
INTRODUCTION
Aneuploidy, the condition of having an imbalanced copy number of chromosomes (DNA content ≠ xn, where n stands for the haploid chromosome set and x ≥ 1), and chromosome instability (CIN), a type of genomic instability in which cells display an elevated rate of whole-chromosome mis-segregations (~1 per 5 cell divisions) and thus frequently change their karyotype [1], are widespread in human tumors [2–5]. Along with this, variations of chromosome number have been linked to cancer progression and aggressiveness [4, 5], as well as therapeutic resistance [6, 7] and poor patient prognosis [8, 9], although their precise impact in tumorigenesis is still debated (for recent reviews refer to [10]).
One prominent mechanism accounting for the generation of aneuploidy in cancer involves a preliminary and unscheduled passage to a tetraploid intermediate (DNA content = 4n) [11–13]. Tetraploid cells are generated by a variation of the canonical G1-S-G2-M cell cycle, such as skipped (i.e., endocycling/endoreplication) or aborted (e.g., endomitosis or cytokinesis failure) mitoses, or by cell membrane fusion [14–18]. According to the current hypotheses, illicitly generated tetraploids are less likely to stably maintain their karyotype across consecutive generations due to intrinsic defects in the machineries involved in DNA replication, DNA repair and/or chromosome segregation [19–21]. These defects might increase the level of CIN, in turn resulting in the generation of aneuploid cells [14, 19, 22]. The evidence supporting this two-step cascade in tumor development includes (1) indirect observations, such as the elevated incidence of tetraploid cells in early stages of tumors and in pre-neoplastic lesions, where tetraploidy appearance often precedes the acquisition of CIN and the subsequent development of aneuploidy [23–26]; (2) mathematical models, which indicate that the gradual loss of chromosomes from a tetraploid intermediate rather than chromosome(s) loss or gain during mitotic divisions of diploid cells accounts for the near-triploid/tetraploid content frequently found in solid tumors [2, 27]; (3) computational studies, inferring that around 37% of all neoplasms have transited through an intermediate tetraploid phase [26, 28–31]; and (4) the experimental demonstration that tetraploid (but not diploid) murine epithelial cells lacking the mouse homologue of tumor protein p53 (TP53, better known as p53) were able to generate chromosomically unstable tumors when injected in the flank of immunodeficient mice [18]. Of note, aneuploid cells generated from tetraploid cells often display elevated tumorigenicity [14, 16, 17, 19, 22].
Programmed changes in ploidy are believed to contribute to the development and homeostasis of a restricted panel of mammalian tissues or organs, including blood, liver, muscle, skin and placenta (reviewed in [12, 32]). In most but not all [33, 34] of these contexts, programmed polyploidy represents a terminally differentiated, non-cycling state [12, 32]. On the contrary, when occurring in proliferating cells, tetraploidy is normally sensed as a danger, leading to the activation of intrinsic, cell-autonomous processes such as cell cycle arrest [35–37], mitotic catastrophe [38], or regulated forms of cell death [39–41]. In addition, non-physiological tetraploidy can be detected and destroyed by the immunosurveillance system [42, 43]. The high incidence of aneuploid genomes in human neoplasms suggests that the barrier limiting the presence of tetraploid cells is bypassed during tumorigenesis. In line with this notion, the loss of tumor suppressor genes, including retinoblastoma 1 (RB1), p53 and adenomatous polyposis coli (APC), as well as the activation of oncogenes, including v-myc avian myelocytomatosis viral oncogene homolog (MYC), results in the generation of tetraploid cells (reviewed in [11]).
Tetraploidy could thus provide a handle by which to selectively eradicate the most aggressive cancer cells [44]. Most anti-tetraploid strategies designed so far are targeting intrinsic characteristics of tetraploid cells, such as their increased dependency upon accurate mitotic machinery, their elevated production of reactive oxygen species (ROS) and their extensive metabolic rewiring. Thus, tetraploids were proven to preferentially or selectively succumb to the inhibition of cell cycle or mitotic regulators, including checkpoint kinase 1 (CHEK1, best known as CHK1) [20], aurora kinase B (AURKB) [45], and kinesin family member 11 (KIF11, best known as EG5) [46], but also to antioxidants [47], and to perturbations of energy metabolism, as those provoked by overactivation of AMP-activated protein kinase (AMPK) [48], the glycolytic inhibitor 2-deoxyglucose [49], and mechanistic target of rapamycin (MTOR) inhibitors in combination with AURKB inhibitors [50, 51].
Increasing CIN by targeting mitotic regulators, including MPS1, a mitotic kinase involved in spindle assembly checkpoint (SAC) [52–55], has emerged as a valid approach to potently and preferentially kill cancer cells [56–63]. Here, we investigated the impact of MPS1 perturbation on the proliferation and survival of diploid versus tetraploid tumor cells, showing that the duplication of an entire set of chromosomes sensitizes cancer cells to MPS1 inhibition or depletion.
RESULTS
Effect of the abrogation of MPS1 function on tetraploid survival
To evaluate the differential impact of MPS1 inhibition on the survival of cancer cells differing in their ploidy, we took advantage of a panel of diploid and tetraploid clones derived from parental human colon carcinoma HCT 116 and RKO cells, which we previously isolated and characterized [41], or from human malignant fibrous histiocytoma MFH152 cells, which we generated in this study by flow cytometry-assisted cloning [41]. These clones were left untreated or were administered with low doses (from 0.05 to 0.30 μM) of reversine, a small molecule that specifically inhibits MPS1 at submicromolar concentrations [64]. At the end of the treatment period, cell death was evaluated by flow cytometry-mediated measurement of well-recognized apoptotic parameters [65, 66], including dissipation of mitochondrial inner transmembrane potential (Δψm), phosphatidylserine (PS) surface exposure and DNA fragmentation (Figure 1 and Supplementary Figure S1). Δψm loss was measured on live cells (excluding the vital dyes propidium iodure, PI, or 4′,6-diamidino-2-phenylindole, DAPI) with either of the two Δψm-sensitive dyes, dihexiloxalocarbocyanine iodide (DiOC6(3)) or tetramethylrhodamine methyl ester (TMRM). PS surface exposure was evaluated in live cells by staining with fluorophore-labeled Annexin V. DNA fragmentation was determined on fixed cells labeled with the DNA intercalating dye PI. As compared to their diploid counterparts, tetraploid HCT 116 (Figure 1A-1F and Supplementary Figure S1), RKO (Supplementary Figure S2A) and MFH152 (Supplementary Figure S2B) clones were particularly sensitive to reversine, as demonstrated by the elevated percentage of dying cells [displaying mitochondrial potential loss (PI−DiOC6(3)low or DAPI−/TMRMlow) or positivity for Annexin V (PI−Annexin V+)], dead cells [i.e., presenting plasma membrane permeabilization with a PI+ or DAPI+ phenotype], and cells with a hypodiploid (subG1) DNA content (Figure 1A–1F and Supplementary Figure S1). In line with these observations, submicromolar doses of reversine significantly reduced the clonogenic potential of tetraploid HCT 116 cells much more than that of diploid controls (surviving fraction of diploids vs tetraploids at 0.3 μM reversine: ~12% vs ~50%) (Figure 1G and 1H).
