
INTRODUCTION
Angiogenesis is a complex process which includes endothelial cells (ECs) proliferation, migration, basement membrane degeneration and new tube formation. It is required for a variety of physiologic processes like development and reproduction. However, angiogenesis also plays a vital role in the growth and spread of cancer as an adequate blood supply, which is necessary for tumor growth and invasion in normal tissues [1–4]. It is known that endothelial cells are normally in a highly quiescent state, and crucial to maintain vascular homeostasis. But they can transform into an active proliferative state if stimulated by angiogenic factors from the tumors, then grow, migrate and form new blood vessels, which offer oxygen and nutrients to the tumors. Inducing angiogenesis is one of the hallmarks of cancer. New blood vessels infiltrate tumors, supplying them with oxygen and nutrients, and offer a route for tumor metastasis. Studies have shown that inhibition of ECs growth can block tumor angiogenesis effectively [5–7]. Moreover, agents that prevent the growth of a tumor’s blood vessels facilitate the regression or dormancy of established tumors and anti-angiogenesis treatment has been an extremely promising form of cancer therapy [8]. Therefore, inhibiting angiogenesis may be an effective method for inhibiting tumor growth and metastasis.
Angiogenesis is tightly controlled by the balance of pro- and anti-angiogenic factors, such as vascular endothelial growth factor (VEGF), platelet-derived growth factor (PDGF), epidermal growth factor (EGF) and angiogenin, etc [9]. Imbalanced pro-angiogenesis stimulation would trigger pathologically angiogenic diseases such as ischemic heart failure as well as cancer progression. Among the pro-angiogenic factors, VEGF is well known to play an important part in tumor biology and more specifically, in the process of tumor angiogenesis, as its expression has been detected in various malignant human tumors, like breast, ovary, kidney, urinary bladder, brain, lung and gastrointestinal tract tumors [10]. VEGF is a family composed of five isoforms denominated VEGF-A, VEGF-B, VEGF-C, VEGF-D and placental growth factor (PlGF). Each of these factors exerts its activity through binding to three high-affinity transmembrane receptors (VEGFR1, VEGFR2 and VEGFR3), and then promote angiogenesis through its ability to stimulate the growth, migration and invasion of endothelial cells [11–15]. Current evidence suggests that the interaction between VEGF and VEGFR-1(Flt1) plays a minor role in angiogenesis, while VEGFR-2 (Flk1/KDR: VEGFR-2 also known as KDR in human and Flk1 in mouse) mediates the major angiogenic function of VEGF [16, 17]. VEGFR-2 is strongly auto-phosphorylated in tumors by elevated VEGF expression [18]. VEGF binds to specific transmembrane receptors on ECs and lymphatic vessels in endothelial cell [19, 20]. VEGFR-2 signaling activation induces the phosphorylation of various downstream signal transduction mediators, including phosphoinositide 3-kinase (PI3K)/AKT/mTOR, and mitogen-activated protein kinase (MAPK), which ultimately control processes critical to angiogenesis such as endothelial cell proliferation, migration and tubulogenesis [21–24]. Therefore, VEGF and VEGFR-2 become potential molecular targets for anti-angiogenic tumor therapy. Several approaches have been taken to block VEGF/VEGFR-2 signaling pathways, such as inhibition of endogenous VEGF release and prevention of VEGF from binding to VEGFR-2 [25].
In recent years, some studies have revealed that natural products which exist in the traditional Chinese medicines [26], have the potential properties in treating different types of cancers. Toad venom (called “Chansu” in China) is a product of the skin gland of toads such as Bufo bufo gargarizans etc [27, 28], which has been widely used as a cardiotonic, diuretic, anodyne and hemostatic agent [29]. Chansu has been used as significant anticancer agents, amending the life quality of cancer patients [30]. Recent experimental studies have demonstrated that Chansu and its active compounds exhibit significant anti-tumor activity via the inhibition of cell proliferation, induction of cell differentiation and apoptosis, disruption of the cell cycle, inhibition of angiogenesis, reversal of multidrug resistance, and regulation of the immune response [31]. A previous study suggests that Chansu could induce apoptosis of cancer cells, such as human gastric cancer and bladder cancer cells [32, 33]. Gamabufotalin (CS-6), a major bufadienolide isolated from Chansu, had the high content of 1.75%-5%, and significant anti-tumor activity. It has been found that it is used for anti-inflammatory, acesodyne and anti-neoplasti. Our previous studies also found that CS-6 suppressed human lung cancer A549 cell proliferation, migration and invasion [34]. However, potential influence of CS-6 in endothelial angiogenic activity is totally unknown.
In this study, we have investigated the effects of gamabufotalin (CS-6) on angiogenesis and characterized the underlying molecular mechanisms. The anti-angiogenesis properties of CS-6 were evaluated in vitro using human umbilical vein endothelial cells (HUVECs) proliferation, migration and tube formation assays, ex vivo by aortic ring assay and in vivo by matrigel plug assay mouse models. Our results exhibited that CS-6 inhibited VEGF-mediated angiogenesis in endothelial cells, which suggesting that CS-6 could be used as a potential anti-angiogenesis agent that targets VEGF/VEGFR-2 signaling pathways.
RESULTS
CS-6 inhibits VEGF-induced cell proliferation of HUVECs
As an importance of proliferation for endothelial angiogenesis and tumor growth [35], we firstly investigated the influence of CS-6 (Fig. 1A) in human endothelial cells proliferation. After treatment for HUVECs with a range of CS-6 (0, 1, 10, 25, 50 and 75 nM) for 12 h and 24 h and cell viability was determined by MTT assay. The results revealed CS-6 has mild inhibition for HUVECs proliferation and showed no obvious cytotoxicity at low concentration (Fig. 1B).
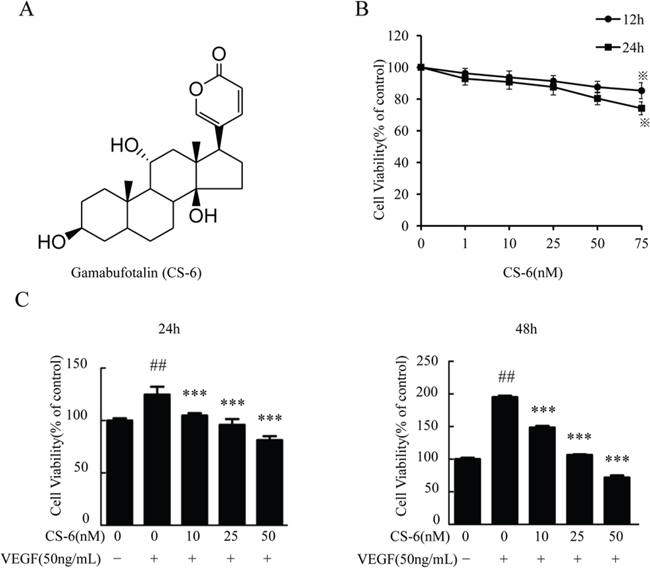
Figure 1: CS-6 inhibits VEGF-induced proliferation of HUVECs. A. Chemical structure of gamabufotalin (CS-6). B. Viability inhibition of CS-6 on HUVECs under normal culture condition. HUVECs were exposed to CS-6 at the indicated doses and times, and viability was measured by MTT assay. Data were represented as percentage of vehicle-treated control. C. CS-6 inhibits the proliferation of VEGF-induced HUVECs. HUVECs were treated with CS-6 with or without 50 ng/mL VEGF for 24 h or 48 h, and viability was measured by MTT assay. Three independent experiments were performed (*p<0.05, CS-6-treated group vs. DMSO group; ##p < 0.01, VEGF-treated group vs. Solvent; ***p < 0.001, VEGF and CS-6-treated group vs. VEGF-treated group).
