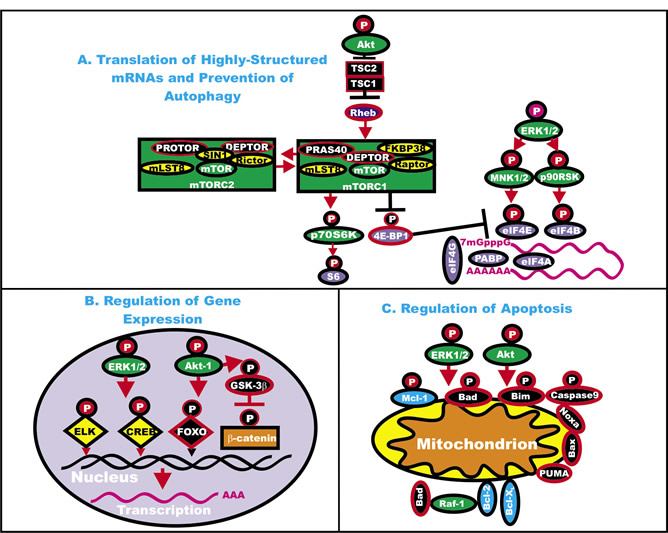
Figure 3: Effects of ERK and Akt on Regulatory Processes. ERK and Akt can phosphorylate many targets which serve to regulate cell proliferation. In Panel A, some of the effects of ERK and Akt on the translation of highly structured mRNAs are indicated. Kinases are indicated in green ovals or rectangles (mTORC1 and mTORC2 complexes). TSC1 and TSC2 are indicated in black squares. Rheb is indicated in a dark blue oval. mTOR interacting proteins which positively regulate mTOR activity are indicated in yellow ovals. mTOR interactiving proteins which negatively regulate mTOR activity are indicated in black ovals. mRNA initiation factors and proteins associated with the ribosome are indicated in purple ovals. In Panel B, some of the effects on the regulation of gene expression by ERK and Akt phosphorylation are indicated. Transcription factors activated by either ERK or Akt phosphorylation are indicated in yellow diamonds. The Foxo transcription factor that is inactivated by Akt phosphorylation is indicated by a black diamond. Beta-catenin is indicated in an orange rectangle. In Panel C, some of the effects of ERK and Akt phosphorylation on apoptotic regulatory molecules are indicated. Molecules such as Mcl-1 which are anti-apoptotic and phosphorylated by ERK and Akt are indicated by blue ovals, other anti-apoptotic molecule are also indicated by blue ovals. Pro-apoptotic molecules are indicated by black ovals. Red arrows indicate activating events in pathways. Black arrows indicate inactivating events in pathway. Activating phosphorylation events are depicted in red circles with Ps with a black outlined circle. Inactivating phosphorylation events are depicted in black circles with Ps with a red outlined circle.
Mutations or Altered Expression of the Ras/Raf/MEK/ERK Pathway Components
In our previous reviews [1,3] we have discussed in detail the frequency of Ras mutations observed in human cancers. Ras mutations have been observed in approximately 20 to 30% of human cancers. Often point mutations are detected in RAS genes in cancer cells from patients which enhance Ras activity. Genome RAS amplification or overexpression of Ras, perhaps due to altered methylation of its promoter region, are also detected in some tumors [1]. The frequency of KRAS mutations is very high (~80%) in advanced pancreatic cancers [1]. Mutations in Ki-Ras will make cells sensitive to HSP90 inhibitors [68,69]. BRAF is mutated frequently in melanomas (50-70%) [5,120], papillary thyroid cancers (40%) [121], Langerhans’-cell histiocytosis (57%) [122]. BRAF is mutated to lesser extent (2-3%) in non small cell lung cancers (NSCLC), [123] and colo-rectal cancers (CRC) (8%) [124]. Recently BRAF has been observed to be frequently mutated (100%) in hairy cell leukemia. [125]. BRAF has been observed to be mutated in 8 of 199 patients (4%) with multiple myeloma and 4 of those were mutant at BRAF V600E [126]. Other B-ALL and peripheral B cell lymphomas have been observed to have low (>3%) frequencies of BRAF mutations, but none of these mutants produced the B-Raf V600E protein [127-130]. Similar mutations were not detected in the Tiacci et al. study with similar leukemias and lymphomas [125]. A recent study detected BRAF mutations in 2/55 (3.6%) of large B-cell lymphoma (DLBCL). The authors postulated that BRAF may be considered driver mutations for those DLBCL [131]. Cancer patients with the BRAF driver mutations are postulated to be sensitive to B-Raf inhibitors such as vemurafenib, dabrafenib, and GDC-0879.
Previously it was thought that the MEK and ERK genes were not frequently mutated in human cancer. More recent analysis has indicated that MEK1 and MEK2 are mutated in certain cancers (e.g., ovarian and lung cancers) and can be driver mutations [49, 131-134]. Mutations at MEK1 are also important in governing the sensitivity/resistance of certain cells to Raf and MEK inhibitors and will be discussed in an accompanying review [134].
Upstream components of this pathway are also mutated or deregulated in human cancer [1-4]. Some common receptors which are altered in human cancer include EGFR, HER2, IGF-1R, PDGFR, VEGF, and FGFR2/3 [1-4].
The Ras/PI3K/PTEN/Akt/mTOR Pathway
Phosphatidylinositol-3-kinase (PI3K) is a heterodimeric protein with an 85-kDa regulatory subunit and a 110-kDa catalytic subunit (PIK3CA) [1-3, 135-138]. PIK3CA is frequently mutated in certain cancers such as: breast, ovarian, colorectal, endometrial and lung [1,14,139] although its role as a driver mutation in these cancers remains controversial [140]. Recent studies have shown in the lung cancers with mutant PIK3CA, there are also mutations at other driver oncogenes, such as EGFR, KRAS, BRAF, MEK, and anaplastic lymphoma kinase (ALK) [141]. Recent studies in melanoma have indicated that some components of the PI3K pathway (PTEN, MTOR, IRS4, PIK3R1, PIK3R4, PIK3R5 and NFKB1) are co-mutated in 17% of BRAF V600E mutant and 9% of NRAS mutant melanomas [5]. An overview of the Ras/PI3K/PTEN/Akt mTOR pathway and the regulator circuits is presented in Figure 4.
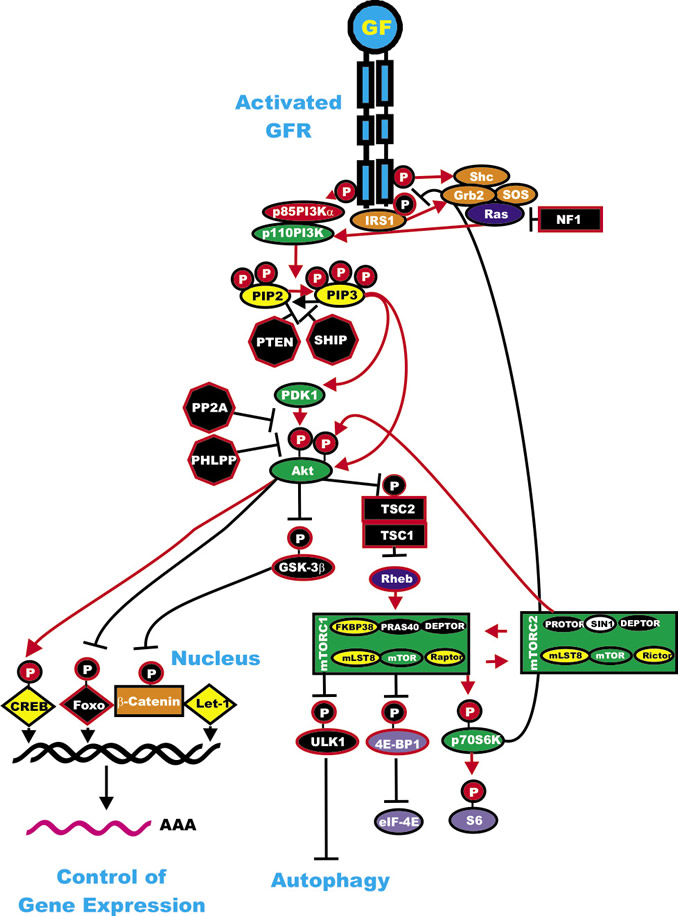
Figure 4: Regulatory Loops in the Ras/PI3K/PTEN/Akt/mTOR Pathway. Some of the regulatory interactions in the Ras/PI3K/PTEN/Akt/mTOR Pathway are indicated. An activated growth factor receptor is indicated in blue. Ras and Rheb are indicated in dark blue ovals. IRS1 is indicated in an orange oval. Kinases are indicated in green ovals with the exception of GSK-3beta which is indicated in a black oval as it is inactivated by Akt phosphorylation. The p85 regulatory subunit of PI3K is indicated in a red oval. Phosphatases are indicated in black octagons. NF1, TSC1 and TSC2 are indicated in black squares. PIP2 and PIP3 are indicated in yellow ovals. Phosphatases are indicated in black octagons. mTOR interacting proteins which positively regulate mTOR activity are indicated in yellow ovals. mTOR interactiving proteins which negatively regulate mTOR activity are indicated in black ovals. Transcription factors activated by either ERK or Akt phosphorylation are indicated in yellow diamonds. The Foxo transcription factor that is inactivated by Akt phosphorylation is indicated by a black diamond. β-catenin is indicated in an orange rectangle. mRNA initiation factors and proteins associated with the ribosome are indicated in purple ovals. mTORC1 phosphorylates the unc-51-like kinase 1 (ULK1) which results in the suppression of autophagy. ULK1 is indicated in a black oval. The mTORC1 inhibitor prevents phosphorylation of ULK1 and autophagy can occur. Red arrows indicate activating events in pathways. Black arrows indicate inactivating events in pathway. Activating phosphorylation events are depicted in red circles with Ps with a black outlined circle. Inactivating phosphorylation events are depicted in black circles with Ps with a red outlined circle.
There are three classes of PI3K, each with distinct substrate specificity and lipid products: I, II, and III [135-138]. In mammals, class I PI3Ks are the best understood PI3Ks and are expressed in all cell types. To date, class I PI3Ks are the most widely implicated in human cancers [2,14,135-138] and for this reason they will be the only PI3Ks discussed in detail in this review. Class I PI3Ks are divided further into A and B subtype. Class IA PI3Ks are dimers comprising a regulatory (p85-alpha, p85-beta, p55-alpha, p55-gamma, p50-alpha) and a catalytic (p110-alpha, p110-beta, p110-delta) subunit. Class IA PI3Ks act downstream of both tyrosine kinase receptors (TKRs) and G protein-coupled receptors (GPCRs). The single class IB PI3K comprise a p110-gamma catalytic subunit which binds one of two related regulatory subunits, p101 and p87. Class IB PI3Ks are activated downstream of GPCRs [2,14,135-138]. PI3K serves to phosphorylate a series of membrane phospholipids including: phosphatidylinositol 4-phosphate and phosphatidylinositol 4,5-bisphosphate, catalyzing the transfer of ATP-derived phosphate to the D-3 position of the inositol ring of membrane phosphoinositides, thereby forming the second messenger lipids phosphatidylinositol 3,4-bisphosphate (PIP2) and phosphatidylinositol 3,4,5-trisphosphate (PIP3) [2,14,135-138]. Most often, PI3K is activated via the binding of a ligand to its cognate receptor, whereby p85 associates with phosphorylated Y residues on the receptor via a Src-homology 2 (SH2) domain. After association with the receptor, the p110 catalytic subunit then transfers phosphate groups to the aforementioned membrane phospholipids [2,14,135-138]. It is these lipids, specifically PIP3, that attract a series of kinases to the plasma membrane thereby initiating the signaling cascade [2,14,135-138,142]. The p85 PI3K subunit also plays key roles in regulating flux through this pathway by controlling both PI3K p110 and PTEN [143].
Downstream of PI3K is the primary effector molecule of the PI3K signaling cascade, Akt/ protein kinase B (PKB) which is a 57 kDa S/T kinase that phosphorylates many targets on RxRxxS/T (R = Arginine) consensus motifs [1-3, 135-138, 142-148]. Driver AKT mutations are detected in some human cancer [10,49].
Akt was discovered originally as the cellular homologue of the transforming retrovirus AKT8. It is a kinase with properties similar to protein kinases A and C [2,144]. Akt contains an amino-terminal pleckstrin homology (PH) domain that serves to target the protein to the membrane for activation [1-3,135-138,142-148]. Within its central region, Akt has a large kinase domain and is flanked on the carboxy-terminus by hydrophobic and proline-rich regions. Akt-1 is activated via phosphorylation of two residues: T308 and S473, Akt-2 and Akt-3 are highly related molecules and have similar modes of activation. Akt-1 and Akt-2 are ubiquitously expressed while Akt-3 exhibits a more restricted tissue distribution. Akt-3 is found abundantly in nervous tissue [144-148].
The phosphotidylinositide-dependent kinases (PDKs) are responsible for activation of Akt. PDK1 is the kinase responsible for phosphorylation of Akt-1 at T308 [144]. Akt-1 is also phosphorylated at S473 by the mammalian target of Rapamycin (mTOR) complex referred to as (Rapamycin-insensitive companion of mTOR/mLST8 complex) mTORC2 [135-138]. Before the discovery of the ability of mTORC2 to phosphorylate S473, the activity responsible for this phosphorylation event was referred to as PDK2. Akt-2 and Akt-3 are phosphorylated in similar fashions. Therefore, phosphorylation of Akt is complicated as it is phosphorylated by a complex that lies downstream of activated Akt itself [1,2,135-138]. Thus, as with the Ras/Raf/MEK/ERK pathway, there are feedback loops that serve to regulate the activity of the Ras/PI3K/PTEN/Akt/mTOR pathway. These events also serve to illustrate that these signal transduction pathways are not really linear, but highly interactive. Once activated, Akt leaves the cell membrane to phosphorylate intracellular substrates. Akt activity is regulated by many mechanisms including the levels of PIP3 which are controlled positively and negatively by PI3K of PTEN respectively, by phosphorylation by PDK1 and mTORC2 as well as ubiquitination [149].
After activation, Akt is able to translocate to the nucleus [1-3, 134-138] where it affects the activity of a number of transcriptional regulators. Some examples of molecules which regulate gene transcription that are phosphorylated by Akt include: CREB [149], E2F [150], nuclear factor kappa from B cells (NF-kappaB) via inhibitor kappa B protein kinase (I-kappaK) [151], the forkhead (FOXO) transcription factors [152], and murine double minute 2 (MDM2) which regulates p53 activity [1-3,134-138]. These are all either direct or indirect substrates of Akt and each can regulate cellular proliferation, survival and some can modulate EMT (e.g., NF-kappaB) [135,151-154]. Besides transcription factors, Akt targets a number of other molecules to affect the survival state of the cell including: the pro-apoptotic molecules Bcl-2-associated death promoter (BAD) and Bcl-2 interacting mediator of cell death (Bim) [153], as well as, glycogen-synthase kinase-3beta (GSK-3beta) (see Figure 3) [164]. GSK-3beta regulates beta-catenin protein stability, which is important in regulation of EMT. When Akt phosphorylates GSK-3beta, it is targeted to the proteasome and beta-catenin is active and able to stimulate gene expression. Hence the PI3K/PTEN/Akt/mTOR pathway is connected to the Wnt/beta-catenin, p53 and many additional pathways including Ras/Raf/MEK/ERK.
Akt has many diverse effects on proliferation, survival, senescence, invasion, metastasis, drug resistance and DNA damage repair and autophagy [155-162]. Akt is involved in cell cycle progression and migration [163]. Akt may also affect the ability of miRNAs to target their respective genes. Akt is a known inhibitor of autophagy and inhibition of Akt by certain tumor suppressors will induce autophagy [164]. A recent study suggests that Akt may regulate the processing of certain miRNAs by post-transcriptional mechanisms regulate miRNAs’ processing or their stability which induces rapid fluctuation in their levels [165]. Akt and its downstream targets (e.g., the Foxo transcription factor) are involved in aging and suppression of Akt activity, which results in increased Foxo activity, by food supplements such as curcumin, prevents aging [166].
Negative regulation of the PI3K pathway is primarily accomplished through the action of the PTEN tumor suppressor protein. PTEN encodes a lipid and protein phosphatase whose primary lipid substrate is PIP3 [1-3,136-138,167-170]. The purported protein substrate(s) of PTEN are more varied, including focal adhesion kinase (FAK), the Shc exchange protein and the transcriptional regulators ETS-2 and Sp1 and the platelet-derived growth factor receptor (PDGFR) [167-171]. Both the lipid and protein phosphatase activities of PTEN are important for prevention of invasion [171].
PTEN has four primary structural domains. In the amino terminus is the lipid and protein phosphatase domain. This is adjacent to the C2 domain that is responsible for lipid binding and membrane localization. Next are two protein sequences rich in proline (P), glutamic acid (E), S, and T (PEST) domains that regulate protein stability. Lastly, PTEN has a PDZ domain [PDZ is an abbreviation for the first three proteins identified to share this domain which are post synaptic density protein 95 (PSD95), Drosophila disc large tumor suppressor (Dlg1), and zonula occludens-1 protein (zo-1)], which helps facilitate protein-protein interactions. Mutations within the phosphatase domain have been reported to nullify the endogenous function of PTEN [1,3,167]. Thus PTEN is an enticing therapeutic target for activation since it is frequently inactivated in many human cancers through point mutations as well as other genetic and biochemical mechanisms [e.g., promoter hypermethylation, gene deletion, expression of various interacting proteins, miRNAs, phosphorylation, acetylation, ubiquitination, oxidization and others]. PTEN inactivation results in elevated Akt activity and abnormal growth regulation [1,3,167,172]. Thus, drugs reactivating PTEN could potentially be useful in the therapy of some types of tumors driven by PTEN inactivation.
Another negative regulator of the PI3K pathway is the PH domain leucine-rich repeat protein phosphatase (PHLPP). PHLPP is a tumor suppressor gene. PHLPP dephosphorylates S473 on Akt-1 which can induce apoptosis and inhibits tumor growth [173]. Two other phosphatases, SHIP-1 and SHIP-2, remove the 5-phosphate from PIP3 to produce PIP2 [174-177]. SHIP1 and SHIP2 are tumor suppressor genes. Mutations in these phosphatases, which eliminate their activity, can lead to tumor progression.
Next we discuss some of the key targets of Akt that can also contribute to abnormal cellular growth and are key therapeutic targets [1-3,135-138,178-185]. Akt-mediated regulation of mTOR activity is a complex, multi-step phenomenon. Akt inhibits tuberous sclerosis 2 (TSC2 or tuberin) function through direct phosphorylation [179]. TSC2 is a GAP that functions in association with TSC1 to inactivate the small G protein Ras homolog enriched in brain (Rheb) [180-182]. TSC1 and TSC2 are both tumor suppressor and gatekeeper genes [186,187]. TSC2 has been recently shown to have other roles, for example when it interacts with transforming acidic coiled-coil-3 (TACC3) a centromere binding protein, it maintains nuclear membrane structure and regulates cell division. [188].
TSC2 phosphorylation by Akt represses GAP activity of the TSC1/TSC2 complex, allowing Rheb to accumulate in a GTP-bound state. Rheb-GTP then activates, through a mechanism not yet fully elucidated, the protein kinase activity of mTOR which complexes with Raptor (Regulatory associated protein of mTOR) adaptor protein, DEP domain containing mTOR-interacting protein (DEPTOR) and mLST8, a member of the Lethal-with-Sec-Thirteen gene family, first identified in yeast, FK506 Binding Protein 38 (FKBP38) and proline-rich Akt substrate 40 kDa protein (PRAS40) [134-138]. Raptor has also recently been shown to have other roles, including interactions with the rDNA transcriptional apparatus in the nucleoli [189].
mTORC1 inhibits Akt via a negative feedback loop which involves, at least in part, p70S6K [181]. This is due to the negative effects that p70S6K has on IRS-1 [134-138]. p70S6K phosphorylates IRS-1 on S312 and/or S636/S639. This targets IRS-1 to the proteasome where it is degraded. Hence PI3K/Akt signaling downstream of IRS-1 is downregulated when p70S6K is active. Rapamycin treatment blocks mTORC1 and p70S6K activation, thus this loop is broken and Akt is activated.
