
INTRODUCTION
MicroRNAs (miRNAs) are a family of highly conserved short non-coding RNAs that can regulate gene expression by base pairing with the 3′-untranslated region (3′-UTR) to enhance mRNA degradation or inhibit post-transcriptional translation [1]. Emerging evidence indicates that miRNAs are abnormally expressed in a variety of human cancers, and the dysregulation of miRNAs contributes to tumor initiation, promotion, and progression [2–4].
Nasopharyngeal carcinoma (NPC) is a malignant tumor with the highest incidence rate in Southern China, especially in Guangdong and Hong Kong [5]. Local recurrence and distant metastasis are two major reasons for treatment failure and NPC-related death [6]. Abnormal expression of miRNAs in NPC has been reported [7–9], and several dysregulated miRNAs, such as miR-29c, miR-34c, miR-93, miR-143, miR-451, miR-504, and miR-744, can regulate NPC cell growth, proliferation, invasion, and metastasis [10–16]. These findings indicate that the dysfunction of miRNAs may be involved in NPC carcinogenesis and progression. Clearly, further investigation is required to clarify the roles of miRNAs in NPC tumorigenesis and to identify miRNAs that may serve as novel treatment targets for NPC patients.
Our previous microarray data indicated that miR-16 expression is significantly decreased in archived NPC tissues [7]. Recent studies have demonstrated that miR-16 acts as tumor suppressor by affecting cell apoptosis, the cell cycle, cell proliferation, and invasion in various types of cancers, such as chronic lymphocytic leukemia, prostate cancer, ovarian cancer, lung cancer, and gastric cancer [17–26]. Investigations have also suggested that miR-16 could serve as prognostic biomarker and enhance chemosensitivity, and the systemic delivery of miR-16 represents a novel treatment approach [27–32]. Fibroblast growth factor (FGF)/fibroblast growth factor receptor (FGFR) signaling axis plays important roles in driving tumor progression [33]. FGF2, which often localizes to the nucleus and/or cytoplasm, belongs to the FGF family and regulates the tumorigenesis and progression of a variety of cancers [34–35].
Here, we investigated the role and mechanism of miR-16 in NPC tumorigenesis and progression. We confirmed that miR-16 was decreased in NPC and that ectopic expression of miR-16 suppressed NPC cell proliferation, migration, and invasion in vitro and in vivo. FGF2 was verified as a direct functional target of miR-16, and both the PI3K/AKT and MAPK signaling pathways were repressed by restoration of miR-16. Taken together, these results suggested that miR-16 suppresses NPC progression by targeting FGF2, thus representing a potential target for miRNA-based therapy for NPC.
RESULTS
miR-16 is decreased in NPC tissues and cell lines
Based on our previous microarray data, miR-16 was found to be significantly decreased in archived NPC tissues. To validate this result, we first detected the miR-16 expression level in 16 freshly frozen NPC and 8 normal nasopharyngeal epithelial tissue samples using quantitative RT-PCR. The overall average expression level of miR-16 in NPC specimens was decreased by 46% compared with the normal nasopharyngeal epithelial tissue samples (fold change = 0.46, p = 0.034, Figure 1A). We then examined the expression level of miR-16 in six NPC cell lines and the immortalized nasopharyngeal epithelial cell line, NP69. Similarly, the miR-16 expression was significantly decreased (0.43 to 0.72 fold) in most NPC cell lines (except for SUNE-1 and HONE-1) compared with the NP69 cell line (Figure 1B). These results suggested that miR-16 is downregulated in NPC and that it may be involved in NPC tumorigenesis and progression.
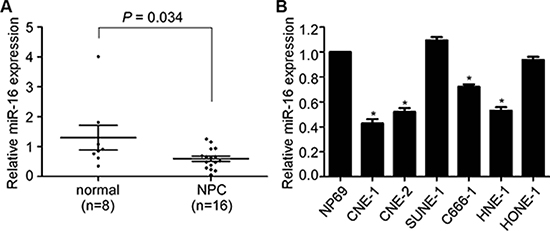
Figure 1: miR-16 is decreased in NPC tissues and cell lines. (A) Relative miR-16 expression in NPC tissues (n = 16) and normal nasopharyngeal epithelial tissues (n = 8). (B) Relative miR-16 expression in six NPC cell lines and the immortalized normal nasopharyngeal epithelial cell, NP69. U6 was used as an endogenous control. The data are presented as the mean ± S.D.; p values were calculated using Student’s t-test.
miR-16 suppresses NPC cell viability, cell proliferation, and tumor growth
To evaluate whether the ectopic expression of miR-16 affects the viability and proliferation of NPC cells, we transiently transfected CNE-1 and CNE-2 cells with miR-16 mimics and performed MTT, colony formation, and anchorage-independent soft-agar assays. Ectopic expression of miR-16 in NPC cells remarkably inhibited the cell growth and colony formation rates, as determined by MTT and colony formation assays, respectively, demonstrating the suppressive effect of miR-16 on cell proliferation, which was further confirmed by anchorage-independent soft-agar assays (Figure 2A–2C). Furthermore, we established CNE-1 cell lines stably expressing miR-16 (lenti-miR-16) or control empty vector (lenti-vector) and examined the effect of miR-16 on tumor growth using a xenograft tumor growth model. As shown in Figure 2D and 2E, the tumors formed by lenti-miR-16 cells had smaller volumes and lower weights than those formed by lenti-vector cells. Meanwhile, the lenti-miR-16 tumors showed lower percentage of Ki67- and PCNA-positive cells compared with the lenti-vector tumors (Figure 2F). These data demonstrated that miR-16 can inhibit NPC cell proliferation and tumor growth.