We further determined whether CS-6 inhibited VEGF-induced HUVECs cell growth using MTT assay. As shown in Fig. 1C, the number of HUVECs stimulated with VEGF for 24 h and 48 h increased about 1.25 folds and 1.9 folds, respectively. These results showed that CS-6 could inhibit VEGF-induced cell growth in a dose-dependent and time-dependent manner; however we observed a greater inhibition by the CS-6 in VEGF stimulated HUVECs proliferation in comparison with absence of VEGF (Fig. 1C). Taken together, our data showed that CS-6 was a potent inhibitor of VEGF-activated endothelial cell proliferation.
CS-6 inhibits VEGF-induced endothelial cell migration and invasion
Cell migration and invasion are essential for endothelial cells during angiogenesis. We next investigated the effects of CS-6 on cell migration and invasion by wound healing assays and Transwell assays, respectively. The results showed that CS-6 significantly inhibited the migrating and invasive properties of VEGF-induced endothelial cells in a dose-dependent manner (Fig. 2A and 2B).
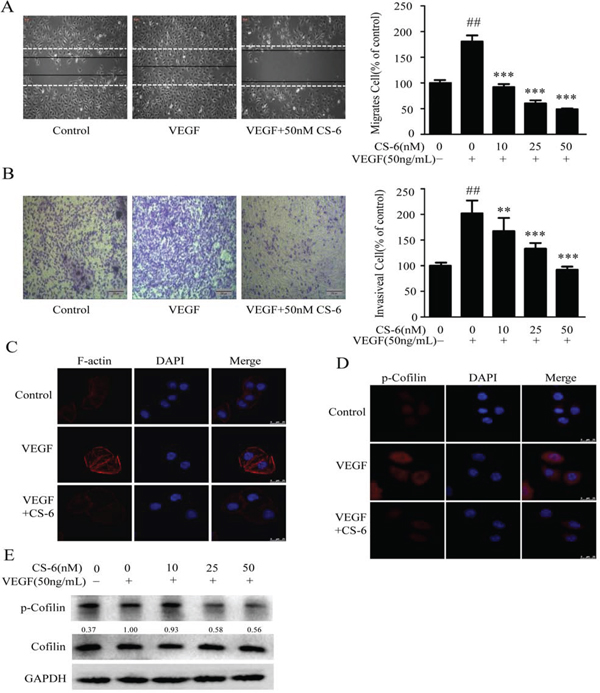
Figure 2: CS-6 inhibits VEGF-induced endothelial cell migration and invasion. A. CS-6 inhibits HUVECs migration induced by VEGF in wound healing assay. HUVECs were plated, scratched and then incubated with CS-6 with or without 50 ng/mL VEGF. Cell migration was measured by manual counting. Original magnification, 100× B. CS-6 inhibits HUVECs invasion in Transwell assay. HUVECs were plated in Transwell pre-coated with matrigel. Cell migrated to the bottom of the membrane were counted by using an inverted microscope. Original magnification, 40× C. CS-6 suppressed VEGF-induced stress fibre formation in endothelial cells. HUVECs were exposed to 50 nM CS-6 for 0.5 h, and then stimulated with or without VEGF for 15 min. F-actin of cells was visualized by dyLightTM 554 palloidin staining and imaged by Leica confocal microscopy. D. CS-6 inhibited VEGF-induced cofilin phosphorylation in HUVECs using immunofluorescences staining with specific antibody for phosphorylated cofilin. E. CS-6 inhibited VEGF-induced cofilin phosphorylation and activation in Western Blotting assay. Three independent experiments were performed (##p < 0.01, VEGF-treated group vs. Solvent; **p < 0.01, VEGF-treated group vs. VEGF and CS-6-treated group; ***p < 0.001, VEGF-treated group vs. VEGF and CS-6-treated group).
As cell cytoskeleton and stress fibre formation are key cellular events involved in cell migration, we further investigated the effects of CS-6 on these aspects. Confocal image analysis of individual cells revealed that VEGF caused a robust induction of stress fibre formation which was inhibited by 50 nM CS-6 (Fig. 2C). As activation of cofilin is an essential component of actin polymerization and depolymerization, and significantly affects cell cytoskeleton reorganization, we examined the effects of CS-6 on VEGF-induced phosphorylated cofilin using immunofluorescence techniques. Results showed in Fig. 2D demonstrate that 50nM CS-6 reduced VEGF-induced cofilin phosphorylation and activation. Furthermore, western blotting further confirmed the phosphorylation and activation of cofilin were reduced by CS-6 in a dose-dependent manner (Fig. 2E). These results suggest that CS-6 inhibited VEGF triggered HUVECs motility by affecting cofilin activity and cell stress fibre formation.
CS-6 suppresses VEGF-induced anti-apoptosis of HUVECs
Consistent with the findings of other investigators that VEGF caused a marked decrease in the apoptosis of HUVECs induced by serum deprivation, as indicated by a VEGF-dependent decrease in cell-surface annexin V binding [36]. We next investigated the effects of CS-6 on VEGF caused anti-apoptosis in HUVECs using Annexin V/propidium iodide assay. As shown in Fig. 3A, the experimental group stimulated with VEGF decreased the apoptosis of HUVECs induced by serum deprivation from 8.6% in control cells to 4.3%, however, co-treated with indicated doses CS-6 and VEGF can dramatically increase cell apoptosis in HUVECs compared with VEGF group, which suggests that CS-6 inhibits VEGF-induced anti-apoptosis. We also detected the expression of three key pro-apoptotic proteins (PARP and caspase-3/9), as well as Bcl-2 and Bax protein by Western blot analysis. CS-6 could markedly increase the expression levels of the cleaved caspase-3/9 and PARP proteins, and reduce the ratio of Bcl-2/Bax as compared with the VEGF group (Fig. 3B).
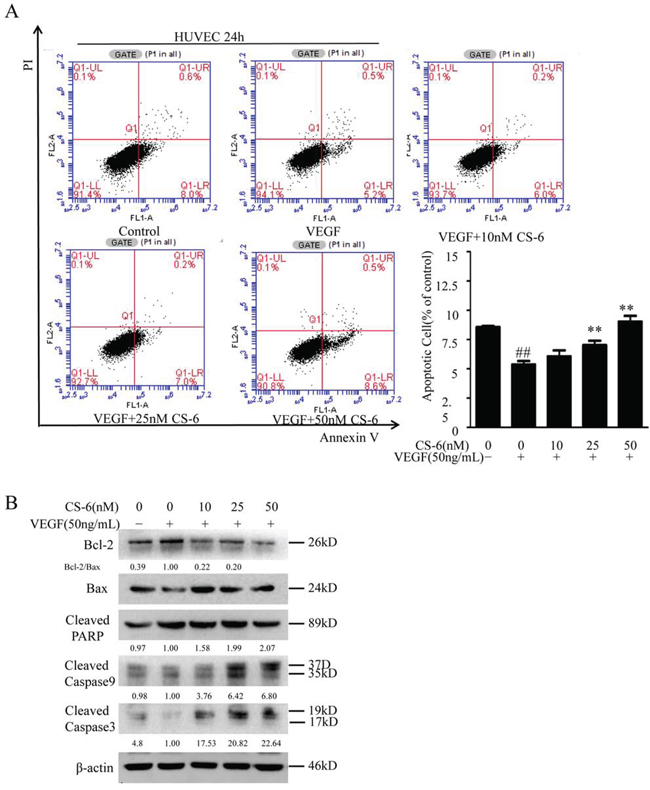
Figure 3: CS-6 suppresses VEGF-induced anti-apoptosis in HUVECs. HUVECs were incubated for 24 h with indicated CS-6 after 24 h of serum starvation, along with or without VEGF. The cell apoptosis rate was determined by a FACS analysis A. and the levels of Bcl-2, Bax, cleaved caspase-3/9 and cleaved PARP proteins were nalyzed by Western blot B. The apoptosis is represented by relative percentages of apoptotic cells versus that in DMSO-treated cells. Three independent experiments were performed (##P < 0.01, VEGF group vs. control group; **P<0.01, ***P < 0.001, VEGF- and CS-6-treated group vs. VEGF-treated group).