Deptor is another component of the mTORC1 complex. DEPTOR may be a tumor suppressor gene as decreased expression of DEPTOR results in increased mTORC1 activity [190].
The mechanism(s) by which Rheb-GTP activates mTORC1 have not been fully elucidated, however it requires Rheb farnesylation and can be blocked by farnesyl transferase (FT) inhibitors. It has been proposed that Rheb-GTP would relieve the inhibitory function of FKBP38 on mTOR, thus leading to mTORC1 activation [182].
As stated previously, TSC1 and TSC2 have important roles in the regulation of mTORC1. An additional molecule important in this regulation is the liver kinase B (LKB1 also known as STK11). LKB1 is an important tumor suppressor and gatekeeper mutations of LKB1 cause the rare Peutz-Jeghers Syndrome (PJS) which is a cancer-prone syndrome [191]. LKB1 is a gatekeeper gene and mutations in LKB1 are involved in the formation of hamartomatous polyps in PJS patients. LKB1 is an upstream activator of 5’AMP-activated protein kinase (AMPK) which activates TSC2 that negatively regulates mTORC1 [192,193]. LKB1 is a critical regulator of cell polarity and energy/metabolism control and exerts it vast effects via diverse effectors [138,194,195].
AMPK is considered a metabolic gatekeeper important in many diseases including diabetes, cancer and neurologic disorders [196-203]. AMPK is activated by the diabetes drug metformin [193]. Hence metformin will indirectly suppress mTORC1 activity. Chronic overfeeding increases mTORC1 activity which in turn promotes adiposity and decreases lifespan and is also believe to enhance cancer growth [204,205]. Inhibiting mTORC1 activity by drugs such as metformin and other drugs (including rapamycin) may not only aid in the treatment of diabetics, but also improve cancer therapies and increase longevity [206-216].
Akt also phosphorylates PRAS40, an inhibitor of mTORC1, and by doing so, it prevents the ability of PRAS40 to suppress mTORC1 signalling (recently reviewed in [135-138]). Thus, this could be yet another mechanism by which Akt activates mTORC1. Moreover, PRAS40 is a substrate of mTORC1 itself, and mTORC1-mediated phosphorylation of PRAS40 prevents inhibition of additional mTORC1 signaling [135-138,181]. Due to its negative regulation of mTORC1, PRAS40 has been proposed to have gatekeeper anti-apoptotic functions [217]. Also Ras/Raf/MEK/ERK signaling positively impinges on mTORC1. Both p90Rsk-1 and ERK 1/2 phosphorylate TSC2, thus suppressing its inhibitory function [135-138,181]. Moreover, mTORC1 inhibition resulted in ERK 1/2 activation, through p70S6K/PI3K/Ras/Raf/MEK [183].
The relationship between Akt and mTOR is further complicated by the existence of the mTOR/Rictor complex (mTORC2), which, in some cell types, displays rapamycin-insensitive activity. mTORC2 is comprised of rapamycin insensitive companion of mTOR (Rictor), mTOR, DEPTOR, mLST8, Stress activated protein kinase INteracting protein 1 (SIN1) and protein observed with Rictor (Protor). mTORC2 phosphorylates Akt on S473 in vitro which facilitates T308 phosphorylation [185]. Thus, mTORC2 can function as the elusive PDK-2 which phosphorylates Akt-1 on S473 in response to growth factor stimulation [186,218]. Akt and mTOR are linked to each other via positive and negative regulatory circuits, which restrain their simultaneous hyperactivation through mechanisms involving p70S6K and PI3K [135-138,181]. Assuming that equilibrium exists between these two complexes, when the mTORC1 complex is formed, it could antagonize the formation of the mTORC2 complex and reduce Akt activity [180,181]. Thus, at least in principle, inhibition of the mTORC1 complex could result in Akt hyperactivation. This is one problem associated with therapeutic approaches using rapamycin or modified rapamycins (rapalogs) that block some, but not all, actions of mTOR.
mTOR is a 289-kDa S/T kinase. mTOR was the first identified member of the phosphatidylinositol 3-kinase-related kinase (PIKK) family [135-138,219]. Recently mTOR has been shown to be cell cycle regulated [220,221]. mTOR has been referred to as the gatekeeper of autophagy. mTOR plays important roles in many biological processes, including; energy control [222-224], insulin resistance [225], diabetes [226], seizures [227,228], protein homeostasis [229], regulation of tRNA expression [230,231], cell cycle arrest [232], cell differentiation [233,234], cell migration [235,236], follicle development [237], DNA damage checkpoint [238], cellular quiescence/senescence [239-248], cancer [249,250], aging [251-260] and Parkinson’s disease [261].
mTORC1 is a repressor of autophagy, a lysosome-dependent degradation pathway which allows cells to recycle damaged or superfluous cytoplasmic content, such as lipids, proteins, and organelles [262-280]. As a consequence, cells produce metabolic precursors for macromolecular biosynthesis or ATP generation. In cancer cells, autophagy fulfills a dual role, as it has both tumor-promoting and tumor-suppressing properties. Functional autophagy prevents necrosis and inflammation, which can lead to genetic instability. However, autophagy might be important for tumor progression by providing energy through its recycling mechanism during unfavorable metabolic circumstances, which are very common in tumors [262-280].
A model has been proposed by Dr. Michael P. Lisanti and colleagues which is called the reverse Warburg Effect. This model proposes that the aerobic glycolysis occurring in the tumor associated fibroblasts and not in the actual epithelial tumor cells [266,270-274]. This results in the transfer of high-energy metabolites (lactate and pyruvate) to adjacent epithelial cancer cells which fuel the cancer cells allowing them to invade and metastize. In addition, oxidative stress generated by the cancer cells induces autophagy of the tumor associated fibroblasts which the cancer cells then recycle and use to fuel their growth. Anti-oxidants (N-acetyl cysteine, NAC), quercetin and the anti-diabetes drug metformin) or autophagy inhibitors (chloroquine) will suppress the destruction of caveolin-1 in stromal fibroblasts and inhibit cancer growth. Caveolin-1 is a key protein at the cell membrane which serves to organize other important signaling molecules into signaling complexes (e.g., Fak, Src). Decreased expression of caveolin-1 is associated with a poorer prognosis of breast and other cancers.
Autophagy is also important in hematopoietic cancer [275-277]. Autophagy can be regulated by epigenetic mechanisms [278]. Autophagy may also become defective in certain drug resistant cells [279]. Defective autophagy may be controlled by the p53 rheostat in cancer [280]. Clearly autophagy is a very important survival process which is regulated in part by mTORC1.
mTOR regulates translation in response to nutrients and growth factors by phosphorylating components of the protein synthesis machinery, including p70S6K and eukaryotic initiation factor (eIF)-4E binding protein-1 (4EBP-1), the latter resulting in release eIF-4E, allowing eIF-4E to participate in the assembly of a translational initiation complex [1-3,135-138]. p70S6K phosphorylates the 40S ribosomal protein S6, (rpS6), leading to translation of “weak” mRNAs [1-3,135-138]. Integration of a variety of signals (mitogens, growth factors, hormones) by mTOR assures cell cycle entry only if nutrients and energy are sufficient for cell duplication [1-3,135-138].
Unphosphorylated 4E-BP1 interacts with the cap-binding protein eIF4E and prevents the formation of the 4F translational initiation complex (eIF4F); by competing for the binding of eukaryotic initiation factor 4G (eIF4G) to eIF4E. 4E-BP1 phosphorylation by mTORC1 results in the release of the eIF4E, which then associates with eIF4G to stimulate translation initiation [1-3,135-138].
eIF4E is a key component for translation of 5’ capped mRNAs, that include transcripts encoding proliferation and survival promoting proteins, such as c-Myc, cyclin D1, cyclin-dependent kinase-2 (CDK-2), signal activator and transducer of transcription-3 (STAT-3), ornithine decarboxylase, survivin, B-cell lymphoma 2 (Bcl) -2, Bcl-xL, myeloid cell leukemia-1 (Mcl-1) and others [1-3,135-138].
The mechanisms which control mTORC2 activity have only begun to be revealed. mTORC2 activation requires PI3K, as inhibition of PI3K decreases mTORC2 activity [138]. mTORC2 phosphorylates Akt-1 on S473 that enhances subsequent Akt phosphorylation on T308 by PDK1. mTORC2 phosphorylates other members of the family of protein kinase A, G, and C (AGC) including as serum/glucocorticoid-regulated kinase (SGK1) [281]. mTORC2 has been shown to phosphorylate certain protein kinase C (PKC) family members [282]. mTORC2 has important roles in regulation of cell growth and it is a critical biological sensor [283]. For mTORC2 activity, it requires association with the ribosome and this may a critical sensor promoting growth when conditions are favorable but hindering growth when conditions are not favorable [284-287]. mTORC2 influences actin cytoskeletal organization [288]. Along these same lines, mTORC2 has been implicated in various aspects of tumor progression including motility, invasion and metastasis [289].
PI3K, Akt, and mTORC1/2 are linked to each other via regulatory feedback loops, which restrain their simultaneous hyperactivation. Negative regulation of Akt activity by mTORC1 is dependent on p70S6K-mediated phosphorylation of IRS-1/2 adapter proteins, downstream of the IR and/or IGF-1R [290-292]. IRS-1 and IRS-2 are normally required to activate class IA PI3Ks after stimulation of IR and IGF-1R tyrosine kinase activity. When mTORC1 is active, p70S6K phosphorylates the IRS-1/2 proteins on serine residues, targeting them for proteasomal degradation [293,294].
Inhibition of mTORC1 signaling by rapamycin/rapalogs removes the previously mentioned negative feedback loop and activates Akt through PI3K. Inhibiting mTORC1 with rapamycin will in some situations activate mTORC2. Recent findings have also highlighted the existence of a rapamycin-sensitive, mTORC1/p70S6K-mediated phosphorylation of Rictor on T1135. This phosphorylation event exerted a negative regulatory effect on the mTORC2-dependent phosphorylation of Akt in vivo [295]. Thus, both mTORC1 and mTORC2 could control Akt activation. PI3K/Akt/mTOR signaling is tightly controlled and negatively regulated by several lipid and protein phosphatases. PTEN removes the 3’-phosphate from PIP3, thereby antagonizing network signalling [296,297]. Two other lipid phosphatases, SHIP-1 and -2, remove the 5-phosphate from PIP3 to yield PIP2 [298].
PP2A downregulates Akt activity directly, by dephosphorylating it at T308 and accumulating evidence indicates that PP2A acts as a tumor suppressor [299]. PP2A is an essential phosphatase critically involved in regulation of cell cycle progression [300] and DNA damage response [301] as well as p53 stability and other important biochemical events.
Recent findings have indicated that there exists an inverse relationship between the levels of the B55-alpha regulatory subunit of the PP2A phosphatase, that functions as an Akt phosphatase [302] and the levels of T308 (but not S473) Akt phosphorylation levels in AML blast cells [303]. This finding suggested that B55-alpha is mediating dephosphorylation of Akt at T308, but not S473, in AML cells [303]. Interestingly, this study reported lower levels of the PP2A B55-alpha regulatory subunit in AML primary cells when compared with CD34+ bone marrow cells from healthy donors. Another report has documented that PP2A activity downregulation is a recurrent event in AML patients [304]. Moreover, the phosphorylated S473 residue on Akt is dephosphorylated by the two isoforms of PHLPP (1 and 2) Decreased PHLPP activity has been linked to specific types of cancers [305,306].
mTOR also controls the translation of hypoxia-inducible transcription factor-1-alpha (HIF-1-alpha) mRNA [2,26,135-138,307]. HIF-1-alpha upregulation leads to increased expression of angiogenic factors such as VEGF and PDGF which are important in many physiological processes including, blood supply, cancer and diabetes [26,308-310]. Moreover, HIF-1-alpha regulates the glycolytic pathway by controlling the expression of glucose-sensing molecules including glucose transporter (Glut) 1 and Glut3 [311,312]. p70S6K and 4E-BP1 also control cell growth and hypertrophy by regulating protein synthesis. Hence targeting the mTOR pathway could have many effects on the regulation of cellular growth.
Mutations or Altered Expression of the Ras/PI3K/PTEN/Akt/mTOR Pathways Can Alter Sensitivity to Therapy
Mutations resulting in activation of the Ras/PI3K/PTEN/Akt/mTOR pathways and play critical roles in EMT, tumor progression and aging [313-319]. Mutations/gene amplification of RAS, PIK3CA, PIK3R1, PIK3R4, PIK3R5, IRS4, PTEN, AKT1, TSC1, TSC2, RHEB, MTOR, and 70S6K are detected in certain cancers [5,320]. Aberrant activation of this pathway may be a contributing factor to transformation of diverse types of cancers [321]. PIK3CA is mutated in approximately 25% of breast, 32% of colorectal, 30% of endometrial, 27% of brain, 25% of gastric, 4% of lung cancers [322-326]. These mutations are clustered in small hot-spot regions within the helical (E542, E545) and kinase (H1047) domains [322-326]. The locations of these mutations have been recently critically evaluated [326]. These mutations frequently result in activation of its kinase activity [326]. Furthermore increased expression of the Ras/PI3K/Akt/mTOR pathway also occurs frequently in some cancers as the PIKC3A gene is amplified in approximately 40% of ovarian cancers [325].
Activation of PI3K/PTEN/Akt/mTOR signaling through mutation, inactivation or silencing of pathway components occurs in various malignancies, including liver cancer [327]. Deregulation of this pathway has clinical importance in hepatocellular carcinoma (HCC). For example, data from genomic sequence of HCC samples identified mutations in PIK3CA in 50% of patients with poor prognosis, survival length < 3 years following partial liver resection, and only 10% of the HCC patients with a good prognosis had mutations in PIK3CA [327]. The identified mutations were restricted to residues H1047 in 61.1%, to E545 in 33.3%, and to E542 in 5.5% of cases, and as a consequence this result in gain of enzymatic function and consequently in oncogenic activity of PI3K [327].
Mutations at PTEN in Human Cancer
Germline PTEN mutations are present in approximately 80% of patients with Cowden syndrome [328]. This disease, which is also known as multiple hamartoma syndrome, is a familial syndrome that includes diverse types of cancer conditions including early onset breast cancer. Mutations have been reported to occur at PTEN in breast cancer in varying frequencies (5-21%) [329,330]. Loss of heterozygosity (LOH) is probably more common (30%) [330]. Mutations at certain residues of PTEN, that are associated with Cowden’s disease, affect the ubiquitination of PTEN and prevent nuclear translocation. These mutations leave the phosphatase activity intact [331]. Inhibition of PTEN activity leads to centromere breakage and chromosome instability [332]. Thus PTEN has diverse activities.
Akt and mTOR phosphorylation are frequently detected in ovarian and endometrial cancers. An early occurrence in endometrial cancer is the loss of functional PTEN activity by mutation or other mechanisms, this occurs in approximately 40-80% of patients [333]. Since the loss of PTEN results in activation of Akt, that in turn up-regulates mTOR activity, cancer cells deficient in PTEN are thought to be major targets of mTOR inhibitors.
Alterations in PTEN expression have also been implicated in HCC. The best evidence that strongly supports the connection between PTEN-suppression and liver carcinogenesis comes from genetic studies. All mice with PTEN-deficient hepatocytes exhibited liver adenomas and 66% of them developed HCC [334]. In these mice, hepatocytes were hyperproliferative and displayed an abnormal activation of Akt [334]. Furthermore, although mutations in the PTEN gene rarely occur in HCC, frequent loss of heterozygosity of PTEN allele has been identified in 20-30% of HCC patients [335-338]. In addition, down-regulation of PTEN expression may be partly due to PTEN promoter methylation [339]. PTEN expression plays a critical role in HCC progression and patient’s outcome. Patients with high expression of PTEN had a significantly better overall survival than patients with low PTEN expression [340]. Hepatitis viruses protect hepatocytes from apoptotic cell death by promoting the activation of Ras/PI3K/Akt/mTOR survival pathway [341,342]. Among the four proteins encoded by HBV genome, HBx has been reported to be involved in hepatocarcinogenesis. It has been reported that HBx expression downregulated PTEN expression in hepatocytes [342]. In contrast, PTEN expression in liver cells downregulated HBx-induced PI3K and Akt activities [343]. Therefore, these studies suggest the possible use of PTEN as a target in therapeutic approaches for the treatment of at least those HCC caused by HBV infection.
Mutations and hemizygous deletions of PTEN have been detected in AML and non Hodgkin’s lymphoma (NHL) and other cancers [344,345]. Although many groups have investigated the PTEN-phosphorylation status in leukemia and lymphoma, its relevance concerning Akt-activation is still not clear [344-348]. PTEN phosphorylation as well as low or absent PTEN expression has been observed in AML.
The level of PTEN expression does not always correlate with the degree of phosphorylation of Akt [344]. Although the picture concerning PTEN-inactivation and corresponding Akt-activation is not clear, in vivo studies indicate, that PTEN dysregulation promotes leukemogenesis. PTEN-deficient hematopoietic stem cells display dysregulated cell cycle progression, and the mice develop a myeloproliferative disease which leads to leukemic transformation [346]. In T-acute lymphoblastic leukemia (T-ALL), PTEN-downregulation is also closely correlated with Akt-activation [347,348]. To discern the role of PTEN for Akt-activation, it may be useful to exclude concomitant causes for Akt-activation such as mutant upstream targets and to include the investigation of regulators of PTEN such as c-Myc and Notch/Hes1 [347,348].
PTEN promoter methylation leads to low PTEN expression [349]. In one study, 26% of primary breast cancers had low PTEN levels that correlated with lymph node metastases and poor prognoses [350].
Other mechanisms important in the regulation of PTEN are miRNAs. Certain miRNAs have been shown to regulate PTEN protein expression. mi-214 induces cell survival and may contribute to oncogenesis and drug resistance (see below) by binding the 3’untranslated region (3’UTR) of PTEN which prevents PTEN mRNA translation and leads of overexpression of downstream Akt [351].
In some cancer settings, PTEN and BRAF mutations appear to interact. Two studies papers have highlighted the hypothesis of mutant BRAF- and PTEN-loss-driven carcinogenesis in mouse models [352,353]. In a study by Dhomen et al., inducible expression of B-RafV600E was sufficient to induce multiple melanocytic lesions including skin hyperpigmentation, dysplastic nevi and melanoma [352]. Tumor cells from these B-RafV600E mice displayed both melanoma growth and melanocyte senescence in this system. Approximately 70% of these mice developed melanomas that exhibited histological and molecular characteristics similar to that of human melanoma and were able to colonize the lungs in nude mice [352]. In contrast, another group of researchers generated mice that conditionally-expressed melanocyte-specific BRAF V600E that were only able to induce benign melanocytic hyperplasias and were unable to progress any further over a 15-20 month period [353]. However, BRAF V600E expression in a PTEN gene-silenced background led to the production of melanomas with 100% establishment, short latency and metastasis to lymph nodes and lungs. This development was prevented by the treatment of mice with either the mTOR inhibitor rapamycin or the MEK1/2 inhibitor (PD0325901). Moreover, while combination treatment with rapamycin or PD0325901 led to the reduction of established tumors, upon termination of drug treatment the melanomas reappeared most likely due to the presence of drug resistant melanoma-initiating cells in these mice. Overall, these two papers further validated the mutated B-Raf/MEK/ERK and the PI3K/Akt/mTOR pathways, as promising therapeutic targets in melanoma.