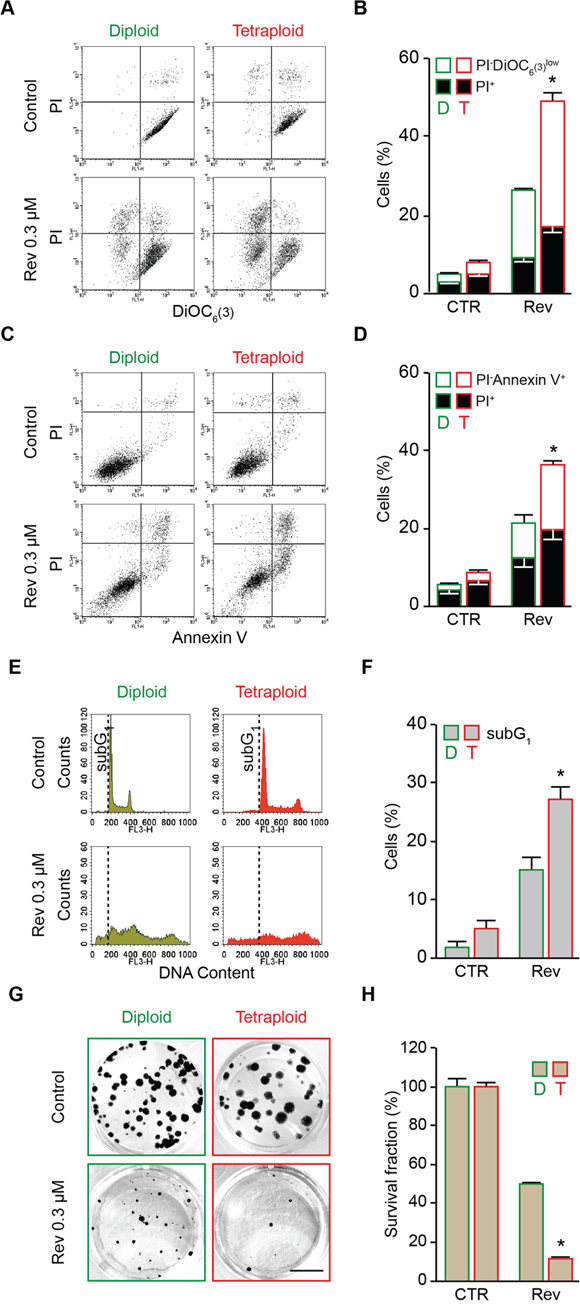
Figure 1: Preferential killing of tetraploid tumor cells by reversine-mediated MPS1 inhibition. A. and B. Diploid and tetraploid human colorectal carcinoma HCT 116 cells (framed in green and red, respectively) were left untreated or treated for 72 hours (h) with 0.3 μM reversine and then co-stained with the vital dye propidium iodure (PI) and the mitochondrial membrane potential (Δψm)-sensing dye DiOC6(3) for the evaluation of cell death–associated parameters by cytofluorometry. Representative plots are showed in panel (A), while quantitative data are represented in panel (B). In panel (B) white and black columns depict the percentage of dying (PI−DiOC6(3)low) and dead (PI+) cells, respectively. C. and D. Diploid and tetraploid HCT 116 cells (framed in green and red, respectively) administered or not with 0.3 μM reversine for 72 h were stained for the cytofluorometric detection of phosphatidylserine exposure with Alexa Fluor 488-conjugated Annexin V. Representative dot plots and quantitative data are reported in panels (C) and (D), respectively. In panel (D) white columns depict the percentage of dying cells (PI−Annexin V+) while black columns illustrate dead cells (PI+). E. and F. Diploid and tetraploid HCT 116 cells (framed in green and red, respectively) left untreated or exposed for 72 h with 0.3 μM reversine were fixed with ethanol and labelled with the DNA dye PI, for the quantification of the hypodiploid, subG1 apoptotic population. Representative plots (E) and quantitative data (F) are reported. G. and H. Diploid and tetraploid HCT 116 cells (framed in green and red, respectively) seeded at low cell density were left untreated or exposed to 0.3 μM reversine for 24 h. Upon drug washout, cells were cultivated for 15 days in fresh, drug-free medium before crystal violet staining and colony counting. Representative images of the plates (scale bar = 1 cm) (G) as well as quantitative data obtained upon normalization to plating efficiency (H) are shown. In panel (B), (D), (F) and (H) data are reported as means ± SEM (n ≥ 3). *p < 0.001 (Mann–Whitney test), as compared with diploid subjected to the same treatment condition. CTR, control; diploid, D; tetraploid, T; reversine, Rev.
A similar preferential anti-tetraploid effect was observed in the HCT 116, RKO and MFH152 diploid/tetraploid pair with an alternative pharmacological inhibitor of MPS1 named AZ 3146 [67] (Figure 2 and Supplementary Figure S2) or in the HCT 116 diploid/tetraploid pair by depleting MPS1 via the transfection of small interfering (si) RNAs specifically directed against this kinase (siMPS1) (Figure 3), thereby ruling out potential off-target effects of reversine.
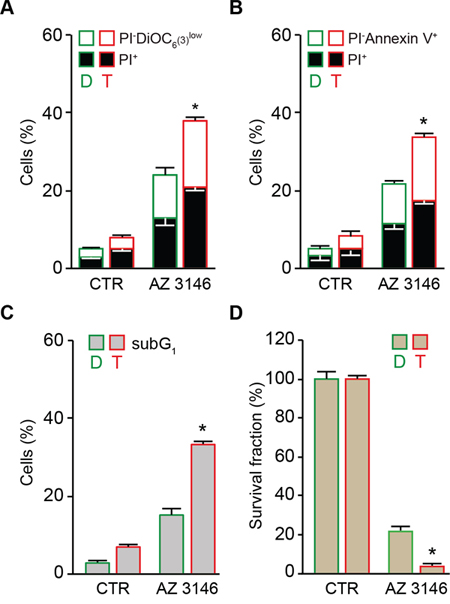
Figure 2: Preferential killing of tetraploid tumor cells by AZ 3146-mediated MPS1 inhibition. A. and B. Diploid and tetraploid human colon carcinoma HCT 116 cells (framed in green and red, respectively) were left untreated or treated with 5 μM AZ 3146 for 72 hours (h) and then either co-stained with the vital dye propidium iodure (PI) and the mitochondrial membrane potential (Δψm)-sensing dye DiOC6(3) (A) or stained with the phosphatidylserine binding protein Annexin V conjugated to Alexa Fluor 488 for the evaluation of cell death–associated parameters by cytofluorometry. In panel (A) white and black columns depict the percentage of dying (PI− DiOC6(3)low) and dead (PI+) cells, respectively. In panel (B) white and black columns illustrate the percentage of dying (PI−Annexin V+) and dead (PI+) cells, respectively. C. Diploid and tetraploid HCT 116 cells (framed in green and red, respectively) left untreated or exposed for 72 h with 5 μM AZ 3146 were fixed with ethanol and labelled with the DNA dye PI, for the quantification of the hypodiploid, subG1 apoptotic population. Quantitative data are reported. D. Diploid and tetraploid HCT 116 cells (framed in green and red, respectively) seeded at low cell density, were left untreated or exposed to 5 μM AZ 3146 for 24 h. Upon drug washout, cells were cultivated for 15 days in fresh, drug-free medium before crystal violet staining and colony counting. Quantitative data obtained upon normalization to plating efficiency are shown. In all the panels data are reported as means ± SEM (n ≥ 3). *p < 0.001 (Mann–Whitney test), as compared with diploid cells subjected to the same treatment condition. Diploid, D; tetraploid, T.