CS-6 inhibits VEGF-induced angiogenesis in vitro and ex vivo
One of the important steps during neo-angiogenesis is the formation and merging of tubes produced by endothelial cells forming a complex network of vessels and capillaries [37, 38]. Furthermore, we examined whether CS-6 inhibited VEGF-induced HUVECs tube formation using an in vitro angiogenesis tube formation assay. As shown in Fig. 4A, HUVECs were placed on the surface of Matrigel, and VEGF (50 ng/mL) significantly enhanced (p<0.001) the endothelial capillary like structures comparing with solvent treated cells. However, the VEGF effects were blocked by CS-6 treatment in the doses indicated. These data indicated that CS-6 would efficiently impair VEGF induced tube formation in HUVECs.
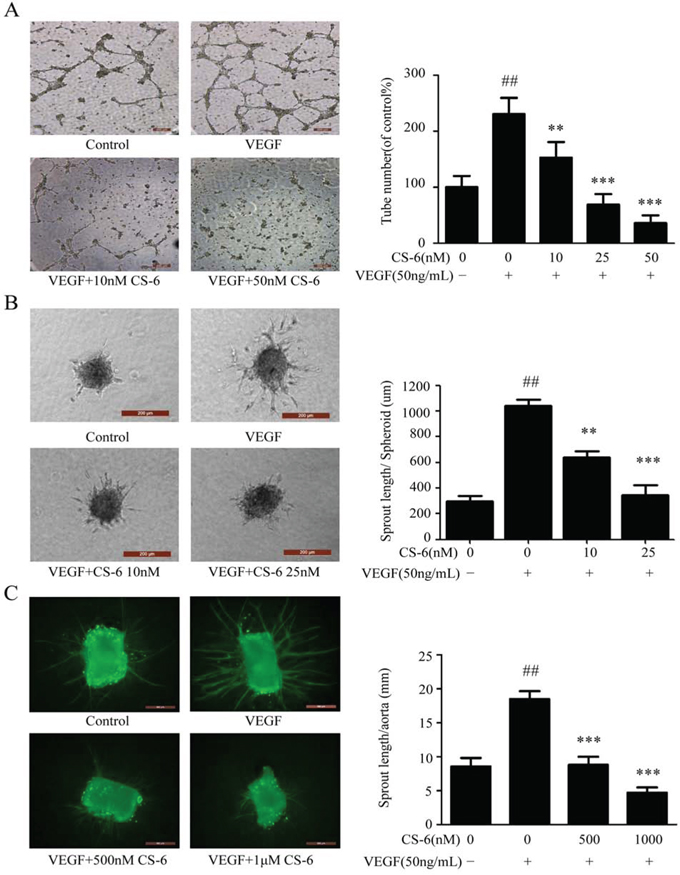
Figure 4: CS-6 inhibits VEGF-induced angiogenesis in vivo and ex vivo. Effect of CS-6 on A. tube formation on Matrigel (Original magnification, 50×) and B. sprouting from modified human endothelial cell spheroids (Original magnification, 200×). Experiments were performed with or without VEGF and indicated CS-6 doses. C. Endothelial sprouting from aortic rings from C57/BL6 mice. Additional VEGF (50 ng/mL) and CS-6 were added and the endothelial sprouts were allowed to develop over 9 days, and then staining with FITC-Lectin (green) Original magnification, 50×. Three independent experiments were performed (##p < 0.01, VEGF-treated group vs. no VEGF-treated group; **p < 0.01, VEGF-treated group vs. VEGF- and CS-6-treated group; ***p < 0.001, VEGF-treated group vs. VEGF- and CS-6-treated group).
We further examined the VEGF also induced endothelial cell sprouting in a modified spheroid assay; however, this response was significantly impaired by CS-6 in endothelial cells (Fig. 4B). Moreover, capillary formation assay was carried out through mouse dorsal aortas in ex vivo. After nine days of culture in matrigel, we observed a significant increase in endothelial sprouts from VEGF treated mouse aorta rings, while the microvessel sprouting was inhibited in a dose-dependent manner by CS-6 treatment (Fig. 4C).
CS-6 inhibits the activation of VEGFR-2 in HUVECs
It is well appreciated the great contribution of VEGF/VEGFR-2 axis in regulating endothelial mitogenesis as well as migration [39]. Previous studies indicated that blockage of VEGFR-2 activity could significantly limit tumoral neo-angiogenesis process [40]. Considering the CS-6 antagonizing VEGF induced angiogenic effects, we assess the influence of CS-6 on VEGF triggered VEGFR2 phosphorylation and as data shown in Fig. 5A. As shown in Fig. 5A, the addition of exogenous VEGF induced VEGFR-2 phosphorylation in two different phosphorylation sites (Tyr951 and Tyr1175), and VEGFR-2 phosphorylation at both Tyr951 and Tyr1175 was specifically suppressed by treatment with CS-6 without affecting the overall VEGFR-2 expression level. As expected, CS-6 limited VEGF-induced phosphorylation of VEGFR-2 in a dose-dependent manner. We hypothesized that CS-6 might bind to VEGFR-2 and subsequently affect the interaction between VEGF and VEGFR-2. To verify this possibility, next, the computer docking simulations of the interaction of CS-6 with VEGFR-2 were carried out. Molecular docking studies predicted that CS-6 would bind at the ATP binding site of VEGFR-2. As shown in Fig. 5C (a), CS-6 forms five hydrogen bonds with the ATP binding pocket of the VEGFR-2 kinase domain. The CO group at the lactonic ring of CS-6 forms a hydrogen bond with the backbone NH of Cys917. The OH group at the C14 position forms strong hydrogen bonds with the backbone at Cxc1045 and Lys 866 simultaneously. Moreover, 3-OH accepts two hydrogen bonds with the CO and NH residues of Phe843. The result of MOLCAD surface modeling indicated that the lactonic ring of CS-6 extends into the deep hydrophobic cavity of the ATP-binding pocket of VEGFR-2 (Fig. 5C, b). Moreover, we carried out Co-IP experiment to further explore whether CS-6 could interfere with the basis for VEGF-VEGFR-2 interaction. As shown in Fig. 5B, both VEGF and VEGFR-2 were detected in the resulting precipitates, indicating that VEGF was associated with VEGFR-2. The expression of phosphorTyr1175-VEGFR-2 was obviously reduced with the treatment of CS-6, which was consistent with the result of Western blotting analysis (Fig. 5B). These results showed that CS-6 had little effect on VEGF-VEGFR-2 interaction, but suppressed the activation of VEGFR-2.