Mutations at SHIP Phosphatase in Human Cancer
The SHIP-1 phosphatase has been implicated as a suppressor of hematopoietic transformation as it basically can prevent Akt-activation [354]. SHIP-1-deficient mice develop a myeloproliferative disease [355] and an inactivating point mutation (SHIP1 V684E) has been observed in approximately one of thirty AML cases [354]. Also another mutation, SHIP1 Q1154L, has been observed in AML, but was even less frequent (1 of 192 cases) [355]. Though some studies confirmed, that SHIP-1 is a leukemia suppressor [354,355] it is unlikely that SHIP1 mutations are a frequent cause of Akt-activation in AML. Disruption of PTEN or SHIP activity by various genetic mechanisms could have vast effects on different processes affecting the sensitivity of different cancers to various therapeutic approaches.
Mutations of AKT in Human Cancer
The roles that Akt plays in cancer are complex. Akt can be activated by genetic mutations, genome amplifications and more commonly by mutations in upstream signaling components. Amplification of Akt-2 was observed in human ovarian carcinomas [356]. Increased levels of Akt are detected in carcinomas of the breast, ovary and prostate and are associated with a poorer prognosis in comparison with tumors that do not display increased levels of expression. AKT is a multi-gene family that consists of AKT1, AKT2 and AKT3. AKT1 has been reported to be mutated in some breast, colorectal, melanoma and ovarian cancers [357-359] (see below). AKT2 is not mutated frequently in human cancer. AKT2 is amplified in certain cancers (e.g., 12.1% ovarian and 2.8% breast carcinomas) [359]. Mutation of AKT3 has been detected in some melanoma samples [360].
AKT1 is mutated in 2 to 8% of breast, 6% of colorectal and 2% of ovarian cancers samples examined in one study [357]. This study documented an Akt mutation that results in an E for a lysine (K) substitution at amino acid 17 (E17K) in the PH domain. Cells with this AKT1 mutation have not been observed to have mutations at PIK3CA; a similar scenario is also frequently observed with RAS and BRAF mutations [361]. This AKT1 mutation alters the electrostatic interactions of Akt-1 which allows it to form new hydrogen bonds with the natural PIP3 ligand [357]. The PH domain mutation confers many different properties to the AKT1 gene. Namely the mutant AKT1 gene has: 1) an altered PH domain conformation, 2) is constitutively-active, 3) has an altered cellular distribution as it is constitutively-associated with the cell membrane, 4) morphologically transforms Rat-1 tissue culture cells and 5) interacts with c-Myc to induce leukemia in E-mu-Myc mice (E-mu = Enhancer of immunoglobulin mu gene, Myc = Myc oncogene originally isolated in avian myelocytomatosis virus) [357]. This PH domain mutated AKT1 gene does not alter its sensitivity to ATP competitive inhibitors, but does alter its sensitivity to allosteric kinase inhibitors [357]. These results demonstrate that targeting the kinase domain of Akt may not be sufficient to suppress the activity of various AKT genes that have mutations in the PH domain. AKT1 and AKT3 E17K mutations in lung cancer have been reported to be rare [362].
Alterations of Akt Expression in Human Cancer.
Akt is often upregulated in cancer cells and its overexpression is associated with a poor prognosis. Increased expression of Akt can result from activating PIK3CA mutations, elimination or decrease in PTEN activity or elevated PKC-epsilon expression. Elevated Akt expression has also been associated with the pathology of pancreatic, glioma and prostate cancers [363-368].
Pancreatic cancer cells have elevated IGF-1R expression and it is well known that Akt regulates IGF-1R expression [369]. This Akt effect on IGF-1R has been suggested to be responsible for the invasiveness of pancreatic cancer cells. Active Src can also activate Akt, and both Src and Akt up-regulate IGF-1R expression in this cancer. It has been demonstrated that IGF-I is expressed in the surrounding stromal cells but not in the cancer cells. This IGF-1 expression may serve as a paracrine growth factor to activate the IGF-1R pathway and the downstream Ras/PI3K/Akt/mTOR pathway in pancreatic cells.
Cyclooxygenase-2 (COX-2) is expressed at high levels in some primary endometrial tumors and is associated with an aggressive phenotype [370]. Akt is elevated and PTEN is often mutated in these cancers which can result in Akt activation. NF-kappaB activation has been shown to have oncogenic effects important in the control of apoptosis, cell cycle, differentiation, cell migration and inflammation [371-383]. Akt may exert its effects through the NF-kappaB pathway and COX-2 is the regulator of this pathway. Akt regulates COX2 gene and protein expression in endometrial cancers. This study was undertaken to examine the involvement of Akt in the regulation of NF- kappaB and COX-2 [370]. The expression of both I-kappaB and phosphorylated I-kappaB were increased in the cells containing mutant PTEN genes. In contrast, there was no difference in NF-kappaB protein abundance between the cell lines, which differed in PTEN gene status. I-kappaB phosphorylation by the PI3K pathway was inhibited by the PI3K inhibitors Wortmannin and LY294002. There was less NF-kappaB nuclear activity, less COX-2 expression and more apoptosis after inhibition of the PI3K pathway. Dominant negative (DN) Akt blocked I-kappaB phosphorylation and decreased COX-2 expression. In contrast, introduction of constitutively-active Akt induced I-kappaB phosphorylation and up-regulated COX-2 expression.
When PTEN is mutated, Akt signals via the NF-kappaB/I-kappaB pathway to induce COX-2 expression in endometrial cancer cells. COX-2 can inhibit apoptosis, increase angiogenesis, and promote invasiveness. COX-2 also promotes inflammation/immunosuppression and conversion of procarcinogens into carcinogens that contribute to tumorigenesis and a malignant phenotype. This study demonstrated that Akt signals via the NF-kappaB/I-kappaB pathway to induce COX2 gene and protein expression in endometrial cancer [370].
Elevated Akt activity can also result in increased phosphorylation of mTOR. mTOR was found to be phosphorylated in AML blasts, along with its two downstream substrates, p70S6K and 4EBP-1, in a PI3K/Akt-dependent fashion [384]. Nevertheless, others failed to detect any relationship between PI3K/Akt signalling upregulation and p70S6K phosphorylation in AML primary cells [385]. This might occur via the Ras/Raf/MEK/ERK pathway activating mTOR via ERK phosphorylation [385]. The Ras/Raf/MEK/ERK pathway is frequently activated in AML [386-388].
Akt is activated in HCC, which results in enhanced resistance to apoptosis through multiple mechanisms [389-392]. As an example, activation of the Akt pathway suppresses transforming growth factor-beta (TGF-beta) induced apoptosis and growth-inhibitory activity of CCAAT/enhancer binding protein alpha (CEBP-alpha). Activation of Akt is a risk factor for early disease recurrence and poor prognosis in patients with HCC [390]. Several mechanisms may be responsible for the activation of Akt. The high frequency of PIK3CA mutations and/or its upregulation in patients with shorter survival might be responsible for the Akt hyperactivation found in HCC with poor prognosis [335-341]. Selective epigenetic silencing of multiple inhibitors of the Ras pathway seems also to be responsible for the activation of Akt found in HCC [327]. Moreover, impaired expression of PTEN is involved in the regulation of Akt activity. Activation of Akt signaling and reduced expression of PTEN has been reported in 40%–60% of human HCC cases [327,335-341]. Some well known risk factors, HBV and HCV seem to utilize the Ras/PI3K/PTEN/Akt/mTOR pathway for the control of hepatocytes survival and viral replication [391,392]. Taken together, these data suggest that Ras/PI3K/Akt/mTOR pathway may represent an important therapeutic target for the treatment of HCC among patients with differing etiologies that lead to the development of this aggressive tumor.
Increased Akt activity due to upstream mutations in growth factor receptor genes or PIK3CA or PTEN may actually render cells and patients sensitive to Akt as well as downstream mTOR inhibitors. The formation of the rapamycin-sensitive mTORC1 complex in certain cancer cells that overexpress activated Akt may be altered in comparison to cells that do not overexpress Akt. In cells that express activated Akt, Akt may phosphorylate TSC2 resulting in its inactivation. In the presence of Akt activation, the mTORC1 complex is formed and downstream p70S6K and 4E-BP1 are phosphorylated, allowing the dissociation of eIF-4E, ribosome biogenesis and protein synthesis. In contrast, in the absence of Akt activation, this complex should not be formed. Rapamycin targets this complex; hence the cells that express elevated levels of activated Akt cells may be more sensitive to rapamycin than the cancer cells that do not express high levels of activated Akt. In the cells that do not express elevated levels of activated Akt, this complex should be transiently assembled after growth factor treatment. In contrast, the assembly of the rapamycin-insensitive mTORC2 complex should be lower in the cells that express elevated levels activated Akt than in those cells that do not as there is equilibrium between the mTORC1 and mTORC2 complexes. The significance of these complex biochemical signaling events is that cancer cells that overexpress activated Akt or lack PTEN/TSC1/TSC2 expression have an Achilles heel with regards to therapeutic intervention as they are highly sensitive to rapamycin treatment.
Mutations of TSC1/TSC2 Genes in Human Cancer.
Mutations in the tumor suppressor genes TSC1 and TSC2 are associated with a dominant genetic disorder, tuberous sclerosis [393,394]. Patients with mutant TSC genes develop benign tumors (hamartomas). In contrast to Cowden’s patients who have germline mutations at PTEN where the patients have a high propensity to develop multiple malignancies, TSC patients rarely develop multiple malignant cancers, and if they do develop malignant cancers they are usually either RCCs or angiomyolipomas [394]. This has been hypothesized to result from a lack of activation of Akt in cells that have mutant TSC1 or TSC2 as mTOR activity is expressed at higher levels which results in inhibition of Akt, perhaps via the effects of p70S6K on IRS1. TSC1 has been shown to be mutated in approximately 15% of urethelial carcinomas (bladder cancers) [394]. RCCs are very sensitive to rapamycin and rapalogs.
Altered Expression of Components Downstream of mTOR in Human Cancer.
mTOR regulates translation by phosphorylating components of the protein synthesis machinery, including p70S6K and 4E-BP1 [395, 396]. p70S6K phosphorylates the 40S ribosomal protein, rpS6, leading to active translation of mRNAs [1-3]. In contrast, 4E-BP1 phosphorylation by mTORC1 on several amino acidic residues (S37; T46; S65; T70) results in the release of the eIF4E [2]. mRNAs differ in their ability to be translated; the length and sequence of the 5’ UTR largely dictates the efficiency with which an mRNA transcript will be translated. Most mRNAs contain short, unstructured GC-poor 5’ UTRs and are efficiently translated. In contrast, long, GC-rich sequences in the 5’ UTR often hinder the ability of the eIF-4E complex to efficiently scan and initiate translation at the start codon. These are called weak mRNAs as previously discussed. Consequently, under normal circumstances these mRNAs are not efficiently translated. However, upon Akt-mediated activation of mTOR, these latter mRNAs are highly and disproportionately translated. Interestingly, many of these weak mRNAs molecules encode oncogenic proteins involved in cell proliferation or survival (e.g., c-Myc, Mcl-1, cyclin-D, VEGF and survivin). These oncogenic mRNAs are therefore tightly regulated at the translation level and their accumulation in cancer cells strongly contributes to the malignant phenotype. These proteins are often subject to the phenomenon of “oncogenic shock” so when an oncogene-addicted cell is treated with a specific inhibitor the expression of these proteins rapidly decays.
Several key proteins are overexpressed as a consequence of mTOR activation including: c-Myc [397-399], cyclin D1 [399], and VEGF [400] and others. Cyclin D1 has been reported to be overexpressed in prostate cancer xenografts and metastases [401], while early stage prostatic lesions possess much lower levels of the protein [402]. A number of reports support the notion that mTOR signaling is a prominent feature of cancer progression and aging, as recurrent tumors have altered expression of a number of molecular targets of rapamycin including the above mentioned genes which encode “weak” mRNAs [403-406]. Hence mTOR inhibitors such as rapamycin may be effective in cancer therapy.
One central molecule involved in cell growth is p70S6K which is regulated by both the Ras/PI3K/PTEN/Akt/mTOR and Ras/Raf/MEK/ERK pathways [2]. The p70S6K gene is amplified in approximately 9% of primary breast cancers and elevated levels of its mRNA transcripts are found in about 41% of the tumors [407,408]. It is known that some PTEN-deficient cells and tumors that are purported to grow in response to activated Akt are hypersensitive to mTOR inhibitors. p70S6K activity is reduced by mTOR inhibitors in PTEN-deficient cells and transgenic PTEN+/- mice [409,410].
Conclusions
In this review, we have discussed the various types of mutations which occur in the Ras/Raf/MEK/ERK and Ras/PI3K/Akt/mTOR pathways and how they can lead to cancer as well as other diseases. We discussed certain classes of genes important in cancer such as oncogenes, tumor suppressor, caretaker and gatekeeper genes. It is obvious that there are many genes which can fit into more than one category. We have introduced the concepts of driver, gatekeeper, passenger, lineage-specific and synthetic lethal mutations so that the reader will have a concept of how these different classes of mutations can contribute to cancer and have been used to identify key interacting genes. We have discussed the concepts of oncogene-addiction, oncogene-bypass and kinase-switching and how they can be important in identifying the key components involved in the growth of the cancer cell and how they may change during treatment with targeted therapy. Mutations at many of the upstream receptor genes or RAS can result in abnormal Raf/MEK/ERK and PI3K/PTEN/Akt/mTOR pathway activation. Hence targeting these cascade components with small-molecule inhibitors may inhibit cell growth. The usefulness of these inhibitors may depend on the mechanism of transformation of the particular cancer. If the tumor exhibits a dependency on the Ras/Raf/MEK/ERK pathway, then it may be sensitive to Raf and MEK inhibitors. In contrast, tumors that do not display enhanced expression of the Ras/Raf/MEK/ERK pathway may not be sensitive to either Raf or MEK inhibitors but if the Ras/PI3K/Akt/mTOR pathway is activated, it may be sensitive to specific inhibitors that target this pathway.
Some scientists and clinicians have considered that the simultaneous targeting of Raf and MEK by individual or dual inhibitors may be more effective in cancer therapy than just targeting Raf or MEK by themselves. This is based in part on the fact that there are intricate feed-back loops from ERK which can inhibit Raf and MEK. For example when MEK1 is targeted, ERK1,2 is inhibited and the negative feed-back loop on MEK is broken and activated MEK accumulates. However, if Raf is also inhibited, it may be possible to completely shut down the pathway. This is a rationale for treatment with both MEK and Raf inhibitors or dual inhibitors. Likewise targeting both PI3K and mTOR may be more effective than targeting either PI3K or mTOR by themselves. If it is a single inhibitor which targets both molecules, such as the new PI3K and mTOR dual inhibitors this becomes a realistic therapeutic option. Finally, an emerging concept is the targeting of two different signal transduction pathways, Raf/MEK/ERK and PI3K/PTEN/Akt/mTOR for example. This has been explored in some preclinical models as well as clinical trials. The rationale for the targeting of both pathways may be dependent on the presence of mutations in either/or both pathways or in upstream Ras in the particular cancer which can activate both pathways. The concepts of targeting these pathways is considered in more detail in an accompanying review [134].
Acknowledgments
MC and GM were supported in part by grants from the Italian “Ministero
dell’Istruzione, dell’Università e della Ricerca (Ministry for Education, Universities and Research) – MIUR” PRIN 2008 and FIRB-MERIT n. RBNE08YYBM. MC was also supported in part by a grant to the CNR from the Italian Ministry of Economy and Finance for
the Project FaReBio di Qualità. LC was supported in part by MIUR-PRIN 2009. ML was supported in part by a grant from the Italian Ministry of Health, Ricerca Finalizzata Stemness 2008 entitled “Molecular Determinants of Stemness and Mesenchymal Phenotype in Breast Cancer”. AMM was supported in part by grants from: MinSan 2008 “Molecular therapy in pediatric sarcomas and leukemias against IGF-IR system: new drugs, best drug-drug interactions, mechanisms of resistance and indicators of efficacy”, MIUR PRIN 2008 (2008THTNLC), and MIUR FIRB 2010 (RBAP10447J-003) and 2011 (RBAP11ZJFA_001). MM was supported in part from the Italian Association for Cancer Research (AIRC), the Cariplo Foundation and the Italian Ministry of Health. AT was supported in part by grants from the Italian “Ministero dell’Istruzione, dell’Università e della Ricerca (Ministry for Education, University and Research) - MIUR - PRIN 2008 and grant from “Sapienza”, University of Rome 2009-11.
Reference
1. Steelman LS, Chappell WH, Abrams SL, Kempf RC, Long J, Laidler P, Mijatovic S, Maksimovic-Ivanic D, Stivala F, Mazzarino MC, Donia M, Fagone P, Malaponte G, Nicoletti F, Libra M, Milella M, et al. Roles of the Raf/MEK/ERK and PI3K/PTEN/Akt/mTOR pathways in controlling growth and sensitivity to therapy-implications for cancer and aging. Aging. 2011; 3: 192-222.
2. Chappell WH, Steelman LS, Long JM, Kempf RC, Abrams SL, Franklin RA, Bäsecke J, Stivala F, Donia M, Fagone P, Malaponte G, Mazzarino MC, Nicoletti F, Libra M, Maksimovic-Ivanic D, Mijatovic S, et al. Ras/Raf/MEK/ERK and PI3K/PTEN/Akt/mTOR inhibitors: Rationale and importance to inhibiting these pathways in human health. Oncotarget. 2011; 2: 135-164.
3. McCubrey JA, Steelman LS, Kempf CR, Chappell W, Abrams SL, Stivala F, Malaponte G, Nicoletti F, Libra M, Bäsecke J, Maksimovic-Ivanic D, Mijatovic S, Montalto G, Cervello M, Cocco L, Martelli AM. Therapeutic resistance resulting from mutations in Raf/MEK/ERK and PI3K/PTEN/Akt/mTOR signaling pathways. J Cell Physiol. 2011; 226: 2762-2781.
4. Little AS, Smith PD, Cook SJ. Mechanisms of acquired resistance to ERK1/2 pathway inhibitors. Oncogene. 2012 May 7. doi: 10.1038/onc.2012.160.
5. Shull AY, Latham-Schwark A, Ramasamy P, Leskoske K, Oroian D, Birtwistle MR, Buckhaults PJ. Novel somatic mutations to PI3K pathway genes in metastatic melanoma. PLoS One. 2012;7:e43369.
6. Muller FL, Colla S, Aquilanti E, Manzo VE, Genovese G, Lee J, Eisenson D, Narurkar R, Deng P, Nezi L, Lee MA, Hu B, Hu J, Sahin E, Ong D, Fletcher-Sananikone E et al. Passenger deletions generate therapeutic vulnerabilities in cancer. Nature. 2012;488:337-342.
7. Blagosklonny MV. Increasing healthy lifespan by suppressing aging in our lifetime: preliminary proposal. Cell Cycle. 2010; 9: 4788-4794.
8. Blagosklonny MV. Progeria, rapamycin and normal aging: recent breakthrough. Aging. 2011; 3: 685-691.
9. Blagosklonny MV. The power of chemotherapeutic engineering: arresting cell cycle and suppressing senescence to protect from mitotic inhibitors. Cell Cycle. 2011; 10: 2295-2298.
10. Negrini S, Gorgoulis VG, Halazonetis TD. Genomic instability--an evolving hallmark of cancer. Nat Rev Mol Cell Biol. 2010; 11: 220-228.
11. Caponigro G, Sellers WR. Advances in the preclinical testing of cancer therapeutic hypotheses. Nat Rev Drug Discov. 2011; 10: 179-187.
12. Torkamani A, Schork NJ. Identification of rare cancer driver mutations by network reconstruction Genome Res. 2009; 19: 1570–1578.