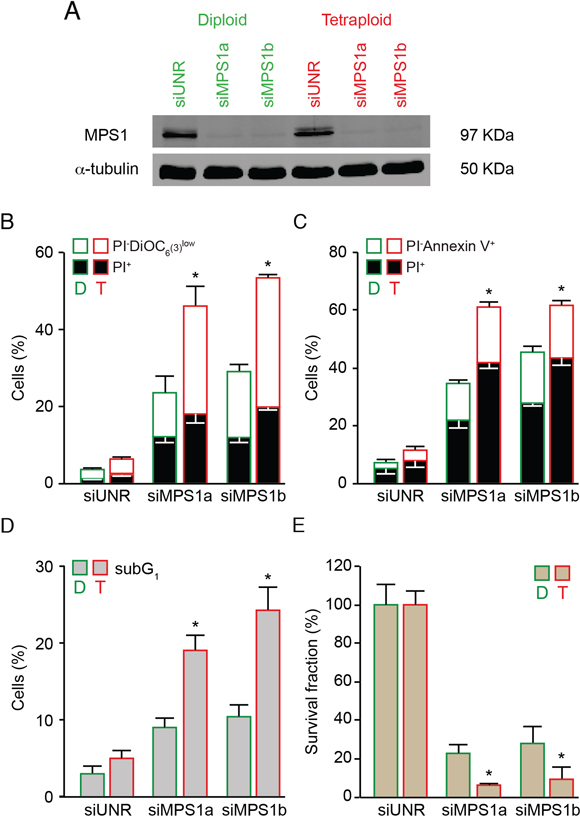
Figure 3: Increased sensitivity of tetraploid tumor cells to MPS1 depletion. A–C. Diploid and tetraploid human colorectal carcinoma HCT 116 cells (framed in green and red, respectively) were transfected with an unrelated small interfering (si) RNA (siUNR) or two specific siRNAs directed against MPS1 (siMPS1a and siMPS1b). Upon 72 hours (h), cells were collected and lysed, then cell lysates were analyzed by western-blot using antibodies directed against MPS1 and α-tubulin (A). Alternatively, cells were subjected to the determination of the cell death–associated parameters by flow cytometry upon co-staining with the propidium iodure (PI) and DiOC6(3) dyes (B) or staining with Alexa Fluor 488-conjugated Annexin V (C). In panel (B) white and black columns illustrate the percentage of dying (PI− DiOC6(3)low) and dead (PI+) cells, respectively. In panel (C) white and black columns illustrate the percentage of dying (PI−Annexin V+) and dead (PI+) cells, respectively. D. Diploid and tetraploid HCT 116 cells (framed in green and red, respectively) transfected for 72 h with siUNR, siMPS1a or siMPS1b were fixed with ethanol and labelled with PI for the quantification of the hypodiploid, subG1 apoptotic population. Quantitative data are reported. E. Clonogenic assay on diploid and tetraploid HCT 116 cells (framed in green and red, respectively) upon transfection with siUNR, siMPS1a and siMPS1b for 48 h followed by washout and cultivation in drug-free medium for further 15 days. Quantitative data obtained upon normalization to plating efficiency are shown. In panel (B), (C), (D) and (E) data are reported as means ± SEM (n ≥ 3). *p < 0.001 (Mann–Whitney test), as compared with diploid clones subjected to the same treatment condition. Diploid, D; tetraploid, T.
Altogether these findings demonstrate that abolishing MPS1 functions is an efficient strategy to preferentially and efficiently kill tetraploid cancer cells.
Targeting MPS1 perturbs tetraploid cell cycle divisions
When analyzing cell cycle profiles by flow-cytometry upon staining of fixed cells with the DNA dye PI, we observed that the inhibition or depletion of MPS1 provoked a dramatic perturbation of diploid and tetraploid cell cycle progression, including a moderate accumulation of cells with a DNA content of 4n (in diploid clones) and 8n (in tetraploid clones) and a major increase in the fraction of polyploid cells (DNA content > 4n and > 8n for diploid and tetraploid clones, respectively) (Figures 1E, 4A and 4B; Supplementary Figure S1C and 1E). Of note, the extent of polyploidization induced by MPS1 depletion was higher in diploid than tetraploid clones (Figure 4B and Supplementary Figure S1B–S1E). We then evaluated the impact of MPS1 abrogation on the regulation and timing of tetraploid mitosis. As illustrated in Figure 4C and 4D, upon MPS1 depletion the percentage of prometaphases plus metaphases displaying the kinetochore localization of the SAC component BUB1 mitotic checkpoint serine/threonine kinase B (BUB1B, better known as BUBR1) dropped drastically close to zero, thus indicating the complete abrogation of SAC function. In line with this evidence, the pharmacological inhibition or RNA interference-mediated depletion of MPS1 prevented the mitotic block imposed by antimitotic agents as demonstrated by the reduction of the fraction of phosphorylated histone H3 (pH3) positive cells (Supplementary Figure S3). To corroborate these findings, we performed videomicroscopy analyses on tetraploid HCT 116 cells engineered to stably express a green fluorescent protein-tagged variant of histone 2B (GFP-H2B), which allows for the intravital visualization of chromatin and chromosomes. The depletion of MPS1 significantly reduced the time spent by tetraploid cells in mitosis (average of 45.8 ± 2.8 minutes when transfected with an unrelated siRNA vs 32.8 ± 5.4 minutes when siMPS1-transfected) (Figure 4E–4G, and Supplementary Movie S1–S4). These results are consistent with the reduction of mitosis duration upon MPS1 inhibition in human osteosarcoma U2OS cell lines [68] and demonstrate that MPS1 contributes to the correct timing and execution of mitosis regardless of the cell ploidy status.