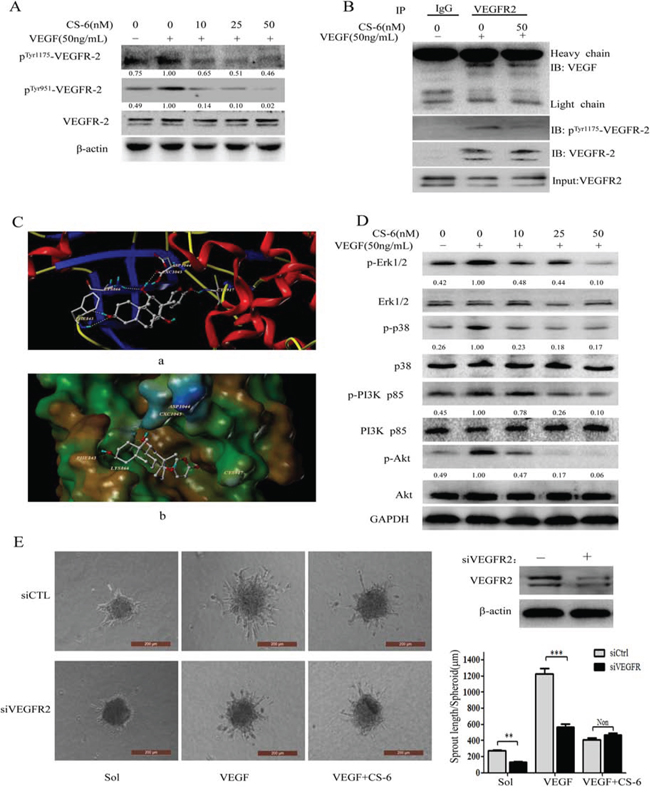
Figure 5: CS-6 inhibits the activation of VEGFR-2 kinase in HUVECs and has no effect on VEGF binding to VEGFR-2. A. CS-6 inhibits the phosphorylation of VEGFR-2. HUVECs were pre-treated with CS-6 for 6 h, and then stimulated with 50 ng/mL VEGF for 1 h. B. CS-6 did not interfere with VEGF binding to VEGFR-2. Whole-cell extracts were collected and analyzed by Co-IP assay and Western blotting using antibodies against VEGF, VEGFR-2 and pTyr1175-VEGFR-2. Three independent experiments were performed (##p < 0.01, VEGF-treated group vs. no VEGF-treated group; **p < 0.01, VEGF-treated group vs. VEGF and CS-6-treated group; ***p < 0.001, VEGF-treated group vs. VEGF and CS-6-treated group). C. The docking stereo view of CS-6 with ATP binding site of VEGFR-2. (a) Interactions of CS-6 and VEGFR-2 are delineated by ribbon structure. (b) MOLCAD surface representation of the ATP binding site of VEGFR-2. Hydrogen bonds are displayed as yellow dashed lines, and the participating amino acid residues are marked. D. CS-6 suppresses activation of VEGFR-2-mediated signaling pathway. HUVECs were pre-treated with CS-6 for 6 h, and then stimulated with 50 ng/mL VEGF for 1 h. Whole-cell extracts were extracted for Western blotting analysis. E. Effect of CS-6 on sprouting from modified human endothelial cell spheroids or VEGFR2-knockdown human endothelial cell spheroids (Original magnification, 200×).
CS-6 suppresses activation of VEGFR-2-mediated signaling pathway
VEGFR-2 activation initiates complex signaling networks with distinct and overlapping functions. We further examined whether CS-6 inhibited VEGFR-2-mediated signaling pathways. As shown in Fig. 5D, CS-6 significantly suppressed the activation of VEGFR-2 downstream signaling molecules such as PI3K/Akt and MAPK pathways, which are critical for VEGF/VEGFR-2-mediated angiogenesis. To be specific, p-Erk1/2 and p-p38 were enhanced by VEGF treatment while the expression level of Erk1/2 and p38 remained unchanged, and CS-6 inhibited the phosphorylation of Erk1/2 and p38 without affecting total Erk1/2 and p38 expression levels. CS-6 also reduces phosphorylation levels of Akt at Ser473, which is a well-known downstream target of VEGFR-2, however, has no effect on total Akt level. Moreover, the action of CS-6 on PI3K p85 was also examined.
Furthermore, in order to verify the anti-angiogenesis effect of CS-6 is via VEGFR2, we next knocked down VEGFR2 by siRNA, and performed endothelial cell spheroids sprouting assay. As shown in Fig. 5E, knockdown of VEGFR2 by siRNA considerably inhibited endothelial cell sprouting in sol group and VEGF group compared with non-siRNA knockdown in a modified spheroid assay, however, there was no obvious difference in CS-6-treatment group. These data indicated that the anti-angiogenesis effect of CS-6 was through VEGFR2 in HUVECs.
CS-6 inhibits VEGF-induced blood vessel formation in mice
To confirm our findings in vivo, we assess the impacts of CS-6 on endothelial angiogenesis in lung cancer xenografts model as well as martrigel plugs. model. Our previous study has shown that CS-6 inhibited the growth of lung cancer xenografts in nude mice [34]. Herein, we further examined the vessel density in lung cancer tissue sections visualized using VE-cadherin staining (Red) under immunofluorescent microscope. We chose VE-cadherin because of its modulation of angiogenesis [41], and its expression could reflect the microvessel density in the tissues. Direct inhibition of VE-cadherin function could impact other signaling functions such as modulation of VEGF receptor signaling. As shown in Fig. 6A, the micro-vessel density identified by VE-cadherin staining was significantly decreased with CS-6 treatment, as compared with the vehicle group. However, we could not exclude possibility that the CS-6 treatment impaired vessel density in the human xenograft was due to the tumor suppressive role of CS-6. To clarify this issue, we further applied matrigel plug assay. After 7 days, matrigel plugs containing VEGF excised from mice were dark red and filled with blood vessels. In contrast, matrigel plugs containing both VEGF and CS-6 were light yellow. The group treated with 5 μM CS-6 was nearly transparent (Fig. 6B). We then used FITC-isolectin and DAPI to stain and quantify the number of functional vessels in the matrigel plugs. As shown in Fig. 6C, fewer vessels were observed in the matrigel plugs treated with both VEGF and CS-6 than in those plugs treated with VEGF alone. These results indicate that CS-6 can significantly inhibit angiogenesis in vivo.
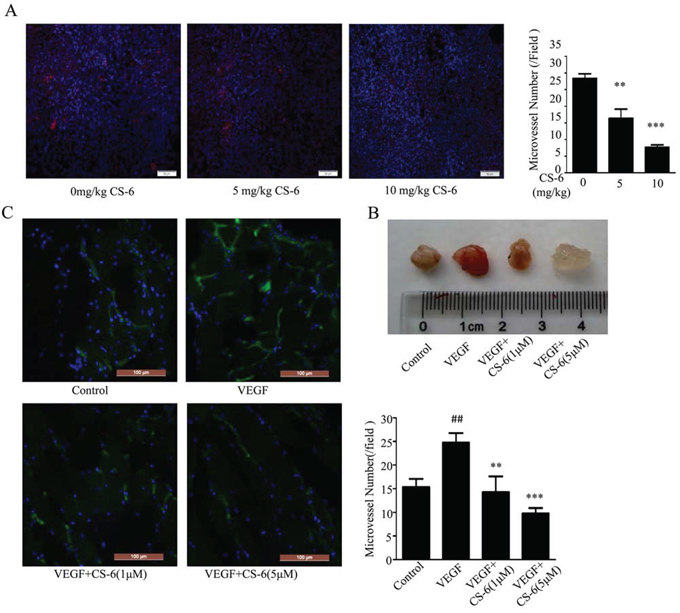
Figure 6: CS-6 inhibited angiogenesis in vivo. A. Neutral formalin fixed tumor samples were prepared from animals and analyzed by immunohistochemical staining with VE-cadherin, and examined under a microscope. (**P < 0.01, ***P < 0.001, significant differences between CS-6 treatment groups and control group. Red is displayed as VE-cadherin protein). B. The axillary fossa of 6-week-old C57/BL/6 mice was injected with 500 mL of matrigel with 250 ng VEGF and 80 unit of heparin. After 9 days, the matrigel plugs were harvested and photographed. C. The matrigel plugs were fixed, sectioned and stained with FITC-Lectin. (##p < 0.01, VEGF-treated group vs. no VEGF-treated group; **p < 0.01, VEGF-treated group vs. VEGF- and CS-6-treated group; ***p < 0.001, VEGF-treated group vs. VEGF- and CS-6-treated group. Green is displayed as FITC-Lectin).