13. Whittaker S, Kirk R, Hayward R, Zambon A, Viros A, Cantarino N, Affolter A, Nourry A, Niculescu-Duvaz D, Springer C, Marais R. Gatekeeper mutations mediate resistance to BRAF-targeted therapies. Sci Transl Med. 2010; 2: 35ra41.
14. Hafsi S, Pezzino FM, Candido S, Ligresti G, Spandidos DA, Soua Z, McCubrey JA, Travali S, Libra M. Gene alterations in the PI3K/PTEN/AKT pathway as a mechanism of drug-resistance. Int J Oncol. 2012, 40: 639-644.
15. Chan DA, Giaccia AJ. Harnessing synthetic lethal interactions in anticancer drug discovery. Nat Rev Drug Discov. 2011; 10: 351-364.
16. Weidle UH, Maisel D, Eick D. Synthetic lethality-based targets for discovery of new cancer therapeutics. Cancer Genomics Proteomics. 2011; 8: 159-171.
17. Janes KA. RUNX1 and its understudied role in breast cancer. Cell Cycle. 2011; 10: 3461-3465.
18. Bauzon F, Zhu L. Racing to block tumorigenesis after pRb loss: an innocuous point mutation wins with synthetic lethality. Cell Cycle. 2010; 9: 2118-2123.
19. Taricani L. Shanahan F, Pierce RH, Guzi TJ, Parry D. Phenotypic enhancement of thymidylate synthetase pathway inhibitors following ablation of Neil1 DNA glycosylase/lyase. Cell Cycle. 2010; 9: 4876-4883.
20. Wellbrock C, Rana S, Paterson H, Pickersgill H, Brummelkamp T, Marais R. Oncogenic BRAF regulates melanoma proliferation through the lineage specific factor MITF. PLoS One. 2008; 3: e2734.
21. Weinstein IB, Joe A. Oncogene Addiction. Cancer Res. 2008; 68: 3077-3080.
22. Engelman JA, Zejnullahu K, Mitsudomi T, Song Y, Hyland C, Park JO, Lindeman N, Gale CM, Zhao X, Christensen J, Kosaka T, Holmes AJ, Rogers AM, Cappuzzo F, Mok T, Lee C et al. MET amplification leads to gefitinib resistance in lung cancer by activating ERBB3 signaling. Science. 2007;316:1039-1043 .
23. Faber AC, Wong KK. Engelman JA. Differences underlying EGFR and HER2 oncogene addiction. Cell Cycle. 2010; 9: 851-852.
24. Lee JT, Shan J, Gu W. Targeting the degradation of cyclin D1 will help to eliminate oncogene addiction. Cell Cycle. 2010; 9: 857-858.
25. Sharma SV, Settleman J. Exploiting the balance between life and death: targeted cancer therapy and “oncogenic shock”. Biochem Pharmacol. 2010; 80: 666-673.
26. Luo J, Solimini NL, Elledge SJ. Principles of cancer therapy: oncogene and non-oncogene addiction. Cell. 2009; 136: 823-837.
27. Carew JS, Kelly KR, Nawrocki ST. Mechanisms of mTOR inhibitor resistance in cancer therapy. Target Oncol. 2011; 6: 17-270.
28. Villanueva J, Vultur A, Lee JT, Somasundaram R, Fukunaga-Kalabis M, Cipolla AK, Wubbenhorst B, Xu X, Gimotty PA, Kee D, Santiago-Walker AE, Letrero R, D’Andrea K, Pushparajan A, Hayden JE, Brown KD et al. Acquired resistance to BRAF inhibitors mediated by a RAF kinase switch in melanoma can be overcome by cotargeting MEK and IGF-1R/PI3K. Cancer Cell.2010;18:683-695 .
29. Simmons GW, Pong WW, Emnett RJ, White CR, Gianino SM, Rodriguez FJ, Gutmann DH. Neurofibromatosis-1 heterozygosity increases microglia in a spatially and temporally restricted pattern relevant to mouse optic glioma formation and growth. J Neuropathol Exp Neurol. 2011; 70: 51-62.
30. Dbouk HA, Backer JM. A beta version of life: p110beta takes center stage. Oncotarget. 2010; 1: 729-733.
31. Fidalgo da Silva E, Ansari SB, Maimaiti J, Barnes EA, Kong-Beltran M, Donoghue DJ, Porter LA. The tumor suppressor tuberin regulates mitotic onset through the cellular localization of cyclin B1. Cell Cycle. 2011; 10: 3129-3139.
32. Searle JS, Li B, Du W. Targeting Rb mutant cancers by inactivating TSC2. Oncotarget. 2010; 1: 228-232.
33. Mavrakis KJ, Leslie CS, Wendel HG. Cooperative control of tumor suppressor genes by a network of oncogenic microRNAs. Cell Cycle. 2011; 10: 2845-2849.
34. Hayashi K, Shibata K, Morita T, Iwasaki K, Watanabe M, Sobue K. Insulin receptor substrate-1/SHP-2 interaction, a phenotype-dependent switching machinery of insulin-like growth factor-I signaling in vascular smooth muscle cells. J Biol Chem. 2004; 279: 40807-40818.
35. Duncan CG, Killela PJ, Payne CA, Lampson B, Chen WC, Liu J, Solomon D, Waldman T, Towers AJ, Gregory SG, McDonald KL, McLendon RE, Bigner DD, Yan H. Integrated genomic analyses identify ERRFI1 and TACC3 as glioblastoma-targeted genes. Oncotarget. 2010; 1: 265-277.
36. Dienstmann R, Martinez P, Felip E. Personalizing therapy with targeted agents in non-small cell lung cancer. Oncotarget. 2011; 2: 165-177.
37. Hart LS, Dolloff NG, Dicker DT, Koumenis C, Christensen JG, Grimberg A, El-Deiry WS. Human colon cancer stem cells are enriched by insulin-like growth factor-1 and are sensitive to figitumumab. Cell Cycle. 2011; 10: 2331-2338.
38. Novosyadlyy R, LeRoith D. Hyperinsulinemia and type 2 diabetes: impact on cancer. Cell Cycle. 2010; 9: 1449-1450.
39. Salzano M, Rusciano MR, Russo E, Bifulco M, Postiglione L, Vitale M. Calcium/calmodulin-dependent protein kinase II (CaMKII) phosphorylates Raf-1 at serine 338 and mediates Ras-stimulated Raf-1 activation. Cell Cycle. 2012; 11: 2100-2106.
40. Nucera C, Lawler J, Hodin R, Parangi S. The BRAFV600E mutation: what is it really orchestrating in thyroid cancer?.Oncotarget. 2010; 1: 751-756.
41. Abraham D, Podar K, Pacher M, Kubicek M, Welzel N, Hemmings BA, Dilworth SM, Mischak H, Kolch W, Baccarini M. Raf-1-associated protein phosphatase 2A as a positive regulator of kinase activation. J Biol Chem. 2000; 275: 22300-22304.
42. Mischak H, Seitz T, Janosch P, Eulitz M, Steen H, Schellerer M, Philipp A, Kolch W. Negative regulation of Raf-1 by phosphorylation of serine 621. Mol Cell Biol. 1996; 10: 5409-5418.
43. Perrotti D, Neviani P. Protein phosphatase 2A (PP2A), a drugable tumor suppressor in Ph1(+) leukemias. Cancer Metastasis Rev. 2008; 27: 159-168.
44. Brennan DF, Dar AC, Hertz NT, Chao WC, Burlingame AL, Shokat KM, Barford D. A Raf-induced allosteric transition of KSR stimulates phosphorylation of MEK. Nature. 2011; 472: 366-369.
45. Ritt DA, Daar IO, Morrison DK. KSR regulation of the Raf-MEK-ERK cascade. Methods Enzymol.2006; 407: 224-237.
46. McKay MM, Ritt DA, Morrison DK. RAF inhibitor-induced KSR1/B-RAF binding and its effects on ERK cascade signaling. Curr Biol. 2011; 21: 563-568.
47. Melissa M McKay, Alyson K Freeman, and Deborah K Morrison Complexity in KSR function revealed by Raf inhibitor and KSR structure studies Small Gtpases. 2011; 2: 276–281.
48. Smith FD, Langeberg LK, Scott JD. Plugging PKA into ERK scaffolds.
Cell Cycle. 2011; 10: 731-732.
49. Pao W, Girard N. New driver mutations in non-small-cell lung cancer. Lancet Oncol. 2011; 12: 175-180.
50. Eblen ST, Slack JK, Weber MJ, Catling AD. Rac-PAK signaling stimulates extracellular signal-regulated kinase (ERK) activation by regulating formation of MEK1-ERK complexes. Mol Cell Biol 2002; 22: 6023-6033.
51, Mack NA. Whalley HJ. Castillo-Lluva S. Malliri A. The diverse roles of Rac signaling in tumorigenesis. Cell Cycle. 2011; 10: 1571-181.
52. Xing J, Ginty DD, Greenberg ME. Coupling of the Ras-MAPK pathway to gene activation by Rsk2, a growth factor regulated CREB kinase. Science. 1996; 273: 959-963.
53. Balan V, Leicht DT, Zhu J, Balan K, Kaplun A, Singh-Gupta V, Qin J, Ruan H, Comb MJ, Tzivion G. Identification of novel in vivo Raf-1 phosphorylation sites mediating positive feedback Raf-1 regulation by extracellular signal-regulated kinase. Mol Biol Cell. 2006; 17: 1141-1153.
54. Dougherty MK, Muller J, Ritt DA, Zhou M, Zhou XZ, Copeland TD, Conrads TP, Veenstra TD, Lu KP, Morrison DK. Regulation of Raf-1 by direct feedback phosphorylation. Mol Cell. 2005; 17: 215-224.
55. Catalanotti, F, Reyes G, Jesenberger V Galabova-Kovacs G, de Matos Simoes R, Carugo O, Baccarini M. A Mek1-Mek2 heterodimer determines the strength and duration of the Erk signal. Nat Struct Mol Biol. 2009; 16: 294-303.
56. Sturm OE, Orton R, Grindlay J, Birtwistle M, Vyshemirsky V, Gilbert D, Calder M, Pitt A, Kholodenko B, Kolch W. The mammalian MAPK/ERK pathway exhibits properties of a negative feedback amplifier. Sci Signal. 2010; 3: ra90.
57. Buday L, Warne PH, Downward J. Downregulation of the Ras activation pathway by MAP kinase phosphorylation of Sos. Oncogene. 1995;11:1327-1331.
58. Li X, Huang Y, Jiang J, Frank SJ. ERK-dependent threonine phosphorylation of EGF receptor modulates receptor downregulation and signaling. Cell Signal. 2008;20:2145-2155.
59. Ekerot M, Stavridis MP, Delavaine L, Mitchell MP, Staples C, Owens DM, Keenan ID, Dickinson RJ, Storey KG, Keyse SM. Negative-feedback regulation of FGF signalling by DUSP6/MKP-3 is driven by ERK1/2 and mediated by Ets factor binding to a conserved site within the DUSP6/MKP-3 gene promoter. Biochem J. 2008;412:287-298.
60. Furukawa T, Sunamura M, Motoi F, Matsuno S, Horii A. Potential tumor suppressive pathway involving DUSP6/MKP-3 in pancreatic cancer. Am J Pathol. 2003;162:1807-1815.
61. Fischer A, Baljuls A, Reinders J, Nekhoroshkova E, Sibilski C, Metz R, Albert S, Rajalingam K, Hekman M, Rapp UR. Regulation of RAF activity by 14-3-3 proteins: RAF kinases associate functionally with both homo- and heterodimeric forms of 14-3-3 proteins. J Biol Chem. 2009; 284: 3183-3194.
62. Schaeffer HJ, Catling AD, Eblen ST, Collier LS, Krauss A, Weber MJ. MP1: a MEK binding partner that enhances enzymatic activation of the MAP kinase cascade. Science. 1998; 281: 1668-1671.
63. Grammatikakis N, Lin JH, Grammatikakis A, Tsichlis PN, Cochran BH. p50(cdc37) acting in concert with Hsp90 is required for Raf-1 function. Mol Cell Biol. 1999; 19: 1661-1672.
64. Goettel JA, Liang D, Hilliard VC, Edelblum KL, Broadus MR, Gould KL, Hanks SK, Polk DB. KSR1 is a functional protein kinase capable of serine autophosphorylation and direct phosphorylation of MEK1 find rest of reference Exp Cell Res. 2011; 317: 452-463.
65. Yeung K, Janosch P, McFerran B, Rose DW, Mischak H, Sedivy JM, Kolch W. Mechanism of suppression of the Raf/MEK/extracellular signal-regulated kinase pathway by the raf kinase inhibitor protein. Mol Cell Biol. 2000; 20: 3079-3085.
66. Mollapour M, Tsutsumi S, Neckers L. Hsp90 phosphorylation, Wee1 and the cell cycle. Cell Cycle. 2010; 9: 2310-2316.
67. Tao YJ. Zheng W. Chaperones and the maturation of steroid hormone receptor complexes. Oncotarget. 2011; 2: 104-106.
68. Sos ML, Michel K, Zander T, Weiss J, Frommolt P, Peifer M, Li D, Ullrich R, Koker M, Fischer F, Shimamura T, Rauh D, Mermel C, Fischer S, Stückrath I, Heynck S, et al. Predicting drug susceptibility of non-small cell lung cancers based on genetic lesions. J Clin Invest. 2009; 119: 1727-1740.
69. Trepel J, Mollapour M, Giaccone G, Neckers L. Targeting the dynamic HSP90 complex in cancer. Nature Reviews Cancer. 2010; 10: 537-549.
70. Rajalingam K, Wunder C, Brinkmann V, Churin Y, Hekman M, Sievers C, Rapp UR, Rudel T. Prohibitin is required for Ras-induced Raf-MEK-ERK activation and epithelial cell migration. Nat Cell Biol. 2005; 7: 837-843.
71. Sanz-Moreno V, Casar B, Crespo P. p38alpha isoform Mxi2 binds to extracellular signal-regulated kinase 1 and 2 mitogen-activated protein kinase and regulates its nuclear activity by sustaining its phosphorylation levels. Mol Cell Biol. 2003; 23: 3079-3090.
72. Casar B, Sanz-Moreno V, Yazicioglu MN, Rodríguez J, Berciano MT, Lafarga M, Cobb MH, Crespo P. Mxi2 promotes stimulus-independent ERK nuclear translocation. EMBO J. 2007; 26: 635-646.
73. Casar B, Rodríguez J, Gibor G, Seger R, Crespo P. Mxi2 sustains ERK1/2 phosphorylation in the nucleus by preventing ERK1/2 binding to phosphatases. Biochem J. 2012; 441: 571-578.
74. Cheng J, Zhang C, Shapiro DJ. A functional serine 118 phosphorylation site in estrogen receptor-alpha is required for down-regulation of gene expression by 17beta-estradiol and 4-hydroxytamoxifen. Endocrinology. 2007; 148: 4634-4641.
75. Chang F, Steelman LS, Lee JT, Shelton JG, Navolanic PM, Blalock WL, Franklin RA, McCubrey JA. Signal transduction mediated by the Ras/Raf/MEK/ERK pathway from cytokine receptors to transcription factors: potential targeting for therapeutic intervention. Leukemia. 2003; 17: 1263-1293.
76. Wang Z, Ge L, Wang M, Carr BI. Phosphorylation regulates Myc expression via prolonged activation of the mitogen-activated protein kinase pathway. J Cell Physiol. 2006; 208: 133-140.
77. Hong J, Zhou J, Fu J, He T, Qin J, Wang L, Liao L, Xu J. Phosphorylation of serine 68 of Twist1 by MAPKs stabilizes Twist1 protein and promotes breast cancer cell invasiveness. Cancer Res. 2011; 71: 3980-3990.
78. Turner FE, Broad S, Khanim FL, Jeanes A, Talma S, Hughes S, Tselepis C, Hotchin NA. Slug regulates integrin expression and cell proliferation in human epidermal keratinocytes. J Biol Chem. 2006; 281: 21321-21331.
79. Chen H, Zhu G, Li Y, Padia RN, Dong Z, Pan ZK, Liu K, Huang S. Extracellular signal-regulated kinase signaling pathway regulates breast cancer cell migration by maintaining slug expression. Cancer Res. 2009; 69: 9228-9235.
80. Nagarajan D, Melo T, Deng Z, Almeida C, Zhao W. ERK/GSK3-beta/Snail signaling mediates radiation-induced alveolar epithelial-to-mesenchymal transition. Free Radic Biol Med. 2012; 52: 983-992.
81. Graham TR, Zhau HE, Odero-Marah VA, Osunkoya AO, Kimbro KS, Tighiouart M, Liu T, Simons JW, O’Regan RM. Insulin-like growth factor-I-dependent up-regulation of ZEB1 drives epithelial-to-mesenchymal transition in human prostate cancer cells. Cancer Res. 2008; 68: 2479-2488.
82. Shin S, Dimitri CA, Yoon SO, Dowdle W, Blenis J. ERK2 but not ERK1 induces epithelial-to-mesenchymal transformation via DEF motif-dependent signaling events. Mol Cell. 2010; 38: 114-1127.
83. Shin S, Blenis J. ERK2/Fra1/ZEB pathway induces epithelial-to-mesenchymal transition. Cell Cycle. 2010; 9: 2483-2484.
84. Smit MA, Peeper DS. Epithelial-mesenchymal transition and senescence: two cancer-related processes are crossing paths. Aging. 2010; 2: 735-741.
85. Browne G, Sayan AE, Tulchinsky E. ZEB proteins link cell motility with cell cycle control and cell survival in cancer. Cell Cycle. 2010; 9: 886-891.
86. Yang DJ, Chung JY, Lee SJ, Park SY, Pyo JH, Ha NC, Yoo MA, Park BJ. Slug, mammalian homologue gene of Drosophila escargot, promotes neuronal-differentiation through suppression of HEB/daughterless. Cell Cycle. 2010; 9: 2789-2802.
87. Jordan NV, Johnson GL, Abell AN. Tracking the intermediate stages of epithelial-mesenchymal transition in epithelial stem cells and cancer. Cell Cycle. 2011; 10: 2865-2873.
88. Baritaki S, Huerta-Yepez S, Sahakyan A, Karagiannides I, Bakirtzi K, Jazirehi A, Bonavida B. Mechanisms of nitric oxide-mediated inhibition of EMT in cancer: inhibition of the metastasis-inducer Snail and induction of the metastasis-suppressor RKIP. Cell Cycle. 2010; 9: 4931-4940.
89. Perez-Losada J, Sanchez-Garcia I. New functions for the Snail family of transcription factors: Two-faced proteins. Cell Cycle. 2010; 9: 2706-2708.
90. Velpula KK, Dasari VR, Tsung AJ, Dinh DH, Rao JS. Cord blood stem cells revert glioma stem cell EMT by down regulating transcriptional activation of Sox2 and Twist1. Oncotarget. 2011; 2: 1028-1042.
91. Gasparotto D, Polesel J, Marzotto A, Colladel R, Piccinin S, Modena P, Grizzo A, Sulfaro S, Serraino D, Barzan L, Doglioni C, Maestro R. Overexpression of TWIST2 correlates with poor prognosis in head and neck squamous cell carcinomas. Oncotarget. 2011; 2: 1165-1175. 92. Eckert MA, Yang J. Targeting invadopodia to block breast cancer metastasis. Oncotarget. 2011; 2: 562-568.
93. Kyriakis JM, Avruch J. Mammalian mitogen-activated protein kinase signal transduction pathways activated by stress and inflammation. Physiol Rev. 2001; 81: 807-869.
94. Roux PP, Blenis J. ERK and p38 MAPK-activated protein kinases: a family of protein kinases with diverse biological functions. Microbiol Mol Biol Rev. 2004; 68: 320-344.
95. Gupta P, Prywes R. ATF1 phosphorylation by the ERK MAPK pathway is required for epidermal growth factor-induced c-jun expression. J Biol Chem. 2002; 277: 50550-50556.