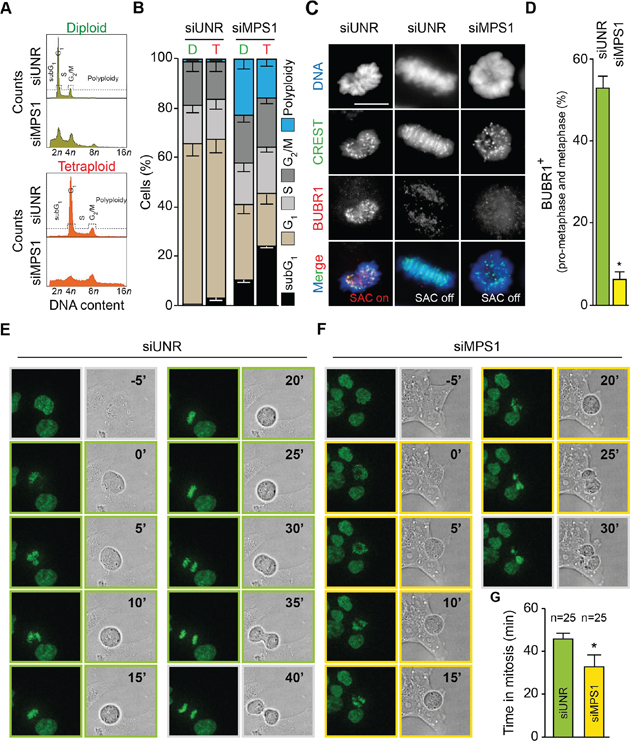
Figure 4: MPS1 abrogation shortens mitosis and abolishes SAC in tetraploid tumor cells. A. and B. Diploid and tetraploid human colorectal carcinoma HCT 116 cells (framed in green and red, respectively) were transfected with an unrelated small interfering (si) RNA (siUNR) or a specific siRNA directed against MPS1 (siMPS1) for 72 hours (h) and then fixed and stained with propidium iodure for the cytofluorometric assessment of cell cycle progression. Cell cycle distribution analyzed by flow cytometry for diploid and tetraploid cells is displayed in panel (A). Quantitative data (means ± SEM; n = 3) of the corresponding flow cytometry profiles are plotted in panel (B). Diploid, D; tetraploid, T. C. and D. Tetraploid HCT 116 cells were transfected with siUNR or siMPS1 for 24 h, treated with 20 μM MG132 for 4 h (a relatively short time to avoid increasing the risk of putative side effects unrelated to mitosis) and then processed for the immunofluorescence-assisted detection of spindle assembly checkpoint (SAC) activation as indicated by the localization of BUBR1 at CREST-labelled kinetochores. In panel (C) representative microphotographs of a normal prometaphase (SAC on), a normal metaphase (SAC off), and an abnormal metaphase (SAC off) in cells transfected with the indicated siRNA are reported. Scale bar = 10 μm. In panel (D) representing quantitative data, columns illustrate the percentage of prometaphases plus metaphases exhibiting SAC activation. Data are reported as means ± SEM (n = 3). *p < 0.01 (two-tailed t test), as compared with siUNR transfected tetraploid cells. E–G. Tetraploid HCT 116 cells expressing a green fluorescent protein-tagged variant of histone 2B (H2B-GFP) chimera were transfected with siUNR or siMPS1 and then monitored by live videomicroscopy for approximately 24 h to determine the time spent in mitosis. Image were taken every 5 minutes. Representative snapshots of siUNR- and siMPS1-transfected tetraploid mitosis are shown in panel (E) and (F), respectively, while quantitative data (means ± SEM) are reported in panel (G) (n = 25). p < 0.01 (two-tailed t test), as compared with siUNR transfected tetraploid cells. Full-length movies are provided as Supplementary Movies 1–4.
To further characterize the mechanism of tetraploid tumor cell killing by MPS1 abrogation, we depleted MPS1 in GFP-H2B diploid and tetraploid clones and followed them by videomicroscopy for 72 hours (h). This analysis confirmed that the knockdown of MPS1 impairs both diploid and tetraploid mitoses (Figure 5A and 5B; Supplementary Movie S5 and S6). In particular, the depletion of MPS1 resulted in aborted cell divisions (1st event depicted in yellow in the single cell fate profiles) generating a single daughter cell with a duplicated genome (Figure 5B). Alternatively, MPS1-depleted cells underwent a bipolar cell division (1st event depicted in green in the single cell fate profiles) generating 2 daughter cells, which either entered an aborted mitosis (2nd event depicted in yellow in the single cell fate profiles) or died during the following interphase (2nd event depicted in dark in the single cell fate profiles) (Figure 5B). By comparing their transgenerational cell fate profiling, a representation in which the destiny of a cellular population is monitored across consecutive generations and events (E1, E2 and E3) are symbolized by a centripetal sequence of concentric ring segments [69], we confirmed that the incidence of abortive mitoses (i.e., the induction of polyploid cells) upon MPS1 depletion was lower in tetraploid than in diploid clones (Figure 5C, see also Figure 4B). Moreover, in the majority of the cases (75%), polyploids generated from diploid clones (depicted in yellow in the inner circle; siMPS1 condition) remained inert or underwent one or even two additional round(s) of aberrant/abortive mitosis (depicted in orange and red in the central and external circle, respectively; siMPS1 condition) with only 25% undergoing cell death (depicted in dark in the central circle; siMPS1 condition) (Figure 5C). On the contrary, a large fraction of polyploids generated from tetraploid clones (depicted in yellow in the inner circle; siMPS1 condition) died during the interphase that followed the first aborted mitosis (depicted in dark in the central circle; siMPS1 condition) (Figure 5C). These findings confirm the antitetraploid effect of MPS1 inhibition underscoring the relative incapability of tetraploid (as compared to diploid) cells to tolerate any further increase in ploidy in the absence of SAC.
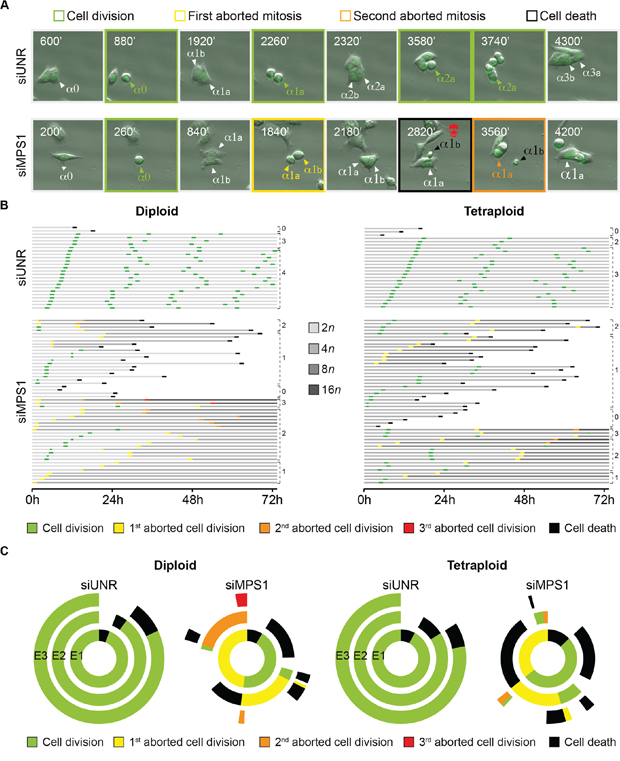
Figure 5: Cell fate profiling of diploid and tetraploid tumor cells depleted of MPS1. A–C. Diploid and tetraploid human colorectal carcinoma HCT 116 cells stably expressing a green fluorescent protein-tagged variant of histone 2B (H2B-GFP) chimera were transfected with an unrelated small interfering (si) RNA (siUNR) or a specific siRNA directed against MPS1 (siMPS1) and then monitored by live videomicroscopy for 72 hours (h). Representative snapshots of tetraploid cells transfected as indicated are shown in panel (A), whereas single cell fate profiles of siUNR- or siMPS1-transfected diploid (n = 25) and tetraploid (n = 50), and transgenerational cell fate profiles of siUNR or siMPS1 transfected cells (n >100) are depicted in panel (B) and (C), respectively. In panel (A) alphas (α) indicate individual cells, whose sequential cell divisions are numbered with “0”, “1”,“2” and “3”. The two daughter cells of a bipolar division are depicted with “a” and “b”; while the single cell derived by an abortive division is indicated by an increase in the size type. The snapshots showing successful cell divisions are framed in green, while those showing the first and second abortive cell divisions are framed in yellow and orange, respectively. The snapshot framed in black highlights the death of one cell following an abortive mitosis. In panel (B) horizontal columns represent single cells over the time, as indicated in hours. The color code depicting successful (green) or aborted (yellow, orange and red) cell divisions, as well as cell death (black) is used as in panel (A). The increase in cell ploidy following abortive cell division is represented by grey darkening, while numbers indicate mitotic events (i.e., successful or abortive cell division). Please note that cell divisions were considered to be successful only when daughter cells were clearly separated. In panel (C) concentric circles depict three consecutive generational events (E), starting from E1 (inner circle), using the same color code as used in panel (A) and (B). Full-length movies are provided as Supplementary Movies 5 and 6.