Taken all together, our results showed that gamabufotalin (CS-6) exerted its anti-angiogenic effect by suppressing VEGFR-2 activation.
DISCUSSION
Our previous study showed that gamabufotalin (CS-6), in the nanomolar range, markedly reduced cell proliferation and migration of NSCLC cells [34]. However, little is known about the inhibitory effects of CS-6 on angiogenesis and associated molecular mechanism. Angiogenesis is important for tumor growth, maintenance and invasion. Tumor growth is initially fed by nearby blood vessels, and new blood vessels are required to support the growth when the tumor size exceeds a certain size [42]. Therefore, Neo-angiogenesis is the critical step in the development and progression of most of the human cancers, and anti-angiogenic therapy of cancer treatment is a new method for cancer treatment.
In previous study, we found that CS-6 reduced cell growth in several human lung cancer cell lines (IC50 was about 55 nM), while had no adverse effect on human normal lung cell line (HLF cells). The interesting observation may suggest that CS-6 would be more favor to affect tumor cell viability, while whether or not it potentially influencing endothelial angiogenesis during tumor development is totally unknown. Herein, we presented in current study that CS-6 showed minor inhibitory effects (IC50 was more than 200 nM) on HUVECs, a similar phenomena observed in previous tests of normal human lung cells; however, CS-6 would remarkably inhibit VEGF induced endothelial growth, suggesting it may interact VEGF signaling as well corresponding endothelial bio-function. Indeed, we found that CS-6 could inhibit angiogenesis in vitro, ex vivo and in vivo, and suppress key steps related to angiogenesis, including proliferation, survival, migration and angiogenesis in endothelial cells. However, we noticed the anti-angiogenic effective doses of CS-6 among cell cultures (10, 25 and 50 nM), aortic tissue cultures (500 and 1000 nM) as well animal levels (1000 and 5000 nM) are different, which may be resulted from varied growth environments or complex background. In order to exclude the possibility that toxicity of CS-6 would give raise to false positive phenomenon on endothelial angiogenesis, we well controlled these issues. For example, the doses of CS-6 used in cell cultures were no clear toxic effects determined by MTT assay; in aortic ring tissue cultures, we did not observed CS-6 increased apoptotic endothelial cells by PI staining. Further investigation showed that CS-6 inhibited angiogenesis via suppressing of VEGF-induced VEGFR-2 phosphorylation and activation by targeting the ATP-binding site of VEGFR-2. Here, to the best of our knowledge, it is the first time to reveal that CS-6 inhibits angiogenesis both in vitro and in vivo.
It has been well documented that Cyclooxygenase-2 (COX-2), as one of the VEGF/VEGFR-2 signaling client proteins, plays a central role in the modulation of angiogenesis, and our previous study found CS-6 suppressed COX-2 expression in non-small cell lung cancer (NSCLC) cells. Therefore, we investigate whether CS-6 inhibit angiogenesis was associated COX-2 expression. We discovered CS-6 would not affect COX-2 expression in HUVECs in basal culture condition; however, it was able to specifically reduce VEGF-induced elevation of COX-2 in endothelial cells (Data not shown). It demonstrated CS-6 performs clearly different mechanism in regulating COX-2 expression in endothelial cell as a downstream event of VEGF-VEGFR2 signaling with the transcriptional regulation in NSCLC.
Tumor growth depends on angiogenesis and anti-angiogenic therapy is a new therapeutic approach for cancer treatment. Previous research indicates that VEGF/VEGFR-2 signaling pathway is the main cascade involved in angiogenesis growth, and Erk1/2 and PI3K/Akt pathways could regulate VEGF expression [43]. VEGFR-2 activation stimulates complex signaling networks with distinct and overlapping functions, including MAPK and PI3K/Akt pathways, which play important roles in vascular endothelial cell growth, survival and migration. In this study, we showed that CS-6 suppressed PI3K/Akt and MAPK signaling pathways through inhibiting VEGFR-2 activation by targeting the ATP-binding site.
VEGF and its high-affinity receptor VEGFR-2 are the most widely studied factors in angiogenesis. Targeting of VEGFR-2 is an intriguing strategy in the anti-angiogenic therapy of tumors [44], which is essential for the functions of vascular endothelial cells. Thus, we studied whether CS-6 could inhibit VEGF-induced VEGFR-2 activation. In the present study, CS-6 was shown to inhibit VEGF-induced tyrosine phosphorylation of VEGFR-2 in a dose-dependent manner. Moreover, computational docking showed that CS-6 occupied the deep hydrophobic pocket in the ATP-binding site of VEGFR-2. In this modeling analysis, five hydroxyl groups bound to the ATP-binding pocket of VEGFR-2 via forming five hydrogen bonds. The CO motif at the lactonic ring of CS-6 forms hydrogen bond with Cys917 of the hinge region of VEGFR-2 [45]. Residue Cys917 is a crucial amino acid for ligand reorganization and binding on the ATP site as reported. The hydroxyl group at the C-14 position of cs6 bound to the ATP-binding pocket of VEGFR-2 via forming two hydrogen bonds with residues Lys866 and Cxc1045 of the polar region. These hydrogen bond interactions were important for improvement the binding affinity of CS-6 to VEGFR-2 [46]. The additional hydrogen bond interactions of CS-6 with Phe843 stabilized its occupation of the hydrophobic pocket of VEGFR-2. These above data suggest that CS-6 may inhibit angiogenesis by targeting VEGFR-2.
MATERIALS AND METHODS
Chemicals and reagents
Gamabufotalin (CS-6) was isolated from ChanSu in our lab, which was secreted from the postauricular and skin glands of Bufo bufo gargarizans Cantor [34]. In our study, CS-6 was dissolved in DMSO as a 100 μM stock solution and diluted in the relevant assay media and kept at −20°C. CS-6 was diluted in culture medium to obtain the desired concentration, which was stable in the dilution with DMSO concentration less than 1‰.
Antibodies and other materials
3-(4, 5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium (MTT), dimethyl sulfoxide (DMSO), M199 and Lectin from Bandeiraea simplicifolia (Griffonia simplicifolia) were purchased from Sigma Chemical Co. (St. Louis, MO). Recombinant human VEGF165 and human basic fibroblast growth factor (bFGF), were obtained from PeproTech Inc. Recombinant human EGF was purchased from ProSpec Company. Matrigel were purchased from BD Bioscience (San Diego, CA). The primary antibodies for VEGF, VEGFR-2, pTyr1175-VEGFR-2, pTyr951-VEGFR-2, p-p38, p-ERK, p38, ERK, Akt, p-Akt, PI3K p110α, PI3K p85, p-PTEN, p-Colifin, Colifin, cleaved-caspase3/9, cleaved-PARP and all the secondary antibodies were acquired from Cell Signaling Technology (Cell Signaling Technology, Inc, USA). The primary antibodies for GAPDH, β-actin, Bax and Bcl-2 were obtained from Proteintech Group (Proteintech Group,Inc., USA). Annexin V-FITC apoptosis detection kit was purchased from Roche (Indianapolis, IN). Control siRNA (5′-uucuuccgaacgugucacgutt-3′) and siRNA against human VEGFR2 (5′-gcggcuaccaguccggauatt-3′) were synthesized by genepharm. Trypsin and DMEM/F12 were obtained from HyClone, and fetal bovine serum (FBS) was obtained from Gibico. M199 Medium was bought from LIFE. siRNA and all other chemicals were purchased from Sigma Chemical Co. unless otherwise specified.
Cell culture
Primary human umbilical vein endothelial cells (HUVECs) were isolated from human umbilical vein as described[47]. HUVECs were cultured in M199 containing 10% fetal bovine serum (FBS), 25 U/mL heparin, 5 ng/mL bFGF and 10 ng/mL EGF, The cells were cultured in a humidified atmosphere of 5% CO2 at 37°C.
siRNA transfection
After one hour serum free starvation in M199 medium with antibiotics, and HUVECs were transfected with control or siRNA against human VEGFR2 were by lipofectamine 3000 (Invitrogen) according to manufactory instruction.