96. Costello PS, Nicolas RH, Watanabe Y, Rosewell I, Treisman R. Ternary complex factor SAP-1 is required for Erk-mediated thymocyte positive selection. Nat Immunol. 2004; 5: 289-298.
97. Costello P, Nicolas R, Willoughby J, Wasylyk B, Nordheim A, Treisman R. Ternary complex factors SAP-1 and Elk-1, but not net, are functionally equivalent in thymocyte development. J Immunol. 2010; 185: 1082-1092.
98. Mylona A, Nicolas R, Maurice D, Sargent M, Tuil D, Daegelen D, Treisman R, Costello P.The essential function for SRF in T cell development reflects its specific coupling to ERK signalling. Mol and Cell Biol. 2010; 31: 267-276.
99. Fuxe J, Vincent T, Garcia de Herreros A. Transcriptional crosstalk between TGF-beta and stem cell pathways in tumor cell invasion: role of EMT promoting Smad complexes. Cell Cycle. 2010; 9: 2363-2374.
100. Arthur JS. MSK activation and physiological roles. Front Biosci. 2008; 13: 5866-5879.
101. Cuevas BD, Abell AN, Witowsky JA, Yujiri T, Johnson NL, Kesavan K, Ware M, Jones PL, Weed SA, DeBiasi RL, Oka Y, Tyler KL, Johnson GL. MEKK1 regulates calpain-dependent proteolysis of focal adhesion proteins for rear-end detachment of migrating fibroblasts. EMBO J. 2003; 22: 3346-3355.
102. Zheng Y, Xia Y, Hawke D, Halle M, Tremblay ML, Gao X, Zhou XZ, Aldape K, Cobb MH, Xie K, He J, Lu Z. FAK Phosphorylation by ERK primes Ras-induced tyrosine dephosphorylation of FAK mediated by PIN1 and PTP-PEST. Mol Cell. 2009; 35: 11-25.
103. Webb DJ, Donais K, Whitmore LA, Thomas SM, Turner CE, Parsons JT, Horwitz AF. FAK-Src signalling through paxillin, ERK and MLCK regulates adhesion disassembly. Nat Cell Biol. 2004; 6: 154-1561.
104. Zheng Y, Yang W, Xia Y, Hawke D, Liu DX, Lu Z. Ras-induced and extracellular signal-regulated kinase 1 and 2 phosphorylation-dependent isomerization of protein tyrosine phosphatase (PTP)-PEST by PIN1 promotes FAK dephosphorylation by PTP-PEST. Mol Cell Biol. 2011; 31: 4258-4269.
105. Abrams SL, Steelman LS, Shelton JG, Wong ET, Chappell WH, Bäsecke J, Stivala F, Donia M, Nicoletti F, Libra M, Martelli AM, McCubrey JA. The Raf/MEK/ERK pathway can govern drug resistance, apoptosis and sensitivity to targeted therapy. Cell Cycle. 2010; 9: 1781-1791.
106. Steelman LS, Abrams SL, Shelton JG, Chappell WH, Bäsecke J, Stivala F, Donia M, Nicoletti F, Libra M, Martelli AM, McCubrey JA. Dominant roles of the Raf/MEK/ERK pathway in cell cycle progression, prevention of apoptosis and sensitivity to chemotherapeutic drugs. Cell Cycle. 2010; 9: 1629-1638.
107. Abrams SL, Steelman LS, Shelton JG, Chappell WH, Bäsecke J, Stivala F, Donia M, Nicoletti F, Libra M, Martelli AM, McCubrey JA. Enhancing therapeutic efficacy by targeting non-oncogene addicted cells with combinations of signal transduction inhibitors and chemotherapy. Cell Cycle. 2010; 9: 1839-46.
108. Kolesnichenko M, Vogt PK. Understanding PLZF: two transcriptional targets, REDD1 and smooth muscle alpha-actin, define new questions in growth control, senescence, self-renewal and tumor suppression. Cell Cycle. 10(5):771-5,
109. Campaner S. Doni M. Verrecchia A. Faga G. Bianchi L. Amati B. Myc, Cdk2 and cellular senescence: Old players, new game. Cell Cycle. 2011; 9: 3655-3661.
110. Bansal R, Nikiforov MA. Pathways of oncogene-induced senescence in human melanocytic cells. Cell Cycle. 2010; 9: 2782-2788.
111. Miller KR, Kelley K, Tuttle R, Berberich SJ. HdmX overexpression inhibits oncogene induced cellular senescence. Cell Cycle. 2010; 9: 3376-3382.
112. Kosar M, Bartkova J, Hubackova S, Hodny Z, Lukas J, Bartek J. Senescence-associated heterochromatin foci are dispensable for cellular senescence, occur in a cell type- and insult-dependent manner and follow expression of p16(ink4a). Cell Cycle. 2011; 10: 457-468.
113. Taylor JR, Lehmann BD, Chappell WH, Abrams SL, Steelman LS, McCubrey JA. Cooperative effects of Akt-1 and Raf-1 on the induction of cellular senescence in doxorubicin or tamoxifen treated breast cancer cells. Oncotarget. 2011; 2: 610-626.
114. Borras C, Monleon D, Lopez-Grueso R, Gambini J, Orlando L, Pallardo FV, Santos E, Vina J, Font de Mora J. RasGrf1 deficiency delays aging in mice. Aging. 2011; 3: 262-276.
115. de Magalhaes JP. A role for Ras signaling in modulating mammalian aging by the GH/IGF1 axis. Aging. 2011; 3: 336-337.
116. Mirisola MG, Longo VD. Conserved role of Ras-GEFs in promoting aging: from yeast to mice. Aging. 2011; 3: 340-343.
117. Ratajczak MZ, Kucia M, Liu R, Shin DM, Bryndza E, Masternak MM, Tarnowski M, Ratajczak J, Bartke A. RasGrf1: genomic imprinting, VSELs, and aging. Aging. 2011; 3: 692-697.
118. Rongo C. Epidermal growth factor and aging: a signaling molecule reveals a new eye opening function. Aging. 2011; 3: 896-905.
119. Qiu W, Sahin F, Iacobuzio-Donahue CA, Garcia-Carracedo D, Wang WM, Kuo CY, Chen D, Arking DE, Lowy AM, Hruban RH, Remotti HE, Su GH. Disruption of p16 and activation of Kras in pancreas increase ductal adenocarcinoma formation and metastasis in vivo. Oncotarget. 2011; 2: 862-873.
120. Davies H, Bignell GR, Cox C, Stephens P, Edkins S, Clegg S, Teague J, Woffendin H, Garnett MJ, Bottomley W, Davis N, Dicks E, Ewing R, Floyd Y, Gray K, Hall S, et al. Mutations of the BRAF gene in human cancer. Nature. 2002; 417: 949-954.
121. Puxeddu E, Moretti S, Elisei R, Romei C, Pascucci R, Martinelli M, Marino C, Avenia N, Rossi ED, Fadda G, Cavaliere A, Ribacchi R, Falorni A, Pontecorvi A, Pacini F, Pinchera A, et al. BRAF(V599E) mutation is the leading genetic event in adult sporadic papillary thyroid carcinomas. J Clin Endocrinol Metab. 2004; 89: 2414-2420.
122. Badalian-Very G, Vergilio JA, Degar BA, MacConaill LE, Brandner B, Calicchio ML, Kuo FC, Ligon AH, Stevenson KE, Kehoe SM, Garraway LA, Hahn WC, Meyerson M, Fleming MD, Rollins BJ. Recurrent BRAF mutations in Langerhans cell histiocytosis. Blood. 2010; 116: 1919-1923.
123. Brose MS, Volpe P, Feldman M, Kumar M, Rishi I, Gerrero R, Einhorn E, Herlyn M, Minna J, Nicholson A, Roth JA, Albelda SM, Davies H, Cox C, Brignell G, Stephens P, et al. BRAF and RAS mutations in human lung cancer and melanoma. Cancer Res. 2002; 62: 6997-7000.
124. Tol J, Nagtegaal ID, Punt CJ. BRAF mutation in metastatic colorectal cancer. N Engl J Med. 2009; 361: 98-99.
125. Tiacci E, Trifonov V, Schiavoni G, Holmes A, Kern W, Martelli MP, Pucciarini A, Bigerna B, Pacini R, Wells VA, Sportoletti P, Pettirossi V, Mannucci R, Elliott O, Liso A, Ambrosetti A, et al. BRAF mutations in hairy-cell leukemia. N Engl J Med. 2011; 364: 2305–2315.
126. Chapman MA, Lawrence MS, Keats JJ, Cibulskis K, Sougnez C, Schinzel AC, Harview CL, Brunet JP, Ahmann GJ, Adli M, Anderson KC, Ardlie KG, Auclair D, Baker A, Bergsagel PL, Bernstein BE, et al. Initial genome sequencing and analysis of multiple myeloma. Nature. 2011; 471: 467-472.
127. Case M, Matheson E, Minto L, Hassan R, Harrison CJ, Bown N, Bailey S, Vormoor J, Hall AG, Irving JA. Mutation of genes affecting the RAS pathway is common in childhood acute lymphoblastic leukemia. Cancer Res. 2008; 68: 6803-6809.
128. Gustafsson B, Angelini S, Sander B, Christensson B, Hemminki K , Kumar R. Mutations in the BRAF and N-ras genes in childhood acute lymphoblastic leukaemia. Leukemia. 2005; 19: 310-312.
129. Davidsson J, Lilljebjorn H, Panagopoulos I, Fioretos T, Johansson B. BRAF mutations are very rare in B- and T-cell pediatric acute lymphoblastic leukemias. Leukemia. 2008; 22: 1619-1621.
130. Lee JW, Yoo NJ, Soung YH, Kim HS, Park WS, Kim SY, Lee JH, Park JY, Cho YG, Kim CJ, Ko YH, Kim SH, Nam SW, Lee JY, Lee SH. BRAF mutations in non-Hodgkin’s lymphoma. Br J Cancer. 2003; 89: 1958-1960.
131. Lohr JG, Stojanov P, Lawrence MS, Auclair D, Chapuy B, Sougnez C, Cruz-Gordillo P, Knoechel B, Asmann YW, Slager SL, Novak AJ, Dogan A, Ansell SM, Link BK, Zou L, Gould J, et al. Discovery and prioritization of somatic mutations in diffuse large B-cell lymphoma (DLBCL) by whole-exome sequencing. Proc Natl Acad Sci U S A. 2012; 109: 3879-3884.
132. Pratilas CA, Hanrahan AJ, Halilovic E, Persaud Y, Soh J, Chitale D, Shigematsu H, Yamamoto H, Sawai A, Janakiraman M, Taylor BS, Pao W, Toyooka S, Ladanyi M, Gazdar A, Rosen N, et al. Genetic predictors of MEK dependence in non-small cell lung cancer. Cancer Res. 2008; 68: 9375-9383.
133. Estep AL, Palmer C, McCormick F, Rauen KA. Mutation Analysis of BRAF, MEK1 and MEK2 in 15 Ovarian Cancer Cell Lines: Implications for Therapy. PLoS ONE. 2007; 2: e1279. doi:10.1371/journal.pone.0001279
134. McCubrey JA, Steelman LS, Chappell WH, Abrams SL, Montalto G, Cervello M, Nicoletti F, Fagone P, Malaponte G, Mazzarino MC, Candido S, Libra M, Bäsecke J, Mijatovic S, Maksimovic-Ivanic D, Cocco L, et al. Ras/Raf/MEK/ERK and PI3K/PTEN/Akt/mTOR Cascade Inhibitors: Targeting these pathways for cancer treatment and overcoming therapy resistance. Oncotarget 2012; In Press.
135. Martelli AM, Evangelisti C, Chappell W, Abrams SL, Bäsecke J, Stivala F, Donia M, Fagone P, Nicoletti F, Libra M, Ruvolo V, Ruvolo P, Kempf CR, Steelman LS, McCubrey JA. Targeting the translational apparatus to improve leukemia therapy: roles of the PI3K/PTEN/Akt/mTOR pathway. Leukemia. 2011; 25: 1064-1079.
136. Martelli AM, Chiarini F, Evangelisti C, Cappellini A, Buontempo F, Bressanin D, Fini M, McCubrey JA. Dual Inhibitors of phosphatidylinositol 3-kinase and mammalian target of rapamycin: a novel therapeutic strategy for acute leukemia treatment? Oncotarget. 2012; 3: 371-394.
137. Cervello M, McCubrey JA, Cusimano A, Lampiasi N, Azzolina A, Montalto G. Targeted therapy for hepatocellular carcinoma: novel agents on the horizon. Oncotarget. 2012; 3: 236-260.
138. Martelli AM, Chiarini F, Evangelisti C, Ognibene A, Bressanin D, Billi AM, Manzoli L, Cappellini A, McCubrey JA. Targeting the Liver Kinase B1/AMP-Dependent Kinase pathway as a Therapeutic Strategy for Hematological Malignancies. Expert Opinion Therapeutic Targets. 2012; 16: 729-742.
139. Adams JR, Schachter NF, Liu JC, Zacksenhaus E, Egan SE. Elevated PI3K signaling drives multiple breast cancer subtypes. Oncotarget. 2011; 2: 435-447.
140. Loi S, Haibe-Kains B, Majjaj S, Lallemand F, Durbecq V, Larsimont D, Gonzalez-Angulo AM, Pusztai L, Symmans WF, Bardelli A, Ellis P, Tutt AN, Gillett CE, Hennessy BT, Mills GB, Phillips WA, et al. PIK3CA mutations associated with gene signature of low mTORC1 signaling and better outcomes in estrogen receptor-positive breast cancer. Proc Natl Acad Sci U S A. 2010; 107: 10208-10213.
141. Chaft JE, Arcila ME, Paik PK, Lau C, Riely GJ, Pietanza MC, Zakowski MF, Rusch V, Sima CS, Ladanyi M, Kris MG. Coexistence of PIK3CA and other oncogene mutations in lung adenocarcinoma-rationale for comprehensive mutation profiling. Mol Cancer Ther. 2012; 11: 485-491.
142. Alessi DR, James SR, Downes CP, Holmes AB, Gaffney PR, Reese CB, Cohen P. Characterization of a 3-phosphoinositide-dependent protein kinase which phosphorylates and activates protein kinase Balpha. Curr Biol. 1997; 7: 261-269.
143. Anderson DH. p85 plays a critical role in controlling flux through the PI3K/PTEN signaling axis through dual regulation of both p110 (PI3K) and PTEN. Cell Cycle. 2010; 9: 2055-2056.
144. Coffer PJ, Woodgett JR. Molecular cloning and characterisation of a novel putative protein-serine kinase related to the cAMP-dependent and protein kinase C families. Eur J Biochem. 1991; 201: 475-481.
145. Brazil DP, Yang ZZ, Hemmings BA. Advances in protein kinase B signalling: AKTion on multiple fronts. Trends Biochem Sci. 2004; 29: 233-242.
146. Meng Q, Xia C, Fang J, Rojanasakul Y, Jiang BH. Role of PI3K and AKT specific isoforms in ovarian cancer cell migration, invasion and proliferation through the p70S6K1 pathway. Cell Signal. 2006; 18: 2262-2271.
147. Gonzalez E, McGraw TE. The Akt kinases: isoform specificity in metabolism and cancer. Cell Cycle. 2009; 8: 2502-2508.
148. Yang WL, Wu CY, Wu J, Lin HK. Regulation of Akt signaling activation by ubiquitination. Cell Cycle. 2010; 9: 487-497.
149. Du K, Montminy M. CREB is a regulatory target for the protein kinase Akt/PKB. J Biol Chem. 1998; 273: 32377-32379.
150. Brennan P, Babbage JW, Burgering BM, Groner B, Reif K, Cantrell DA. Phosphatidylinositol 3-kinase couples the interleukin-2 receptor to the cell cycle regulator E2F. Immunity. 1997; 7: 679-689.
151. Kane LP, Shapiro VS, Stokoe D, Weiss A. Induction of NF-kappaB by the Akt/PKB kinase. Curr Biol. 1999; 9: 601-604.
152. Buitenhuis M, Coffer PJ. The role of the PI3K-PKB signaling module in regulation of hematopoiesis. Cell Cycle. 2009; 8: 560-566.
153. del Peso L, Gonzalez-Garcia M, Page C, Herrera R, Nuñez G. Interleukin-3-induced phosphorylation of BAD through the protein kinase Akt. Science. 1997;278: 687-689.
154. Cross DA, Alessi DR, Cohen P, Andjelkovich M, Hemmings BA. Inhibition of glycogen synthase kinase-3 by insulin mediated by protein kinase B. Nature. 1995; 378: 785-789.
155. Steelman LS, Navolanic P, Chappell WH, Abrams SL, Wong EWT, Martelli AM, Cocco L, Stivala F, Libra M, Nicoletti F, Drobot LB, Franklin RA, McCubrey JA. Involvement of Akt and mTOR in chemotherapeutic- and hormonal-based drug resistance and response to radiation in breast cancer cells. Cell Cycle. 2011; 10; 3003-3015.
156. Sokolosky ML, Stadelman KM, Chappell WH, Abrams SL, Martelli AM, Stivala F, Libra M, Nicoletti F, Drobot LB, Franklin RA, Steelman LS, McCubrey JA. Involvement of Akt-1 and mTOR in Sensitivity of Breast Cancer to Targeted Therapy. Oncotarget. 2011; 2: 538-550.
157. Ambs S, Glynn SA. Candidate pathways linking inducible nitric oxide synthase to a basal-like transcription pattern and tumor progression in human breast cancer. Cell Cycle. 2011; 10: 619-624.
158. Nowicki TS, Zhao H, Darzynkiewicz Z, Moscatello A, Shin E, Schantz S, Tiwari RK, Geliebter J. Downregulation of uPAR inhibits migration, invasion, proliferation, FAK/PI3K/Akt signaling and induces senescence in papillary thyroid carcinoma cells. Cell Cycle. 2011; 10: 100-107.
159. Hayashi M. Yoo YY. Christensen J. Huang LE. Requirement of evading apoptosis for HIF-1alpha-induced malignant progression in mouse cells. Cell Cycle. 2011; 10: 2364-2372.
160. Erol A. Genotoxic stress-mediated cell cycle activities for the decision of cellular fate. Cell Cycle. 2011; 10: 3239-3248.
161. Khalil A, Morgan RN, Adams BR, Golding SE, Dever SM, Rosenberg E, Povirk LF, Valerie K. ATM-dependent ERK signaling via AKT in response to DNA double-strand breaks. Cell Cycle. 2011; 10: 481-491.
162. Fraser M, Harding SM, Zhao H, Coackley C, Durocher D, Bristow RG. MRE11 promotes AKT phosphorylation in direct response to DNA double-strand breaks. Cell Cycle. 2011; 10: 2218-2232.
163. Wani R, Bharathi NS, Field J, Tsang AW, Furdui CM. Oxidation of Akt2 kinase promotes cell migration and regulates G1-S transition in the cell cycle. Cell Cycle. 2011; 10: 3263-3268.
164. Kandouz M, Haidara K, Zhao J, Brisson ML, Batist G. The EphB2 tumor suppressor induces autophagic cell death via concomitant activation of the ERK1/2 and PI3K pathways Cell Cycle. 2010; 9: 398-407.
165. Sayed D, Abdellatif M. AKT-ing via microRNA. Cell Cycle. 2010; 9: 3213-3217.
166. Xiang L, Nakamura Y, Lim YM, Yamasaki Y, Kurokawa-Nose Y, Maruyama W, Osawa T, Matsuura A, Motoyama N, Tsuda L. Tetrahydrocurcumin extends life span and inhibits the oxidative stress response by regulating the FOXO forkhead transcription factor. Aging. 2011; 3: 1098-1109.