Altogether these results indicate that targeting MPS1 preferentially kills tetraploid tumor cells by abolishing SAC function, eventually triggering an uncontrolled and lethal polyploidization program.
Molecular mechanisms underlying the antitetraploid effect of MPS1 abrogation
We thus investigated the mechanisms involved in the execution of mitotic catastrophe induced by MPS1 inhibition or depletion in tetraploid cells. As shown by videomicroscopic analyses, the death of tetraploid tumor cells succumbing upon MSP1 inhibition was always preceded by chromatin condensation (pyknosis) and nuclear fragmentation (karyorrhexis), two morphological hallmarks of apoptosis [66] (Figures 5A and 6A, and Supplementary Movie S6). Moreover, tetraploid cell death triggered by MPS1 inhibition or depletion displayed the classical features of the intrinsic pathway of apoptosis, including the release of the pro-apoptotic factor cytochrome c from mitochondria to the cytosol and the activation of caspase-3 (Figure 6A and 6B). Of note, the percentage of both cytochrome c release and caspase-3 activation was higher in tetraploid clones than in their diploid counterparts, a result that is in line with the preferential sensitivity of tetraploid cells to MPS1 inhibition/depletion. Further confirming the role of caspases in the execution of apoptosis, tetraploid cells (but less so their diploid counterparts) responding to reversine or siMPS1 manifested the apoptosis-associated cleavage of poly (ADP-ribose) polymerase (PARP) (Figure 6C). Moreover, the pre-administration of the broad-spectrum caspase inhibitor Z-Val-Ala-Asp-fluoromethylketone (Z-VAD-fmk) significantly reduced the death of tetraploid cells responding to the inhibition or depletion of MPS1 (Figure 6D). Recently, we and others reported some cooperations between polo-like kinase 1 (PLK1) and MPS1 in SAC initiation [70, 71]. In line with this evidence, not only the depletion of MPS1 but also that of PLK1 had a preferential cytotoxic effect on tetraploid as compared to diploid cancer cells (Supplementary Figure S4). Of note, the co-depletion of MPS1 and PLK1 did not further increase the level of cell death, while the knock-down of MPS1 sensitized tumor cells to sublethal doses of paclitaxel independently of the basal ploidy level (Supplementary Figure S4). Altogether, these results indicate that, in tetraploid cells, MPS1 inhibition triggers mitotic catastrophe that is executed by the intrinsic pathway of apoptosis.
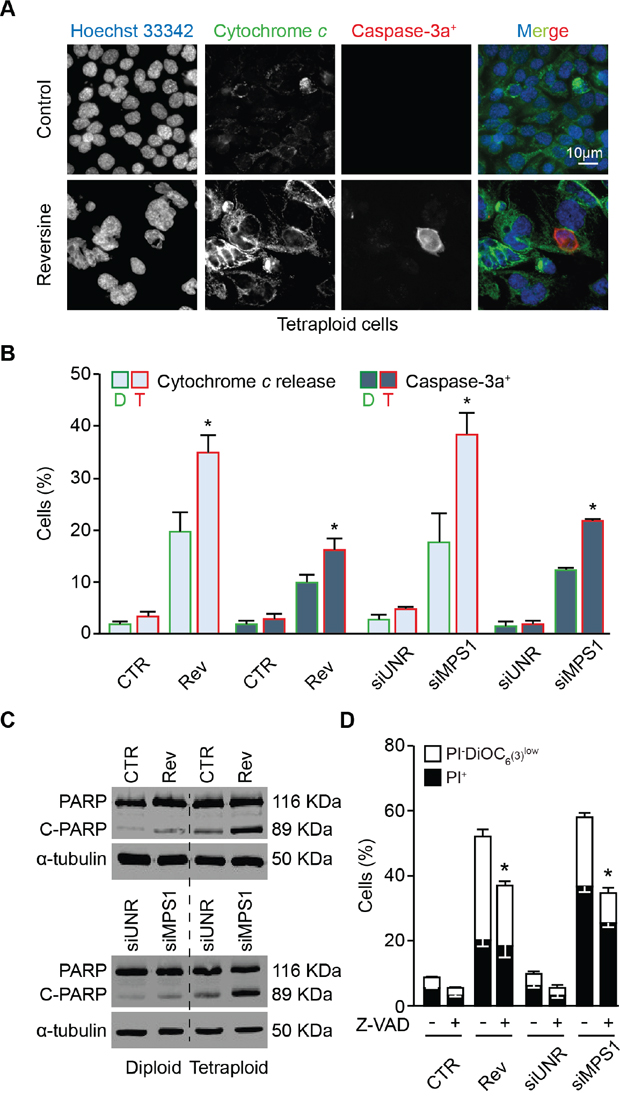
Figure 6: Mechanisms of cell death induced by MPS1 abrogation. A. and B. Diploid and tetraploid human colorectal carcinoma HCT 116 cells (framed in green and red, respectively) treated or not with 0.3 μM reversine (A and B) or transfected with an unrelated small interfering (si)RNA (siUNR) or a specific siRNA directed against MPS1 (siMPS1) for 72 hours (h) (B) were stained to visualize cytochrome c (green fluorescence), activated caspase-3 (caspase-3a+, red fluorescence) and nuclei (Hoechst 33342, blue fluorescence) and analyzed by fluorescence microscopy. The percentage of cells exhibiting diffuse (as opposed to punctuate) cytochrome c staining or caspase-3 activation was determined as described in Materials and Methods. Representative fluorescence microphotographs of tetraploid cells (A) and quantitative results (means ± SEM, n = 3) for both diploid and tetraploid clones (B) are shown. *p < 0.001 (Mann–Whitney test), as compared with diploid cells subjected to the same treatment or transfection condition. C. Western-blot analysis of protein extracts from diploid and tetraploid HCT 116 cells treated or not with 0.3 μM reversine or transfected with siUNR or siMPS1 for 72 h using an antibody directed against poly (ADP-ribose) polymerase (PARP) and recognizing the cleaved (C-PARP, 89 KDa) and uncleaved (PARP, 116 KDa) forms. Alpha-tubulin was used to verify equal loading. D. Tetraploid HCT 116 cells were treated or not for 72 h with 0.3 μM reversine or transfected with the indicated siRNAs alone or in combination with 25 μM Z-Val-Ala-Asp-fluoromethylketone (Z-VAD-fmk, abbreviated as Z-VAD), followed by co-staining with DiOC6(3)/propidium iodure (PI) and cytofluorometric analysis. White and black columns illustrate the percentage of dying (PI−DiOC6(3)low) and dead (PI+) cells, respectively. Data are reported as means ± SEM (n = 3). *p < 0.001 (Mann–Whitney test), as compared with cells subjected to the same treatment or transfection condition but not exposed to Z-VAD. CTR, control; diploid, D; tetraploid, T; reversine, Rev.
DISCUSSION
In this study we developed a novel strategy for targeting tetraploid tumor cells based on the abrogation of the mitotic kinase MPS1. By employing different pairs of tumor clones generated from the same parental cell lines and displaying distinct levels of ploidy (i.e., diploid vs. tetraploid) we provided strong evidence that the depletion or inhibition of MPS1 potently kills tetraploid cancer cells (and to a lesser extent diploid cancer cells) via a mechanism involving the induction of mitotic catastrophe following aberrant or aborted cell divisions, and the activation of a mitochondrion- and caspase-dependent pathway of regulated cell death.
It is becoming increasingly clear that measures inducing an exaggerated level of CIN or an unscheduled hyper-polyploidization program may constitute valid antineoplastic strategies, potentially sparing non-tumor cells and/or selectively targeting cancers according to their genetic background [58–61, 63, 72–74]. The approaches developed so far are based on the rationale that tumor cells (1) frequently display defects in cell cycle checkpoints and thus could progress in their cell cycle even in the presence of gross alterations as those provoked by mitosis-perturbing and/or CIN-inducing agents, and (2) may survive only until a certain threshold level of CIN and/or ploidy. Here we demonstrated that MPS1 inhibition kills both diploid and tetraploid cancer cells, though with a preferential action on tumor cells that have undergone whole-genome duplication. The tumorigenic potential of tetraploid cells arises from their intrinsic high level of genomic instability [12, 22], and/or their capability to tolerate and survive the burden of continuous karyotype changes [26]. In this context, we surmise that the peculiar sensitivity of tetraploid tumors to MPS1 inhibition is not necessarily linked to their basal level of CIN but rather arises from (1) the elevated dependency of tetraploids on the activity of the SAC, which is functional and overactivated in these cells and whose abrogation affects their survival more potently than the survival of their diploid counterpart [20], and (2) the incapability of tetraploids to tolerate a further increase in ploidy in absence of SAC functions, a condition that de facto exaggerates CIN to a level incompatible with life. These findings are in line with the lack of correlation between the pattern of genomic instability (i.e., microsatellite instability vs. CIN) and the sensitivity to MPS1 inhibitors, and with the involvement of an obligatory step of tetraploidization in cancer killing induced by MPS1 inhibitors [59]. Emerging evidence ascribes non-mitotic functions to SAC components [75, 76]. Given our results, it is tempting to speculate that SAC activity may play a role in the increased tolerance to chromosome aberrations of tetraploids, which has been recently associated to tumor evolution [26]. Videomicroscopic analyses demonstrated that, in the absence of SAC, newly-generated octaploid cells died during the interphase that follows the aborted mitosis of tetraploid cells. Further studies are required to elucidate whether higher-order ploidy (i.e., more-than-tetraploidy) intrinsically triggers a specific apoptotic program and, if so, whether the absence of SAC function may be the signal responsible for the activation of this cascade.
MPS1 is considered among the most promising targets for cancer therapy for multiple reasons, including: (1) the low frequency of mutations of MPS1 detected in tumors (reviewed by [52]); (2) the upregulation of MPS1 found in a variety of human cancers [55, 57, 77–83] often correlated to CIN/aneuploidy [4, 9, 84–86] or high tumor grade and aggressiveness [58, 87, 88]; (3) the pleiotropic roles of MPS1 in distinct phases of the cell cycle [52, 62, 89–100]; (4) the potential selectivity of MPS1 inhibitors toward malignant versus normal or immortalized cells [56–58, 60–62]; and (5) the synergy between MPS1 inhibitors and conventional anticancer agents, including antimitotics [59, 61, 87] and radiotherapy [89].
Here, we provide an extra point in support of the therapeutic utility of MPS1 inhibitors, showing that MPS1 depletion or inhibition kills tetraploid cancer cells more efficiently than diploid cancer cells. This may be relevant in the context of cancer therapy given that tetraploid cells are believed to promote tumorigenesis and also display high resistance to conventional anticancer regimens such as DNA damaging agents.
Some clinical trials have been launched to assess the safety and therapeutic profile of specific MPS1 inhibitors in cancer patients (source: https://clinicaltrials.gov/). It appears thus of interest to confirm the potential antitetraploid effect of these compounds either in mouse models of spontaneous tetraploidization-driven tumorigenesis [48] or in clinical samples, by evaluating the ploidy status of tumors responding to MPS1 inhibition.
MATERIALS AND METHODS
Cell lines and culture conditions
Diploid and tetraploid clones derived from human colon carcinoma HCT 116 cells were routinely maintained in McCoy's 5A medium supplemented with 10% fetal calf serum (FCS), 10 mM HEPES buffer, 100 units/mL penicillin G sodium and 100 μg/mL streptomycin sulfate (all provided from Thermo Fisher Scientific-Gibco, Waltham, MA). Human malignant fibrous histiocytomas MFH152 cells as well as diploid and tetraploid clones derived from these cell lines were cultured in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% FCS and 100 units/mL penicillin G sodium (Thermo Fisher Scientific-Gibco). Diploid and tetraploid HCT 116 and RKO clones transfected with a cDNA coding for a histone 2B-green fluorescent protein (H2B-GFP) (from BD Biosciences-PharMingen, San Jose, CA) were grown in McCoy's 5A medium supplemented as above plus 20 μg/mL blasticidine (Thermo Fisher Scientific-Gibco). Cells were seeded onto the appropriate supports (6-, 12-, 24- or 96-well plates) 24 h before the beginning of experiments.
Chemicals
MG132, paclitaxel, nocodazole, and reversine were purchased from Sigma-Aldrich (St. Louis, MO) and stocked as a 10 mM solution in dimethyl sulfoxide (DMSO). AZ 3146 was obtained from Tocris (Bristol, United Kingdom) and stocked as a 10 mM solution in DMSO. The pan-caspase inhibitor z-VAD-fmk was obtained from Bachem Bioscience (Bubendorf, Switzerland) and stocked as a 50 mM solution in N,N-dimethylformamide (DMF). The appropriate amount of DMSO and/or DMF was always employed for negative control conditions.
RNA interference
Diploid and tetraploid HCT 116 cells were seeded at low density in 6-, 12- or 96-well plates and after 24 h transfected with an unrelated siRNA (siUNR), two specific siRNAs directed against MPS1 mRNAs (siMPS1a and siMPS1b) (all purchased from Eurogentec, Liege, Belgium) or a specific siRNA directed against PLK1 mRNAs (siPLK1, ON-TARGETplus SMARTpool, L-003290–00-0005; GE Dharmacon, Lafayette, CO) by means of oligofectamine RNAiMAX transfection reagent (Thermo Fisher Scientific-Invitrogen), according to the manufacturer's instructions. The following siRNAs were used: 5′-GCCGGUAUGCCGGUUAAGUdTdT-3′(siUNR), 5′-UGGUUGAGUUUGUUGCUCAUUdTdT-3′(siMPS1a), and 5′-CCCAGAGGACUGGUUGAGUd TdT-3′(siMPS1b).
Cytofluorometric studies
For the quantification of apoptotic features, the assessment of cell cycle distribution, and the simultaneous measurement of DNA content and phosphorylated histone H3 (pH3) levels were performed as reported [48, 73]. The following antibodies were used: primary anti-phosphorylated histone H3 antibody (rabbit polyclonal IgG1 #06–570; Millipore-Chemicon International, Temecula, CA) and Alexa Fluor 488 goat anti-rabbit secondary antibody (Thermo Fisher Scientific). Phosphatidylserine exposure was quantified using the Alexa Fluor 488 Annexin V/Dead Cell Apoptosis Kit (Thermo Fisher Scientific-Invitrogen). Cytofluorometric acquisitions were performed by means of a FACSCalibur (BD Biosciences) or a FACSCanto (BD Biosciences) cytofluorometer, while data analysis was conducted using the CellQuestTM software (BD Biosciences). Only the events characterized by normal forward scatter (FSC) and side scatter (SSC) parameters were gated for inclusion in the statistical analysis.
Clonogenic survival assay
The clonogenic assay was performed and analyzed as reported above [73]. Briefly, cells were seeded at low concentrations and were left untreated, treated with MPS1 inhibitors or transfected with siUNR or siMPS1 for further 24 h followed by washout and culture in standard conditions for up to 15 days. Colonies were then fixed/stained with aqueous crystal violet and counted.
Immunoblotting
For the detection of protein levels, cells were harvested, washed with PBS and lysed for 30 min on ice in a buffer prepared in 50 mM Tris (pH 7.4) and containing 250 mM NaCl, 0.1% NP-40, 0.1 mM phenylmethylsulfonyl fluoride (PMSF), aprotinine at 10 mg/mL, leupeptine at 10 mg/mL, and 100 mM NaF. Cell lysates were then centrifuged for 10 min at 13 000 rpm and the concentration of soluble proteins in supernatant was measured by the Bradford method. Equal amount of proteins (30 μg) were resolved by SDS/PAGE and electro-transferred onto nitrocellulose membrane, which was then incubated overnight with the appropriate primary antibody. Thereafter, membranes were incubated for 1 h at room temperature with the appropriate DyLight 800 conjugate secondary antibody (Thermo Fisher Scientific-Pierce antibodies) and revealed with the LI-COR Odyssey® scanner and software (LI-COR Biosciences, Lincoln, NE). The following antibodies were used: α-tubulin (mouse monoclonal IgG1, #T9026; Sigma-Aldrich), MPS1 (mouse monoclonal IgG1 #ab11108; Abcam, Cambridge, UK) and PARP (mouse monoclonal IgG1 #9542; Cell Signaling Technology, Billerica, MA).
Immunofluorescence microscopy
For detection of apoptotic markers and SAC activation, cells were fixed with 4% PFA (in PBS), permeabilized with 0.1% Triton X-100 (in PBS), and immunostained with antibodies directed against cytochrome c (1/100 dilution; #6H2.B4, BD Biosciences), cleaved caspase-3 (Asp175) (1/200 dilution; #9661, Merck Millipore-Cell Signaling Technology), BUBR1 (1/200 dilution; mouse monoclonal IgG1 #612502, BD Biosciences) or human anti-nuclear-centromere CREST (Europa Bioproducts, Cambridge, UK). Revelation was performed with the appropriate Alexa Fluor conjugated secondary antibodies (Thermo Fisher Scientific-Invitrogen). Hoechst 33342 (1 μg/mL; BD Biosciences) was used for nuclear counterstaining. Images were captured with a Zeiss AxioimagerZ1 motorized microscope (Zeiss, Oberkochen, Germany) driven by Axiovision software (Zeiss) and analyzed with the open source software Image J (freely available from the National Institute of Health, http://rsb.info.nih.gov/ij/).
Videomicroscopy
For videomicroscopy, diploid and tetraploid H2B-GFP HCT 116 cells were grown in appropriate plates. The recording of the images started at the beginning of the treatment (t = 0) and images were taken every 5 min (Supplementary Movies S1–S4) for 24 h using a confocal spinning disk CSU-X1 Andor Nikonor (Andor Technology, Belfast, UK) coupled with Ti Eclipse microscope (Nikon, Tokyo, Japan) and driven by iQ3 software (Andor) or every 20 min (Supplementary Movies S5 and S6) for 72 h with a Leica DMIRE2 automated live cell microscope with a LMC 20 × 0.4 lens and appropriate filters (Leica, Wetzlar, Germany). Both transmitted light and fluorescence imaging were used to detect the cells. Images were analyzed with the open-source software Image J.
Statistical procedures
Unless otherwise specified, all experiments were performed and independently repeated at least three times. In order to evaluate if the differences observed between diploid and tetraploid cell lines subjected to a given tratment were significantly larger than those observed in control (i.e., untreated) conditions, results from diploid and tetraploid clones were randomly paired to compute differences. Thereafter, these differences were compared between treatment and control conditions using one-tail Wilcoxon-Mann-Whitney test. One hundred random pairings were performed and the 100 obtained p-values were corrected for multiple testing using Benjamini-Hochberg method. Then, the number of corrected p-values lower than 5% (N) was computed. The higher this number the more probable the difference between treatment and control. The significance of N was then assessed using a resampling method allowing to estimate its distribution under the null hypothesis (that is “differences between diploid and tetraploid were equivalent between treatment and control”) and to compare the N value to this distribution. Hence, the computation of an empirical p-value allowed to identify situations where differences observed between diploid and tetraploid cell lines are significantly larger when applying a given treatment than in conditions. A significance threshold of 5% has been chosen but all significant tests exhibited p-values lower than 0.1%. Data were analyzed using “R” software (R Foundation for Statistical Computing, Vienna, Austria; HYPERLINK “https://www.r-project.org/” http://www.R-project.org/). Two-tailed t-tests were performed with GraphPad Prism (GraphPad Software, Inc. La Jolla, CA) when conditions (treated/untreated) could be compared within a single cell line (Figure 3). Calculations exhibiting p-value < 0.01 were considered as statistically significant and indicated by an asterisk (*). Means ± SEM are represented.
ACKNOWLEDGMENTS AND FUNDING
The experiments reported in this paper were performed within the France-BioImaging national research infrastructure, at MRI facility, Montpellier. France-BioImaging is supported by the French National Research Agency through the “Investments for the Future” program (ANR-10-INSB-04). MJ is supported by the Ligue Nationale contre le Cancer. GM is funded by Associazione Italiana per la Ricerca sul Cancro (AIRC: Triennal Fellowship “Antonietta Latronico”, 2014). GL is supported by the Fondation pour la Recherche Médicale (FRM “engineer funding” granted for the team of AA). IV is funded by the Associazione Italiana per la Ricerca sul Cancro. IV is supported by the Associazione Italiana per la Ricerca sul Cancro (AIRC: MFAG 2013 #14641), Ministero Italiano della Salute (RF_GR–2011–02351355), and the Programma per i Giovani Ricercatori “Rita Levi Montalcini” 2011. GK is supported by the Ligue contre le Cancer (équipe labelisée); Agence National de la Recherche (ANR) – Projets blancs; ANR under the frame of E-Rare-2, the ERA-Net for Research on Rare Diseases; Association pour la recherche sur le cancer (ARC); Cancéropôle Ile-de-France; Institut National du Cancer (INCa); Institut Universitaire de France; Fondation pour la Recherche Médicale (FRM); the European Commission (ArtForce); the European Research Council (ERC); the LabEx Immuno-Oncology; the SIRIC Stratified Oncology Cell DNA Repair and Tumor Immune Elimination (SOCRATE); the SIRIC Cancer Research and Personalized Medicine (CARPEM); and the Paris Alliance of Cancer Research Institutes (PACRI). AA and NM salaries are provided by INSERM (French Medical Health and Research Institute) and research in their lab is supported by ANR grant from the French Research Ministry and from the “Fondation de France”. We thank V. Baldin and S. Desagher for kindly providing antibodies, D. Fesquet, J. Espeut, K. Hached and V. Georget for critical discussions and comments.
Abbreviations
CIN, chromosome instability; FCS, fetal calf serum; GFP, green fluorescent protein; H2B, histone 2B; MPS1, monopolar spindle 1; PARP, poly (ADP-ribose) polymerase 1; pH3, phosphorylated H3; PLK1, polo-like kinase 1; PS, phosphatidylserine; SAC, spindle assembly checkpoint; siRNA, small interfering RNA; UNR, unrelated; Z-VAD-fmk, Z-Val-Ala-Asp-fluoromethylketone.
CONFLICTS OF INTEREST
No potential conflicts of interest were disclosed.
REFERENCES
1. Thompson SL, Bakhoum SF and Compton DA. Mechanisms of chromosomal instability. Curr Biol. 2010; 20:R285–295.
2. Mitelman F, Johansson B and Mertens F. Mitelman Database of Chromosome Aberrations and Gene Fusions in Cancer (2015). Mitelman F, Johansson B and Mertens F (Eds.), “http://cgap.nci.nih.gov/Chromosomes/Mitelman”.
3. Zasadil LM, Britigan EM and Weaver BA. 2n or not 2n: Aneuploidy, polyploidy and chromosomal instability in primary and tumor cells. Semin Cell Dev Biol. 2013; 24:370–379.
4. Gordon DJ, Resio B and Pellman D. Causes and consequences of aneuploidy in cancer. Nat Rev Genet. 2012; 13:189–203.
5. Holland AJ and Cleveland DW. Boveri revisited: chromosomal instability, aneuploidy and tumorigenesis. Nat Rev Mol Cell Biol. 2009; 10:478–487.
6. Lee AJ, Endesfelder D, Rowan AJ, Walther A, Birkbak NJ, Futreal PA, Downward J, Szallasi Z, Tomlinson IP, Howell M, Kschischo M and Swanton C. Chromosomal instability confers intrinsic multidrug resistance. Cancer Res. 2011; 71:1858–1870.
7. Swanton C, Nicke B, Schuett M, Eklund AC, Ng C, Li Q, Hardcastle T, Lee A, Roy R, East P, Kschischo M, Endesfelder D, Wylie P, Kim SN, Chen JG, Howell M, et al. Chromosomal instability determines taxane response. Proc Natl Acad Sci U S A. 2009; 106:8671–8676.
8. Bakhoum SF, Danilova OV, Kaur P, Levy NB and Compton DA. Chromosomal instability substantiates poor prognosis in patients with diffuse large B-cell lymphoma. Clin Cancer Res. 2011; 17:7704–7711.
9. Carter SL, Eklund AC, Kohane IS, Harris LN and Szallasi Z. A signature of chromosomal instability inferred from gene expression profiles predicts clinical outcome in multiple human cancers. Nat Genet. 2006; 38:1043–1048.
10. Varetti G, Pellman D and Gordon DJ. Aurea mediocritas: the importance of a balanced genome. Cold Spring Harb Perspect Biol. 2014; 6:a015842.
11. Vitale I, Galluzzi L, Senovilla L, Criollo A, Jemaa M, Castedo M and Kroemer G. Illicit survival of cancer cells during polyploidization and depolyploidization. Cell Death Differ. 2011; 18:1403–1413.
12. Davoli T and de Lange T. The causes and consequences of polyploidy in normal development and cancer. Annu Rev Cell Dev Biol. 2011; 27:585–610.
13. Ganem NJ, Storchova Z and Pellman D. Tetraploidy, aneuploidy and cancer. Curr Opin Genet Dev. 2007; 17:157–162.
14. Davoli T and de Lange T. Telomere-driven tetraploidization occurs in human cells undergoing crisis and promotes transformation of mouse cells. Cancer Cell. 2012; 21:765–776.
15. Davoli T, Denchi EL and de Lange T. Persistent telomere damage induces bypass of mitosis and tetraploidy. Cell. 2010; 141:81–93.
16. Nguyen HG, Makitalo M, Yang D, Chinnappan D, St Hilaire C and Ravid K. Deregulated Aurora-B induced tetraploidy promotes tumorigenesis. FASEB J. 2009; 23:2741–2748.
17. Duelli DM, Padilla-Nash HM, Berman D, Murphy KM, Ried T and Lazebnik Y. A virus causes cancer by inducing massive chromosomal instability through cell fusion. Curr Biol. 2007; 17:431–437.
18. Fujiwara T, Bandi M, Nitta M, Ivanova EV, Bronson RT and Pellman D. Cytokinesis failure generating tetraploids promotes tumorigenesis in p53-null cells. Nature. 2005; 437:1043–1047.
19. Ganem NJ, Godinho SA and Pellman D. A mechanism linking extra centrosomes to chromosomal instability. Nature. 2009; 460:278–282.
20. Vitale I, Galluzzi L, Vivet S, Nanty L, Dessen P, Senovilla L, Olaussen KA, Lazar V, Prudhomme M, Golsteyn RM, Castedo M and Kroemer G. Inhibition of Chk1 kills tetraploid tumor cells through a p53-dependent pathway. PLoS One. 2007; 2:e1337.
21. Storchova Z, Breneman A, Cande J, