
Introduction
Metastasis is the main reason for many solid tumors to be life-threatening. The metastatic cascade involves several steps, ranging from dissemination from the primary tumor to growth at a secondary site. The acquisition of metastatic capability by tumor cells can be associated with Epithelial-Mesenchymal Transition (EMT). Upon EMT, tumor cells are able to invade through the basement membrane of the primary tissue and stroma, and to enter the blood circulation. They often become anoikis resistant, which allows them to survive in the absence of attachment. Finally, they associate with the endothelium and extravasate to a secondary tissue. For outgrowth at secondary sites, the newly formed tumor foci need to induce angiogenesis [1, 2]. Metastases are often difficult to cure because they can be widespread, affecting tissue function, and they are usually resistant to conventional therapies. Furthermore, intervention of metastatic cancer progression is rarely efficient due to lack of early detection methods. Therefore, it is crucial to predict metastatic potential of disease and to target metastasis.
One of the well-known regulators of metastasis is the Activator Protein 1 (AP-1) complex. AP-1 is a family of transcription factors regulating a broad spectrum of cellular processes including proliferation, migration and invasion [3]. AP-1 dimers are formed by Fos (c-FOS, FOSB, Fra-1, Fra-2), Jun (c-JUN, JUNB, JUND), ATF and MAF protein families. AP-1 members are encoded by immediate early genes that are rapidly activated and deactivated in response to a wide range of stimuli. Although some AP-1 components have been reported to act as tumor suppressors, AP-1 complexes are mostly known for their ability to induce oncogenic transformation among other processes such as proliferation, apoptosis, invasion and angiogenesis [4]. c-Fos, c-Jun and Fra-1 are among the AP-1 components whose overexpression correlate with poor prognosis in several types of malignancies including ovarian, lung, and breast cancers [5-7].
AP-1 is regulated by the Ras/Raf/MEK/ERK [8], [9] and the Wnt [10] pathways. The Wnt pathway is often deregulated in colon cancer as a result of activating mutations in beta-catenin (CTNNB1) or inactivating mutations in adenomatous polyposis coli (APC), which is a negative regulator of beta-catenin. Wnt signaling is not only critical for developmental and oncogenic characteristics like proliferation, survival, and differentiation but also drives metastasis-related processes such as migration and cell polarity [11]. Previous reports have shown that the Wnt pathway negatively regulates Fos and FosB expression, whereas it increases Fra-1 mRNA levels in mouse epithelial cells [12]. Moreover, non-canonical Wnt signaling activates AP-1 through TCF binding to c-Jun in human colon cancer cells [13].
Fra-1 is one of the AP-1 transcription factors; it lacks a transactivation domain and has therefore a weak transforming activity. It forms heterodimers with Jun family members in order to activate target gene transcription. We and others have shown that Fra-1 promotes metastasis through various molecules: ADORA2B [7] in breast cancer, MMPs in breast cancer [14] and in lung epithelial cells [15], CD44 in mesothelioma [16], AXL in bladder cancer [17], FAK and EZH2 in colon cancer [18], [19].
Colorectal cancer (CRC) is among the most common cancers and one of the leading causes of cancer-related deaths worldwide. Traditional classification divides CRC into four main stages based on the local extent of the tumor, with three subtypes of stage III tumors based on the number of cancer-positive nodes [20]. However, CRC is more heterogeneous than the categories used in the clinic with regard to progression, recurrence, metastasis and therapy response[21]. In the present study, we investigated the role of Fra-1 in colon cancer progression in vivo and the clinical impact of Fra-1 on disease outcome.
Results
Fra-1 is not critically required for proliferation of colon cancer cells in vitro
As we have previously shown that Fra-1 is largely dispensable for human breast cancer cell growth in vitro but crucial for their ability to metastasize in vivo [7], we decided to investigate whether Fra-1 has a similar role in human colon cancer. Fra-1 was stably depleted in HT29, HCT116 and DLD-1 cells by two independent shRNAs (Figure 1A). There was no difference in proliferation rates of Fra-1-depleted cells and control cells on 2D culture plates (Figure 1B). However, we found a 30-50% decrease in the number of cells surviving under anoikis-inducing conditions (Figure 1C) and a three-fold decrease in the number of colonies formed by Fra-1 deficient HT29 cells in soft agar (Figure 1D). Colo205 cells, which have low endogenous Fra-1 expression levels, successfully formed colonies in soft agar and survived in anoikis-inducing conditions; Fra-1 overexpression caused a mild but significant increase in the number cells in both cases (Suppl. Fig. 1A-1C). Thus, Fra-1 is neither critically required for 2D nor 3D proliferation in vitro.
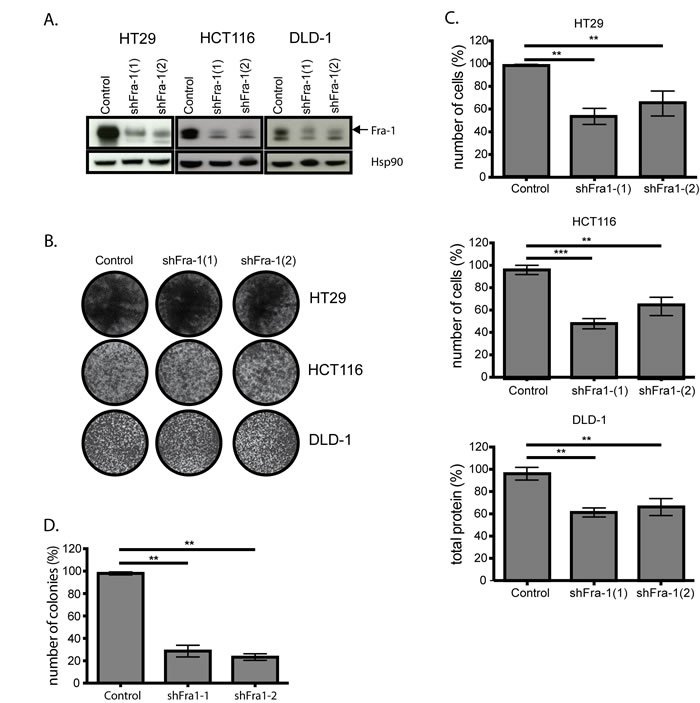
Figure 1: Fra-1 is not critically required for proliferation of colon cancer cells in vitro. A. Fra-1 depletion upon lentiviral transduction of two independent shRNAs. B. HT29 and HCT116 cells with or without Fra-1 knockdown were seeded into 6-well plates (30000 cells/well). The plates were stained with crystal violet after 7 days. C. HT29 cells with or without Fra-1 knockdown were seeded in duplicates in 0,3% agar suspension on top of a 1% agar base in 6-well plates at 24000 cells/well. After three weeks, colonies were stained with crystal violet and counted by Image J software (n=3). D. 0.4*106 cells were seeded into 6-well ultra-low-attachment plates in duplicate. The cells were harvested at day 6, trypsinized, resuspended and counted. Results presented are the combination of three experiments. Error bars represent SEM. Statistics: One-Way ANOVA. * p < 0.05, ** p < 0.01, *** p < 0.001.
Fra-1 is largely dispensable for primary colon tumor growth in vivo
In order to assess the role of Fra-1 in in vivo tumor growth, we next injected control and Fra-1-depleted HT29 cells subcutaneously into severely immune-compromised (NOD/SCID IL2gamma, NSG) mice. Fra-1-depleted tumors grew approximately two-fold slower than control tumors (Figure 2A-2B). Immunohistochemistry staining and western blots showed that Fra-1 levels were still low in these tumors at the end of the experiment (Figure 2C-D), indicating that there is no selective pressure to lose Fra-1 shRNAs during tumor progression. These data show that although Fra-1 contributes somewhat to the expansion of colon cancer tumors in vivo, it is not strictly required.
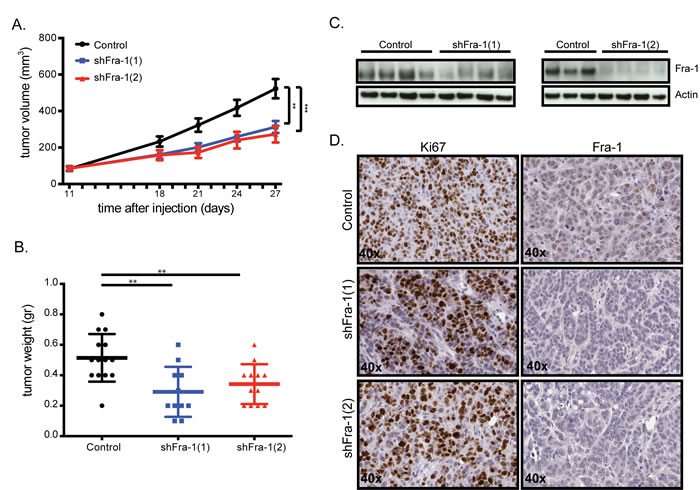
Figure 2: Fra-1 is largely dispensable for primary colon tumor growth in vivo. A. 0,5*106 HT29 cells with or without Fra-1 knockdown were mixed 1:1 with matrigel and injected subcutaneously into severely immune compromised (NOD/SCID IL2γ) mice. Tumor growth was measured by a caliper at indicated time points (ncontrol=13, nshFra1-1=12, nshFra1-2=13). B. The weight of tumors harvested at the end time point of two independent experiments combined. C. Western blot showing Fra-1 levels in tumors harvested from mice injected with control or Fra-1-depleted HT29 cells. D. Tumors harvested at the end of the experiment were analyzed for the expression of Ki67 and Fra-1 by immunostaining (40X). Error bars represent SEM. Statistics: One-Way ANOVA. * p < 0.05, ** p < 0.01, *** p < 0.001.
Fra-1 is crucial for efficient metastatic spread of colon cancer cells
We and others have implicated Fra-1 as an important determinant of the metastatic capacity of cancer cells, which is associated with its ability to induce EMT and with clinical outcome [7, 22]. In order to determine the role of Fra-1 in colon cancer metastasis in vivo, we injected Fra-1-depleted HT29 cells intravenously into NSG mice and monitored tumor expansion in time via a luciferase-dependent non-invasive in vivo imaging system. Whereas mice injected with cells carrying a control construct showed a substantial number of tumor foci distributed all over the body, tumor burden was sharply reduced in mice injected with Fra-1-depleted cells (Figure 3A-3B). 29 days after injection, control mice had a saturated luciferase signal accompanied by severe weight loss (Suppl. Fig. 2A). At this time point, the average difference between control mice and mice injected with Fra-1-depleted cells was 206-fold.
At autopsy, multiple macroscopic tumors were observed on the subcutaneous skin and peritoneal wall as well as several organs of the control mice such as lung, spine, kidneys, ovaries, lymph nodes, skin and muscles in the extremities (Suppl. Fig. 2B). Immunohistochemical staining further showed foci in the liver, bones and brain. Much fewer tumors were observed in the mice injected with Fra-1-depleted cells, both macroscopically and by immunostaining (Figure 3C-3D). Importantly, and in contrast to our observations for primary tumor growth, in a great majority of the cases, the tumor foci formed by Fra-1-depleted cells were positive for Fra-1, sometimes in a heterogeneous fashion (Suppl. Fig. 2C, 2D, 2E). We observed a similar pattern in HCT116 cells, which metastasize preferentially to the liver: Fra-1- depleted HCT116 cells formed significantly fewer and smaller foci in the liver upon intravenous injection (Suppl. Fig. 1D). Together, these results demonstrate that Fra-1 is critical for the metastatic spread of colon cancer cells in vivo, yet expendable for primary tumor growth.
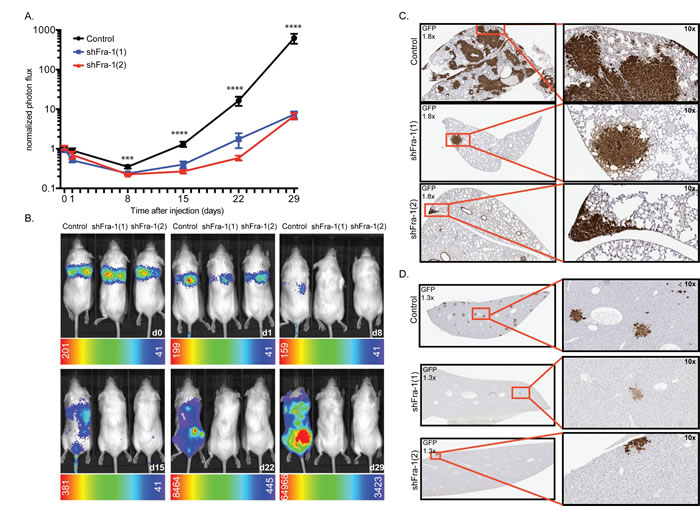
Figure 3: Fra-1 is crucial for efficient metastatic spread of colon cancer cells. A. 0,5*106 HT29 cells were injected intravenously into NSG mice. Tumor expansion was followed by IVIS from day0 onwards. Photon flux from two independent experiments was combined. n = 6 mice/group/experiment. Error bars represent SEM. Statistics: One-Way ANOVA * p < 0.05, **p < 0.005,****p < 0.0001 B. Representative images of mice at each time point. C.-D. GFP staining showing tumor foci in lungs and liver.
Acute Fra-1 depletion impairs growth of established metastatic foci
The results obtained with cells lacking Fra-1 expression suggest an important contribution of Fra-1 to the metastatic potential of colon cancer cells. From a clinical point of view, it would be more relevant to determine the impact of Fra-1 depletion on tumors that have already been established, rather than to prevent outgrowth. Therefore, we decided to investigate whether acute loss of Fra-1 affects the growth of established tumor foci. This system also allowed us to exclude the potential bias where one group of cells may not survive the injection procedure or the mechanical stress caused by the blood circulation. We used an inducible tet-on system enabling us to deplete Fra-1 on doxycycline administration via the drinking water of the mice (Suppl. Fig 3A). To ensure a homogeneous level of downregulation of Fra-1 upon doxycycline treatment, we generated a cell clone (HT29-C25) harboring the tet-on construct. The mice were injected intravenously with control or HT29-C25 cells and each group was randomized into two sub-groups at the day of injection. One group continuously received doxycycline in drinking water from day 0 onwards after the inoculation of tumor cells, whereas the other was mock-treated. The total tumor burden was reduced 56-times in HT29-C25-injected mice upon doxycycline treatment (Figure 4A), whereas mice injected with control cells had no difference in luciferase signal until the end of the experiment (Suppl. Fig. 3B). This result indicates that when Fra-1 knockdown is induced after the initial seeding of tumor cells upon intravenous inoculation, Fra-1 is required for tumor outgrowth.
Next, Fra-1 depletion was induced when the tumor burden started increasing (after an initial drop as judged by luciferase imaging). Twenty days after treatment, Fra-1 depletion caused an eight-fold reduction in tumor burden (Figure 4B). Once again, the rate of tumor development was the same in the mice injected with control cells regardless of doxycycline treatment (Suppl. Fig. 3C). Similar to the previous experiment (Figure 3), mice injected with control cells developed tumors in a broad range of organs, but there were very few macroscopically detectable tumors in the HT29-C25-injected mice on doxycycline treatment. Also similar to the previous experiment, tumors harvested at the end of the experiment showed varying levels of Fra-1 suggesting that some tumors were formed by Fra-1-proficient cells (Suppl. Fig. 3D). As assessed by the luciferase signal, doxycycline-treated mice have approximately five times less burden in their lungs compared to mock-treated mice (Figure 4C-4D). These mice had not only fewer but also smaller tumor foci in their lungs (Figure 4E). Altogether, these data suggest that Fra-1 is essential also for growth and expansion of established (micro)metastases of colon cancer cells.
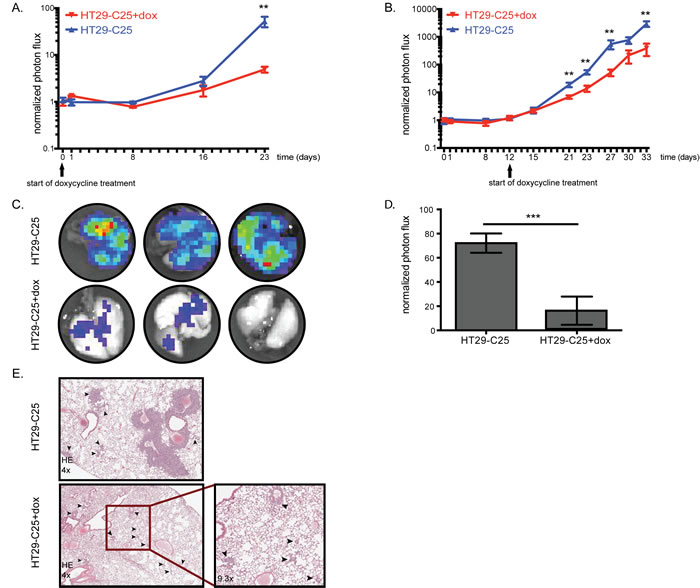
Figure 4: Acute Fra-1 depletion impairs growth of established metastatic foci. A.-B. 0,5*106 HT29-C25 cells were intravenously injected into NSG mice. Growth curves of intravenously injected HT29-C25 cells with or without doxycycline treatment starting at day0 or day12. C.-D. The mice in the experiment shown in B were injected with 15mg/kg luciferin five minutes before sacrifice. The lungs were harvested and imaged in luciferin-containing PBS and quantified. E. Hematoxylin and eosin staining of representative lung sections from mice of experiment shown in B, treated with doxycycline or not. The arrowheads point to the tumor foci. Error bars represent SEM. Statistics: Non-parametric student’s t-test. **p < 0.005,***p < 0.001.
Fra-1-regulated gene signature is a prognostic classifier in colon cancer
Based on these findings, which are consistent with, and extend, those of others [18], [23], [24], Fra-1 acts as an important pro-metastatic factor in colon cancer. Since metastatic relapse is a major reason of cancer-related deaths, we asked whether we could stratify colon cancer patients based on Fra-1 expression levels, similar to what we have shown recently for breast cancer [7]. The prognostic value of Fra-1 was assessed by correlating FOSL1 mRNA levels (encoding Fra-1) in colon cancer patient samples to disease-free survival in five gene expression datasets. We observed that patients with tumors showing FOSL1 expression higher than median levels had a significantly worse prognosis in the first five years after treatment or surgery (Figure 5A).
However, Fra-1 is not an ideal drug target due to the absence of a catalytic site that can be readily targeted by a small molecule. The lack of an available inhibitor against Fra-1 prompted us to search for critical downstream targets of Fra-1 that are involved in metastasis. We compared the expression profiles of control and Fra-1-depleted HT29 cells by RNA sequencing and selected the genes that are significantly regulated by Fra-1. This classifier contains a total of 199 genes, 88 of which are positively regulated by Fra-1 and 111 negatively (Suppl. Table 1).
According to non-negative matrix factorization (NMF) analysis, colon cancer patients could be divided into three prognostic groups based on the expression levels of Fra-1-regulated genes. The heat map demonstrates that the genes that were positively regulated by Fra-1 are overexpressed in patients in subtype 1 and not in subtypes 2 and 3. On the other hand, genes that were negatively regulated by Fra-1 have lower expression levels in patients in cluster 1 (Figure 5B), independently of tumor stage or dataset (Suppl. Fig. 4). A Kaplan-Meier analysis showed that subtypes 2 and 3 are good prognosis groups. They only slightly differ from each other in the initial survival rates but in the long term have a similarly good prognosis. subtype 1, on the other hand, has a significantly worse disease-free survival compared to the other two groups as well as poorer disease-specific and overall survival (Figure 5C, Suppl. Fig. 5). In a Cox proportional hazards model stratified for gender and stage, subtypes 2 and 3 showed significantly better disease-free survival (HR = 0.43, p = 4.42*10-5 and HR = 0.51, p = 0.001) than subtype 1. Analyzing each stage separately, we found similar effects for each stage albeit with different effect size (Table 1). A comparable pattern was observed with disease-specific and overall survival analysis with the exception of stage 3 patients in subtype 3 in case of overall survival (Suppl. Table 2a, 2b). These data suggest that overexpression of genes positively regulated by Fra-1 is correlated with poor outcome, whereas the expression of genes negatively regulated by Fra-1 is associated with better outcome. Therefore, our Fra-1 classifier has prognostic power to predict the clinical outcome of colon cancer patients.
Table1: Cox proportional hazards model estimating hazard ratios for disease-free survival for the subtypes stratified for gender and tumor stage
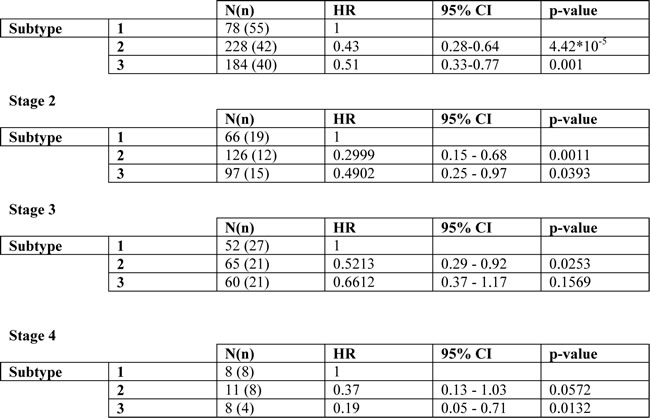
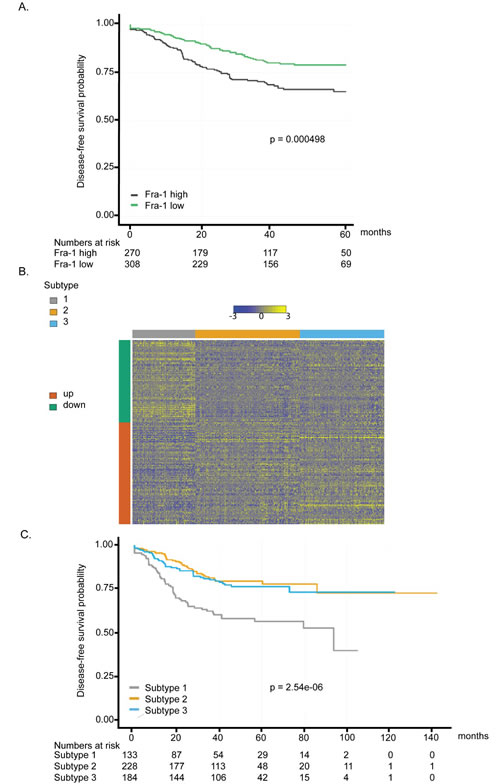
Figure 5: Fra-1-regulated gene signature is a prognostic classifier in colon cancer. A. Disease free survival (DFS) analysis based on FOSL1 expression. Samples were split according to lower or higher than average expression of FOSL1. Patients with low expression of FOSL1 exhibited significantly longer DFS than patients with higher expression. B. Heat map of the gene expression of the Fra-1 signature. Gene expression is shown as color gradient from blue (low expression) to yellow (high expression). The color bar on the left side indicates direction of regulation in Fra-1 knock-down cells. Color bars on top of the heat map show sample stage, source data set and Fra-1KD signature cluster, in this order. C. Disease-free survival curve for the three subtypes resulting from hierarchical clustering with the Fra-1 signature. Subtype 1 has significantly shorter DFS than subtypes 2 and 3, which show no survival difference.
Fra-1 regulates the Wnt pathway
The influence of focal adhesions on motility and invasiveness and focal adhesion pathway regulation by Fra-1 in colon cancer cells are previously reported mechanisms of the pro-metastatic activity of Fra-1 [18], [25]. Consistently, Fra-1 knockdown in colon cancer cells decreased the expression of a panel of focal adhesion genes, indicating that our classifier is relevant and a reliable indicator of the aggressiveness of colon cancer (Figure 6A).