Cell proliferation assay
HUVECs viability was measured by MTT assay. Briefly, HUVECs were seeded at 8 × 103 cells/well in 96-well plates, and allowed to adhere to obtain 80% confluent monolayer. The medium was replaced with normal growth medium containing CS-6 (0, 1, 10, 25, 50 and 75 nM). After 12 h or 24 h incubation, the growth of cells was measured at 490 nm using an EnSpire® Multimode Plate Reade (Perkin Elmer, USA). The effect of CS-6 on VEGF-induced cell viability was measured by MTT assay. HUVECs (6 × 103 cells/well) were respectively treated with or without VEGF (50 ng/mL) or various concentration of CS-6 (0, 10, 25 and 50 nM) for 24 h and 48 h, and then cell growth was measured. In control cells, equivalent amount of DMSO was added as vehicle. The number of viable cells was presented relative to untreated controls.
Wound healing migration assay
HUVECs were allowed to grow into full confluence in 6-well plates. A line of HUVECs was then scraped away in each well using a pipette tip after 6h of serum starvation. Subsequently, cells were washed twice to remove detached cells. Fresh M199 medium containing different concentrations of CS-6 (0, 10, 25 and 50 nM) or vehicle, with or without 50 ng/mL VEGF was added to the scratched monolayers. Images were taken using a Leica DM 14000B microscope after 12 h incubation. The migrated cells were observed from three randomly chosen fields and quantified by manual counting. Inhibition percentage was expressed as percentage of the untreated cells (100%).
Endothelial cell transwell invasion assay
The motility of HUVECs was performed in 24-well transwell plates [48]. The upper surface of polycarbonate filters with 8 μm pores was coated with 75 μL Matrigel (Matrigel: M199=1:3, without growth factors) and incubated for 0.5 h at 37°C for gelling. Then, Cells were seeded into the upper chambers at a density of 5 × 104 cells per chamber, the bottom chamber were filled with 600 μL M199 with 10% FBS supplemented with VEGF (50 ng/mL) or vehicle. Both top and bottom chamber contained the same concentrations of CS-6. After 24 h incubation, non-invasive cells on the upper membrane surfaces were removed by wiping with cotton swabs. Invaded cells were fixed with methanol and stained with 0.1% Crystal Violet Staining Solution. The membrane was dried in the air. Images were taken using a Leica DM 14000B microscope. Cell invasion counted in five independent areas per membrane. The results were the means calculated from five replicates of each experiment.
Tube formation assay
HUVECs [1 × 104 in 50 μL M199 medium with 0.1% BSA containing CS-6 (0, 10, 25, 50 nM)] were seeded on Ibitreat angiogenesis slides (Ibidi, Martinsried, Germany) pre-coated with 10 μL Matrigel, and the formation of tubular like structure was recorded by a Leica DM14000B microscope at 6 hours or 24 hours.
Endothelial cell spheroids sprouting assay
Spheroids were generated by gravity as described [47]. Subsequently, spheroids were harvested by 5 min centrifugation at 1000 rpm and embedded into a collagen gel (containing collagen (1 mg/mL); 1x M199 (sigma); 0.22% NaHCO3) with pH at 7.4 adjusted by 0.1 N NaOH. The spheroid-containing gel was rapidly transferred into a 24-well plate and allowed to polymerize at 37°C incubators for 30 min, M199 medium with or without VEGF and CS-6 was applied on top of the gel. Spheroids sprouts were evaluated by measuring the cumulative length of all capillary like sprouts using a Leica DM14000B microscope. At least 5 randomly selected spheroids per experimental group and experiment were analyzed. And sprout length was measured with Image J software.
Aortic ring assay
Aorta from C57/BL6 mice were removed, cleaned and approximate 1mm aortic rings were embedded in a collagen gel (collagen type I, BD Biosciences) in a 48 well plate, After gel polymerization, DMEM/F12 medium (HyClone) supplemented with 2.5% mouse serum. Additional VEGF (50 ng/mL) and CS-6 (500 nM and 1000 nM) were added and the endothelial sprouts were allowed to develop over 9 days. Thereafter, the samples were fixed (4% paraformaldehyde) and endothelial cells were visualized using FITC-Lectin (Sigma) staining under immunofluorescent microscope. The results were the means calculated from five replicates of each experiment.
Matrigel plug assay
All animals were maintained, and animal experiments were done in SPF Laboratory Animal Center at Dalian Medical University. The animals used in this research were 6 weeks old male C57/BL6 mice (three mice per group). Matrigel (0.5 mL/plug) containing 250 ng VEGF and 80 units heparin with various concentrations of CS-6 (1 or 5 μM) were injected (S.C.) into near the axillary fossa of mice. Matrigel mixed with medium alone was used as a negative control. After 9 days of implantation, the matrigel plugs were removed and the surrounding tissues were trimmed. The matrigel plugs were embedded with O.C.T. Compound (Sakura Finetek USA, Inc.). Ten-micron sections were stained by FITC-Lectin (Sigma) staining. The number of blood vessels in high power field (HPF, magnification) was counted under immunofluorescent microscope. The results were the means calculated from five replicates of each experiment.
Microvessel density assay by VE-cadherin staining
Human lung xenografts samples were from our previous study [34]. 4μm sections were stained with VE-cadherin (1:200, Santa Cruz, sc-6458) and DAPI (1:5000). The endothelial staining was photographed under immunofluorescent microscope and vessel density was evaluated by Image J software. The results were shown as Mean ± SD from at least 5 tumor samples for each group. The tumor sections were from three treatment groups that each contained five mice.
Molecular modeling
Docking studies were performed to explore the potential binding interactions between CS-6 and VEGFR-2. The compound CS-6 was optimized using the semi-empirical PM3 method with the Polak-Ribie’re conjugate gradient algorithm with an RMS gradient of 0.001 kcal mol−1 Å−1 as convergence criterion. The optimized structure was further docked into the active site of VEGFR-2 (PDB Code: 1YWN). The crystallographic ligand was extracted from the active site, and the residues within a 6.5 Å radius around the VEGFR-2 molecule were defined as the active site. The Surflex-Dock program was used for the docking calculations with default parameters. MOLCAD surfaces were generated for visualizing the binding mode of the docked protein–ligand complexes.
Co-immunoprecipitation (Co-IP)
In order to determine the effect of CS-6 on the interaction between VEGFR-2 and VEGF, HUVECs were incubated in M199 with 0.5% FBS for 6 h followed by treating with CS-6 for 4 h, followed by 50 ng/mL VEGF165 treatments for 1 h. Briefly, cell extract proteins (500 μg) were pre-cleared by adding 1 μg normal rabbit IgG and 40 μL protein A/G-Agarose beads. Together, they were incubated at 4°C for 1 h with gentle agitation and centrifuge. The supernatants were collected and incubated with a specific rabbit anti-VEGFR-2 antibody at 4°C overnight with gentle rotary agitation. The immune complexes were pulled down by protein A/G agarose beads (Santa Cruz Biotechnology) and washed with ice-cold PBS buffer containing proteinase inhibitor for 3 times. After final wash, the immune complexes were released by boiling in 2 × electrophoresis sample buffer for 5 min, followed by western blotting analysis with rabbit anti-VEGFR-2, rabbit anti-phosphoTyr1175-VEGFR-2 and mouse anti-VEGF antibodies.
Cell apoptosis analysis
Cell apoptosis analyses were carried out by fluorescence-activated cell sorter (FACS). After treatment, the cells were harvested by 0.25% trypsin and washed by PBS buffer and then stained simultaneously with FITC-labeled annexin V and PI. Stained cells were analyzed using FACS Accuri C6.
F-actin staining and immunofluorescence analysis
HUVECs were seeded onto coverslips in a 6-well plate. The cells were treated with or without 50 μM CS-6 for 1h and then were stimulated with 50 ng/mL VEGF for 15min. After that, the cells were fixed in 4% paraformaldehyde for 20 min, and then permeabilized with 0.2% Triton X-100 for 5 min at room temperature. Actin filaments were stained by phalloidin-FITC for 20 min, and nuclei were detected by DAPI. For phosphorcofilin immunofluorescence, cells were blocked with 5% BSA in PBS for 1 h. After washing three times with PBS, cells were incubated with antibody against phospho-cofilin overnight at 4°C, and then with secondary antibodies for another 1 h at room temperature. Next, cell nuclei were stained by DAPI for 5 min. The slides were examined with a Leica DM 14000B confocal microscope.
Western blotting
To determine the effects of CS-6 on VEGFR-2-dependent signaling cascade, starved HUVECs were treated with CS-6 for 6 h, followed by 50ng/mL VEGF165 treatments for 1 h. Cells were lysed with an appropriate cold lysis buffer (Beyotime Biotechnology, China) supplemented with proteinase inhibitor cocktail (Sigma, USA). Protein concentrations were determined using a BCA protein assay kit (Beyotime Biotechnology, China). Proteins (50 μg) were applied to 8% to 12% SDS-PAGE gels and transferred onto a PVDF membrance (Millipore, USA). The membranes were incubated with specific primary antibodies. Protein bands were detected using an enhanced chemiluminescence method. Bands were normalized with GAPDH or β-actin as an internal control. Similar experiments were performed at least three times.
Statistical analysis
Data are represented as mean ± SD obtained for at least three independent experiments. Statistical significance was determined by one-way ANOVA. P<0.05 was considered to be a statistically significant difference. SPSS 17.0 software was used for all statistical analysis. The mean density of the interest bands (where mentioned) was determined using Scion Image software.
CONCLUSION
We demonstrated for the first time that CS-6 as a natural bufadienolide, exhibited the significant anti-angiogenesis effect by inhibiting proliferation, migration, invasion, tube formation and enhancing apoptosis of human umbilical vein endothelial cells in a dose-dependent manner. Moreover, its molecular mechanisms inhibiting angiogenesis are mainly mediated through suppressing the VEGF/VEGFR-2-mediated signaling cascades by targeting ATP binding site, inactivating PI3K/Akt and MAPK pathways. From the above, these findings strongly demonstrate that CS-6 could serve as a potential candidate for anti-angiogenic therapy and a potent inhibitor of VEGFR-2-activated.
ACKNOWLEDGMENTS
This work was supported by the National Natural Science Foundation of China (No. 81274047, 81473334, 81503201 and 81303146), Education Department of Liaoning Province (LR2014025 and L2014352), Distinguished professor of Liaoning Province, Outstanding Youth Science and Technology Talents of Dalian (2014J11JH132), and Innovation Team of Dalian Medical University.
CONFLICTS OF INTEREST
The authors declare that they have no competing interests.
Author contributions
ZY, LS and XM contributed to the conception or design of the work. NT, LS, ZY, PD, CW, XH, BZ, SH, SD and XM contributed to the acquisition, analysis or interpretation of data for the work. NT and ZY drafted the manuscript. LS, KL, TM, XW, LW and XM revised the manuscript. All authors have read and approved submission of this manuscript and take full responsibility for the manuscript.
REFERENCES
1. Folkman J. Angiogenesis in cancer, vascular, rheumatoid and other disease. Nature medicine. 1995; 1:27-31.
2. Carmeliet P and Jain RK. Angiogenesis in cancer and other diseases. Nature. 2000; 407:249-257.
3. Dvorak HF. Angiogenesis: update 2005. Journal of thrombosis and haemostasis : JTH. 2005; 3:1835-1842.
4. Ivy SP, Wick JY and Kaufman BM. An overview of small-molecule inhibitors of VEGFR signaling. Nature reviews Clinical oncology. 2009; 6:569-579.
5. Zhao Y and Adjei AA. Targeting Angiogenesis in Cancer Therapy: Moving Beyond Vascular Endothelial Growth Factor. The oncologist. 2015; 20:660-673.
6. Turner HE, Harris AL, Melmed S and Wass JA. Angiogenesis in endocrine tumors. Endocrine reviews. 2003; 24:600-632.
7. Folkman J. Angiogenesis: an organizing principle for drug discovery? Nature reviews Drug discovery. 2007; 6:273-286.
8. Shojaei F. Anti-angiogenesis therapy in cancer: current challenges and future perspectives. Cancer letters. 2012; 320:130-137.
9. Ambasta RK, Sharma A and Kumar P. Nanoparticle mediated targeting of VEGFR and cancer stem cells for cancer therapy. Vascular cell. 2011; 3:26.
10. Senger DR, Van de Water L, Brown LF, Nagy JA, Yeo KT, Yeo TK, Berse B, Jackman RW, Dvorak AM and Dvorak HF. Vascular permeability factor (VPF, VEGF) in tumor biology. Cancer metastasis reviews. 1993; 12:303-324.
11. Stefanini MO, Wu FT, Mac Gabhann F and Popel AS. A compartment model of VEGF distribution in blood, healthy and diseased tissues. BMC systems biology. 2008; 2:77.
12. Romon R, Adriaenssens E, Lagadec C, Germain E, Hondermarck H and Le Bourhis X. Nerve growth factor promotes breast cancer angiogenesis by activating multiple pathways. Molecular cancer. 2010; 9:157.
13. Greenberg S and Rugo HS. Triple-negative breast cancer: role of antiangiogenic agents. Cancer journal. 2010; 16:33-38.
14. Delli Carpini J, Karam AK and Montgomery L. Vascular endothelial growth factor and its relationship to the prognosis and treatment of breast, ovarian, and cervical cancer. Angiogenesis. 2010; 13:43-58.
15. Otrock ZK, Makarem JA and Shamseddine AI. Vascular endothelial growth factor family of ligands and receptors: review. Blood cells, molecules & diseases. 2007; 38:258-268.
16. Ferrara N, Gerber HP and LeCouter J. The biology of VEGF and its receptors. Nature medicine. 2003; 9:669-676.
17. Ferrara N. Vascular endothelial growth factor: basic science and clinical progress. Endocrine reviews. 2004; 25:581-611.
18. Xia Y, Song X, Li D, Ye T, Xu Y, Lin H, Meng N, Li G, Deng S, Zhang S, Liu L, Zhu Y, Zeng J, Lei Q, Pan Y, Wei Y, et al. YLT192, a novel, orally active bioavailable inhibitor of VEGFR2 signaling with potent antiangiogenic activity and antitumor efficacy in preclinical models. Scientific reports. 2014; 4:6031.
19. Wiesmann C, Fuh G, Christinger HW, Eigenbrot C, Wells JA and de Vos AM. Crystal structure at 1.7 A resolution of VEGF in complex with domain 2 of the Flt-1 receptor. Cell. 1997; 91:695-704.
20. Lu D, Kussie P, Pytowski B, Persaud K, Bohlen P, Witte L and Zhu Z. Identification of the residues in the extracellular region of KDR important for interaction with vascular endothelial growth factor and neutralizing anti-KDR antibodies. The Journal of biological chemistry. 2000; 275:14321-14330.
21. van der Meel R, Symons MH, Kudernatsch R, Kok RJ, Schiffelers RM, Storm G, Gallagher WM and Byrne AT. The VEGF/Rho GTPase signalling pathway: a promising target for anti-angiogenic/anti-invasion therapy. Drug discovery today. 2011; 16:219-228.
22. Kranenburg O, Gebbink MF and Voest EE. Stimulation of angiogenesis by Ras proteins. Biochimica et biophysica acta. 2004; 1654:23-37.
23. Meadows KN, Bryant P, Vincent PA and Pumiglia KM. Activated Ras induces a proangiogenic phenotype in primary endothelial cells. Oncogene. 2004; 23:192-200.
24. Meadows KN, Bryant P and Pumiglia K. Vascular endothelial growth factor induction of the angiogenic phenotype requires Ras activation. The Journal of biological chemistry. 2001; 276:49289-49298.
25. Li M, Wu S, Liu Z, Zhang W, Xu J, Wang Y, Liu J, Zhang D, Tian H, Li Y and Ye W. Arenobufagin, a bufadienolide compound from toad venom, inhibits VEGF-mediated angiogenesis through suppression of VEGFR-2 signaling pathway. Biochemical pharmacology. 2012; 83:1251-1260.
26. May BH, Lu C, Bennett L, Hugel HM and Xue CC. Evaluating the traditional Chinese literature for herbal formulae and individual herbs used for age-related dementia and memory impairment. Biogerontology. 2012; 13:299-312.
27. Xin XL, Liu J, Ma XC, Wei Q, Lv L, Wang CY, Yao JH and Cui J. Preparative separation of four major bufadienolides from the Chinese traditional medicine, Chansu, using high-speed counter-current chromatography. Natural product communications. 2010; 5:1031-1034.
28. Ma XC, Xin X, Zhang BJ, Deng S, Yao JH, Wang CY, Cui J, Tian Y and Liu KX. Efficient isolation and purification of five products from microbial biotransformation of cinobufagin by high-speed counter-current chromatography. Journal of separation science. 2010; 33:2272-2277.
29. Kamano Y, Kotake A, Hashima H, Inoue M, Morita H, Takeya K, Itokawa H, Nandachi N, Segawa T, Yukita A, Saitou K, Katsuyama M and Pettit GR. Structure-cytotoxic activity relationship for the toad poison bufadienolides. Bioorganic & medicinal chemistry. 1998; 6:1103-1115.
30. Qin TJ, Zhao XH, Yun J, Zhang LX, Ruan ZP and Pan BR. Efficacy and safety of gemcitabine-oxaliplatin combined with huachansu in patients with advanced gallbladder carcinoma. World journal of gastroenterology. 2008; 14:5210-5216.
31. Qi F, Li A, Inagaki Y, Kokudo N, Tamura S, Nakata M and Tang W. Antitumor activity of extracts and compounds from the skin of the toad Bufo bufo gargarizans Cantor. International immunopharmacology. 2011; 11:342-349.
32. Li D, Qu X, Hou K, Zhang Y, Dong Q, Teng Y, Zhang J and Liu Y. PI3K/Akt is involved in bufalin-induced apoptosis in gastric cancer cells. Anti-cancer drugs. 2009; 20:59-64.
33. Ko WS, Park TY, Park C, Kim YH, Yoon HJ, Lee SY, Hong SH, Choi BT, Lee YT and Choi YH. Induction of apoptosis by Chan Su, a traditional Chinese medicine, in human bladder carcinoma T24 cells. Oncology reports. 2005; 14:475-480.
34. Yu Z, Guo W, Ma X, Zhang B, Dong P, Huang L, Wang X, Wang C, Huo X, Yu W, Yi C, Xiao Y, Yang W, Qin Y, Yuan Y, Meng S, et al. Gamabufotalin, a bufadienolide compound from toad venom, suppresses COX-2 expression through targeting IKKbeta/NF-kappaB signaling pathway in lung cancer cells. Molecular cancer. 2014; 13:203.
35. Xu JL, Lai R, Kinoshita T, Nakashima N and Nagasaka T. Proliferation, apoptosis, and intratumoral vascularity in multiple myeloma: correlation with the clinical stage and cytological grade. Journal of clinical pathology. 2002; 55:530-534.
36. Abu-Ghazaleh R, Kabir J, Jia H, Lobo M and Zachary I. Src mediates stimulation by vascular endothelial growth factor of the phosphorylation of focal adhesion kinase at tyrosine 861, and migration and anti-apoptosis in endothelial cells. The Biochemical journal. 2001; 360:255-264.
37. Patan S. Vasculogenesis and angiogenesis. Cancer treatment and research. 2004; 117:3-32.
38. Saunders WB, Bohnsack BL, Faske JB, Anthis NJ, Bayless KJ, Hirschi KK and Davis GE. Coregulation of vascular tube stabilization by endothelial cell TIMP-2 and pericyte TIMP-3. The Journal of cell biology. 2006; 175:179-191.
39. Uchida C and Haas TL. Evolving strategies in manipulating VEGF/VEGFR signaling for the promotion of angiogenesis in ischemic muscle. Current pharmaceutical design. 2009; 15:411-421.
40. Guo S, Colbert LS, Fuller M, Zhang Y and Gonzalez-Perez RR. Vascular endothelial growth factor receptor-2 in breast cancer. Biochimica et biophysica acta. 2010; 1806:108-121.
41. Carmeliet P, Lampugnani MG, Moons L, Breviario F, Compernolle V, Bono F, Balconi G, Spagnuolo R, Oosthuyse B, Dewerchin M, Zanetti A, Angellilo A, Mattot V, Nuyens D, Lutgens E, Clotman F, et al. Targeted deficiency or cytosolic truncation of the VE-cadherin gene in mice impairs VEGF-mediated endothelial survival and angiogenesis. Cell. 1999; 98:147-157.
42. Cook KM and Figg WD. Angiogenesis inhibitors: current strategies and future prospects. CA: a cancer journal for clinicians. 2010; 60:222-243.
43. Zhang S, Cao Z, Tian H, Shen G, Ma Y, Xie H, Liu Y, Zhao C, Deng S, Yang Y, Zheng R, Li W, Zhang N, Liu S, Wang W, Dai L, et al. SKLB1002, a novel potent inhibitor of VEGF receptor 2 signaling, inhibits angiogenesis and tumor growth in vivo. Clinical cancer research. 2011; 17:4439-4450.
44. Sandur SK, Ichikawa H, Sethi G, Ahn KS and Aggarwal BB. Plumbagin (5-hydroxy-2-methyl-1,4-naphthoquinone) suppresses NF-kappaB activation and NF-kappaB-regulated gene products through modulation of p65 and IkappaBalpha kinase activation, leading to potentiation of apoptosis induced by cytokine and chemotherapeutic agents. The Journal of biological chemistry. 2006; 281:17023-17033.
45. Miyazaki Y, Matsunaga S, Tang J, Maeda Y, Nakano M, Philippe RJ, Shibahara M, Liu W, Sato H, Wang L and Nolte RT. Novel 4-amino-furo[2,3-d]pyrimidines as Tie-2 and VEGFR2 dual inhibitors. Bioorganic & medicinal chemistry letters. 2005; 15:2203-2207.
46. Chen J, Sheng C-Q, Zhang W-N, Zheng C-H, Zhou Y-J, Li Y-W and Zhu J. Study of Properties of VEGFR2 Active Site and Binding Mode of VEGFR2 and Its Inhibitors. Acta Chimica Sinica. 2007; 65, 2007(No. 6):6.
47. Shi L, Fisslthaler B, Zippel N, Fromel T, Hu J, Elgheznawy A, Heide H, Popp R and Fleming I. MicroRNA-223 antagonizes angiogenesis by targeting beta1 integrin and preventing growth factor signaling in endothelial cells. Circulation research. 2013; 113:1320-1330.
48. Agarwal C, Singh RP and Agarwal R. Grape seed extract induces apoptotic death of human prostate carcinoma DU145 cells via caspases activation accompanied by dissipation of mitochondrial membrane potential and cytochrome c release. Carcinogenesis. 2002; 23:1869-1876.