167. Chalhoub N, Baker SJ. PTEN and the PI3-kinase pathway in cancer. Ann Rev Pathol Mech Dis. 2009; 4: 127-150.
168. Diaz-Meco MT, Abu-Baker S. The Par-4/PTEN connection in tumor suppression. Cell Cycle. 2009; 8: 2518-2522.
169. Mahimainathan L, Choudhury GG. Inactivation of platelet-derived growth factor receptor by the tumor suppressor PTEN provides a novel mechanism of action of the phosphatase. J Biol Chem. 2004; 279: 15258-15268.
170. Gupta A, Yang Q, Pandita RK, Hunt CR, Xiang T, Misri S, Zeng S, Pagan J, Jeffery J, Puc J, Kumar R, Feng Z, Powell SN, Bhat A, Yaguchi T, Wadhwa R, et al. Cell cycle checkpoint defects contribute to genomic instability in PTEN deficient cells independent of DNA DSB repair. Cell Cycle. 2009; 8: 2198-2210.
171. Poon JS, Eves R, Mak AS. Both lipid- and protein-phosphatase activities of PTEN contribute to the p53-PTEN anti-invasion pathway. Cell Cycle. 2010; 9: 4450-4454.
172. Silva A, Yunes JA, Cardoso BA, Martins LR, Jotta PY, Abecasis M, Nowill AE, Leslie NR, Cardoso AA, Barata JT. PTEN posttranslational inactivation and hyperactivation of the PI3K/Akt pathway sustain primary T cell leukemia viability. J Clin Invest. 2008; 118: 3762-3774.
173. Gao T, Furnari F, Newton AC. PHLPP: a phosphatase that directly dephosphorylates Akt, promotes apoptosis, and suppresses tumor growth. Mol Cell. 2005; 18: 13-24.
174. Damen JE, Liu L, Rosten P, Humphries RK, Jefferson AB, Majerus PW, Krystal G. The 145-kDa protein induced to associate with Shc by multiple cytokines is an inositol tetraphosphate and phosphatidylinositol 3,4,5-triposphate 5-phosphatase. Proc Natl Acad Sci USA. 1996; 93: 1689-1693.
175. Lioubin MN, Algate PA, Tsai S, Carlberg K, Aebersold A, Rohrschneider LR. p150Ship, a signal transduction molecule with inositol polyphosphate-5-phosphatase activity. Genes & Devel. 1996; 10: 1084-1095.
176. Muraille E, Pesesse X, Kuntz C, Erneux C. Distribution of the src-homology-2-domain-containing inositol 5-phosphatase SHIP-2 in both non-haemopoietic and haemopoietic cells and possible involvement in SHIP-2 in negative signaling of B-cells. Biochem J. 1999; 342: 697-705.
177. Taylor V, Wong M, Brandts C, Reilly L, Dean NM, Cowsert LM, Moodie S, Stokoe D. 5’phospholipid phosphatase SHIP-2 causes protein kinase B inactivation and cell cycle arrest in glioblastoma cells. Mol Cell Biol. 2000; 20: 6860-6871.
178. Simioni C, Neri LM, Tabellini G, Ricci F, Bressanin D, Chiarini F, Evangelisti C, Cani A, Tazzari PL, Melchionda F, Pagliaro P, Pession A, McCubrey JA, Capitani S, Martelli AM. Cytotoxic activity of the novel Akt inhibitor, MK-2206, in T-cell acute lymphoblastic leukemia. Leukemia. 2012; May 22. doi: 10.1038/leu.2012.136
179. Krymskaya VP, Goncharova EA. PI3K/mTORC1 activation in hamartoma syndromes: therapeutic prospects. Cell Cycle. 2009; 8: 403-413.
180. Vazquez-Martin A, Oliveras-Ferraros C, Lopez-Bonet E, Menendez JA. AMPK: Evidence for an energy-sensing cytokinetic tumor suppressor. Cell Cycle. 2009; 8: 3679-3683.
181. Tamburini J, Green AS, Chapuis N, Bardet V, Lacombe C, Mayeux P, Bouscary D. Targeting translation in acute myeloid leukemia: a new paradigm for therapy? Cell Cycle. 2009; 8: 3893-3899.
182. Sato T, Nakashima A, Guo L, Tamanoi F. Specific activation of mTORC1 by Rheb G-protein in vitro involves enhanced recruitment of its substrate protein. J Biol Chem. 2009; 284: 12783-12791.
183. Carracedo A, Ma L, Teruya-Feldstein J, Rojo F, Salmena L, Alimonti A, Egia A, Sasaki AT, Thomas G, Kozma SC, Papa A, Nardella C, Cantley LC, Baselga J, Pandolfi PP. Inhibition of mTORC1 leads to MAPK pathway activation through a PI3K-dependent feedback loop in human cancer. J Clin Invest. 2008; 118: 3065-3074.
184. Martelli AM, Tabellini G, Ricci F, Evangelisti C, Chiarini F, Bortul R, McCubrey JA, Manzoli FA. PI3K/AKT/mTORC1 and MEK/ERK signaling in T-cell acute lymphoblastic leukemia: New options for targeted therapy. Adv Enzyme Regul. 2011; In Press.
185. Hresko RC, Mueckler M. mTOR.RICTOR is the Ser473 kinase for Akt/protein kinase B in 3T3-L1 adipocytes. J Biol Chem. 2005; 280: 40406-40416.
186. Gomez-Baldo L, Schmidt S, Maxwell CA, Bonifaci N, Gabaldon T, Vidalain PO, Senapedis W, Kletke A, Rosing M, Barnekow A, Rottapel R, Capella G, Vidal M, Astrinidis A, Piekorz RP, Pujana MA. TACC3-TSC2 maintains nuclear envelope structure and controls cell division. Cell Cycle. 2010; 9: 1143-1155.
187. Bhatia B, Nahle Z, Kenney AM. Double trouble: when sonic hedgehog signaling meets TSC inactivation. Cell Cycle. 2010; 9: 456-459.
188. Kofman AE. McGraw MR. Payne CJ. Rapamycin increases oxidative stress response gene expression in adult stem cells. Aging. 2012;4: 279-289.
189. Vazquez-Martin A, Cufi S, Oliveras-Ferraros C, Menendez JA. Raptor, a positive regulatory subunit of mTOR complex 1, is a novel phosphoprotein of the rDNA transcription machinery in nucleoli and chromosomal nucleolus organizer regions (NORs). Cell Cycle. 2011; 10: 3140-3152.
190. Peterson TR, Laplante M, Thoreen CC, Sancak Y, Kang SA, Kuehl WM, Gray NS, Sabatini DM. DEPTOR is an mTOR inhibitor frequently overexpressed in multiple myeloma cells and required for their survival. Cell. 2009; 137: 873-886.
191. Jansen M, Ten Klooster JP, Offerhaus GJ, Clevers H. LKB1 and AMPK family signaling: the intimate link between cell polarity and energy metabolism. Physiol Rev. 2009; 89: 777-798.
192. Shaw RJ, Bardeesy N, Manning BD, Lopez L, Kosmatka M, DePinho RA, Cantley LC. The LKB1 tumor suppressor negatively regulates mTOR signaling. Cancer Cell. 2004; 6: 91–99.
193. Shaw RJ, Lamia KA, Vasquez D, Koo SH, Bardeesy N, Depinho RA, Montminy M, Cantley LC. The kinase LKB1 mediates glucose homeostasis in liver and therapeutic effects of metformin. Science. 2005; 310: 1642-1660.
194. van der Velden YU, Haramis AP. Insights from model organisms on the functions of the tumor suppressor protein LKB1: zebrafish chips in. Aging. 2011; 3: 363-367.
195. Godlewski J, Bronisz A, Nowicki MO, Chiocca EA, Lawler S. microRNA-451: A conditional switch controlling glioma cell proliferation and migration. Cell Cycle. 2010; 9: 2742-2748.
196. Chen V, Shtivelman E.CC3/TIP30 regulates metabolic adaptation of tumor cells to glucose limitation. Cell Cycle. 2010; 9: 4941-4953.
197. Chiacchiera F, Simone C. The AMPK-FoxO3A axis as a target for cancer treatment. Cell Cycle. 2010; 9: 1091-1096.
198. Guo D, Cloughesy TF, Radu CG, Mischel PS. AMPK: A metabolic checkpoint that regulates the growth of EGFR activated glioblastomas. Cell Cycle. 2010; 9: 211-212.
199. Saha AK, Xu XJ, Balon TW, Brandon A, Kraegen EW, Ruderman NB. Insulin resistance due to nutrient excess: is it a consequence of AMPK downregulation?. Cell Cycle. 2011; 10: 3447-3451.
200. Amato S, Man HY. Bioenergy sensing in the brain: the role of AMP-activated protein kinase in neuronal metabolism, development and neurological diseases. Cell Cycle. 2011; 10: 3452-3460.
201. Malik SA, Marino G, BenYounes A, Shen S, Harper F, Maiuri MC, Kroemer G. Neuroendocrine regulation of autophagy by leptin.Cell Cycle. 2011; 10: 2917-2923.
202. Mounier R, Lantier L, Leclerc J, Sotiropoulos A, Foretz M, Viollet B. Antagonistic control of muscle cell size by AMPK and mTORC1. Cell Cycle. 2011; 10: 2640-2646.
203. Vazquez-Martin A, Oliveras-Ferraros C, Cufi S, Menendez JA. Polo-like kinase 1 regulates activation of AMP-activated protein kinase (AMPK) at the mitotic apparatus. Cell Cycle. 2011; 10: 1295-1302.
204. Polak, P, Cybulski N, Feige JN, Auwerx J, Rüegg MA, Hall MN. Adipose-specific knockout of raptor results in lean mice with enhanced mitochondrial respiration. Cell Metab. 2008; 8: 399–410.
205. Harrison DE, Strong R, Sharp ZD, Nelson JF, Astle CM, Flurkey K, Nadon NL, Wilkinson JE, Frenkel K, Carter CS, Pahor M, Javors MA, Fernandez E, Miller RA. Rapamycin fed late in life extends lifespan in genetically heterogeneous mice. Nature. 2009; 460: 392–395.
206. Evans JM, Donnelly LA, Emslie-Smith AM, Alessi DR, Morris AD. Metformin and reduced risk of cancer in diabetic patients. BMJ. 2005;330:1304–1305.
207. Shackelford DB, Shaw RJ. The LKB1-AMPK pathway: metabolism and growth control in tumour suppression. Nat Rev Cancer. 2009; 9: 563-575.
208. Miller RA, Harrison DE, Astle CM, Baur JA, Boyd AR, de Cabo R, Fernandez E, Flurkey K, Javors MA, Nelson JF, Orihuela CJ, Pletcher S, Sharp ZD, Sinclair D, Starnes JW, Wilkinson JE, et al. Rapamycin, but not resveratrol or simvastatin, extends life span of genetically heterogeneous mice. J Gerontol A Biol Sci Med Sci. 2011; 66: 191-201.
209. Liu B. Fan Z. Edgerton SM. Yang X, Lind SE, Thor AD. Potent anti-proliferative effects of metformin on trastuzumab-resistant breast cancer cells via inhibition of erbB2/IGF-1 receptor interactions. Cell Cycle. 2011; 10: 2959-2966.
210. Green AS, Chapuis N, Lacombe C, Mayeux P, Bouscary D, Tamburini J. LKB1/AMPK/mTOR signaling pathway in hematological malignancies: from metabolism to cancer cell biology. Cell Cycle. 2011; 10: 2115-2120.
211. Vazquez-Martin A, Oliveras-Ferraros C, Cufi S, Martin-Castillo B, Menendez JA. Metformin activates an ataxia telangiectasia mutated (ATM)/Chk2-regulated DNA damage-like response. Cell Cycle. 2011; 10: 1499-1501.
212. Mashhedi H, Blouin MJ, Zakikhani M, David S, Zhao Y, Bazile M, Birman E, Algire C, Aliaga A, Bedell BJ, Pollak M. Metformin abolishes increased tumor (18)F-2-fluoro-2-deoxy-D-glucose uptake associated with a high energy diet. Cell Cycle. 2011; 10: 2770-2778.
213.Oliveras-Ferraros C, Cufi S, Vazquez-Martin A, Torres-Garcia VZ, Del Barco S, Martin-Castillo B, Menendez JA. Micro(mi)RNA expression profile of breast cancer epithelial cells treated with the anti-diabetic drug metformin: induction of the tumor suppressor miRNA let-7a and suppression of the TGFbeta-induced oncomiR miRNA-181a. Cell Cycle. 2011; 10: 1144-1451.
214. Martin-Castillo B. Vazquez-Martin A. Oliveras-Ferraros C. Menendez JA. Metformin and cancer: doses, mechanisms and the dandelion and hormetic phenomena. Cell Cycle. 2010; 9: 1057-1064.
215. Cufi S, Vazquez-Martin A, Oliveras-Ferraros C, Martin-Castillo B, Joven J, Menendez JA. Metformin against TGFβ-induced epithelial-to-mesenchymal transition (EMT): from cancer stem cells to aging-associated fibrosis. Cell Cycle. 2010; 9: 4461-4468.
216. Anisimov VN, Egormin PA, Piskunova TS, Popovich IG, Tyndyk ML, Yurova MN, Zabezhinski MA, Anikin IV, Karkach AS, Romanyukha AA. Metformin extends life span of HER-2/neu transgenic mice and in combination with melatonin inhibits growth of transplantable tumors in vivo. Cell Cycle. 2010; 9: 188-197.
217. Fuchs C, Rosner M, Dolznig H, Mikula M, Kramer N, Hengstschläger M. Tuberin and PRAS40 are anti-apoptotic gatekeepers during early human amniotic fluid stem-cell differentiation. Hum Mol Genet. 2012; 21: 1049-1061.
218. Oh WJ, Jacinto E. mTOR complex 2 signaling and functions. Cell Cycle. 2011; 10: 2305-2316.
219. Memmott RM, Dennis PA. Akt-dependent and -independent mechanisms of mTOR regulation in cancer. Cell Signal. 2009; 21: 656-664.
220. Rosner M, Hengstschlager M. mTOR protein localization is cell cycle-regulated.Cell Cycle. 2011; 10: 3608-3610.
221. Jiang Y. mTOR goes to the nucleus. Cell Cycle. 2010; 9: 868.
222. Panieri E, Toietta G, Mele M, Labate V, Ranieri SC, Fusco S, Tesori V, Antonini A, Maulucci G, De Spirito M, Galeotti T, Pani G. Nutrient withdrawal rescues growth factor-deprived cells from mTOR-dependent damage. Aging. 2010; 2: 487-503.
223. Lisse TS, Hewison M. Vitamin D: a new player in the world of mTOR signaling. Cell Cycle. 2011; 10: 1888-1889.
224. Blagosklonny MV. Calorie restriction: decelerating mTOR-driven aging from cells to organisms (including humans). Cell Cycle. 2010; 9: 683-638.
225. Rodriguez A. Catalan V. Gomez-Ambrosi J. Fruhbeck G. Aquaglyceroporins serve as metabolic gateways in adiposity and insulin resistance control. Cell Cycle. 2011; 10: 1548-1556.
226. Habib SL. Tuberin and mTOR, a key apoptotic pathway in diabetes. Cell Cycle. 2011; 10: 2237-2238.
227. Zeng LH, McDaniel S, Rensing NR, Wong M. Regulation of cell death and epileptogenesis by the mammalian target of rapamycin (mTOR): a double-edged sword? Cell Cycle. 2010; 9: 2281-2285.
228. D’Arcangelo G. Rapamycin treatment suppresses epileptogenic activity in conditional Pten knockout mice. Cell Cycle. 2010; 9: 2487-2488.
229. Conn CS, Qian SB. mTOR signaling in protein homeostasis: less is more?. Cell Cycle. 2011; 10: 1940-1947.
230. Tsang CK, Liu H, Zheng XF. mTOR binds to the promoters of RNA polymerase I- and III-transcribed genes. Cell Cycle. 2010; 9: 953-957.
231. Kantidakis T, White RJ. A feedback loop between mTOR and tRNA expression?. Cell Cycle. 2010; 9: 2934-2935.
232. Dalvai M, Schubart K, Besson A, Matthias P. Oct1 is required for mTOR-induced G1 cell cycle arrest via the control of p27(Kip1) expression. Cell Cycle. 2010; 9: 3933-3944.
233. Salmond RJ, Zamoyska R. How does the mammalian target of rapamycin (mTOR) influence CD8 T cell differentiation? Cell Cycle. 2010; 9: 2952-2957.
234. Rao RR, Li Q, Shrikant PA. Fine-tuning CD8(+) T cell functional responses: mTOR acts as a rheostat for regulating CD8(+) T cell proliferation, survival and differentiation? Cell Cycle. 2010; 9: 2996-3001.
235. Woods TC. Regulation of cell migration by mTOR is mediated through changes in p27Kip1 phosphorylation. Cell Cycle. 2010; 9: 2057-2058.
236. Godlewski J, Bronisz A, Nowicki MO, Chiocca EA, Lawler S. microRNA-451: A conditional switch controlling glioma cell proliferation and migration. Cell Cycle. 2010; 9: 2742-2748.
237. Adhikari D, Liu K. mTOR signaling in the control of activation of primordial follicles. Cell Cycle. 2010; 9: 1673-1674.
238. Rodriguez-Jimenez FJ, Moreno-Manzano V, Mateos-Gregorio P, Royo I, Erceg S, Murguia JR, Sanchez-Puelles JM. FM19G11: A new modulator of HIF that links mTOR activation with the DNA damage checkpoint pathways. Cell Cycle. 2010; 9: 2803-2813.
239. Leontieva OV, Blagosklonny MV. DNA damaging agents and p53 do not cause senescence in quiescent cells, while consecutive re-activation of mTOR is associated with conversion to senescence. Aging. 2010; 2: 924-935.
240. Serrano M. Shifting senescence into quiescence by turning up p53. Cell Cycle. 2010; 9: 4256-4257.
241. Romanov VS, Abramova MV, Svetlikova SB, Bykova TV, Zubova SG, Aksenov ND, Fornace AJ Jr, Pospelova TV, Pospelov VA. p21(Waf1) is required for cellular senescence but not for cell cycle arrest induced by the HDAC inhibitor sodium butyrate. Cell Cycle. 2010; 9: 3945-3955.
242. Dulic V. Be quiet and you’ll keep young: does mTOR underlie p53 action in protecting against senescence by favoring quiescence? Aging. 2011; 3: 3-4.
243. Wang C, Maddick M, Miwa S, Jurk D, Czapiewski R, Saretzki G, Langie SA, Godschalk RW, Cameron K, von Zglinicki T. Adult-onset, short-term dietary restriction reduces cell senescence in mice. Aging. 2010; 2: 555-566.
244. Darzynkiewicz Z. Another “Janus paradox” of p53: induction of cell senescence versus quiescence. Aging. 2010; 2: 329-330.
245. Schug TT. mTOR favors senescence over quiescence in p53-arrested cells. Aging. 2010; 2: 327-328.
246. Martins I, Galluzzi L, Kroemer G. Hormesis, cell death and aging. Aging. 2011; 3: 821-828.
247. Galluzzi L, Kepp O, Kroemer G. Aging. TP53 and MTOR crosstalk to regulate cellular senescence. 2010; 2: 535-537.
248. Narita M. Quality and quantity control of proteins in senescence. Aging. 2010; 2: 311-314.
249. Sahin M. Eph receptor and mTOR pathway crosstalk: implications for cancer.
Cell Cycle. 2010; 9: 2053-2054.
250. Gruppuso PA, Boylan JM, Sanders JA. The physiology and pathophysiology of rapamycin resistance: implications for cancer. Cell Cycle. 2011; 10: 1050-1058.
251. Blagosklonny MV. Why the disposable soma theory cannot explain why women live longer and why we age. Aging. 2010; 2: 884-887.
252. Wesierska-Gadek J. mTOR and its link to the picture of Dorian Gray - re-activation of mTOR promotes aging. Aging. 2010; 2: 892-893.
253. Zhao C, Vollrath D. mTOR pathway activation in age-related retinal disease. Aging. 2011; 3: 346-347.
254. Maki CG. Decision-making by p53 and mTOR .Aging. 2010; 2: 324-326.
255. Blagosklonny MV. Why men age faster but reproduce longer than women: mTOR and evolutionary perspectives. Aging. 2010; 2: 265-273.
256. Blagosklonny MV. Why human lifespan is rapidly increasing: solving “longevity riddle” with “revealed-slow-aging” hypothesis. Aging. 2010; 2: 177-182.
257. Blagosklonny MV. Rapamycin and quasi-programmed aging: four years later. Cell Cycle. 2010; 9: 1859-1862.
258. Blagosklonny MV. Increasing healthy lifespan by suppressing aging in our lifetime: preliminary proposal. Cell Cycle. 2010; 9: 4788-4794.
259. Foster DA. Reduced mortality and moderate alcohol consumption: the phospholipase D-mTOR connection. Cell Cycle. 2010; 9: 1291-1294.
260. Pani G. From growing to secreting: new roles for mTOR in aging cells. Cell Cycle. 2011; 10: 2450-2453.
261. Santini E, Valjent E, Fisone G. mTORC1 signaling in Parkinson’s disease and L-DOPA-induced dyskinesia: A sensitized matter. Cell Cycle. 2010; 9: 2713-2718.
262. Rambold AS, Lippincott-Schwartz J. Starved cells use mitochondria for autophagosome biogenesis. Cell Cycle. 2010; 9: 3633-3634.
263. Wu JJ, Quijano C, Wang J, Finkel T. Metabolism meets autophagy.
Cell Cycle. 2010; 9: 4780-4781.
264. Ciavarra G, Zacksenhaus E. Multiple pathways counteract cell death induced by RB1 loss: implications for cancer. Cell Cycle. 2011; 10: 1533-1539.
265. Castello-Cros R, Bonuccelli G, Molchansky A, Capozza F, Witkiewicz AK, Birbe RC, Howell A, Pestell RG, Whitaker-Menezes D, Sotgia F, Lisanti MP. Matrix remodeling stimulates stromal autophagy, “fueling” cancer cell mitochondrial metabolism and metastasis. Cell Cycle. 2011; 10: 2021-2034.
266. Martinez-Outschoorn UE, Pavlides S, Whitaker-Menezes D, Daumer KM, Milliman JN. Chiavarina B, Migneco G, Witkiewicz AK, Martinez-Cantarin MP, Flomenberg N, Howell A, Pestell RG, Lisanti MP, Sotgia F. Tumor cells induce the cancer associated fibroblast phenotype via caveolin-1 degradation: implications for breast cancer and DCIS therapy with autophagy inhibitors. Cell Cycle. 2010; 9: 2423-2433.
267. Iozzo RV. Autophagic tumor stroma: a biofuel for cancer growth. Cell Cycle. 2011; 10: 3231-3232.
268. Richardson AD, Scott DA. Reversing the Warburg effect through stromal autophagy. Cell Cycle. 2011; 10: 2830-2831.
269. Korkaya H, Wicha MS. Inflammation and autophagy conspire to promote tumor growth. Cell Cycle. 2011; 10: 2623-2624.
270. Martinez-Outschoorn UE, Whitaker-Menezes D, Pavlides S, Chiavarina B, Bonuccelli G, Casey T, Tsirigos A, Migneco G, Witkiewicz A, Balliet R, Mercier I, Wang C, Flomenberg N, Howell A, Lin Z, Caro J, et al. The autophagic tumor stroma model of cancer or “battery-operated tumor growth”: A simple solution to the autophagy paradox. Cell Cycle. 2010; 9: 4297-4306.
271. Pavlides S, Tsirigos A, Migneco G, Whitaker-Menezes D, Chiavarina B, Flomenberg N, Frank PG, Casimiro MC, Wang C, Pestell RG, Martinez-Outschoorn UE, Howell A, Sotgia F, Lisanti MP. The autophagic tumor stroma model of cancer: Role of oxidative stress and ketone production in fueling tumor cell metabolism. Cell Cycle. 2010; 9: 3485-3505.
272. Chiavarina B, Whitaker-Menezes D, Migneco G, Martinez-Outschoorn UE, Pavlides S, Howell A, Tanowitz HB, Casimiro MC, Wang C, Pestell RG, Grieshaber P, Caro J, Sotgia F, Lisanti MP. HIF1-alpha functions as a tumor promoter in cancer associated fibroblasts, and as a tumor suppressor in breast cancer cells: Autophagy drives compartment-specific oncogenesis. Cell Cycle. 2010; 9: 3534-3551.
273. Martinez-Outschoorn UE, Trimmer C, Lin Z, Whitaker-Menezes D, Chiavarina B, Zhou J, Wang C, Pavlides S, Martinez-Cantarin MP, Capozza F, Witkiewicz AK, Flomenberg N, Howell A, Pestell RG, Caro J, Lisanti MP, et al. Autophagy in cancer associated fibroblasts promotes tumor cell survival: Role of hypoxia, HIF1 induction and NFkappaB activation in the tumor stromal microenvironment.Cell Cycle. 2010; 9: 3515-3533.
274. Bonuccelli G, Tsirigos A, Whitaker-Menezes D, Pavlides S, Pestell RG, Chiavarina B, Frank PG, Flomenberg N, Howell A, Martinez-Outschoorn UE, Sotgia F, Lisanti MP. Ketones and lactate “fuel” tumor growth and metastasis: Evidence that epithelial cancer cells use oxidative mitochondrial metabolism. Cell Cycle. 2010; 9: 3506-3514.
275. Puissant A, Robert G, Auberger P. Targeting autophagy to fight hematopoietic malignancies. Cell Cycle. 2010; 9: 3470-3478.
276. Watson AS, Mortensen M, Simon AK. Autophagy in the pathogenesis of myelodysplastic syndrome and acute myeloid leukemia. Cell Cycle. 2011; 10: 1719-1725.
277. Cluzeau T, Robert G, Puissant A, Jean-Michel K, Cassuto JP, Raynaud S, Auberger P. Azacitidine-resistant SKM1 myeloid cells are defective for AZA-induced mitochondrial apoptosis and autophagy. Cell Cycle. 2011; 10: 2339-2343.
278. Boehrer S, Lainey E, Kroemer G. Coordinated epigenetic regulation of autophagy and apoptosis. Cell Cycle. 2011; 10: 2832-2833.
279. Shen S, Kepp O, Martins I, Vitale I, Souquere S, Castedo M, Pierron G, Kroemer G. Defective autophagy associated with LC3 puncta in epothilone-resistant cancer cells.
Cell Cycle. 2010; 9: 377-383.
280. Galluzzi L, Morselli E, Kepp O, Maiuri MC, Kroemer G. Defective autophagy control by the p53 rheostat in cancer. Cell Cycle. 2010; 9: 250-255.
281. García-Martínez JM, Alessi DR. mTOR complex 2 (mTORC2) controls hydrophobic motif phosphorylation and activation of serum-and glucocorticoid-induced protein kinase 1 (SGK1). Biochem. J. 2008; 416: 375-385.
282. Ikenoue T, Inoki K, Yang Q, Zhou X, Guan KL. Essential function of TORC2 in PKC and Akt turn motif phosphorylation, maturation and signalling. EMBO J. 2008; 27: 1919-1931.
283. Mamane Y, Petroulakis E, LeBacquer O, Sonenberg N. mTOR, translation initiation and cancer. Oncogene. 2006; 25: 6416-6422.
284. Zinzalla V, Stracka D, Oppliger W, Hall MN. Activation of mTORC2 by association with the ribosome. Cell. 2011; 144; 757-768.
285. Oh WJ, Wu CC, Kim SJ, Facchinetti V, Julien LA, Finlan M, Roux PP, Su B, Jacinto E. mTORC2 can associate with ribosomes to promote cotranslational phosphorylation and stability of nascent Akt polypeptide. EMBO J. 2010; 29: 3939-3951.
286. Oh WJ, Jacinto E. mTOR complex 2 signaling and functions. Cell Cycle. 2011; 10: 2305-2316.
287. Benjamin D, Colombi M, Moroni C, Hall MN. Rapamycin passes the torch: a new generation of mTOR inhibitors. Nat Rev Drug Discov. 2011; 10: 868-880.
288. Sarbassov DD, Ali SM, Kim DH, Guertin DA, Latek RR, Erdjument-Bromage H, Tempst P, Sabatini DM. Rictor, a novel binding partner of mTOR, defines a rapamycin-insensitive and raptor-independent pathway that regulates the cytoskeleton. Curr Biol. 2004; 14: 1296-1302.
289. Zhou H, Huang S. Role of mTOR signaling in tumor cell motility, invasion and metastasis. Curr Protein Pept Sci. 2011; 12: 30-42.
290. Shah OJ, Wang Z, Hunter T. Inappropriate activation of the TSC/Rheb/mTOR/S6K cassette induces IRS1/2 depletion, insulin resistance, and cell survival deficiencies. Curr Biol. 2004; 14: 1650-1656.
291. Lang SA, Hackl C, Moser C, Fichtner-Feigl S, Koehl GE, Schlitt HJ, Geissler EK, Stoeltzing O. Implication of RICTOR in the mTOR inhibitor-mediated induction of insulin-like growth factor-I receptor (IGF-IR) and human epidermal growth factor receptor-2 (Her2) expression in gastrointestinal cancer cells. Biochim Biophys Acta. 2010; 1803: 435-442.
292. Bhaskar PT, Hay N. The two TORCs and Akt. Dev Cell. 2007; 12: 487-502.
293. Xu X, Sarikas A, Dias-Santagata DC, Dolios G, Lafontant PJ, Tsai SC, Zhu W, Nakajima H, Nakajima HO, Field LJ, Wang R, Pan ZQ. The CUL7 E3 ubiquitin ligase targets insulin receptor substrate 1 for ubiquitin-dependent degradation. Mol Cell. 2008; 30: 403-414.
294. Sriburi R, Jackowski S, Mori K, Brewer JW. XBP1: a link between the unfolded protein response, lipid biosynthesis, and biogenesis of the endoplasmic reticulum. J Cell Biol. 2004; 167: 35-41.
295. Dibble CC, Asara JM, Manning BD. Characterization of Rictor phosphorylation sites reveals direct regulation of mTOR complex 2 by S6K1. Mol Cell Biol. 2009; 29: 5657-5670.
296. Keniry M, Parsons R. The role of PTEN signaling perturbations in cancer and in targeted therapy. Oncogene. 2008; 27: 5477-5485.
297. Stiles BL. Phosphatase and tensin homologue deleted on chromosome 10: extending its PTENtacles. Int J Biochem Cell Biol. 2009; 41: 757-761.
298. Kalesnikoff J, Sly LM, Hughes MR, Buchse T, Rauh MJ, Cao LP, Lam V, Mui A, Huber M, Krystal G. The role of SHIP in cytokine-induced signaling. Rev Physiol Biochem Pharmacol. 2003; 149: 87-103.
299. Eichhorn PJ, Creyghton MP, Bernards R. Protein phosphatase 2A regulatory subunits and cancer. Biochim Biophys Acta. 2009; 1795: 1-15.
300. Nolt JK, Rice LM, Gallo-Ebert C, Bisher ME, Nickels JT. PP2A (Cdc)55 is required for multiple events during meiosis. Cell Cycle. 2011; 10: 1420-134.
301. Lee HJ, Hwang HI, Jang YJ. Mitotic DNA damage response: Polo-like kinase-1 is dephosphorylated through ATM-Chk1 pathway. Cell Cycle. 2010; 9: 2389-2398.
302. Kuo YC, Huang KY, Yang CH, Yang YS, Lee WY, Chiang CW. Regulation of phosphorylation of Thr-308 of Akt, cell proliferation, and survival by the B55a regulatory subunit targeting of the protein phosphatase 2A holoenzyme to Akt. J Biol Chem. 2008; 283: 1882-1892.
303. Ruvolo PP, Qui YH, Coombes KR, Zhang N, Ruvolo VR, Borthakur G, Konopleva M, Andreeff M, Kornblau SM. Low expression of PP2A regulatory subunit B55a is associated with T308 phosphorylation of AKT and shorter complete remission duration in acute myeloid leukemia patients. Leukemia. 2011; 25: 1711-1717.
304. Cristobal I, Garcia-Orti L, Cirauqui C, Alonso MM, Calasanz MJ, Odero MD. PP2A impaired activity is a common event in acute myeloid leukemia and its activation by forskolin has a potent anti-leukemic effect. Leukemia. 2011; 25: 606-614.
305. Brognard J, Newton AC. PHLiPPing the switch on Akt and protein kinase C signaling. Trends Endocrinol Metab. 2008; 19: 223-230.
306. Hirano I, Nakamura S, Yokota D, Ono T, Shigeno K, Fujisawa S, Shinjo K, Ohnishi K. Depletion of Pleckstrin homology domain leucine-rich repeat protein phosphatases 1 and 2 by Bcr-Abl promotes chronic myelogenous leukemia cell proliferation through continuous phosphorylation of Akt isoforms. J Biol Chem. 2009; 284: 22155-2165.
307. Demidenko ZN, Blagosklonny MV. The purpose of the HIF-1/PHD feedback loop: to limit mTOR-induced HIF-1alpha. Cell Cycle. 2011; 10: 1557-1562.
308. Rehn M, Cammenga J. (H)if blood stem cells gO2 sleep: what can wake them up?. Cell Cycle. 2011; 10: 3419-3420.
309. Gubin MM, Calaluce R, Davis JW, Magee JD, Strouse CS, Shaw DP, Ma L, Brown A, Hoffman T, Rold TL, Atasoy U. Overexpression of the RNA binding protein HuR impairs tumor growth in triple negative breast cancer associated with deficient angiogenesis. Cell Cycle. 2010; 9: 3337-3346.
310. Thangarajah H, Vial IN, Grogan RH, Yao D, Shi Y, Januszyk M, Galiano RD, Chang EI, Galvez MG, Glotzbach JP, Wong VW, Brownlee M, Gurtner GC. HIF-1alpha dysfunction in diabetes. Cell Cycle. 2010; 9: 75-79.
311. Mobasheri A, Richardson S, Mobasheri R, Shakibaei M, Hoyland JA. Hypoxia inducible factor-1 and facilitative glucose transporters GLUT1 and GLUT3: putative molecular components of the oxygen and glucose sensing apparatus in articular chondrocytes. Histol Histopathol. 2005; 20: 1327-1338.
312. Ousset M, Bouquet F, Fallone F, Biard D, Dray C, Valet P, Salles B, Muller C. Loss of ATM positively regulates the expression of hypoxia inducible factor 1 (HIF-1) through oxidative stress: Role in the physiopathology of the disease. Cell Cycle. 2010; 9: 2814-2822.
313. Liu Y, Dean DC. Tumor initiation via loss of cell contact inhibition versus Ras mutation: do all roads lead to EMT? Cell Cycle. 2010; 9: 897-900.
314. Wang J, Liu Y, Tan LX, Lo JC, Du J, Ryu MJ, Ranheim EA, Zhang J. Distinct requirements of hematopoietic stem cell activity and Nras G12D signaling in different cell types during leukemogenesis. Cell Cycle. 2011; 10: 2836-2839.
315. Wang C, Lisanti MP, Liao DJ. Reviewing once more the c-myc and Ras collaboration: converging at the cyclin D1-CDK4 complex and challenging basic concepts of cancer biology. Cell Cycle. 2011; 10: 57-67.
316. Webb JD, Simon MC. Novel insights into the molecular origins and treatment of lung cancer. Cell Cycle. 2010; 9: 4098-4105.
317. Shahbazian D, Parsyan A, Petroulakis E, Hershey J, Sonenberg N. eIF4B controls survival and proliferation and is regulated by proto-oncogenic signaling pathways. Cell Cycle. 2010; 9: 4106-4109.
318. de Magalhaes JP. A role for Ras signaling in modulating mammalian aging by the GH/IGF1 axis. Aging. 2011; 3: 336-337.
319. Borras C, Monleon D, Lopez-Grueso R, Gambini J, Orlando L, Pallardo FV, Santos E, Vina J, Font de Mora J. RasGrf1 deficiency delays aging in mice. Aging. 2011; 3: 262-276.
320. Zhao L, Vogt PK. Hot-spot mutations in p110alpha of phosphatidylinositol 3-kinase (PI3K): differential interactions with the regulatory subunit p85 and with RAS. Cell Cycle. 2010; 9: 596-600.
321. Adams JR, Schachter NF, Liu JC, Zacksenhaus E, Egan SE. Elevated PI3K signaling drives multiple breast cancer subtypes. Oncotarget. 2011; 2: 435-447.
322. Samuels Y, Wang Z, Bardelli A, Silliman N, Ptak J, Szabo S, Yan H, Gazdar A, Powell SM, Riggins GJ, Willson JK, Markowitz S, Kinzler KW, Vogelstein B, Velculescu VE. High frequency of mutations of the PIK3CA gene in human cancers. Science. 2004; 304: 554.
323. Huang CH, Mandelker D, Gabelli SB, Amzel LM. Insights into the oncogenic effects of PIK3CA mutations from the structure of p110alpha/p85alpha. Cell Cycle 2008; 7: 1151-1156.
324. Lee JW, Soung YH, Kim SY, Lee HW, Park WS, Nam SW, Kim SH, Lee JY, Yoo NJ, Lee SH. PIK3CA gene is frequently mutated in breast carcinomas and hepatocellular carcinomas. Oncogene. 2005; 24: 1477-1480.
325. Shatyesteh L, Lu Y, Kuo WL, Baldocchi R, Godfrey T, Collins C, Pinkel D, Powell B, Mills GB, Gray JW. PIK3CA is implicated as an oncogene in ovarian cancer. Nat Genet. 1999; 21: 99-102.
326. Ligresti G, Militello L, Steelman LS, Cavallaro A, Basile F, Nicoletti F, Stivala F, McCubrey JA, Libra M. PIK3CA mutations in human solid tumors. Cell Cycle. 2009: 8: 1352-1358.
327. Calvisi DF, Ladu S, Gorden A, Farina M, Lee JS, Conner EA, Schroeder I, Factor VM, Thorgeirsson SS. Mechanistic and prognostic significance of aberrant methylation in the molecular pathogenesis of human hepatocellular carcinoma. J Clin Invest. 2007; 117: 2713-2722.
328. Steelman LS, Bertrand FE, McCubrey JA. The complexity of PTEN: mutation, marker and potential target for therapeutic intervention. Expert Opinion Ther Targets. 2004; 8: 537-550.
329. Rhei E, Kang L, Bogomolniy F, Federici MG, Borgen PI, Boyd J. Mutation analysis of the putative tumor suppressor gene PTEN/MMAC1 in primary breast carcinomas. Cancer Res. 1997; 57: 3657-3659.
330. Singh B, Ittmann MM, Krolewski JJ. Sporadic breast cancers exhibit loss of heterozygosity on chromosome segment 10q23 close to the Cowden disease locus. Genes Chromosome Can. 1998; 21: 166-171.
331. Trotman LC, Wang X, Alimonti A, Chen Z, Teruya-Feldstein J, Yang H, Pavletich NP, Carver BS, Cordon-Cardo C, Erdjument-Bromage H, Tempst P, Chi SG, Kim HJ, Misteli T, Jiang X, Pandolfi PP. Ubiquitination regulates PTEN nuclear import and tumor suppression. Cell. 2007; 128: 141-156.