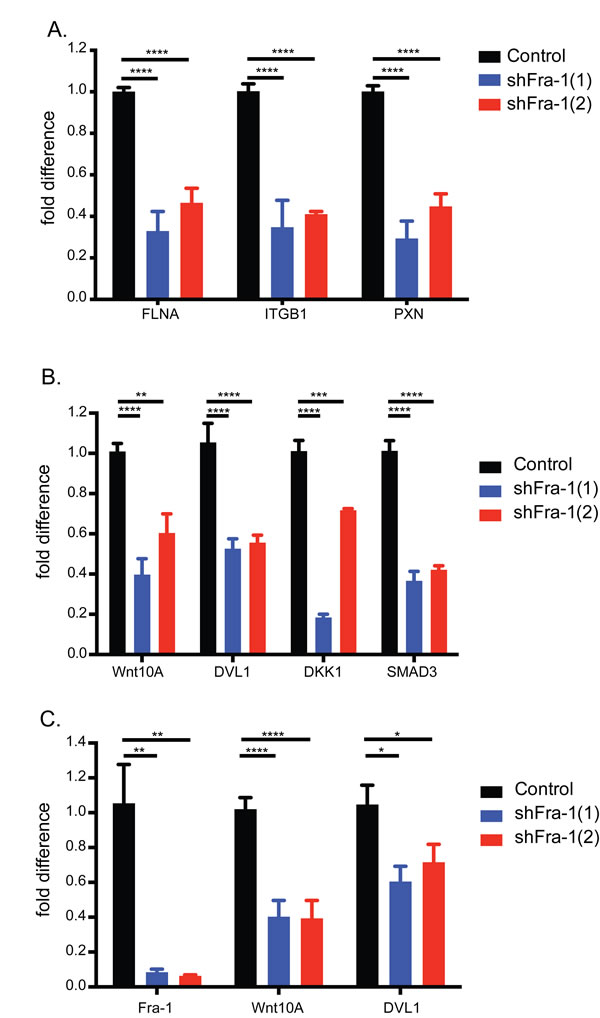
Figure 6: Fra-1 regulates the expression of Wnt pathway components. A. Validation of the focal adhesion pathway by qRT-PCR upon Fra-1 depletion in HT29 cells. B.-C. qRT-PCR analysis of Wnt pathway components in the classifier in HT29 and HCT15 cells. Error bars represent SEM. Statistics: One_Way ANOVA * p < 0.05, ** p < 0.01, *** p < 0.001, ****p < 0.0001.
On the other hand, regulation of the Wnt pathway by Fra-1 is an unexplored phenomenon. The Wnt pathway is significantly represented by seven genes in the classifier: whereas Wnt10A, SMAD3, DKK1 and DVL1 were downregulated upon Fra-1 knockdown, BAMBI, ROCK2 and PLCB4 were upregulated (Table 2). Notably, Wnt10A is the most abundantly expressed Wnt gene in HT29 cells (Suppl. Fig. 6). We validated Wnt10A, SMAD3, DKK1 and DVL1 dowregulation by Fra-1 depletion in HT29, and Wnt10A and DVL1 in HCT15 cells (Figure 6B-6C). We also examined by a luciferase reporter assay whether Fra-1 depletion modified beta-catenin activity. HT29, HCT15 and DLD-1 cells with or without a Fra-1 knockdown were transfected with the TOP/FOP constructs to measure transcriptional activity of beta-catenin upon loss of Fra-1 expression. We observed an effective reduction in the beta-catenin-mediated transcription between control and Fra-1-depleted cells (Figure 7). These data suggest that Fra-1 regulates the canonical Wnt signaling by modulating the expression of Wnt pathway components and the transcriptional activity of beta-catenin.
Table2: KEGG pathway analysis on the classifier genes reveals Wnt pathway as one of the significantly regulated pathways by Fra-1.
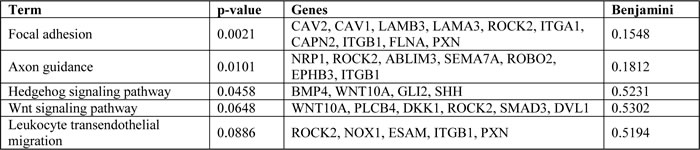
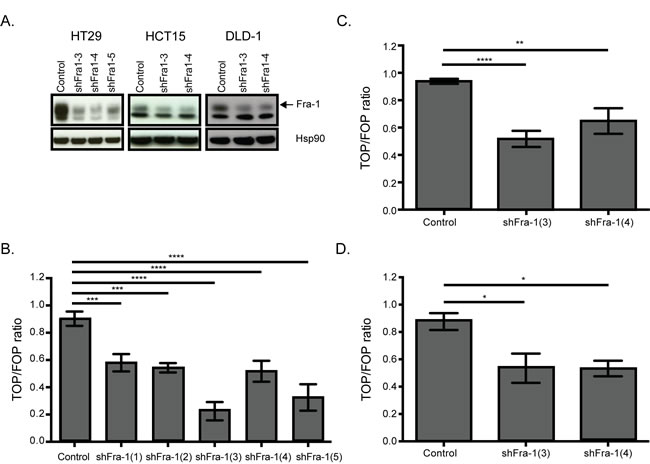
Figure 7: Fra-1 regulates beta-catenin activity. A. Western blots showing efficient depletion of Fra-1 in HT29, HCT15, and DLD-1 cells. B.-D. Beta-catenin reporter assay on HT29, HCT15 and DLD-1 cells upon Fra-1 depletion. Error bars represent SEM. Statistics: One_Way ANOVA * p < 0.05, ** p < 0.01, *** p < 0.001, ****p < 0.0001.
Discussion
The high lethality rate of colon cancer is mainly due to recurrence and distant metastasis. It therefore is crucial to better understand and predict these outcomes in order to take appropriate action with regard to treatment options. In this report, we demonstrate that Fra-1 is a critical biological determinant of colon cancer metastasis, as judged by two main observations. First, Fra-1 depletion severely impaired metastatic foci formation of colon cancer cells in vivo. Second, gene expression analysis by RNA sequencing of metastatic colon cancer cells revealed that a Fra-1 classifier comprising genes significantly regulated by Fra-1 is a strong predictor of disease-free survival.
Others have previously shown that Fra-1 is responsible for migration of colon cancer cells in vitro [18]. We found that Fra-1 is critical for the metastatic spread of colon cancer cells, even after establishment, yet largely dispensable for primary tumor growth. The growth rate of subcutaneous xenografts mirrors the results of 3D colony formation assays in vitro, showing a significant three-fold decrease in growth and colony number, respectively. These results, combined with the fact that Fra-1 knockdown was retained until the end of the experiment, indicate that Fra-1 is not critically required for primary tumor growth, since the tumors still grow in the absence of Fra-1. This differs, for example, from our recent observations for DDR kinases, for which shRNAs were commonly lost during tumor expansion [26]. In contrast, in an experimental metastasis model where the cells are injected intravenously, Fra-1-depleted colon cancer cells show a stark defect in their ability to form metastatic foci in the mice. The observation that the few tumor foci that could be found in the lungs of mice injected with Fra-1 depleted cells were largely Fra-1-positive indicate that the tumor burden is mainly caused by Fra-1-proficient cells. We did not observe such a negative selection pressure against, nor a similar growth disadvantage of, Fra-1-depleted cells in primary tumor growth. These data together suggest that Fra-1 has predominantly a metastasis-related role in colon cancer.
