
Introduction
Liver fibrosis, a major cause of morbidity and mortality worldwide, represents the common responses of the liver to infectious, toxic or metabolic agents and is a big medical problem [1]. Liver fibrosis is characterized by an excessive deposition of extracellular matrix (ECM) proteins in liver, mainly synthesized by activated hepatic stellate cells (HSCs). The excess deposition of ECM disrupts the normal architecture of the liver resulting in pathophysiological damage to the organ, which eventually undergoes the liver fibrosis-cirrhosis [2]. During liver injury, quiescent HSCs are exposed to inflammatory and profibrogenic factors, and transdifferentiate into myofibroblast-like cells that are characterized by expression of alpha-smooth muscle actin (α-SMA), contributing to the progression of liver fibrosis [3]. Therefore, HSCs, the major mesenchymal cells in liver, are widely accepted as playing a critically important role in liver fibrosis [4]. However, the underlying molecular mechanisms responsible for the proliferation and activation of HSCs are still not completely understood.
Aberrant Wnt/β-catenin signaling has been shown to be involved in the development of organ fibrosis including liver fibrosis [5, 6]. Sustained Wnt/β-catenin pathway activation contributes to HSC activation and mediates HSC proliferation, resolution, and ECM accumulation [5, 7, 8]. It has been demonstrated that Wnt antagonists such as secreted Frizzled-related proteins (sFRPs), Wnt inhibitory factor 1 (WIF1) and secreted Dickkopf (DKK) family (DKK1-4) can attenuate hepatic fibrosis via inhibiting Wnt/β-catenin pathway, suggesting Wnt/β-catenin pathway may be a novel therapeutic target in liver fibrosis [9, 10].
MicroRNAs (miRNAs) are short 20-22 nucleotides and act as negative regulators of gene expression by inhibiting protein translation or inducing mRNA degradation [11]. Growing evidence has demonstrated that miRNAs are involved in the control of a wide range of biological functions and processes such as development, differentiation, metabolism, carcinogenesis, and immune response [12]. HSCs could be activated or suppressed by miRNAs, suggesting that miRNAs act as HSC regulators in liver fibrosis. For instance, over-expression of miR-146a suppresses transforming growth factor-beta (TGF-β)-induced HSC proliferation, and increases HSC apoptosis via its target Smad4 [4]. Recently, miR-17-5p, a member of the miR-17-92 cluster, is often up-regulated in many malignancies including hepatocellular carcinoma (HCC) and functions as an oncogenic miRNA [13, 14]. MiR-17-5p is not only involved in cell functions such as proliferation and migration but also a key regulator of the G1/S phase cell cycle transition [15]. Our previous study demonstrated that over-expression of miR-17-5p promotes HSC proliferation and activation [16].
In China, traditional Chinese medicine is often used to treat hepatic fibrosis because of patients’ trust in traditional Chinese medicine. For example, Fuzheng Huayu recipe (FZHY), a formula with anti-hepatic fibrosis activity, is often used as an anti-liver fibrotic product in China [17, 18]. It has been demonstrated that Radix Salviae miltiorrhizae is the main effective herb in FZHY. Salvianolic acid B (Sal B), one of the water soluble components from Radix Salviae miltiorrhizae, has good action against hepatic fibrosis in animal model and patients with chronic hepatitis B [19, 20]. But so far, the mechanisms for the anti-fibrotic effects of Sal B have not been well elucidated.
Results
Sal B inhibited liver fibrosis in vivo and in vitro
It has been demonstrated that Sal B can inhibit liver fibrosis [20, 21]. To confirm the effects of Sal B on rat liver fibrosis induced by carbon tetrachloride (CCl4), the degree of rat liver fibrosis was determined by hematoxylin and eosin (H&E) staining and Masson staining. As indicated by H&E and Masson staining, CCl4 caused prominent hepatic steatosis, necrosis, and formation of regenerative nodules in rat liver tissues, which was ameliorated by Sal B (Figure 1A). Immunohistochemical results demonstrated that the increased α-SMA levels in CCl4-treated rats were reduced by Sal B (P < 0.05, Figure 1B). Next, the effects of Sal B on the mRNA and protein levels of α-SMA and type I collagen were analyzed in rat liver tissues by real-time PCR and western blotting, respectively. Our results showed that the increased levels of α-SMA and type I collagen caused by CCl4 were inhibited by Sal B (Figure 1C and Figure 1D). To further investigate the anti-fibrotic effects of Sal B in vitro, the levels of α-SMA and type I collagen were analyzed in HSC-T6 cells and primary HSCs after Sal B treatment. Compared with the control, the levels of α-SMA and type I collagen were reduced by Sal B in HSC-T6 cells (Figure 2D and Figure 2E). Similar results were also observed in primary HSCs. As shown in the MTT assay, the growth rate was reduced to 53.8% and 65.6%, respectively, in Sal B-treated primary HSCs and HSC-T6 cells compared to untreated cells (Figure 2F). These data confirmed that Sal B could inhibit liver fibrosis.
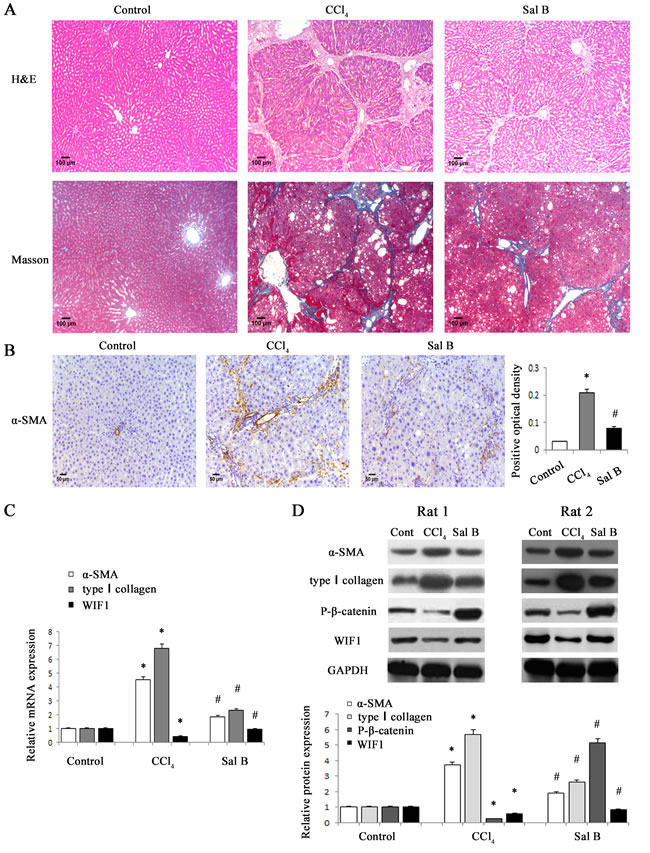
Figure 1: Sal B significantly ameliorated CCl4-induced liver fibrosis in rats. A. H&E staining (×100) and Masson staining (×100) for assessing liver fibrosis. B. The levels of α-SMA were analyzed by immunohistochemistry in CCl4-treated rats after Sal B treatment. Representative views from each group are presented (original magnification, ×10). C. The mRNA levels of α-SMA, type I collagen and WIF1 were detected in CCl4-treated rats after Sal B treatment. D.The protein levels of α-SMA, type I collagen, P-β-catenin and WIF1 were detected in CCl4-treated rats after Sal B treatment. GAPDH was used as internal control. Each value is the mean ± SD of three experiments. *P < 0.05 compared with the control and #P < 0.05 compared with the CCl4 group.