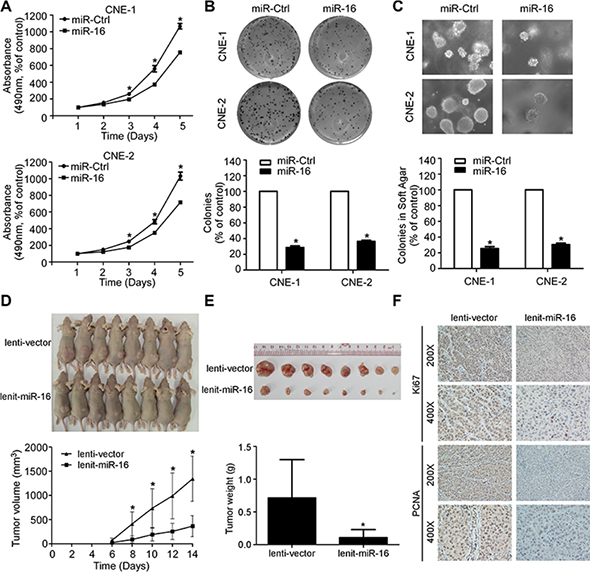
Figure 2: miR-16 suppresses NPC cell viability, cell proliferation, and tumor growth. (A) Cell viability of the indicated NPC cells was examined using MTT assays. (B–C) Representative images and quantification of colonies of the indicated cells tested by colony formation (B) and anchorage-independent soft-agar assays (C). (D–F) Xenograft tumor growth models in nude mice were constructed, and representative images of the formed tumors and the growth curves of tumor volumes (D) as well as of the excised tumors and tumor weights (E) are shown. Immunohistochemistry staining showed that miR-16 overexpression suppressed NPC cell proliferation in vivo as indicated by the expression of Ki67 and PCNA (F). The data are presented as the mean ± S.D.; p values were calculated using Student’s t-test.
miR-16 inhibits NPC cell migration, cell invasion, and lung metastatic colonization
To further determine whether the ectopic overexpression of miR-16 affects the migratory and invasive abilities of NPC, we performed wound healing, transwell migration, and invasion assays. The wound healing assay showed that both the CNE-1 and CNE-2 cell lines transfected with miR-16 mimics migrated more slowly compared with those transfected with miR-Ctrl (Figure 3A). In addition, ectopic overexpression of miR-16 in NPC cells significantly reduced the number of migratory and invasive cells as determined by transwell migration and invasion assays (Figure 3B and 3C). Furthermore, we constructed a lung metastatic colonization model to examine the effect of miR-16 on lung metastatic colonization formation ability. As shown in Figure 3D and 3E, the macroscopic and microscopic tumor nodules formed in the lungs by lenti-miR-16 cells were significantly fewer and smaller than those formed by lenti-vector cells. These results indicated that miR-16 overexpression can suppress NPC cell migration and invasion as well as lung metastatic colonization and growth.
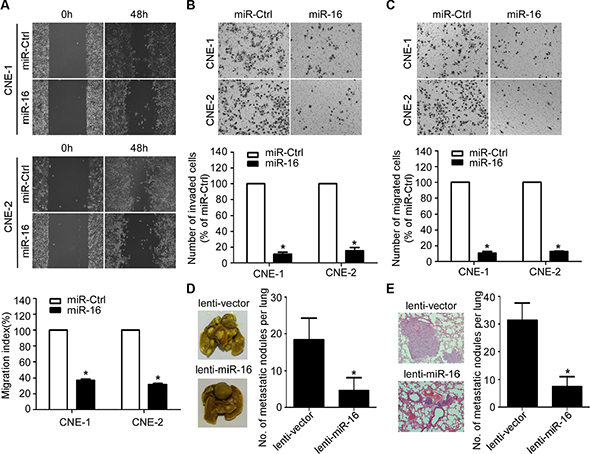
Figure 3: miR-16 inhibits NPC cell migration, invasion, and lung metastatic colonization. (A) Representative images and quantification of wound healing assays in the indicated cells. (B–C) Representative images and quantification of the indicated cells determined by Transwell migration (B) and invasion assays (C). (D–E) Lung metastatic colonization models in nude mice were constructed. Representative images and quantification of macroscopic tumor nodules formed on the lung surface (D); as well as of microscopic tumor nodules formed in the lung tissue sections stained with hematoxylin and eosin (E) are shown. The data are presented as the mean ± S.D.; p values were calculated using Student’s t-test.
FGF2 is a direct transcriptional target of miR-16 in NPC
Recently, FGF2 was demonstrated to promote tumor cell proliferation, migration, and invasion [35]. Based on bioinformatics analysis of three online databases (TargetScan, DIANA, and miRanda), the complementary sequence of miR-16 was found in two sites of the 3′-UTR of FGF2 mRNA (Figure 4A). To validate the transcriptional regulation of miR-16 on FGF2 expression, we cloned the FGF2 3′-UTR regions containing miR-16 binding sites or corresponding mutant sites into a luciferase reporter vector, and we performed luciferase reporter assays in CNE-1 and CNE-2 cells. miR-16 overexpression significantly reduced the luciferase activity of the FGF2 wild-type reporter plasmids compared with miR-Ctrl, and this inhibition was not observed in FGF2 mutant reporter plasmids (Figure 4B). Furthermore, miR-16 overexpression significantly decreased FGF2 expression at both the mRNA and protein levels (Figure 4C–4E). In addition, xenograft tumors formed in mice by lenti-miR-16 cells expressed higher levels of miR-16 and lower levels of FGF2 mRNA compared with tumors formed in mice by lenti-vector cells (Figure 4F and 4G). These findings suggested that miR-16 can transcriptionally regulate FGF2 expression in NPC cell lines.
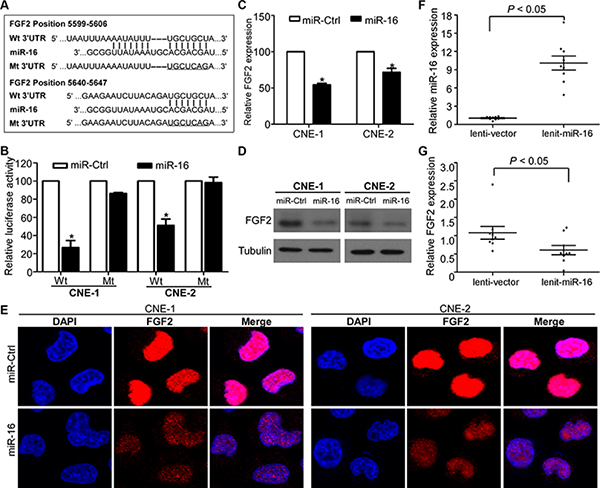
Figure 4: FGF2 is a direct transcriptional target of miR-16 in NPC. (A) Wild-type and mutant miR-16 target sequences of the FGF2 mRNA 3′-UTR. (B) Relative luciferase activities of the indicated cells quantified using luciferase reporter assays. (C–E) Quantification of FGF2 mRNA and protein expression using quantitative RT-PCR (C), western blotting (D), and immunofluorescent staining (E). The data are presented as the mean ± S.D.; p values were calculated using Student’s t-test.
Silencing of miR-16 promotes NPC cell viability, proliferation, migration, and invasion
To explore whether inhibition of miR-16 affects the NPC cell viability, proliferation, migratory, and invasive abilities, we transiently transfected SUNE-1 and HONE-1 cells with miR-16 inhibitors and performed MTT, colony formation, wound healing, and transwell invasion assays. As determined by the MTT and colony formation assays, silencing of miR-16 significantly increased the NPC cell growth and colony formation rates (Figure 5A and 5B). The wound healing assay showed that both SUNE-1 and HONE-1 cells transfected with miR-16 inhibitors migrated more quickly than anti-miR-Ctrl (Figure 5C). The transwell invasion assay also showed that inhibition of miR-16 in NPC cells remarkably increased the number of invasive cells (Figure 5D). Furthermore, inhibition of miR-16 remarkably increased the luciferase activity of the FGF2 wild-type reporter plasmids, and this increasing was not observed in FGF2 mutant reporter plasmids (Figure 5E). In addition, silencing of miR-16 obviously increased FGF2 expression at both the mRNA and protein levels (Figure 5F and 5G).
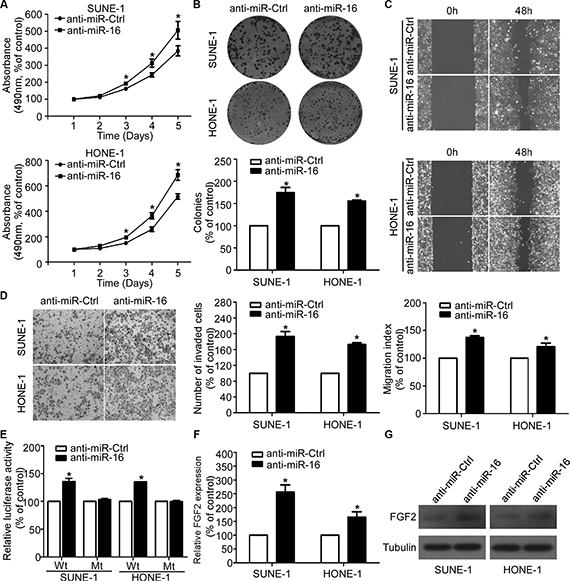
Figure 5: Silencing of miR-16 promotes NPC cell viability, proliferation, migration, and invasion. (A–D) Effects of the silencing of miR-16 on NPC cell viability, proliferation, migration and invasion. Representative results of the MTT (A), colony formation (B), wound healing (C), and transwell invasion assays (D). (E) Relative luciferase activities of the indicated cells quantified using luciferase reporter assays. (F–G) Quantification of FGF2 mRNA and protein expression using quantitative RT-PCR (F), and western blotting (G). The data are presented as the mean ± S.D.; p values were calculated using Student’s t-test.
miR-16 suppresses the PI3K/AKT and MAPK signaling pathways
FGF2 is one of the members of the FGF family and, it binds to FGF receptors (FGFRs), thus constituting the FGF/FGFR signaling pathway, which can activate several downstream signaling pathways, including the phosphoinositide-3-kinase/AKT (PI3K/AKT) and the mitogen-activated protein kinase (MAPK) pathways [35]. Because our findings suggested that miR-16 might inhibit NPC cell proliferation, migration and invasion by targeting FGF2, we assessed the effects of miR-16 overexpression on the MAPK and PI3K/AKT signaling pathways to determine the mechanism by which FGF2 contributed to NPC progression. Ectopic expression or silencing of miR-16 suppressed or promoted the expression of p-AKT and p-ERK, but not total AKT and ERK, in NPC cell lines (Figure 6). These results demonstrated that miR-16 may target FGF2 to inhibit NPC cell proliferation and invasion via the MAPK and PI3K/AKT signaling pathways.
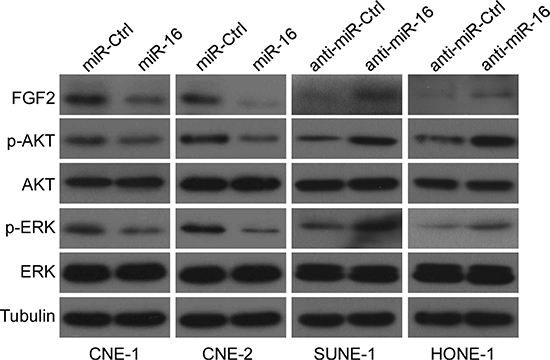
Figure 6: miR-16 suppresses the PI3K/AKT and MAPK signaling pathways. Western blotting was used to determine the expression levels of p-AKT, AKT, p-ERK, and ERK in CNE-1 and CNE-2 cells transfected with miR-16 mimics or inhibitor.
FGF2 mediates the effect of miR-16 on NPC cell proliferation and invasion
To further elucidate whether the tumor suppressive effects of miR-16 in NPC was directly mediated by FGF2, we cotransfected CNE-1 and CNE-2 cells with miR-16 mimics or miR-Ctrl together with either the empty vector or the FGF2 plasmid, which encoded the full-length coding sequence of FGF2 lacking its 3′-UTR, and we performed MTT, colony formation, anchorage-independent soft-agar, wound healing, and transwell migration/invasion assays. Western blotting validated the expression of FGF2 in the cotransfected cells (Figure 7A). Forced expression of FGF2 abrogated the suppressive effects of miR-16 on cell viability (Figure 7B), proliferation (Figure 7C), migration (Figure 7D and 7E), and invasion (Figure 7F). These results suggested that FGF2 is a direct and functional target of miR-16 in NPC.
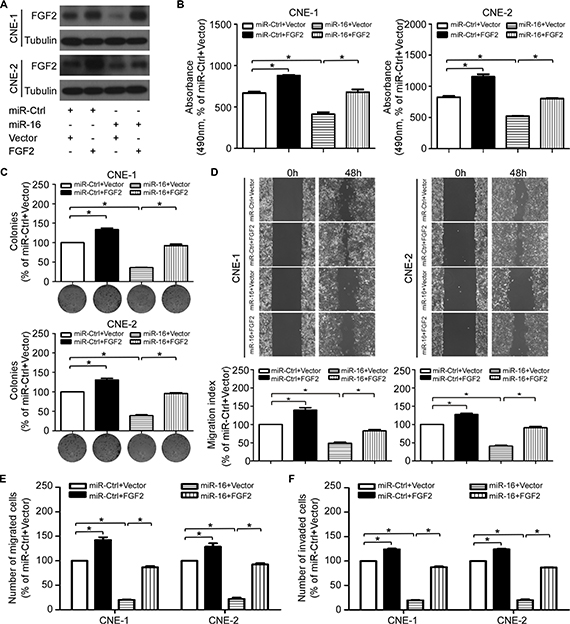
Figure 7: FGF2 mediates the effect of miR-16 on NPC cell proliferation and invasion. (A) The FGF2 protein expression of the indicated cells was determined with western blotting. (B–F) Effects of the restoration of FGF2 on NPC cell viability, proliferation, migration and invasion. Representative results of the MTT (B), colony formation (C), wound healing (D), transwell migration (E) and invasion assays (F). The data are presented as the mean ± S.D.; p values were calculated using Student’s t-test.
DISCUSSION
Local recurrence and distant metastasis are the major treatment failure patterns in patients with NPC. Thus, better understanding the mechanisms involved in NPC development and progression are important strategies for improving prognosis. In our current study, we verified that miR-16 is decreased in NPC and that miR-16 targets FGF2 to inhibit NPC cell proliferation and invasion via the MAPK and PI3K/AKT signaling pathways.
Previous studies have demonstrated that miRNAs are dysregulated in NPC tissues [7–9], and several dysregulated miRNAs are involved in regulating NPC proliferation, migration and invasion [10–16]. Studies have also suggested that miRNAs can serve as prognostic biomarkers for NPC patients [36]. Our previous microarray data indicated that miR-16 expression was decreased in archived NPC tissue specimens. We confirmed this finding in our present study, and found that miR-16 was downregulated in NPC cell lines except for SUNE-1 and HONE-1, which may due to biological heterogeneity of cancer. We further investigated its biological functions by transient or stable overexpression of miR-16 in NPC cells. Functional investigations showed that forced expression of miR-16 suppressed NPC cell proliferation, migration and invasion in vitro, and it inhibited tumor growth and lung metastatic colonization in vivo. Interestingly, during the preparation of this manuscript, a study reported that miR-16 could inhibit CNE-2 cell proliferation and increase the apoptosis and radiosensitivity by regulating Bcl-2 expression [37]. Taken together, these findings indicate that miR-16 plays an important role in NPC development and progression.
miR-16 has been identified as a tumor suppressor in human cancers, and its downregulation may affect tumor cell apoptosis, the cell cycle, cell proliferation, and invasion [38]. miR-16 was firstly reported to be decreased in CLL due to the deletion in chromosome 13q14, and could inhibit apoptosis through targeting Bcl-2 [17]. miR-16 is decreased in prostate cancer, whereas the expression levels of its targets, namely Bcl-2, CCND1 and WNT3A, are inversely increased. Reconstitution of miR-16 expression results in growth arrest, apoptosis, and marked regression of tumor xenograft [18]. In ovarian cancer, Bmi-1 protein expression has been shown to be regulated by miR-16 and result in ovarian cancer cell proliferation and tumor growth [21]. In non-small cell lung cancer, miR-16 is frequently deleted and downregulated, and miR-16 has been shown to regulate the cell cycle of lung cancer cells by regulating CCND1 [22]. Here, our in vitro and in vivo functional study enriched the tumor suppressive role of miR-16 in various types of cancer.
miRNAs exert their function by base pairing to the 3′-UTR of their target genes [1]. Several oncogenes, including Bcl-2, CCND1, CCNE1, WNT3A, Bmi-1, and YAP1, have been identified and confirmed to be targets of miR-16 [17–26]. Each individual miRNA can regulate multiple target genes that harbor target sequence in their 3′-UTR [39]. Therefore, in our present study, we predicted FGF2 to be a potential target of miR-16 using three online databases, and we then confirmed this prediction using a luciferase reporter assay, quantitative RT-PCR and western blotting. FGF2, as a member of the FGF family, has also been verified as a target of miR-16 in colorectal carcinoma [40]. FGF2 has also been demonstrated to be regulated by several other miRNAs, including miR-101, miR-152, miR-497, miR-503, and miR646 [41–45]. FGF2 binds to FGF receptors, thus constituting FGF/FGFR signaling, which is involved in regulating the tumorigenesis and progression of a variety of cancers [33]. A recent study has also reported that FGFR1 signaling can be activated by EBV-encoded latent membrane protein 1 (LMP1) and is involved in the EBV-driven pathogenesis of NPC [46]. Our present investigation demonstrated that miR-16 targets FGF2 to regulate NPC proliferation and invasion via the MAPK and PI3K/AKT pathways, thereby enriching the understanding of the FGF/FGFR signaling pathway involvement in NPC tumorigenesis.
In conclusion, our findings revealed that miR-16, which functions as a tumor suppressor, was decreased in NPC. Ectopic overexpression of miR-16 inhibited NPC cell proliferation, migration, and invasion through targeting FGF2 to inactivate the MAPK and PI3K/AKT signaling pathways. These findings demonstrated that the downregulated expression of miR-16 in NPC contributes to the development and progression of NPC. Therefore, miR-16 may serve as a potential therapeutic target for patients with NPC.
MATERIALS AND METHODS
Ethics statement
Investigations were performed according to the ethical standards of the Declaration of Helsinki. All research protocols involving patient samples or animals were approved by the Institutional Review Boards of Sun Yat-sen University Cancer Center. Written informed consent was obtained from each patient, and the animal experiments were performed in accordance with the guidelines of the Experimental Animal Care and Use Committee.
Cell lines and clinical samples
The human immortalized nasopharyngeal epithelial cell line NP69, and six human NPC cell lines (CNE-1, CNE-2, SUNE-1, C666–1, HNE-1, and HONE-1) were available from Sun Yat-sen University Cancer Center (Guangzhou, China). The NP69 cell line was maintained in KSFM (Invitrogen, Grand Island, NY, USA) supplemented with bovine pituitary extract (BD Biosciences, San Diego, CA, USA). The NPC cell lines were cultured in RPMI-1640 medium (Invitrogen) supplemented with 10% FBS (Gibco, Grand Island, NY, USA). All cell lines were incubated in a humidified atmosphere at 37°C with 5% CO2. A collection of 16 freshly frozen NPC and 8 normal nasopharyngeal epithelial tissue specimens were obtained from the tissue bank of Sun Yat-sen University Cancer Center (Guangzhou, China).
RNA isolation and quantitative RT-PCR
Total RNA isolation from NPC cell lines and tissue samples was performed with TRIzol Reagent (Invitrogen) following the manufacturer’s instruction. cDNA was synthesized using M-MLV reverse transcriptase (Promega, Madison, WI, USA) and amplified with Platinum SYBR Green qPCR SuperMix-UDG reagents (Invitrogen) using the CFX96 sequence detection system (Bio-Rad, Hercules, CA, USA). The Bulge-Loop miRNA-specific primers (RiboBio, Guangzhou, China) were used for the detection of miR-16. The following primers were used for the detection of FGF2: forward, 5′-ACTGGCTTCTAAATGTGTTACG-3′; and reverse, 5′-TTGGATCCAAGTTTATACTGCC-3′). U6 and GAPDH were used as controls.
Transient and stable transfection
The miR-16, inhibitor, or controls (Genepharma, Suzhou, China) were transfected into NPC cells using Lipofectamine 2000 reagent (Invitrogen). The FGF2 plasmid or empty vector (FulenGen, Guangzhou, China) together with miR-16 mimics or miR-Ctrl was used to cotransfect CNE-1 and CNE-2 cells using Lipofectamine 2000 reagent (Invitrogen). The cells were harvested for assays 48 h after transient transfection. The precursor sequence of miR-16 was synthesized and cloned into the pSin-EF2-puromycin lentiviral plasmid (Addgene, Cambridge, MA, USA) to construct a vector expressing miR-16 (named lenti-miR-16). The empty pSin-EF2 vector (lenti-vector) was used as a control. CNE-1 cells were transfected with lenti-miR-16 or lenti-vector and then selected using puromycin.
MTT, colony formation, and anchorage-independent soft-agar assays
For the MTT assay, cells (1 × 103) were seeded into 96-well plates and incubated for 0–4 days. On the indicated days, the cells were stained with MTT dye (Sigma, St. Louis, MO, USA), and the absorbance at 490 nm for each well was read on a spectrophotometer. For the colony formation assay, cells (0.5 × 103) were seeded into 6-well plates and cultured for 7 to 12 days. Colonies were fixed with paraformaldehyde, stained with crystal violet, and counted. For the soft-agar assay, cells (2.5 × 104) were suspended in complete medium containing 0.66% agar (Sigma) and then placed on top of a layer of complete medium containing 1% agar in 6-well plates. Colonies were counted under an inverted microscope after 7 to 12 days.
Wound healing, transwell migration and invasion assays
For the wound healing assay, cells were seeded into 6-well plates and starved for 24 h. Artificial wounds were then created in the cell monolayer with a sterile 200-μl tip, and images were captured at 0 and 48 h with an inverted microscope. For the migration and invasion assays, cells (5 × 104 or 1 × 105) were harvested, resuspended in serum-free medium, and plated into the upper chambers (Corning, Steuben, NY, USA) coated without or with Matrigel (BD Biosciences) on the upper surface of the 8 μm pore size membrane. Medium supplemented with 10% FBS was placed into the lower chambers. After a 16 to 24 h incubation, the cells that had migrated or invaded through the membrane were fixed with paraformaldehyde, stained with crystal violet, and counted under an inverted microscope.
Tumor models, immunohistochemistry, and hematoxylin and eosin staining
BALB/c-nu mice (4–6 weeks old) were purchased from the Medical Experimental Animal Center of Guangdong Province (Guangzhou, China) and housed in barrier facilities on a 12 h light/dark cycle. For the xenograft tumor growth model, lenti-miR-16 or lenti-vector CNE-1 cells (1 × 106) were subcutaneously inoculated in the right dorsal flank of the mice. Tumor size was measured every 3 days, and tumor volumes were calculated. After two weeks, the mice were sacrificed, and the tumors were excised, weighed, and paraffin-embedded. Sections were cut and subjected to immunohistochemistry with anti-Ki67 (1:400; Proteintech, Wuhan, China) and anti-PCNA (1:400; Proteintech) antibodies. For the lung metastatic colonization model, lenti-miR-16 or lenti-vector CNE-1 cells (1 × 106) were intravenously injected via the tail lateral vein. Five weeks later, the mice were sacrificed, and the lung tissues were excised, paraffin-embedded, cut into slices, and stained with hematoxylin and eosin. The number of macroscopic and microscopic lung tumor nodules was counted.
Luciferase reporter assay
The FGF2 3′-UTR regions containing predicted miR-16 binding sites and corresponding mutant sites were amplified and then inserted downstream of the luciferase gene in the psiCHECK™ vector (Promega). The FGF2 wild-type or mutant 3′-UTR luciferase reporter plasmids, the p-TK Renilla plasmid (Promega), plus miR-16 mimics, inhibitor, or controls, were used to cotransfect NPC cells using Lipofectamine 2000 (Invitrogen) in accordance with the manufacturer’s recommendation. Renilla and firefly luciferase signals were measured 24 h after transfection using the Dual Luciferase Reporter Assay System (Promega).
Western blotting and immunofluorescent staining
For western blotting, equal amounts of proteins were separated and transferred to PVDF membranes (Millipore, Billerica, MA, USA). The membranes were then incubated with the following primary antibodies: anti-FGF2 (1:1000; Millipore); anti-p-Akt (1:1000; Thr 473; Cell Signaling Technology, Beverly, MA, USA); anti-Akt (1:1000; Cell Signaling Technology); anti-p-ERK (1:1000; Thr 202/Tyr204; Cell Signaling Technology); and anti-ERK (1:1000; Cell Signaling Technology). An anti-α-tubulin antibody (1:1000; Sigma) was used as a protein loading control. For immunofluorescent staining, cells were grown on coverslips (Thermo Fisher Scientific, Rochester, NY, USA) for 24 h and then incubated with an anti-FGF2 antibody (1:200; Millipore) followed by incubation with an Alexa Fluor 594 IgG antibody (Life Technologies, Carlsbad, CA, USA). The cells were then counterstained with DAPI, and images were captured using a confocal laser scanning microscope (Olympus FV1000, Olympus, Tokyo, Japan).
Statistical analyses
All of the statistical analyses were performed using SPSS 13.0 (SPSS Inc., Chicago, IL, USA). All data shown are representative results of at least 3 independent experiments, and the data are expressed as the mean ± standard deviation (S.D.). Significant differences between two groups were analyzed using two-tailed unpaired Student’s t-test, and a probability value of p < 0.05 was considered significant.
GRANT SUPPORT
This work was supported by grants from the National Natural Science Foundation of China (81402516), the Pearl River Science and Technology New Star of Guangzhou, China (201506010070), the Planned Science and Technology Project of Guangdong Province (2014A020212082), the Health & Medical Collaborative Innovation Project of Guangzhou City, China (201400000001), and the National Science & Technology Pillar Program during the Twelfth Five-year Plan Period (2014BAI09B10).
CONFLICTS OF INTEREST
The authors declare no conflicts of interest.
REFERENCES
1. He L, Hannon GJ. MicroRNAs: small RNAs with a big role in gene regulation. Nat Rev Genet. 2004; 5:522–531.
2. Calin GA, Croce CM. MicroRNA signatures in human cancers. Nat Rev Cancer. 2006; 6:857–866.
3. Ventura A, Jacks T. MicroRNAs and cancer: short RNAs go a long way. Cell. 2009; 136:586–591.
4. Kong YW, Ferland-McCollough D, Jackson TJ, Bushell M. microRNAs in cancer management. Lancet Oncol. 2012; 13:e249–258.
5. Jia WH, Luo XY, Feng BJ, Ruan HL, Bei JX, Liu WS, Qin HD, Feng QS, Chen LZ, Yao SY, Zeng YX. Traditional Cantonese diet and nasopharyngeal carcinoma risk: a large-scale case-control study in Guangdong, China. BMC Cancer. 2010; 10:446.
6. Lai SZ, Li WF, Chen L, Luo W, Chen YY, Liu LZ, Sun Y, Lin AH, Liu MZ, Ma J. How does intensity-modulated radiotherapy versus conventional two-dimensional radiotherapy influence the treatment results in nasopharyngeal carcinoma patients? Int J Radiat Oncol Biol Phys. 2011; 80:661–668.
7. Liu N, Chen NY, Cui RX, Li WF, Li Y, Wei RR, Zhang MY, Sun Y, Huang BJ, Chen M, He QM, Jiang N, Chen L, et al. Prognostic value of a microRNA signature in nasopharyngeal carcinoma: a microRNA expression analysis. Lancet Oncol. 2012; 13:633–641.
8. Chen HC, Chen GH, Chen YH, Liao WL, Liu CY, Chang KP, Chang YS, Chen SJ. MicroRNA deregulation and pathway alterations in nasopharyngeal carcinoma. Br J Cancer. 2009; 100:1002–1011.
9. Sengupta S, den Boon JA, Chen IH, Newton MA, Stanhope SA, Cheng YJ, Chen CJ, Hildesheim A, Sugden B, Ahlquist P. MicroRNA 29c is down-regulated in nasopharyngeal carcinomas, up-regulating mRNAs encoding extracellular matrix proteins. Proc Natl Acad Sci U S A. 2008; 105:5874–5878.