Metastasis is a stepwise process in which the cells must first disseminate from the primary tumor, join the blood stream or the lymphatic system by digesting through the stroma and the basement membrane, extravasate at a secondary site, and grow out [27], [1]. Since our experimental metastasis model bypasses the initial steps of metastasis and the cells directly enter the blood circulation upon injection, the difference between the metastatic ability of Fra-1-deficient and Fra-1-proficient cells is most likely because Fra-1 deficient cells fail at survival in the blood stream or at extravasation at a secondary site. By using an inducible system, and therefore giving equal chances of survival after inoculation, we tested whether the cells would still suffer from an acute loss of Fra-1 after intravenous injection and establishment of (micro)metastases. Extravasation and micrometastasis formation have been shown to occur within the first 24 hours after inoculation [28-30]. In the absence of the support of other cancer cells or a stromal mimic like matrigel, HT29 cells depleted of Fra-1 were 56 times less successful in forming metastatic tumor foci, resulting in significantly fewer and smaller tumor foci in the lungs of the mice. Heterogeneous Fra-1 levels in the tumor foci were commonly observed, with many cells showing restoration of Fra-1 levels, suggesting a negative pressure against Fra-1 knockdown cells. These results demonstrate further that Fra-1 is critical for the metastatic growth of colon cancer cells.
Fra-1 is overexpressed in several cancers [31] and we show that its expression correlates with a poor 5-year survival chance of colon cancer patients. It has proven difficult to develop inhibitors against transcription factors, making Fra-1 an unlikely drug target, even though this could aid in improving the treatment options of colon cancer patients. Furthermore, expression levels of transcription factors do not necessarily reflect the level of their activity. It has been suggested that in a data-driven approach, targets acting downstream of a transcription factor, rather than the transcription factor itself, possess better distinguishing features, because they reflect the activity of the transcription factor [32]. For these reasons, we compared the RNA expression profiles of Fra-1 proficient and deficient HT29 cells. We found a total of 199 genes significantly regulated by Fra-1. Our Fra-1 classifier is able to stratify colon cancer patients into three groups based on their disease outcome: two good (subtypes 2 and 3) and one poor (subtype 1) prognosis groups. Consistent with the role of Fra-1 in metastasis, in patients with a poor prognosis, genes positively regulated by Fra-1 are overexpressed while genes negatively regulated by Fra-1 have a low expression. Based on this classifier, disease-free survival rates of patients in subtype 1 are significantly lower than those in subtypes 2 and 3. The same pattern is observed for disease-specific and overall survival rates, however with less significance most likely owing to the fact that this information is only available for a subset of patient samples.
Fra-1 is a transcription factor functioning in heterodimers with other components of the AP-1 family. We and others have previously shown that Fra-1 down-regulation restores epithelial characteristics, including an epithelial-like morphology from a mesenchymal-like one, in breast cancer cells [7], [22] and colon cancer cells [33]. Moreover, several attempts to classify CRC based on gene expression data identified an EMT-related subtype associated with poor prognosis [34-37]. However, in colon cancer cell lines, we did not observe any change in the morphology, nor in E-cadherin or Vimentin protein levels upon Fra-1 depletion. We also failed to find any EMT genes significantly regulated in our RNA sequencing data. Furthermore, our colon cancer Fra-1 classifier has minimal or no overlap with other prognostic classifiers [35-39] nor with our Fra-1 breast cancer classifier [7]. Since Fra-1 has hundreds if not thousands of target genes, it is conceivable that it regulates several oncogenic processes via different mechanisms in different contexts. Furthermore, the basal expression pattern of Fra-1 target genes is conceivably not identical among different (cancer) tissues. On the other hand, earlier studies comparing metastatic to primary tumors to identify prognostic metastasis genes failed to identify Fra-1 [40-42], although Fra-1 was found to be upregulated in cancer cells compared to normal colon [40].
KEGG pathway analysis on this list of genes in the classifier revealed the focal adhesion and the Wnt pathways as overrepresented. Focal adhesions are known to be regulated by several AP-1 components, including Fra-1 [18], [43]. We confirmed the reliability of our results by validating the Fra-1-mediated regulation of several genes involved in focal adhesions by qRT-PCR. While expression of AP-1 components has been reported to be regulated by the Wnt pathway [10], [12], a reciprocal regulation between the AP-1 transcription factor complex and Wnt signaling has only been shown in an RNA profiling study [33] and awaits further validation. Here, we validated that Wnt pathway genes such as DKK-1, DVL-1 and Wnt10A are indeed positively regulated by Fra-1 in both HT29 and HCT15 colon cancer cell lines. Wnt10A plays an oncogenic role in renal cell carcinoma by activating the canonical Wnt pathway [44], and has been found to be highly expressed especially in the invasive fronts of esophageal cancer [45]. DVL-1 is a scaffolding protein that interacts with the Wnt receptor upon ligand binding and prevents the destruction of beta-catenin, allowing it to be transported to the nucleus and to form a transcription factor complex with TCF/LEF [46]. Despite some conflicting reports about DKK-1 promoting migration and invasion [47], it is recognized as a tumor suppressor and an inhibitor of Wnt signaling [48]. In this context, one would expect DKK-1 levels to increase upon depletion of Fra-1. However, since DKK-1 expression is regulated by beta-catenin [49], reduced beta-catenin activity results in reduced DKK-1 levels in Fra-1-depleted colon cancer cells. Although we did not see beta-catenin being directly regulated by Fra-1 at the RNA level, reporter assays showed that the activity of beta-catenin is decreased upon Fra-1 depletion. Because beta-catenin is known to regulate EMT and metastasis, it is plausible that the pro-metastatic function of Fra-1 is partially dependent on beta-catenin activity, which is tightly regulated by the Wnt pathway.
In conclusion, we find that Fra-1 is a critical factor in driving metastasis of human colon cancer cells in vivo. Furthermore, we show that a Fra-1 classifier is a highly significant predictor of patient outcome, independent of disease stage. We propose that Fra-1-regulated genes may be explored as therapeutic targets for colorectal cancer.
Materials and Methods
Cell culture
HEK293T and colon cancer cell lines (HT29, HCT116, HCT15, DLD-1, Colo205) were cultured in DMEM supplemented with 2mM glutamine and 9% fetal bovine serum (Gibco). For lentivirus production, HEK293T cells were refreshed with complete medium containing 25 mM chloroquine, transfected with 8 μg of lentiviral construct and 4 μg of pMDLglpRRE, pHCMV-G, and pRSVrev and refreshed in complete medium after 6-8 hours. The lentivirus containing supernatant was used to transduce cell lines, followed by antibiotic selection when applicable. shRNA sequences were as follows: shFra-1(1): GTAGATCCTTAGAGGTCCT, shFra-1(2): GGCCTGTGCTTGAACCTGA, shFra-1(3): TRCN0000019539, shFra-1(4): TRCN0000019541, shFra1-(5): TRCN0000019542 (TRC Library, Sigma). The shRNAs 1 and 2 against Fra-1 were custom designed and cloned into KH vector. The others were cloned in the pLKO-puro vector. Fra-1 overexpression plasmid was from the CCSB-Broad lentiviral expression library in pLX204-Blast-V5 vector (Thermo Scientific). Luciferase expressing cells were generated by HIV-CS-CG-luc construct. shFra1-2 sequence was cloned into pLKO-teton vector to generate the inducible shFra1 construct. Cells harboring an empty pLKO-teton vector were used as control. pRL, TOP and FOP constructs for the beta-catenin reporter assay were a kind gift from Emile Voest, NKI. 2D proliferation assays were carried out by seeding 3*104 cells/well in 6-well plates (Corning) and stained with crystal violet after 7 days. For 3D proliferation assays, 2.4*104 cells were seeded in 0,4% low-melting-point agarose (Sigma) on 6-well plates (Corning) coated with a 1% agarose layer. The plates were stained with crystal violet after 3 weeks and colonies were counted by Image J software. Anoikis resistance experiments were performed on ultra-low-attachment 6-well plates (Corning). 0.4*106 cells were seeded in complete medium. After 6 days, the cells were collected, washed, trypsinized, resuspended in complete medium and counted, or were collected and lysed for total protein measurement.
Beta-catenin reporter assay
The cells were seeded on 24-well plates (Costar) at 1*105 cells/well and co-transfected with 100ng of TOP or FOP constructs and 10ng of pRL using Lipofectamine 3000 reagent (Invitrogen) following manufacturers instructions. 48 hours after transfection, the cells were lysed and luciferase signals were measured in triplicates by dual-luciferase reporter assay (Promega). Transfection efficiencies were normalized by dividing the firefly luciferase signal by renilla luceriferase signal for each well. TOP/FOP ratio was calculated by dividing luciferase signal from TOP-transfected cells to FOP-transfected cells.
Immunoblot analysis and antibodies
Cells were harvested by scraping in cold PBS and the pellets were lysed in RIPA buffer (50 mM TRIS pH 8.0, 150 mM NaCl, 1% Nonidet P40, 0.5% sodium deoxycholate, 0.1% SDS, complete protease inhibitor cocktail (Roche), and phosphatase inhibitors 10 mM NaF, 1 mM Na3VO4, 1 mM sodium pyrophosphate, 10 mM beta-glycerophosphate). After centrifugation the protein concentrations were determined by Bio-Rad protein assay (Bio-Rad). Immunoblot analysis was performed by using 4-12% Bis-Tris polyacrylamide-SDS gels (NuPAGE) and transferring these on to nitrocellulose membranes (Amersham). Membranes were blocked in 4% skimmed milk powder dissolved in 0,2% Tween-containing PBS and incubated with primary antibodies followed by secondary antibodies (Invitrogen). Primary antibodies used were Fra-1 (sc-605, Santa Cruz Biotechnology), human-specific Fra-1 (sc-28310, Santa Cruz Biotechnology), beta-actin (A5316, Sigma), Hsp90 (sc-7947, Santa Cruz Biotechnology).
qRT-PCR primers
Total RNA was isolated by harvesting the cells in Trizol (Invitrogen) 6 days after lentivirus transduction and extracting RNA by subsequent chloroform, isopropanol and ethanol treatments. Following DNase treatment for 1 hour at 37°C, cDNA was prepared by a reverse transcriptase kit (Invitrogen). The average values obtained from two independent experiments are presented.
The primer sequences are as follows:
| Wnt10A |
forward |
GGAGACTCGCAACAAGATCC |
| reverse |
AAAGCGCTCTCTCGGAAAC |
|
| DKK-1 |
forward |
CCTTGGATGGGTATTCCAGA |
| reverse |
CCGGAGACAAACAGAACCTT |
|
| DVL-1 |
forward |
GAGCTTGAGTCCAGCAGCTT |
| reverse |
CGGATGAGTCTGGATGAGGT |
|
| SMAD3 |
forward |
ACACCAAGTGCATCACCATC |
| reverse |
GCGGCAGTAGATGACATGAG |
|
| PXN |
forward |
GCACAATCCTTGACCCCTTA |
| reverse |
GAGCCGTACACAGGTGATGA |
|
| FLNA |
forward |
AGCCTCAACGTCACCTATGG |
| reverse |
ACTTGACCTTGGACGCATCT |
|
| ITGB2 |
forward |
TTCTCCAGAAGGTGGTTTCG |
| reverse |
AGCAGCCGTGTAACATTCCT |
Survival analysis
From the Gene Expression Omnibus, we downloaded five publicly available data sets (GSE17536 [41], GSE17537 [41], GSE14333 [42], GSE33113 [50] and GSE37892) with gene expression data from primary CRC samples. Samples contained in both GSE14333 and GSE17536 were removed from GSE14333. Disease-free survival and staging information was available for a total of 578 tumor samples contained in these data sets. Disease-specific survival and overall survival were only available for datasets GSE17536 and GSE17537, totaling 232 tumors. Differences in survival times were analyzed using the Mantel-Cox log-rank test as implemented in the survival package. We performed survival analysis combining all stages stratified by stage and gender, and stage-specific survival analysis.
RNA sequencing and generation of the Fra-1 classifier
Fra-1 was depleted from HT29 cells with two independent short hairpins. RNA was isolated by using Trizol and sequenced on a HiSeq 2000 System (Illumina). Data are available at NCBI Gene Expression Omnibus with the accession number GSE69415 (http://www.ncbi.nlm.nih.gov). Data were analyzed using the R statistical environment [49]. Illumina sequencing data was processed using DESeq version 1.12 [49]. We derived a Fra-1 knockdown (Fra-1KD) gene expression signature comparing RNA sequencing data from HT29 cell lines without and with shRNA knockdown of Fra-1. Knockdown was performed using two different hairpins in triplicate. In total, three samples of the wild type cell line and two knockdown samples (each with a different hairpin) were sequenced. Genes were selected as differentially expressed if they were differentially expressed between wild type and Fra-1KD cells but not between replicates. Nominal p-values were corrected for multiple testing using the Benjamini-Hochberg procedure and corrected p-values < 0.1 were regarded as significant. We applied the Fra-1KD signature to the independent data set consisting of 578 samples described above. More specifically, we used non-negative matrix factorization (NMF) as implemented in the NMF package for R (http://dx.doi.org/10.1186/1471-2105-11-367) to cluster the samples into three subtypes. We compared diseases free survival between these clusters as described above.
Tumor xenografts and bioluminescence analysis
All animal work was done in accordance with a protocol approved by the Netherlands Cancer Institute Animal Experiment Ethics Committee. Female NOD/SCIDIL2gamma mice aged 5-8 weeks were used for all in vivo experiments. 0,5*106 cells were injected into the lateral tail vein in 150ul PBS or subcutaneously into both flanks in a 100ul 1:1 mixture of growth factor reduced matrigel and complete medium. Subcutaneous tumors were manually measured twice weekly by a caliper. For bioluminescence imaging, the mice were intraperitoneally injected with 15mg/kg D-luciferin (Caliper Life Sciences) 15 minutes prior to imaging. The mice were anaesthetized and imaged with 60 seconds of exposure time (binning=8). Tumor burden in individual organs were quantified by injecting the mice with D-luciferin five minutes prior to sacrifice, harvesting the organs and imaging in a PBS-luciferin mixture. The data was analyzed by Living Image software. For shFra-1 induction, mice were treated with 2mg/ml doxycycline in drinking water containing 10mg/ml sucrose.
Immunohistochemistry
Histological sections and hematoxylin-eosin staining were performed using standard procedures. Paraffin sections were deparaffinized, rehydrated, pretreated in 0.1 mM sodium citrate pH 6.0, washed and incubated with peroxide. The tissue was incubated with primary antibodies for Fra-1 (1:200, sc-28310, Santa Cruz Biotechnology) or GFP (1:2000, Abcam). Secondary antibody was PowerVision (DPVB-999HRP, ImmunoLogic). Peroxidase activity was detected with Liquid DAB (K3468, DAKO).
Statistical analysis
Comparisons of two experimental groups were analyzed with two-tailed student’s t-test. One-Way ANOVA corrected for multiple comparisons (Holm-Sidak) was used to compare more than two experimental groups (Prism; GraphPad Software). Error bars represent standard error of the mean (SEM).
Multivariate analysis was performed by fitting a Cox proportional hazards model to estimate hazard ratios for the subtypes by stratifying for gender and tumor stage.
Acknowledgments
The authors would like to thank Prof. Dr. Emile Voest for sharing constructs, Dr. Wilbert Zwart for his useful suggestions, and all the members of the Peeper Laboratory for their valuable input. We would also like to acknowledge the Animal Pathology Department for the immunohistochemistry stainings, Dr. Ji-Ying Song for her help interpreting the immunohistochemistry stainings, the Sequencing Core Facility for the generation of RNAseq data and Animal Caretakers for their assistance with the mouse experiments.
This work was financially supported by a grant from the Dutch Cancer Society (2009-4552) to SI and DP.
conflicts of interests
Authors do not disclose any potential conflicts.
Financial support
Dutch Cancer Society (2009-4552).
References
1. Geiger TR, Peeper DS. Metastasis mechanisms. BBA - Reviews on Cancer. 2009;1796(2):293–308. doi:10.1016/j.bbcan.2009.07.006
2. Nguyen DX, Bos PD, Massagué J. Metastasis: from dissemination to organ-specific colonization. Nat Rev Cancer. 2009;9(4):274–84. doi:10.1038/nrc2622
3. Ozanne BW, Spence HJ, McGarry LC, Hennigan RF. Transcription factors control invasion: AP-1 the first among equals. Oncogene. 2006;26(1):1–10. doi:10.1038/sj.onc.1209759
4. Eferl R, Wagner EF. AP-1: a double-edged sword in tumorigenesis. Nat Rev Cancer. 2003;3(11):859–68. doi:10.1038/nrc1209
5. Mahner S, Baasch C, Schwarz J, Hein S, Wölber L, Jänicke F, Milde-Langosch K. C-Fos expression is a molecular predictor of progression and survival in epithelial ovarian carcinoma. British Journal of Cancer. 2008;99(8):1269–75. doi:10.1038/sj.bjc.6604650
6. Volm M, Drings P, Wodrich W. Prognostic significance of the expression of c-fos, c-jun and c-erbB-1 oncogene products in human squamous cell lung carcinomas. J Cancer Res Clin Oncol. 1993;119:507–510.
7. Desmet CJ, Gallenne T, Prieur A, Reyal F, Visser NL, Ben S Wittner, Smit MA, Geiger TR, Laoukili J, Iskit S, Rodenko B, Zwart W, Evers B, et al. Identification of a pharmacologically tractable Fra-1/ADORA2B axis promoting breast cancer metastasis. Proceedings of the National Academy of Sciences. 2013;110(13):5139–5144. doi:10.1073/pnas.1222085110
8. Young MR, Nair R, Bucheimer N, Tulsian P, Brown N, Chapp C, Hsu TC, Colburn NH. Transactivation of Fra-1 and Consequent Activation of AP-1 Occur Extracellular Signal-Regulated Kinase Dependently. Molecular and Cellular Biology. 2002;22(2):587–598. doi:10.1128/mcb.22.2.587–598.2002
9. Vial E. Elevated ERK-MAP kinase activity protects the FOS family member FRA-1 against proteasomal degradation in colon carcinoma cells. Journal of Cell Science. 2003;116(24):4957–4963. doi:10.1242/jcs.00812
10. Mann B, Gelos M, Siedow A, Hanski ML, Gratchev A, Ilyas M, Bodmer WF, Moyer MP, Riecken EO, Buhr HJ, Hanski C. Target genes of beta-catenin-T cell-factor/lymphoid-enhancer-factor signaling in human colorectal carcinomas. 1999;96:1063–1068.
11. Anastas JN, Moon RT. WNT signalling pathways as therapeutic targets in cancer. Nat Rev Cancer. Nature Publishing Group; 2013;11–26. doi:10.1038/nrc3419
12. Tice DA, Soloviev I, Polakis P. Activation of the Wnt Pathway Interferes with Serum Response Element-driven Transcription of Immediate Early Genes. Journal of Biological Chemistry. 2001;277(8):6118–6123. doi: 10.1074/jbc.M111255200
13. Nateri AS, Spencer-Dene B, Behrens A. Interaction of phosphorylated c-Jun with TCF4 regulates intestinal cancer development. Nat Cell Biol. 2005;437(7056):281–285. doi:10.1038/nature03914
14. Belguise K, Kersual N, Galtier F, Chalbos D. FRA-1 expression level regulates proliferation and invasiveness of breast cancer cells. Oncogene. 2004;41(16):2449–2461. doi:10.1016/j.ejca.2005.08.008
15. Adiseshaiah P, Vaz M, Machireddy N, Kalvakolanu DV, Reddy SP. A Fra-1-dependent, matrix metalloproteinase driven EGFR activation promotes human lung epithelial cell motility and invasion. J Cell Physiol. 2008;216(2):405–412. doi: 10.1002/jcp.21410
16. Ramos-Nino ME, Blumen SR, Pass H, Mossman BT. Fra-1 governs cell migration via modulation of CD44 expression in human mesotheliomas. Mol Cancer. 2007;6(81):1-14. doi:10.1186/1476-4598-6-81
17. Sayan AE, Stanford R, Vickery R, Grigorenko E, Diesch J, Kulbicki K, Edwards R, Pal R, Greaves P, Jariel-Encontre I, Piechaczyk M, Kriajevska M, Mellon JK, Dhillon AS, et al. Fra-1 controls motility of bladder cancer cells via transcriptional upregulation of the receptor tyrosine kinase AXL. Oncogene. 2011;31(12):1493–1503. doi:10.1038/onc.2011.336
18. Vial E, Sahai E, Marshall CJ. ERK-MAPK signaling coordinately regulates activity of Rac1 and RhoA for tumor cell motility. Cancer Cell. 2003;4(1):67–79.
19. Ferraro A, Mourtzoukou D, Kosmidou V, Avlonitis S, Kontogeorgos G, Zografos G, Pintzas A. EZH2 is regulated by ERK/AKT and targets integrin alpha2 gene to control Epithelial–Mesenchymal Transition and anoikis in colon cancer cells. International Journal of Biochemistry and Cell Biology. Elsevier Ltd; 2013;45(2):243–254. doi:10.1016/j.biocel.2012.10.009
20. Compton CC, Greene FL. The Staging of Colorectal Cancer: 2004 and Beyond. CA Cancer J Clin. 2004;54:295–308.
21. Linnekamp JF, Wang X, Medema JP, Vermeulen L. Colorectal Cancer Heterogeneity and Targeted Therapy: A Case for Molecular Disease Subtypes. Cancer Research. 2015;75(2):245–249. doi:10.1158/0008-5472.can-14-2240
22. Tam WL, Lu H, Buikhuisen J, Soh BS, Lim E, Reinhardt F, Wu ZJ, Krall JA, Bierie B, Guo W, Chen X, Liu XS, Brown M, et al. Protein Kinase C α Is a Central Signaling Node and Therapeutic Target for Breast Cancer Stem Cells. Cancer Cell. 2013;24:347–364. doi:10.1016/j.ccr.2013.08.005
23. Leupold JH, Asangani I, Maurer GD, Lengyel E, Post S, Allgayer H. Src Induces Urokinase Receptor Gene Expression and Invasion/Intravasation via Activator Protein-1/p-c-Jun in Colorectal Cancer. Molecular Cancer Research. 2007;5(5):485–496. doi:10.1158/1541-7786.mcr-06-0211
24. Andreolas C, Kalogeropoulou M, Voulgari A, Pintzas A. Fra-1 regulates vimentin during Ha-RAS-induced epithelial mesenchymal transition in human colon carcinoma cells. Int J Cancer. 2007;122:1745–1756. doi:10.1002/ijc.23309
25. Pollock CB, Shirasawa S, Sasazuki T, Kolch W, Dhillon AS. Oncogenic K-RAS Is Required to Maintain Changes in Cytoskeletal Organization, Adhesion, and Motility in Colon Cancer Cells. Cancer Research. 2005;65(4):1244–1250.
26. Possik PA, Müller J, Gerlach C, Kenski JCN, Huang X, Shahrabi A, Krijgsman O, Song J-Y, Smit MA, Gerritsen B, Lieftink C, Kemper K, Michaut M, et al. Parallel In Vivo and In Vitro Melanoma RNAi Dropout Screens Reveal Synthetic Lethality between Hypoxia and DNA Damage Response Inhibition. Cell Reports. 2014;9:1375–1386. doi:10.1016/j.celrep.2014.10.024
27. Fidler IJ. Critical determinants of metastasis. Seminars in Cancer Biology. 2002;12:89–96. doi:10.1006/scbi.2001.0416
28. Stoletov K, Kato H, Zardouzian E, Kelber J, Yang J, Shattil S, Klemke R. Visualizing extravasation dynamics of metastatic tumor cells. Journal of Cell Science. 2010;123:2332–2341. doi:10.1242/jcs.069443
29. Martin MD, Kremers G-J, Short KW, Rocheleau JV, Xu L, Piston DW, Matrisian LM, Gorden DL. Rapid extravasation and establishment of breast cancer micrometastases in the liver microenvironment. Mol Cancer Res. 2010;8(10):1319–1327. doi: 10.1158/1541-7786.mcr-09-0551
30. Wolf MJ, Hoos A, Bauer J, Boettcher S, Knust M, Weber A, Simonavicius N, Schneider C, Lang M, Stürzl M, Croner RS, Konrad A, Manz MG, et al. Endothelial CCR2 Signaling Induced by Colon Carcinoma Cells Enables Extravasation via the JAK2-Stat5 and p38MAPK Pathway. Cancer Cell. 2012;22:91–105. doi:10.1016/j.ccr.2012.05.023
31. Young MR, Colburn NH. Fra-1 a target for cancer prevention or intervention. Gene. 2006;379:1–11. doi:10.1016/j.gene.2006.05.001
32. van ‘t Veer LJ, Bernards R. Enabling personalized cancer medicine through analysis of gene-expression patterns. Nature. 2008;452(7187):564–570. doi:10.1038/nature06915
33. Diesch J, Sanij E, Gilan O, Love C, Tran H, Fleming NI, Ellul J, Amalia M, Haviv I, Pearson RB, Tulchinsky E, Mariadason JM, Sieber OM, et al. Widespread FRA1-Dependent Control of Mesenchymal Transdifferentiation Programs in Colorectal Cancer Cells. Schneider G, editor. PLoS ONE. 2014;9(3):1-11. doi:10.1371/journal.pone.0088950
34. Loboda A, Nebozhyn MV, Watters JW, Buser CA, Shaw PM, Huang PS, Veer LV, Tollenaar RA, Jackson DB, Agrawal D, Dai H, Yeatman TJ. EMT is the dominant program in human colon cancer. BMC Medical Genomics. 2011;4(9):1-10. doi:10.1186/1755-8794-4-9
35. Schlicker A, Beran G, Chresta CM, McWalter G, Pritchard A, Weston S, Runswick S, Davenport S, Heathcote K, Castro DA, Orphanides G, French T, Wessels LF. BMC Medical Genomics. 2012;5(66):1–15. doi:10.1186/1755-8794-5-66
36. Roepman P, Schlicker A, Tabernero J, Majewski I, Tian S, Moreno V, Snel MH, Chresta CM, Rosenberg R, Nitsche U, Macarulla T, Capella G, Salazar R, et al. Colorectal cancer intrinsic subtypes predict chemotherapy benefit, deficient mismatch repair and epithelial-to-mesenchymal transition. Int J Cancer. 2013;134:552–562. doi:10.1002/ijc.28387
37. De Sousa E Melo F, Wang X, Jansen M, Fessler E, Trinh A, de Rooij LPMH, de Jong JH, de Boer OJ, van Leersum R, Bijlsma MF, Rodermond H, van der Heijden M, et al. Poor-prognosis colon cancer is defined by a molecularly distinct subtype and develops from serrated precursor lesions. Nat Med. 2013;19(5): 614–821. doi:10.1038/nm.3174
38. Budinska E, Popovici V, Tejpar S, D’Ario G, Lapique N, Sikora KO, Di Narzo AF, Yan P, Hodgson JG, Weinrich S, Bosman F, Roth A, Delorenzi M. Gene expression patterns unveil a new level of molecular heterogeneity in colorectal cancer. J Pathol. 2013;231:63–76. doi: 10.1002/path.4212
39. Marisa L, de Reyniès A, Duval A, Selves J, Gaub MP, Vescovo L, Etienne-Grimaldi M-C, Schiappa R, Guenot D, Ayadi M, Kirzin S, Chazal M, Fléjou J-F, et al. Gene Expression Classification of Colon Cancer into Molecular Subtypes: Characterization, Validation, and Prognostic Value. PLoS Med. 2013;10(5):1–13. doi:10.1371/journal.pmed.1001453
40. Koehler A, Bataille F, Schmid C, Ruemmele P, Waldeck A, Blaszyk H, Hartmann A, Hofstaedter F, Dietmaier W. Gene expression profiling of colorectal cancer and metastases divides tumours according to their clinicopathological stage. J Pathol. 2004;204:65–74. doi: 10.1002/path.1606
41. Smith JJ, Deane NG, Wu F, Merchant NB, Zhang B, Jiang A, Lu P, Johnson JC, Schmidt C, Bailey CE, Eschrich S, Kis C, Levy S, et al. Experimentally Derived Metastasis Gene Expression Profile Predicts Recurrence and Death in Patients With Colon Cancer. YGAST. 2010;138:958–968. doi:10.1053/j.gastro.2009.11.005
42. Jorissen RN, Gibbs P, Christie M, Prakash S, Lipton L, Desai J, Kerr D, Aaltonen LA, Arango D, Kruhoffer M, Orntoft TF, Andersen CL, Gruidl et al. Metastasis-Associated Gene Expression Changes Predict Poor Outcomes in Patients with Dukes Stage B and C Colorectal Cancer. Clinical Cancer Research. 2009;15(24):7642–7651. doi: 10.1158/1078-0432.CCR-09-1431
43. Tkach V, Tulchinsky E, Lukanidin E, Vinson C, Bock E, Berezin V. Role of the Fos family members, c-Fos, Fra-1 and Fra-2, in the regulation of cell motility. Oncogene. 2003;22:5045–54. doi:10.1038/sj.onc.1206570
44. Hsu R-J, Ho J-Y, Cha T-L, Yu D-S, Wu C-L, Huang W-P, Chu P, Chen Y-H, Chen J-T, Yu C-P. WNT10A Plays an Oncogenic Role in Renal Cell Carcinoma by Activating WNT/β-catenin Pathway. PLoS ONE. 2012;7(10):1-16. doi:10.1371/journal.pone.0047649
45. Long A, Giroux V, Whelan KA, Hamilton KE, Tétreault M-P, Tanaka K, Lee J-S, Klein-Szanto AJ, Nakagawa H, Rustgi AK. WNT10A promotes an invasive and self-renewing phenotype in esophageal squamous cell carcinoma. Carcinogenesis. 2015; 36(5):598–606. doi:10.1093/carcin/bgv025
46. Najdi R, Holcombe R, Waterman M. Wnt signaling and colon carcinogenesis: Beyond APC. J Carcinog. 2011;10(5):1-15. doi: 10.4103/1477-3163.78111
47. Chen L, Li M, Li Q, Wang C-J, Xie S-Q. DKK1 promotes hepatocellular carcinoma cell migration and invasion through β-catenin/MM7 signaling pathways. Mol Cancer. 2013;12(157):1–14. doi:10.1186/1476-4598-12-157
48. Pendás-Franco N, Aguilera O, Pereira F, González-Sancho JM, Muñoz A. Vitamin D and Wnt/B-catenin Pathway in Colon Cancer: Role and Regulation on DICKKOPF Genes. Anticancer Research. 2008;28:2613-2624.
49. González-Sancho JM, Aguilera O, García JM, Pendás-Franco N, Peña C, Cal S, de Herreros AG, Bonilla F, Muñoz A. The Wnt antagonist DICKKOPF-1 gene is a downstream target of β-catenin/TCF and is downregulated in human colon cancer. Oncogene. 2004;24:1098–1103. doi:10.1038/sj.onc.1208303
50. De Sousa E Melo F, Colak S, Buikhuisen J, Koster J, Cameron K, de Jong JH, Tuynman JB, Prasetyanti PR, Fessler E, van den Bergh SP, Rodermond H, Dekker E, et al. Methylation of Cancer-Stem-Cell-Associated Wnt Target Genes Predicts Poor Prognosis in Colorectal Cancer Patients. Stem Cell. 2011;9:476–485. doi:10.1016/j.stem.2011.10.008
51. Gentleman RC, Carey VJ, Bates DM, Bolstad B, Dettling M, Dudoit S, Ellis B, Gautier L, Ge Y, Gentry J, Hornik K, Hothorn T, Huber W, et al. Bioconductor: open software development for computational biology and bioinformatics. Genome Biology. 2004;5(R80):1–16.
52. Anders S, Huber W. Differential expression analysis for sequence count data. Genome Biology. 2010;11(R106):1-12.