332. Gupta A, Yang Q, Pandita RK, Hunt CR, Xiang T, Misri S, Zeng S, Pagan J, Jeffery J, Puc J, Kumar R, Feng Z, Powell SN, Bhat A, Yaguchi T, Wadhwa R, et al. Cell cycle checkpoint defects contribute to genomic instability in PTEN deficient cells independent of DNA DSB repair. Cell Cycle. 2009; 8: 2198-2210.
333. Kappes H, Goemann C, Bamberger AM, Löning T, Milde-Langosch K. PTEN expression in breast and endometrial cancer: correlation with steroid hormone receptor status. Pathobiology. 2001; 69: 136-142.
334. Suzuki A, Nakano T, Mak TW, Sasaki T. Portrait of PTEN: messages from mutant mice. Cancer Sci. 2008; 99: 209-213.
335. Yao YJ, Ping XL, Zhang H, Chen FF, Lee PK, Ahsan H, Chen CJ, Lee PH, Peacocke M, Santella RM, Tsou HC. PTEN/MMAC1 mutations in hepatocellular carcinomas. Oncogene. 1999; 18: 3181-3185.
336. Yeh KT, Chang JG, Chen YJ, Chen ST, Yu SY, Shih MC, Perng LI, Wang JC, Tsai M, Chang CP. Mutation analysis of the putative tumor suppressor gene PTEN/MMAC1 in hepatocellular carcinoma. Cancer Invest. 2000; 18: 123-129.
337. Kawamura N, Nagai H, Bando K, Koyama M, Matsumoto S, Tajiri T, Onda M, Fujimoto J, Ueki T, Konishi N, Shiba T, Emi M. PTEN/MMAC1 mutations in hepatocellular carcinomas: somatic inactivation of both alleles in tumors. Jpn J Cancer Res. 1999; 90: 413-418.
338. Fujiwara Y, Hoon DS, Yamada T, Umeshita K, Gotoh M, Sakon M, Nishisho I, Monden M. PTEN/MMAC1 mutation and frequent loss of heterozygosity identified in chromosome 10q in a subset of hepatocellular carcinomas. Jpn J Cancer Res. 2000; 91: 287-292.
339. Wang L, Wang WL, Zhang Y, Guo SP, Zhang J, Li QL. Epigenetic and genetic alterations of PTEN in hepatocellular carcinoma. Hepatol Res. 2007; 37: 389-396.
340. Rahman MA, Kyriazanos ID, Ono T, Yamanoi A, Kohno H, Tsuchiya M, Nagasue N. Impact of PTEN expression on the outcome of hepatitis C virus-positive cirrhotic hepatocellular carcinoma patients: possible relationship with COX II and inducible nitric oxide synthase. Int J Cancer. 2002; 100: 152-157.
341. Hu TH, Wang CC, Huang CC, Chen CL, Hung CH, Chen CH, Wang JH, Lu SN, Lee CM, Changchien CS, Tai MH. Down-regulation of tumor suppressor gene PTEN, overexpression of p53, plus high proliferating cell nuclear antigen index predict poor patient outcome of hepatocellular carcinoma after resection. Oncol Rep. 2007; 18: 1417-1426.
342. Chung TW, Lee YC, Ko JH, Kim CH. Hepatitis B Virus X protein modulates the expression of PTEN by inhibiting the function of p53, a transcriptional activator in liver cells. Cancer Res. 2003; 63: 3453-3458.
343. Kang-Park S, Im JH, Lee JH, Lee YI. PTEN modulates hepatitis B virus-X protein induced survival signaling in Chang liver cells. Virus Res. 2006; 122: 53-60.
344. Butler MP, Wang SI, Chaganti RS, Parsons R, Dalla-Favera R. Analysis of PTEN mutations and deletions in B-cell non-Hodgkin‘s lymphomas. Genes Chromosomes Cancer. 1999; 24: 322-327.
345. Sakai A, Thieblemont C, Wellmann A, Jaffe ES, Raffeld M. PTEN gene alterations in lymphoid neoplasms. Blood. 1998; 92: 3410-3415.
346. Uddin S, Hussain A, Al Hussein K, Platanias LC, Bhatia KG. Inhibition of phosphatidylinositol 3‘-kinase induces preferentially killing of PTEN-null T leukemias through AKT pathway. Biochem Biophys Res Commun. 2004; 320: 932-938.
347. Palomero T, Sulis ML, Cortina M, Real PJ, Barnes K, Ciofani M, Caparros E, Buteau J, Brown K, Perkins SL, Bhagat G, Agarwal AM, Basso G, Castillo M, Nagase S, Cordon-Cardo C, et al. Mutational loss of PTEN induces resistance to NOTCH1 inhibition in T-cell leukemia. Nat Med. 2007; 13: 1203-1210.
348. Palomero T, Dominguez M, Ferrando AA. The role of the PTEN/AKT Pathway in NOTCH1-induced leukemia. Cell Cycle. 2008; 7: 965-970.
349. Garcia JM, Silva J, Pena C, Garcia V, Rodríguez R, Cruz MA, Cantos B, Provencio M, España P, Bonilla F. Promoter methylation of the PTEN gene is a common molecular change in breast cancer. Genes Chromosomes Cancer. 2004; 41: 117-124.
350. Tsutsui S, Inoue H, Yasuda K, Suzuki K, Higashi H, Era S, Mori M. Reduced expression of PTEN protein and its prognostic implications in invasive ductal carcinoma of the breast. Oncology. 2005; 68: 398-404.
351.Yang H, Kong W, Zhao JJ, O‘Donnell JD, Wang J. MicroRNA expression profiling in human ovarian cancer: miR-274 induces cell survival and cisplatin resistance by targeting PTEN. Cancer Res. 2008; 68: 425-433.
352. Dhomen N, Reis-Filho JS, da Rocha Dias S, Hayward R, Savage K, Delmas V, Larue L, Pritchard C, Marais R. Oncogenic Braf induces melanocyte senescence and melanoma in mice. Cancer Cell. 2009; 15: 294-303.
353. Dankort D, Curley DP, Cartlidge RA, Nelson B, Karnezis AN, Damsky WE Jr, You MJ, DePinho RA, McMahon M, Bosenberg M. Braf(V600E) cooperates with PTEN loss to induce metastatic melanoma. Nat Genet. 2009; 41: 544-552.
354. Luo JM, Yoshida H, Komura S, Ohishi N, Pan L, Shigeno K, Hanamura I, Miura K, Iida S, Ueda R, Naoe T, Akao Y, Ohno R, Ohnishi K. Possible dominant-negative mutation of the SHIP gene in acute myeloid leukemia. Leukemia. 2003; 17: 1-8.
355. Metzner A, Horstmann MA, Fehse B, Ortmeyer G, Niemeyer CM, Stocking C, Mayr GW, Jücker M. Gene transfer of SHIP-1 inhibits proliferation of juvenile myelomonocytic leukemia cells carrying KRAS2 or PTPN11 mutations. Gene Ther. 2007; 14: 699-703.
356.Cheng JQ, Godwin AK, Bellacosa A, Taguchi T, Franke TF, Hamilton TC, Tsichlis PN, Testa JR. AKT2, a putative oncogene encoding a member of a subfamily of protein-serine/threonine kinases, is amplified in human ovarian carcinomas. Proc Natl Acad Sci USA. 1992; 89: 9267-9271.
357. Carpten JD, Faber AL, Horn C, Donoho GP, Briggs SL, Robbins CM, Hostetter G, Boguslawski S, Moses TY, Savage S, Uhlik M, Lin A, Du J, Qian YW, Zeckner DJ, Tucker-Kellog G, et al. A transforming mutation in the pleckstrin homology domain of AKT1 in cancer. Nature. 2007; 448: 439-444.
358. Stemke-Hale K, Gonzalez-Angelo M, Lhuch A Neve RM, Kuo WL, Davies M, Carey M, Hu Z, Guan Y, Sahin A, Symmans WF, Pusztai L, Nolden LK, Horlings H, Berns K, Hung MC, et al. An integrative genomic and proteomic analysis of PIK3CA, PTEN and Akt mutations in breast cancer. Cancer Res. 2008; 68: 6084-6091.
359. Bellacosa A, DeFeo D, Godwin AK, Bell DW, Cheng JQ, Altomare DA, Wan M, Dubeau L, Scambia G, Masciullo V, Ferrandina G, Benedetti Panici P, Mancuso S, Neri G, Testa JR. Molecular alterations of the Akt oncogene in breast cancer. Int J Cancer. 1995; 64: 280-285.
360. Davies MA, Stemke-Hale K, Tellez C, Calderone TL, Deng W, Prieto VG, Lazar AJ, Gershenwald JE, Mills GB. A novel Akt3 mutation in melanoma tumours and cell lines. Brit J Cancer. 2008; 99: 1265-1268.
361. Greenman C, Stephens P, Smith R, Dalgliesh GL, Hunter C, Bignell G, Davies H, Teague J, Butler A, Stevens C, Edkins S, O’Meara S, Vastrik I, Schmidt EE, Avis T, Barthorpe S, et al. Patterns of somatic mutation in human cancer genomes. Nature. 2007; 446:153-158.
362. Do H, Salemi R, Murone C, Mitchell PL, Dobrovic A. Rarity of AKT1 and AKT3 E17K mutations in squamous cell carcinoma of lung. Cell Cycle. 2010; 9: 4411-4412.
363. Karst AM, Dai DL, Cheng JQ, Li G. Role of p53 up-regulated modulator of apoptosis and phosphorylated Akt in melanoma cell growth, apoptosis and patient survival. Cancer Res. 2006; 66: 9221-9226.
364. Slipicevic A, Holm R, Nguyen MTP, Bøhler PJ, Davidson B, Flørenes VA. Expression of activated Akt and PTEN in malignant melanomas: Relationship with clinical outcome. Am J Clin Pathol. 2005; 124: 528-536.
365. Balsara BR, Pei J, Mitsuuchi Y, Page R, Klein-Szanto A, Wang H, Unger M, Testa JR. Frequent activation of AKT in non-small cell lung carcinomas and preneoplastic bronchial lesions. Carcinogenesis. 2004; 25: 2053-2059.
366. Benavides F, Blando J, Perez CJ, Garg R, Conti CJ, DiGiovanni J, Kazanietz MG. Transgenic overexpression of PKCepsilon in the mouse prostate induces preneoplastic lesions. Cell Cycle. 2011; 10: 268-277.
367. Basu A. PKCepsilon paves the way for prostate cancer. Cell Cycle. 2011; 10: 378.
368. Knudsen KE. A tale of three PKCs: epsilon emerges as a driver of pre-neoplastic phenotypes. Cell Cycle. 2011; 10: 379.
369. Tanno S, Tanno S, Mitsuuchi Y, Altomare DA, Xiao GH, Testa JR. AKT activation up-regulates insulin-like growth factor I receptor expression and promotes invasiveness of human pancreatic cancer cells. Cancer Res. 2001; 61: 589-593.
370. St-Germain ME, Gagnon V, Parent S, Asselin E. Regulation of COX-2 protein expression by Akt in endometrial cancer cells is mediated through NF-kB/IkB pathway. Mol Cancer. 2004; 3: 7.
371. Chefetz I, Holmberg JC, Alvero AB, Visintin I, Mor G. Inhibition of Aurora-A kinase induces cell cycle arrest in epithelial ovarian cancer stem cells by affecting NFkB pathway. Cell Cycle. 2011; 10: 2206-2214.
372. Iwai K. Linear polyubiquitin chains: a new modifier involved in NFkappaB activation and chronic inflammation, including dermatitis. Cell Cycle. 2011; 10: 3095-3104.
373. Melvin A, Mudie S, Rocha S. Further insights into the mechanism of hypoxia-induced NFkappaB. Cell Cycle. 2011; 10: 879-882.
374. Madge LA, May MJ. The NFkappaB paradox: RelB induces and inhibits gene expression. Cell Cycle. 2011; 10: 6-7.
375. Oren M, Cooks T. NFkappaB and p53: A life and death affair. Cell Cycle. 2010; 9: 1027.
376. Barre B, Perkins ND. Phosphorylation of the p52 NF-kappaB subunit.
Cell Cycle. 2010; 9: 4774-4775.
377. Barre B, Coqueret O, Perkins ND. Regulation of activity and function of the p52 NF-kappaB subunit following DNA damage. Cell Cycle. 2010; 9: 4795-4804.
378. Lamprecht B, Bonifer C, Mathas S. Repeat-element driven activation of proto-oncogenes in human malignancies. Cell Cycle. 2010; 9: 4276-4281.
379. Martin AG. NFkappaB anti-apoptotic or pro-apoptotic, maybe both. Cell Cycle. 2010; 9: 3131-3132.
380. O’Prey J, Crighton D, Martin AG, Vousden KH, Fearnhead HO. Ryan KM. p53-mediated induction of Noxa and p53AIP1 requires NFkappaB. Cell Cycle. 2010; 9: 947-952.
381. Vaughan S, Jat PS. Deciphering the role of nuclear factor-kappaB in cellular senescence. Aging. 2011; 3: 913-919.
382. Moskalev A, Shaposhnikov M. Pharmacological inhibition of NF-kappaB prolongs lifespan of Drosophila melanogaster. Aging. 2011; 3: 391-394.
383. Hofseth LJ, Singh UP, Singh NP, Nagarkatti M, Nagarkatti PS. Taming the beast within: resveratrol suppresses colitis and prevents colon cancer. Aging. 2010; 2: 183-184.
384. Chow S, Minden MD, Hedley DW. Constitutive phosphorylation of the S6 ribosomal protein via mTOR and ERK signaling in the peripheral blasts of acute leukemia patients. Exp Hematol. 2006; 34: 1183-1191.
385. Grandage VL, Gale RE, Linch DC, Khwaja A. PI3-kinase/Akt is constitutively active in primary acute myeloid leukaemia cells and regulates survival and chemoresistance via NF-kappaB, Mapkinase and p53 pathways. Leukemia. 2005; 19: 586-594. 386. Ricciardi MR, McQueen T, Chism D, Milella M, Estey E, Kaldjian E, Sebolt-Leopold J, Konopleva M, Andreeff M. Quantitative single cell determination of ERK phosphorylation and regulation in relapsed and refractory primary acute myeloid leukemia. Leukemia. 2005; 19: 1543-1549.
387. McCubrey JA, Milella M, Tafuri A, Martelli AM, Lunghi P, Bonati A, Bonati A, Cervello M, Lee JT, Steelman LS. Targeting the Raf/MEK/ERK pathway with small molecule inhibitors. Current Opinion Investigational Drugs. 2008; 9: 614-630.
388. McCubrey JA, Steelman LS, Abrams SL, Chappell WH, Russo S, Ove R, Milella M, Tafuri A, Lunghi P, Bonati A, Stivala F, Nicoletti F, Libra M, Martelli AM, Montalto G, Cervello M. Emerging MEK Inhibitors. Exp Opin Emerging Drugs. 2010; 15: 203-223.
389. Yun C, Cho H, Kim SJ, Lee JH, Park SY, Chan GK, Cho H. Mitotic aberration coupled with centrosome amplification is induced by hepatitis B virus X oncoprotein via the Ras-mitogen-activated protein/extracellular signal-regulated kinase-mitogen-activated protein pathway. Mol Cancer Res. 2004; 2: 159-169.
390. Hu TH, Huang CC, Lin PR, Chang HW, Ger LP, Lin YW, Changchien CS, Lee CM, Tai MH. Expression and prognostic role of tumor suppressor gene PTEN/MMAC1/TEP1 in hepatocellular carcinoma. Cancer. 2003; 97: 1929-1940.
391.Lee YI, Kang-Park S, Do SI, Lee YI. The hepatitis B virus-X protein activates a phosphatidylinositol 3-kinase-dependent survival signaling cascade. J Biol Chem. 2001; 276: 16969-16977.
392. Mannová P, Beretta L. Activation of the N-Ras-PI3K-Akt-mTOR pathway by hepatitis C virus: control of cell survival and viral replication. J Virol. 2005; 79: 8742-8749.
393. Krymskaya VP, Goncharova EA. PI3K/mTORC1 activation in hamartoma syndromes: therapeutic prospects. Cell Cycle. 2009; 8: 403-413.
394. Knowles MA, Platt FM, Ross RL, Hurst CD. Phosphatidylinositol 3-kinase (PI3K) pathway activation in bladder cancer. Cancer Metastasis Rev. 2009; 28: 305-316.
395. Graff JR, Zimmer SG. Translational control and metastatic progression: enhanced activity of the mRNA cap-binding protein eIF-4E selectively enhances translation of metastasis-related mRNAs. Clin Exp Metastasis. 2003; 20: 265-273.
396. Zimmer SG, DeBenedetti A, Graff JR. Translational control of malignancy: the mRNA cap-binding protein, eIF-4E, as a central regulator of tumor formation, growth, invasion and metastasis. Anticancer Res. 2000; 20: 1343-1351.
397. Galmozzi E, Casalini P, Iorio MV, Casati B, Olgiati C, Menard S. HER2 signaling enhances 5’UTR-mediated translation of c-Myc mRNA. J Cell Physiol. 2004; 200: 82-88.
398. Podar K, Anderson KC. A therapeutic role for targeting c-Myc/Hif-1-dependent signaling pathways. Cell Cycle. 2010; 9: 1722-1728.
399. Gera JF, Mellinghoff IK, Shi Y, Rettig MB, Tran C, Hsu JH, Sawyers CL, Lichtenstein AK. AKT activity determines sensitivity to mammalian target of rapamycin (mTOR) inhibitors by regulating cyclin D1 and c-myc expression. J Biol Chem. 2004; 279: 2737-2746.
400. Chung J, Bachelder RE, Lipscomb EA, Shaw LM, Mercurio AM. Integrin (alpha 6 beta 4) regulation of eIF-4E activity and VEGF translation: a survival mechanism for carcinoma cells. J Cell Biol. 2002; 158: 165-174.
401. Drobnjak M, Osman I, Scher HI, Fazzari M, Cordon-Cardo C. Overexpression of cyclin D1 is associated with metastatic prostate cancer to bone. Clin Cancer Res. 2000; 6: 1891-1895.
402. Dunsmuir WD, Gillett CE, Meyer LC, Young MP, Corbishley C, Eeles RA, Kirby RS. Molecular markers for predicting prostate cancer stage and survival. BJU Int. 2000; 6: 869-878.
403. Gallant P, Steiger D. Myc’s secret life without Max. Cell Cycle. 2009; 8: 3848-3853.
404. Hydbring P, Larsson LG. Cdk2: a key regulator of the senescence control function of Myc. Aging. 2010; 2: 244-250.
405. Jin J, Wang GL, Timchenko L, Timchenko NA. GSK3beta and aging liver. Aging. 2009; 1: 582-585.
406. Gan B, DePinho RA. mTORC1 signaling governs hematopoietic stem cell quiescence. Cell Cycle. 2009; 8: 1003-1006.
407. Lluis F, Cosma MP. Somatic cell reprogramming control: signaling pathway modulation versus transcription factor activities. Cell Cycle. 2009; 8: 1138-1144.
408. Wu GJ, Sinclair CS, Paape J, Ingle JN, Roche PC, James CD, Couch FJ. 17q23 amplifications in breast cancer involve the PAT1, RAD51C, PS6K and SIGma1B genes. Cancer Res. 2000; 60: 5371-5375.
409. Barlund M, Forozan F, Kononen J, Bubendorf L, Chen Y, Bittner ML, Torhorst J, Haas P, Bucher C, Sauter G, Kallioniemi OP, Kallioniemi A. Detecting activation of ribosomal protein S6 kinase by complementary DNA and tissue microarray analysis. J Natl Cancer Inst. 2000; 92: 1252-1259.
410. Neshat MS, Mellinghoff IK, Tran C, Stiles B, Thomas G, Petersen R, Frost P, Gibbons JJ, Wu H, Sawyers CL. Enhanced sensitivity of PTEN-deficient tumors to inhibition of FRAP/mTOR. Proc Natl Acad Sci USA. 2001; 98: 10314-10319.