10. Liu N, Tang LL, Sun Y, Cui RX, Wang HY, Huang BJ, He QM, Jiang W, Ma J. MiR-29c suppresses invasion and metastasis by targeting TIAM1 in nasopharyngeal carcinoma. Cancer Lett. 2013; 329:181–188.
11. Li YQ, Ren XY, He QM, Xu YF, Tang XR, Sun Y, Zeng MS, Kang TB, Liu N, Ma J. MiR-34c suppresses tumor growth and metastasis in nasopharyngeal carcinoma by targeting MET. Cell Death Dis. 2015; 6:e1618.
12. Xu YF, Mao YP, Li YQ, Ren XY, He QM, Tang XR, Sun Y, Liu N, Ma J. MicroRNA-93 promotes cell growth and invasion in nasopharyngeal carcinoma by targeting disabled homolog-2. Cancer Lett. 2015; 363:146–155.
13. Xu YF, Li YQ, Guo R, He QM, Ren XY, Tang XR, Jia WH, Kang TB, Zeng MS, Sun Y, Ma J, Liu N. Identification of miR-143 as a tumour suppressor in nasopharyngeal carcinoma based on microRNA expression profiling. Int J Biochem Cell Biol. 2015; 61:120–128.
14. Liu N, Jiang N, Guo R, Jiang W, He QM, Xu YF, Li YQ, Tang LL, Mao YP, Sun Y, Ma J. MiR-451 inhibits cell growth and invasion by targeting MIF and is associated with survival in nasopharyngeal carcinoma. Mol Cancer. 2013; 12:123.
15. Zhao L, Tang M, Hu Z, Yan B, Pi W, Li Z, Zhang J, Zhang L, Jiang W, Li G, Qiu Y, Hu F, Liu F, et al. miR-504 mediated down-regulation of nuclear respiratory factor 1 leads to radio-resistance in nasopharyngeal carcinoma. Oncotarget. 2015; 6:15995–16018. doi:10.18632/oncotarget.4138.
16. Fang Y, Zhu X, Wang J, Li N, Li D, Sakib N, Sha Z, Song W. MiR-744 functions as a proto-oncogene in nasopharyngeal carcinoma progression and metastasis via transcriptional control of ARHGAP5. Oncotarget. 2015; 6:13164–13175. doi:10.18632/oncotarget.3754.
17. Cimmino A, Calin GA, Fabbri M, Iorio MV, Ferracin M, Shimizu M, Wojcik SE, Aqeilan RI, Zupo S, Dono M, Rassenti L, Alder H, Volinia S, et al. miR-15 and miR-16 induce apoptosis by targeting BCL2. Proc Natl Acad Sci U S A. 2005; 102:13944–13949.
18. Bonci D, Coppola V, Musumeci M, Addario A, Giuffrida R, Memeo L, D’Urso L, Pagliuca A, Biffoni M, Labbaye C, Bartucci M, Muto G, Peschle C, et al. The miR-15a-miR-16–1 cluster controls prostate cancer by targeting multiple oncogenic activities. Nat Med. 2008; 14:1271–1277.
19. Linsley PS, Schelter J, Burchard J, Kibukawa M, Martin MM, Bartz SR, Johnson JM, Cummins JM, Raymond CK, Dai H, Chau N, Cleary M, Jackson AL, et al. Transcripts targeted by the microRNA-16 family cooperatively regulate cell cycle progression. Mol Cell Biol. 2007; 27:2240–2252.
20. Liu Q, Fu H, Sun F, Zhang H, Tie Y, Zhu J, Xing R, Sun Z, Zheng X. miR-16 family induces cell cycle arrest by regulating multiple cell cycle genes. Nucleic Acids Res. 2008; 36:5391–5404.
21. Bhattacharya R, Nicoloso M, Arvizo R, Wang E, Cortez A, Rossi S, Calin GA, Mukherjee P. MiR-15a and MiR-16 control Bmi-1 expression in ovarian cancer. Cancer Res. 2009; 69:9090–9095.
22. Bandi N, Zbinden S, Gugger M, Arnold M, Kocher V, Hasan L, Kappeler A, Brunner T, Vassella E. miR-15a and miR-16 are implicated in cell cycle regulation in a Rb-dependent manner and are frequently deleted or down-regulated in non-small cell lung cancer. Cancer Res. 2009; 69:5553–5559.
23. Ofir M, Hacohen D, Ginsberg D. MiR-15 and miR-16 are direct transcriptional targets of E2F1 that limit E2F-induced proliferation by targeting cyclin E. Mol Cancer Res. 2011; 9:440–447.
24. Lezina L, Purmessur N, Antonov AV, Ivanova T, Karpova E, Krishan K, Ivan M, Aksenova V, Tentler D, Garabadgiu AV, Melino G, Barlev NA. miR-16 and miR-26a target checkpoint kinases Wee1 and Chk1 in response to p53 activation by genotoxic stress. Cell Death Dis. 2013; 4:e953.
25. Yang TQ, Lu XJ, Wu TF, Ding DD, Zhao ZH, Chen GL, Xie XS, Li B, Wei YX, Guo LC, Zhang Y, Huang YL, Zhou YX, et al. MicroRNA-16 inhibits glioma cell growth and invasion through suppression of BCL2 and the nuclear factor-kappaB1/MMP9 signaling pathway. Cancer Sci. 2014; 105:265–271.
26. Kang W, Tong JH, Lung RW, Dong Y, Zhao J, Liang Q, Zhang L, Pan Y, Yang W, Pang JC, Cheng AS, Yu J, To KF. Targeting of YAP1 by microRNA-15a and microRNA-16–1 exerts tumor suppressor function in gastric adenocarcinoma. Mol Cancer. 2015; 14:52.