Sal B induced the inactivation of Wnt/β-catenin pathway and the up-regulation of WIF1 expression
Aberrant Wnt/β-catenin pathway is involved in the development of organ fibrosis [22, 23]. To explore whether Wnt/β-catenin pathway is involved in the effects of Sal B on liver fibrosis, the protein levels of P-β-catenin were detected in vivo and in vitro after Sal B treatment. The results showed that Sal B caused a significant increase in the phosphorylation of β-catenin in CCl4-treated rats, HSC-T6 cells and primary HSCs (Figure 1D and Figure 2E). Sal B treatment also resulted in a reduced TCF activity in HSC-T6 and primary HSCs (Figure 2C). To further study whether the suppression of Wnt signaling caused by Sal B is associated with the increased expressions of Wnt signaling inhibitors, the mRNA expressions of Wnt signaling inhibitors including WIF1, sFRP1, sFRP2 and DKK1-4, were detected in CCl4-treated rats, HSC-T6 cells and primary HSCs after Sal B treatment. It was found that the mRNA level of WIF1 was increased by Sal B whereas others not (Figure 1C, Figure 2A and Supporting Information Figure S1). Immunoblot analysis further confirmed that Sal B caused an increase in WIF1 protein in vivo and in vitro (Figure 1D and Figure 2B). Therefore, WIF1 was chosen for the next experiments. These results indicated that Sal B caused the suppression of Wnt/β-catenin pathway with an increase in the levels of P-β-catenin and WIF1.
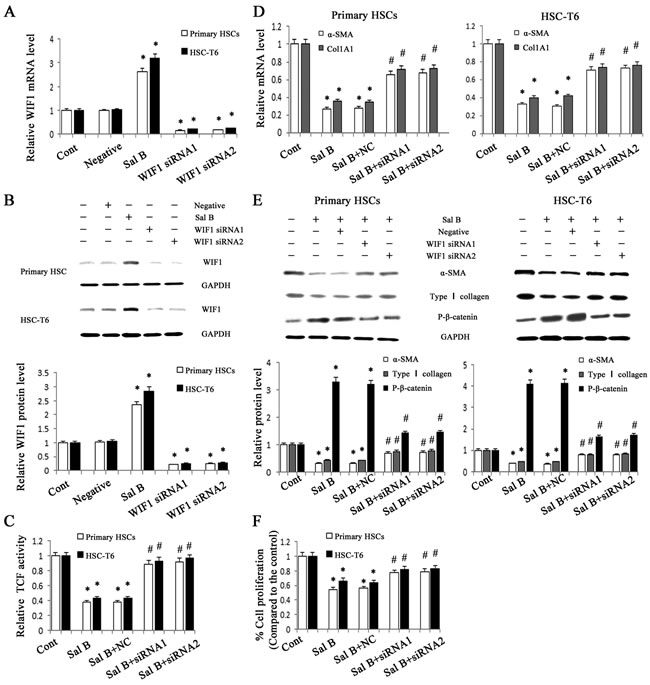
Figure 2: Roles of WIF1 in the anti-fibrotic effects induced by Sal B. HSC-T6 cells and primary HSCs were transfected with WIF1 siRNA for 48 h and/or treated with Sal B for 48 h. A. The mRNA expression of WIF1 was analyzed by real-time PCR. B. The protein expression of WIF1 was analyzed by western blotting. GAPDH was used as internal control. C. Reduced TCF activity by Sal B was restored by WIF1 siRNA. D. The decreased mRNA levels of α-SMA and Col1A1 induced by Sal B were blocked down by WIF1 siRNA. E. The effects of Sal B on the protein levels of α-SMA, type I collagen and P-β-catenin were attenuated by WIF1 siRNA. GAPDH was used as internal control. F. The reduction of cell proliferation induced by Sal B was inhibited by WIF1 siRNA. Each value is the mean ± SD of three experiments. *P < 0.05 compared with the control and #P < 0.05 compared with the Sal B group.
WIF1 up-regulation caused by Sal B suppressed HSC activation
Previous studies have reported that WIF1 is a potent Wnt pathway antagonist [9]. To explore whether WIF1 is involved in the anti-fibrotic effect induced by Sal B, WIF1 siRNA was transfected into HSC-T6 cells and primary HSCs to silent WIF1. The blockade of WIF1 with WIF1 siRNA successfully inhibited the mRNA and protein levels of WIF1 in HSC-T6 cells and primary HSCs (Figure 2A and Figure 2B). Next, the TCF activity and the levels of P-β-catenin in Sal B-treated cells with WIF1 siRNA were examined. TCF reporter activity assay showed that the TCF activity decreased by Sal B was reversed by WIF1 siRNA in HSC-T6 cells and primary HSCs (Figure 2C). Moreover, the enhanced P-β-catenin levels caused by Sal B were reversed by WIF1 siRNA (Figure 2E). Of note, the effects of Sal B on the mRNA and protein levels of α-SMA and type I collagen in HSCs were blocked down by WIF1 siRNA (Figure 2D and Figure 2E). The reduction of cell proliferation caused by Sal B treatment was also blocked down by the silencing of WIF1 expression (Figure 2F). These findings demonstrated that WIF1 was involved in the anti-fibrotic effect induced by Sal B.
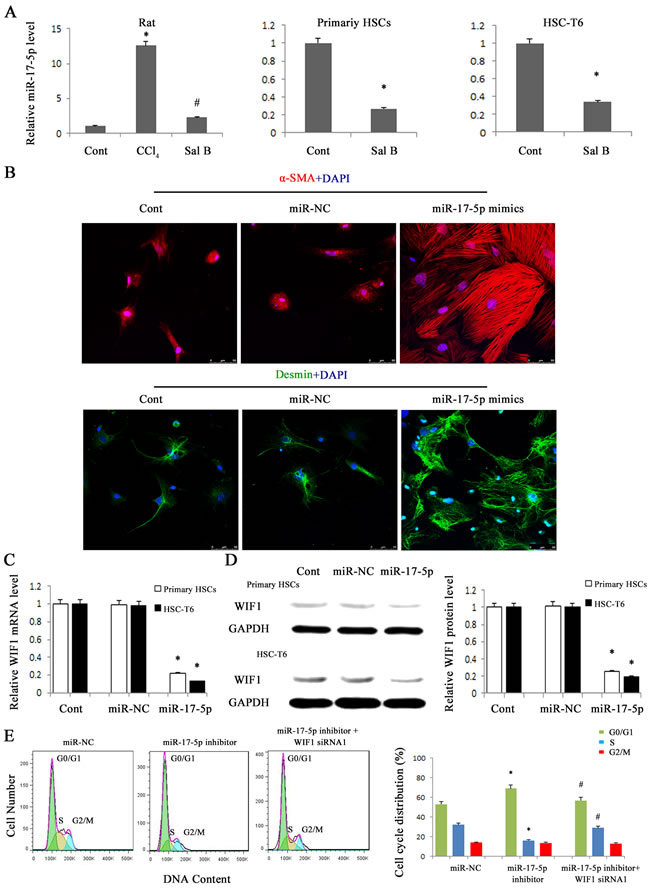
Figure 3: miR-17-5p over-expression promoted the activation of HSCs and contributed to the reduction of WIF1 levels. HSC-T6 cells and primary HSCs were transfected with miR-17-5p mimics or miR-NC for 48 h. A. The down-regulation of miR-17-5p expression was found in CCl4-treated rats, HSC-T6 cells and primary HSCs after Sal B treatment. B. Immunofluorescence staining for α-SMA (red) and desmin (green) were evaluated by confocal laser microscopy. DAPI stained nuclei blue. Scale bar, 50 μm. C. The mRNA expression of WIF1 was analyzed by real-time PCR in HSCs treated with miR-17-5p mimics. D. The protein expression of WIF1 was analyzed by western blotting in HSCs treated with miR-17-5p mimics. GAPDH was used as internal control. E. Effect of WIF1 siRNA on the cell cycle in HSCs transfected with miR-17-5p inhibitor. The primary HSCs were transfected with miR-17-5p inhibitor for 48 h and then transfected with WIF1 siRNA for an additional 48 h. Each value is the mean ± SD of three experiments. *P < 0.05 compared with the control or miR-NC. #P < 0.05 compared with miR-17-5p inhibitor group.
MiR-17-5p was involved in Sal B effects and promoted the activation of HSCs
To determine whether WIF1 expression is inhibited by miRNAs, we searched for predictable miRNAs that could bind with 3′untranslated region (3′UTR) of rat WIF1 mRNA using microRNA.org (http://www.microrna.org/microrna/home.do). As a result, miR-16, miR-17-5p, miR-20a, miR-20b-5p, miR-21 and miR-199-5p were extracted as candidates. The results showed that only miR-17-5p was reduced in vivo and in vitro after Sal B treatment (Figure 3A and Supporting Information Figure S1). Therefore, miR-17-5p was selected for further experiments. To investigate the effects of miR-17-5p over-expression on the activation of HSCs, immunofluorescence staining for α-SMA and the HSC marker desmin was examined in primary HSCs. We found that the levels of desmin (green) and α-SMA (red) were increased by miR-17-5p over-expression (Figure 3B). These data indicated that miR-17-5p promoted the activation of HSCs. Given that WIF1 was predicted as a putative target of miR-17-5p (Figure 5A and Figure 5B), we hypothesized that miR-17-5p might promote hepatic fibrosis via inhibiting WIF1 expression. Next, the mRNA and protein levels of WIF1 were examined in cells transfected with miR-17-5p mimics. The results showed that both the mRNA and protein levels of WIF1 were reduced by miR-17-5p mimics (Figure 3C and Figure 3D). We also examined the effects of miR-17-5p inhibitor and the silencing of WIF1 on cell cycle. Cell cycle analysis revealed that compared with the control, miR-17-5p inhibitor suppressed a proportion of cells in the S phase and increased the number of cells in the G0/G1 phase, suggesting that miR-17-5p inhibitor contributed to the inactivation of HSCs (Figure 3E). However, the effects of miR-17-5p inhibitor on cell cycle were blocked down by WIF1 siRNA (Figure 3E). Notably, as shown in H&E and Masson staining, miR-17-5p inhibitor treatment significantly suppressed liver fibrosis caused by CCl4 (Figure 4A). Immunohistochemical results additionally confirmed that the levels of α-SMA increased in CCl4-treated rats were attenuated by miR-17-5p inhibitor treatment (P < 0.05, Figure 4B). All these results suggested that miR-17-5p could promote the activation of HSCs via the down-regulation of WIF1.
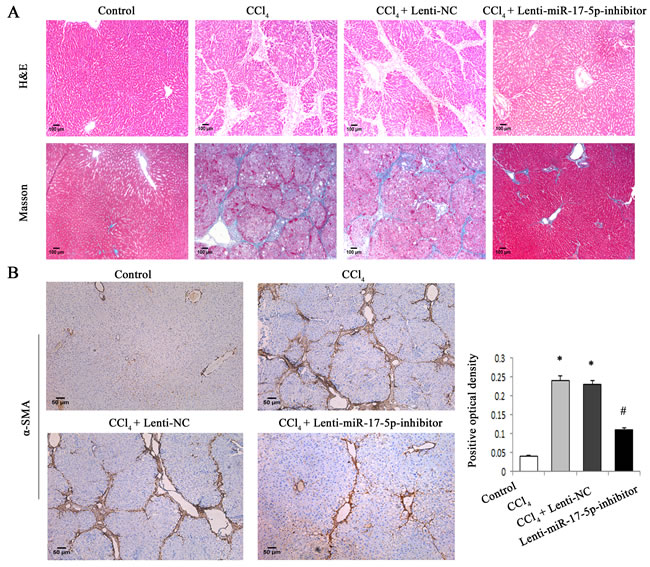
Figure 4: MiR-17-5p inhibitor treatment significantly suppressed rat liver fibrosis caused by CCl4. A. H&E staining (×100) and Masson staining (×100) for assessing liver fibrosis. B. The levels of α-SMA were analyzed by immunohistochemistry in CCl4-treated rats after miR-17-5p inhibitor treatment. Representative views from each group are presented (original magnification, ×10). Each value is the mean ± SD of three experiments. *P < 0.05 compared with the control and #P < 0.05 compared with the CCl4 group.
WIF1 is a target of miR-17-5p
To further investigate whether WIF1 is the direct downstream target of miR-17-5p, the sequence of 3′UTR of WIF1 mRNA target region was cloned into pMIR-REPORTTM Luciferase plasmid (Figure 5A and Figure 5B). The construct was cotransfected into HSCs along with miR-17-5p precursor or miRNA negative control (miR-NC). β-gal reporter control plasmid was cotransfected to monitor transfection efficiency. Our results showed that miR-17-5p precursor significantly reduced luciferase activity driven by the wild-type 3′UTR of WIF1 compared to miR-NC in HSCs. Meanwhile, the luciferase activities of mutated type WIF1 3′UTR and empty vector were not inhibited by miR-17-5p precursor (Figure 5C). These results confirmed that WIF1 was a direct target of miR-17-5p.
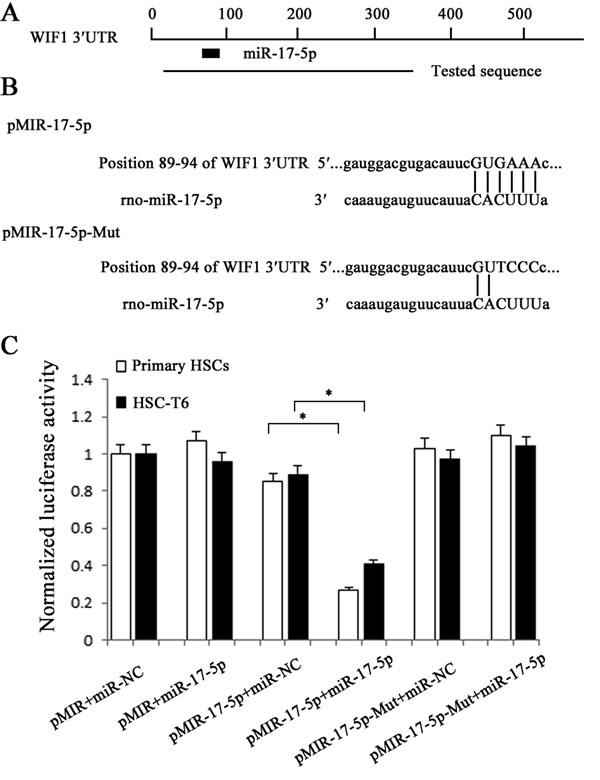
Figure 5: Interaction of miR-17-5p with the 3’UTR of WIF1. A. Schematic indication of the miRNA binding sites in the 3’UTR of WIF1 mRNA based on microRNA.org. Black box indicates miR-17-5p and a tested sequence indicates the region that was inserted into the luciferase reporter vector. B. Predicted consequential pairing of the target region and miR-17-5p. According to the pairing sites, the corresponding luciferase reporter vectors were named pMIR-17-5p and pMIR-17-5p-Mut. C. The HSCs were transfected with pMIR (empty vector), pMIR containing miR-17-5p targeting sequence (pMIR-17-5p) and pMIR with miR-17-5p mutated target sequence (pMIR-17-5p-Mut). The graph shows luciferase activity in cells transfected with pMIR-17-5p or pMIR-17-5p-Mut. It also shows cotransfection of miR-17-5p precursor or miR-NC. *P < 0.05.
MiR-17-5p activated HSCs via Wnt/β-catenin pathway
Our previous study showed that WIF1 was a direct target of miR-17-5p and the levels of miR-17-5p were reduced by Sal B in HSCs. To further investigate whether miR-17-5p is involved in the anti-fibrotic effects of Sal B, we transfected miR-17-5p mimics into Sal B-treated cells. The effects of Sal B on the levels of P-β-catenin, α-SMA and type I collagen were attenuated by miR-17-5p mimics (Figure 6A and Figure 6B). Notably, increased WIF1 levels in Sal B-treated cells were additionally reversed by miR-17-5p mimics. Moreover, the reduction of TCF activity and cell proliferation rate induced by Sal B were almost blocked down by miR-17-5p mimics (Figure 6C and Figure 6D). All these results indicated that the reduction of miR-17-5p level in the treatment of Sal B contributed to the suppression of activated HSCs and miR-17-5p promoted the activation of Wnt/β-catenin pathway via inhibiting WIF1 level.
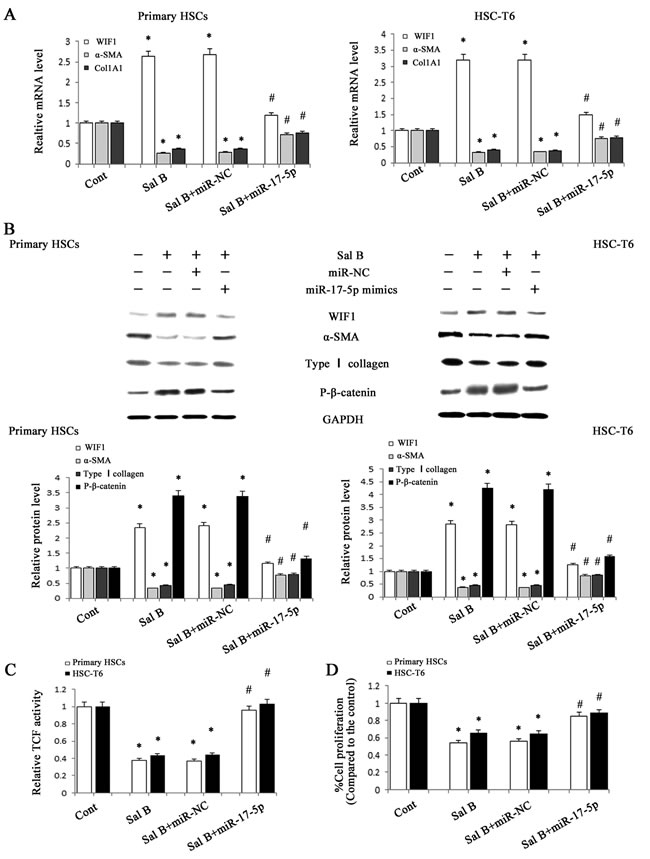
Figure 6: Roles of miR-17-5p in the anti-fibrotic effects induced by Sal B. HSC-T6 cells and primary HSCs were transfected with miR-17-5p mimics for 48 h and treated with Sal B for additional 48 h. A. The mRNA expressions of WIF1, α-SMA and Col1A1 were analyzed by real-time PCR. B. The protein expressions of WIF1, α-SMA, type I collagen and P-β-catenin were analyzed by western blotting. GAPDH was used as internal control. C. The reduction of TCF activity induced by Sal B was restored by miR-17-5p mimics. D. The reduction of cell proliferation induced by Sal B was restored by miR-17-5p mimics. Each value is the mean ± SD of three experiments. *P < 0.05 compared with the control and #P < 0.05 compared with the Sal B group.
Discussion
Activation of HSCs to myofibroblast-like cells is the pivotal event in the initiation and progression of hepatic fibrosis [24]. HSC activation is characterized by enhanced cell proliferation, over-production of ECM, and de novo synthesis of α-SMA [25]. In this study, we found that Wnt/β-catenin pathway was attenuated by Sal B via restoration of WIF1 and inhibition of miR-17-5p, with a reduction in TCF activity and an increase in P-β-catenin level. Owing to the suppression of Wnt/β-catenin pathway, HSC activation was inhibited, leading to the reduction of HSC proliferation, ECM proteins and α-SMA expression. The silencing of WIF1 blocked down the anti-fibrotic effects of Sal B and WIF1 was a direct target of miR-17-5p. These data revealed that Sal B suppresses HSC activation, at least in part, through inhibiting miR-17-5p-activated Wnt/β-catenin pathway.
It has been reported that HSC activation can be suppressed by Sal B treatment. Wang et al. showed that Sal B prevents the progression of liver angiogenesis and alleviates liver fibrosis via NF-κB signaling [26]. Li et al. reported that Sal B may exert an anti-hepatic fibrosis effect via down-regulating angiotensin II signaling in HSC activation [20]. Another study demonstrated that Sal B prevents HSC activation through TGF-β signaling pathway, i.e. inhibiting TGF-β1 expression, activity of TβR-I kinase and Smads phosphorylation [27]. In our study, it was found that Sal B could inhibit HSC activation, at least in part, via suppressing Wnt/β-catenin pathway. To our knowledge, this is the first report to show that miR-17-5p-activated Wnt/β-catenin pathway is involved in the effects of Sal B.
MiR-17-5p was described as an oncogenic miRNA in cancers. For example, Li et al. found that miR-17-5p is able to enhance cell proliferation by promoting G1/S transition of the cell cycle and suppressing apoptosis in ovarian cancer cell lines [28]. Moreover, emerging studies show that miR-17-5p over-expression promotes HCC development and the inhibition of miR-17-5p results in the suppression of HCC proliferation and migration [29, 30]. Notably, elevated serum miR-17-5p has also been reported to correlate with the poor prognosis in HCC patients [31]. In this study, miR-17-5p over-expression increased the levels of α-SMA and desmin in primary HSCs. Furthermore, miR-17-5p was involved in the regulation of cell cycle and inhibited the effects of Sal B on cell proliferation. Notably, the inhibition of miR-17-5p suppressed CCl4-induced liver fibrosis. All the data suggest that miR-17-5p over-expression contributes to the activation of HSCs, which is also consistent with our previous study [16]. We previously showed that miR-17-5p promotes HSC proliferation and activation, at least in part, via reduction of Smad7 [16]. Herein, we demonstrated that WIF1 is a new target of miR-17-5p. In addition, it was found that all the anti-fibrotic effects of Sal B could be blocked down by miR-17-5p mimics. Taken together, our data suggested that the activation of HSCs was suppressed by Sal B via miR-17-5p and WIF1 (Figure 7). However, the mechanism of the direct regulation of miR-17-5p by Sal B still remains unclear and further studies are warranted.
In conclusion, we demonstrate that Sal B down-regulates miR-17-5p expression, leading to the restoration of WIF1 and the inhibition of Wnt/β-catenin signaling, which contributes to the suppression of activated HSCs. Our results not only provide a new insight of the role of miRNA-activated Wnt/β-catenin signaling in liver fibrosis, but also show a new anti-fibrotic mechanism of Sal B.
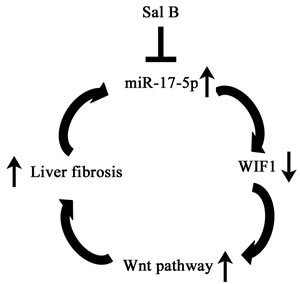
Figure 7: The signaling pathway was discovered in activated HSCs after Sal B treatment. Sal B induces miR-17-5p down-regulation, WIF1 up-regulation, the inactivation of Wnt pathway, resulting in the inhibition of the activated HSCs.
Materials and methods
Materials
CCl4 was obtained from Sigma (St Louis, MO, USA). Antibodies against type I collagen, WIF1 and P-β-catenin were obtained from Abcam (Cambridge, MA, USA). Antibodies against α-SMA and GAPDH were purchased from Santa Cruz Biotechnology (Santa Cruz, CA, USA).
Cell culture
Rat HSC-T6 cell line was obtained from Research of the Chinese Academy of Medical Sciences (Beijing, China). Cells were grown in DMEM containing 10% fetal bovine serum (FBS) (Gibco, Carlsbad, CA) and maintained in a 37°C incubator with 5% CO2. HSC-T6 cells and primary rat HSCs were both treated with 10 μmol/L Sal B for 48 h, which is a safe dose [27].
Isolation and culture of rat HSCs
Adult male Sprague-Dawley rats (body weight, 400-500 g) were used for HSC isolation as described previously [32]. The primary HSCs were studied at day 3 after isolation throughout all the studies. The purity of cultures was confirmed by immunocytochemical staining for α-SMA and the purity reached > 98%.
CCl4 liver injury model
Adult male Sprague-Dawley (180-220 g) rats were provided by the Experimental Animal Center of Wenzhou Medical University. Liver fibrosis was induced by administration of 2 ml of CCl4/olive oil (1:1, v/v)/kg body weight by intraperitoneal injection twice weekly for up to 6 weeks [33]. Thirty rats were randomly divided into three groups, including olive oil, CCl4 plus oral phosphate-buffered saline (PBS) and CCl4 plus oral Sal B (10 mg /kg), respectively. The experimental protocol was approved by the Institutional Animal Committee of Wenzhou Medical College. All animals received care in accordance with ‘Guide for the Care and Use of Laboratory Animals’. Rats were sacrificed under anesthesia at the end of six weeks and the livers were removed for further analysis. The liver tissues were used for H&E staining and Masson staining by fixation with 10 % formalin.
Lentivirus production and transfection
The lentiviral vector containing negative control (Lenti-NC) and lentiviral miR-17-5p inhibitor (Lenti-miR-17-5p-inhibitor) were obtained from Shanghai GeneChem. Rats were treated with olive oil (n = 6), CCl4 (n = 6), CCl4 plus Lenti-NC (n = 6) and CCl4 plus Lenti-miR-17-5p-inhibitor (n = 6). Lenti-miR-17-5p-inhibitor or Lenti-NC was injected via the tail vein only once at three weeks after CCl4 injection (1×109 transducing unit/rat). After the following 3-weeks CCl4 treatment, the rats were sacrificed.
RNA interference analysis
RNA interference experiments were performed before the treatment of Sal B using Lipofectamine 2000 (Invitrogen Carlsbad, CA, USA) in accordance with the manufacturer’s instructions. siRNA oligonucleotides against WIF1 or scrambled sequences (Table. S1) were synthesized by Gene Pharma (Shanghai, China) and transfected in HSC-T6 cell and primary HSCs for 48 h.
miRNA transfection
Cells were seeded in a 6-well plate at a density of 1×106 cells per well. Then, medium was replaced with Opti-MEM (Invitrogen, USA) and cells were transfected with miR-17-5p mimics (60 nM) and miR-NC (60 nM) (GenePharma, China) using Lipofectamine 2000 for 48 h. After 6 h of transfection, the medium was replaced with DMEM containing 10 % FBS.
Immunofluorescence microscopy
Cells were seeded on 18-mm cover glasses and fixed in an acetic acid: ethanol (1:3) solution for 5 min at -20°C. Nonspecific binding was blocked with 5% goat and horse serum/PBS for 1 h at room temperature. Then, cells were incubated with primary antibodies against α-SMA or desmin (Abcam), followed by fluorescein-labeled secondary antibody (1:50 dilution; Dianova) [34]. The nuclei were stained with 4,6-diamidino-2-phenylindole (DAPI). The slides were washed twice with PBS, covered with DABCO (Sigma-Aldrich), and examined with confocal laser scanning microscopy (Olympus, Tokyo, Japan) at 488 and 568 nm.
Immunohistochemistry
Immunohistochemical staining was performed on the sections (3 μm thick) from the liver tissues, as described previously [35, 36]. Briefly, after deparaffinization, hydration, and antigen retrieval, samples were incubated overnight at 4°C with a primary antibody against α-SMA (1:100) and then with a biotinylated secondary antibody. α-SMA expression was visualized by 3,3’-diaminobenzidine tetrahydrochloride (DAB) staining. Slides were counterstained with hematoxylin before dehydration and mounting α-SMA-positive areas within the fibrotic region were then observed. Quantitative analysis was calculated from five fields for each liver slice.
Quantitative real-time PCR
Total RNA was extracted from tissues and cells using miRNeasy Mini kit (Qiagen, Valencia, CA, USA) according to manufacturer’s instructions. Gene expression (Table. S1) was measured by real-time PCR using SYBR Green real-time PCR Master Mix (Toyobo, Osaka, Japan). Moreover, the primers of alpha-1 (I) collagen (Col1A1), α-SMA, GAPDH and U6 were designed as described previously [37]. Expression of mature miRNAs was detected using TaqMan MicroRNA Assay (Applied Biosystems, Foster City, CA). The GAPDH and U6 snRNA levels were used to normalize the relative abundance of mRNAs and miRNAs [38], respectively.
Western blot analysis
Tissues and cells were lysed with ice-cold lysis buffer (50 mM Tris-HCl, pH 7.4, 100 mM 2-Mercaptoethanol, 2% w/v SDS, 10% glycerol). Total proteins were quantified and separated by SDS-PAGE. Then western blot assay was performed as described previously [39]. The levels of protein were normalized to total GAPDH.
Cell proliferation assay
Cells were seeded in a 96-well plate at a density of 1×103 cells per well, then cells were transfected with miR-17-5p mimics and miR-NC as described above. Cell proliferation was determined by MTT assay according to the instructions of a MTT cell proliferation assay kit (Beyotime Biotechnology, Jiangsu, China). The optical density was measured at 570 nm on a microplate reader (Bio-Rad 550, USA).
TCF reporter activity assay
Cells were transiently transfected with TOPFLASH and FOPFLASH (Upstate Biotechnology Inc., Lake Placid, NY, USA) using Lipofectamine 2000. Twenty-four hours after transfection, the cells were harvested and luciferase and Renilla luminescence were measured using the Dual-Luciferase Reporter Assay System (Promega, Wisconsin, WI, USA) on a luminometer (BioTek Instruments, Winooski, VT, USA). TCF reporter activity was presented as the ratio of firefly-to-Renilla luciferase activity.
Cell cycle analysis
For cell cycle analysis, we performed Cell Cycle Analysis Kit (Beyotime, China). Cells were fixed in 70 % ethanol in PBS at -20°C for 24 h and then labeled with 0.5 ml propidium iodide (PI) staining buffer containing 200 mg/ml RNase A and 50 μg/ml PI at 37°C for 40 min in the dark. Analyses were performed on a BD LSR flow cytometer (BD Biosciences) and experiments repeated at least three times.
Luciferase activity assay
The 3’UTR region of WIF1 gene was cloned into the pMIR-REPORT™ Luciferase plasmid (Applied Biosystems) to generate pMIR-17-5p and pMIR-17-5p-Mut vectors. WIF1 3’UTR for miR-17-5p forward, 5’-TCGAGTTACGCCGAGTTCAC-3’ and reverse, 5’-GTTTCGCCTCTCTAGGGCTC-3’. Transfection was performed with Lipofectamine 2000 according to the manufacturer’s recommendations [40]. pMIR-REPORT β-gal control plasmid was used for transfection normalization. Luciferase values were measured using Dual-Light System (Applied Biosystems).
Statistical analysis
Data from at least three independent experiments were expressed as the mean ± SD. Statistical analysis was performed using Student’s t -test and P < 0.05 was considered significant. All statistical analyses were performed with SPSS software (version 13; SPSS, Chicago, IL).
Acknowledgments
The project was supported by the National Natural Science Foundation of China (No.81000176/H0317, 81100292/H0317 and 81500458/H0317), Zhejiang Provincial Natural Science Foundation of China (No.Y2090326, Y2110634 and LY16H030012), Wenzhou Municipal Science and Technology Bureau (No.Y20110033 and Y20120127), the Wang Bao-En Liver Fibrosis Foundation (No.20100002 and 20120127), the incubator program of the First Affiliated Hospital of Wenzhou Medical University (HFY2014045) and the key disciplines in Colleges and Universities of Zhejiang Province.
Conflicts of interest
The authors declared no conflicts of interest.
References
1. Hernandez-Gea V and Friedman SL. Pathogenesis of liver fibrosis. Annu Rev Pathol. 2011; 6:425-456.
2. Wei J, Feng L, Li Z, Xu G and Fan X. MicroRNA-21 activates hepatic stellate cells via PTEN/Akt signaling. Biomed Pharmacother. 2013; 67:387-392.
3. Tu X, Zhang H, Zhang J, Zhao S, Zheng X, Zhang Z, Zhu J, Chen J, Dong L and Zang Y. MicroRNA-101 suppresses liver fibrosis by targeting the TGFbeta signalling pathway. J Pathol. 2014; 234:46-59.
4. He Y, Huang C, Sun X, Long XR, Lv XW and Li J. MicroRNA-146a modulates TGF-beta1-induced hepatic stellate cell proliferation by targeting SMAD4. Cell Signal. 2012; 24:1923-1930.
5. Cheng JH, She H, Han YP, Wang J, Xiong S, Asahina K and Tsukamoto H. Wnt antagonism inhibits hepatic stellate cell activation and liver fibrosis. Am J Physiol Gastrointest Liver Physiol. 2008; 294:G39-49.
6. Kim KK, Wei Y, Szekeres C, Kugler MC, Wolters PJ, Hill ML, Frank JA, Brumwell AN, Wheeler SE, Kreidberg JA and Chapman HA. Epithelial cell alpha3beta1 integrin links beta-catenin and Smad signaling to promote myofibroblast formation and pulmonary fibrosis. J Clin Invest. 2009; 119:213-224.
7. Kordes C, Sawitza I and Haussinger D. Canonical Wnt signaling maintains the quiescent stage of hepatic stellate cells. Biochem Biophys Res Commun. 2008; 367:116-123.
8. Jiang F, Parsons CJ and Stefanovic B. Gene expression profile of quiescent and activated rat hepatic stellate cells implicates Wnt signaling pathway in activation. J Hepatol. 2006; 45:401-409.
9. Bovolenta P, Esteve P, Ruiz JM, Cisneros E and Lopez-Rios J. Beyond Wnt inhibition: new functions of secreted Frizzled-related proteins in development and disease. J Cell Sci. 2008; 121:737-746.
10. Mao B, Wu W, Davidson G, Marhold J, Li M, Mechler BM, Delius H, Hoppe D, Stannek P, Walter C, Glinka A and Niehrs C. Kremen proteins are Dickkopf receptors that regulate Wnt/beta-catenin signalling. Nature. 2002; 417:664-667.
11. Croce CM and Calin GA. miRNAs, cancer, and stem cell division. Cell. 2005; 122:6-7.
12. Ghildiyal M and Zamore PD. Small silencing RNAs: an expanding universe. Nat Rev Genet. 2009; 10:94-108.
13. Yang X, Du WW, Li H, Liu F, Khorshidi A, Rutnam ZJ and Yang BB. Both mature miR-17-5p and passenger strand miR-17-3p target TIMP3 and induce prostate tumor growth and invasion. Nucleic Acids Res. 2013; 41:9688-9704.
14. Shan SW, Fang L, Shatseva T, Rutnam ZJ, Yang X, Du W, Lu WY, Xuan JW, Deng Z and Yang BB. Mature miR-17-5p and passenger miR-17-3p induce hepatocellular carcinoma by targeting PTEN, GalNT7 and vimentin in different signal pathways. J Cell Sci. 2013; 126:1517-1530.
15. Cloonan N, Brown MK, Steptoe AL, Wani S, Chan WL, Forrest AR, Kolle G, Gabrielli B and Grimmond SM. The miR-17-5p microRNA is a key regulator of the G1/S phase cell cycle transition. Genome Biol. 2008; 9:R127.
16. Yu F, Guo Y, Chen B, Dong P and Zheng J. MicroRNA-17-5p activates hepatic stellate cells through targeting of Smad7. Lab Invest. 2015; 95:781-789.
17. Liu C, Liu P, Liu CH, Zhu XQ and Ji G. Effects of Fuzhenghuayu decoction on collagen synthesis of cultured hepatic stellate cells, hepatocytes and fibroblasts in rats. World J Gastroenterol. 1998; 4:548-549.
18. Liu C, Jiang C, Liu P and Hu Y. [Intervention of Fuzhenghuayu decoction on autocrine activation of hepatic stellate cells]. Zhonghua Gan Zang Bing Za Zhi. 2001; 9:282-284.
19. Liu P, Hu YY, Liu C, Zhu DY, Xue HM, Xu ZQ, Xu LM, Liu CH, Gu HT and Zhang ZQ. Clinical observation of salvianolic acid B in treatment of liver fibrosis in chronic hepatitis B. World J Gastroenterol. 2002; 8:679-685.
20. Li S, Wang L, Yan X, Wang Q, Tao Y, Li J, Peng Y, Liu P and Liu C. Salvianolic Acid B Attenuates Rat Hepatic Fibrosis via Downregulating Angiotensin II Signaling. Evid Based Complement Alternat Med. 2012; 2012:160726.
21. Zhao JF, Liu CH, Hu YY, Xu LM, Liu P and Liu C. Effect of salvianolic acid B on Smad3 expression in hepatic stellate cells. Hepatobiliary Pancreat Dis Int. 2004; 3:102-105.
22. Guo Y, Xiao L, Sun L and Liu F. Wnt/beta-catenin signaling: a promising new target for fibrosis diseases. Physiol Res. 2012; 61:337-346.
23. Kim TH, Kim SH, Seo JY, Chung H, Kwak HJ, Lee SK, Yoon HJ, Shin DH, Park SS and Sohn JW. Blockade of the Wnt/beta-catenin pathway attenuates bleomycin-induced pulmonary fibrosis. Tohoku J Exp Med. 2011; 223:45-54.
24. He Y, Huang C, Zhang SP, Sun X, Long XR and Li J. The potential of microRNAs in liver fibrosis. Cell Signal. 2012; 24:2268-2272.
25. Lin J and Chen A. Activation of peroxisome proliferator-activated receptor-gamma by curcumin blocks the signaling pathways for PDGF and EGF in hepatic stellate cells. Lab Invest. 2008; 88:529-540.
26. Wang R, Yu XY, Guo ZY, Wang YJ, Wu Y and Yuan YF. Inhibitory effects of salvianolic acid B on CCl(4)-induced hepatic fibrosis through regulating NF-kappaB/IkappaBalpha signaling. J Ethnopharmacol. 2012; 144:592-598.
27. Tao YY, Wang QL, Shen L, Fu WW and Liu CH. Salvianolic acid B inhibits hepatic stellate cell activation through transforming growth factor beta-1 signal transduction pathway in vivo and in vitro. Exp Biol Med (Maywood). 2013; 238:1284-1296.
28. Li L, He L, Zhao JL, Xiao J, Liu M, Li X and Tang H. MiR-17-5p up-regulates YES1 to modulate the cell cycle progression and apoptosis in ovarian cancer cell lines. J Cell Biochem. 2015; 116:1050-1059.
29. Yang F, Yin Y, Wang F, Wang Y, Zhang L, Tang Y and Sun S. miR-17-5p Promotes migration of human hepatocellular carcinoma cells through the p38 mitogen-activated protein kinase-heat shock protein 27 pathway. Hepatology. 2010; 51:1614-1623.
30. Peng H, Ishida M, Li L, Saito A, Kamiya A, Hamilton JP, Fu R, Olaru AV, An F, Popescu I, Iacob R, Dima S, Alexandrescu ST, Grigorie R, Nastase A, Berindan-Neagoe I, et al. Pseudogene INTS6P1 regulates its cognate gene INTS6 through competitive binding of miR-17-5p in hepatocellular carcinoma. Oncotarget. 2015; 6:5666-5677. Doi: 10.18632/oncotarget.3290.
31. Zheng J, Dong P, Gao S, Wang N and Yu F. High expression of serum miR-17-5p associated with poor prognosis in patients with hepatocellular carcinoma. Hepatogastroenterology. 2013; 60:549-552.
32. Weiskirchen R and Gressner AM. Isolation and culture of hepatic stellate cells. Methods Mol Med. 2005; 117:99-113.
33. Yao QY, Xu BL, Wang JY, Liu HC, Zhang SC and Tu CT. Inhibition by curcumin of multiple sites of the transforming growth factor-beta1 signalling pathway ameliorates the progression of liver fibrosis induced by carbon tetrachloride in rats. BMC Complement Altern Med. 2012; 12:156.
34. Chan LK, Chiu YT, Sze KM and Ng IO. Tensin4 is up-regulated by EGF-induced ERK1/2 activity and promotes cell proliferation and migration in hepatocellular carcinoma. Oncotarget. 2015; 6: 20964-76. Doi: 10.18632/oncotarget.4122.
35. Zheng J, Wu C, Xu Z, Xia P, Dong P, Chen B and Yu F. Hepatic stellate cell is activated by microRNA-181b via PTEN/Akt pathway. Mol Cell Biochem. 2015; 398:1-9.
36. Kawahara T, Kashiwagi E, Ide H, Li Y, Zheng Y, Miyamoto Y, Netto GJ, Ishiguro H and Miyamoto H. Cyclosporine A and tacrolimus inhibit bladder cancer growth through down-regulation of NFATc1. Oncotarget. 2015; 6:1582-1593. Doi: 10.18632/oncotarget.2750.
37. Zheng J, Wu C, Lin Z, Guo Y, Shi L, Dong P, Lu Z, Gao S, Liao Y, Chen B and Yu F. Curcumin up-regulates phosphatase and tensin homologue deleted on chromosome 10 through microRNA-mediated control of DNA methylation - a novel mechanism suppressing liver fibrosis. FEBS J. 2014; 281:88-103.
38. Schmittgen TD and Livak KJ. Analyzing real-time PCR data by the comparative C(T) method. Nat Protoc. 2008; 3:1101-1108.
39. Hua HW, Jiang F, Huang Q, Liao Z and Ding G. MicroRNA-153 promotes Wnt/beta-catenin activation in hepatocellular carcinoma through suppression of WWOX. Oncotarget. 2015; 6:3840-3847. Doi: 10.18632/oncotarget.2927.
40. Kushwaha D, Ramakrishnan V, Ng K, Steed T, Nguyen T, Futalan D, Akers JC, Sarkaria J, Jiang T, Chowdhury D, Carter BS and Chen CC. A genome-wide miRNA screen revealed miR-603 as a MGMT-regulating miRNA in glioblastomas. Oncotarget. 2014; 5:4026-4039. Doi: 10.18632/oncotarget.1974.