27. Kaddar T, Chien WW, Bertrand Y, Pages MP, Rouault JP, Salles G, Ffrench M, Magaud JP. Prognostic value of miR-16 expression in childhood acute lymphoblastic leukemia relationships to normal and malignant lymphocyte proliferation. Leuk Res. 2009; 33:1217–1223.
28. Gao X, Zhang R, Qu X, Zhao M, Zhang S, Wu H, Jianyong L, Chen L. MiR-15a, miR-16–1 and miR-17–92 cluster expression are linked to poor prognosis in multiple myeloma. Leuk Res. 2012; 36:1505–1509.
29. Huang N, Wu J, Qiu W, Lyu Q, He J, Xie W, Xu N, Zhang Y. MiR-15a and miR-16 induce autophagy and enhance chemosensitivity of Camptothecin. Cancer Biol Ther. 2015; 16:941–948.
30. Chatterjee A, Chattopadhyay D, Chakrabarti G. MiR-16 targets Bcl-2 in paclitaxel-resistant lung cancer cells and overexpression of miR-16 along with miR-17 causes unprecedented sensitivity by simultaneously modulating autophagy and apoptosis. Cell Signal. 2015; 27:189–203.
31. Takeshita F, Patrawala L, Osaki M, Takahashi RU, Yamamoto Y, Kosaka N, Kawamata M, Kelnar K, Bader AG, Brown D, Ochiya T. Systemic delivery of synthetic microRNA-16 inhibits the growth of metastatic prostate tumors via downregulation of multiple cell-cycle genes. Mol Ther. 2010; 18:181–187.
32. Reid G, Pel ME, Kirschner MB, Cheng YY, Mugridge N, Weiss J, Williams M, Wright C, Edelman JJ, Vallely MP, McCaughan BC, Klebe S, Brahmbhatt H, et al. Restoring expression of miR-16: a novel approach to therapy for malignant pleural mesothelioma. Ann Oncol. 2013; 24:3128–3135.
33. Turner N, Grose R. Fibroblast growth factor signalling: from development to cancer. Nat Rev Cancer. 2010; 10:116–129.
34. Zhang L, Yu H, Badzio A, Boyle TA, Schildhaus HU, Lu X, Dziadziuszko R, Jassem J, Varella-Garcia M, Heasley LE, Kowalewski AA, Ellison K, Chen G, et al. Fibroblast growth factor receptor 1 and related ligands in small-cell lung cancer. J Thorac Oncol. 2015; 10:1083–1090.
35. Sharpe R, Pearson A, Herrera-Abreu MT, Johnson D, Mackay A, Welti JC, Natrajan R, Reynolds AR, Reis-Filho JS, Ashworth A, Turner NC. FGFR signaling promotes the growth of triple-negative and basal-like breast cancer cell lines both in vitro and in vivo. Clin Cancer Res. 2011; 17:5275–5286.
36. Liu N, Cui RX, Sun Y, Guo R, Mao YP, Tang LL, Jiang W, Liu X, Cheng YK, He QM, Cho WC, Liu LZ, Li L, et al. A four-miRNA signature identified from genome-wide serum miRNA profiling predicts survival in patients with nasopharyngeal carcinoma. Int J Cancer. 2014; 134:1359–1368.
37. Zhang C, Fang X, Li W, Shi Q, Wu L, Chen X, Huang Z, Wu P, Wang Z, Liao Z. Influence of recombinant lentiviral vector encoding miR-15a/16–1 in biological features of human nasopharyngeal carcinoma CNE-2Z cells. Cancer Biother Radiopharm. 2014; 29:422–427.
38. Aqeilan RI, Calin GA, Croce CM. miR-15a and miR-16–1 in cancer: discovery, function and future perspectives. Cell Death Differ. 2010; 17:215–220.
39. Bartel DP. MicroRNAs: genomics, biogenesis, mechanism, and function. Cell. 2004; 116:281–297.
40. Xue G, Yan HL, Zhang Y, Hao LQ, Zhu XT, Mei Q, Sun SH. c-Myc-mediated repression of miR-15–16 in hypoxia is induced by increased HIF-2alpha and promotes tumor angiogenesis and metastasis by upregulating FGF2. Oncogene. 2015; 34:1393–1406.
41. Kottakis F, Polytarchou C, Foltopoulou P, Sanidas I, Kampranis SC, Tsichlis PN. FGF-2 regulates cell proliferation, migration, and angiogenesis through an NDY1/KDM2B-miR-101-EZH2 pathway. Mol Cell. 2011; 43:285–298.
42. Cheng Z, Ma R, Tan W, Zhang L. MiR-152 suppresses the proliferation and invasion of NSCLC cells by inhibiting FGF2. Exp Mol Med. 2014; 46:e112.
43. Xu J, Wang T, Cao Z, Huang H, Li J, Liu W, Liu S, You L, Zhou L, Zhang T, Zhao Y. MiR-497 downregulation contributes to the malignancy of pancreatic cancer and associates with a poor prognosis. Oncotarget. 2014; 5:6983–6993. doi:10.18632/oncotarget.2184.
44. Zhou B, Ma R, Si W, Li S, Xu Y, Tu X, Wang Q. MicroRNA-503 targets FGF2 and VEGFA and inhibits tumor angiogenesis and growth. Cancer Lett. 2013; 333:159–169.
45. Sun XH, Geng XL, Zhang J, Zhang C. miRNA-646 suppresses osteosarcoma cell metastasis by downregulating fibroblast growth factor 2 (FGF2). Tumour Biol. 2015; 36:2127–2134.
46. Lo AK, Dawson CW, Young LS, Ko CW, Hau PM, Lo KW. Activation of the FGFR1 signalling pathway by the Epstein-Barr virus-encoded LMP1 promotes aerobic glycolysis and transformation of human nasopharyngeal epithelial cells. J Pathol. 2015; doi:10.1002/path.4575.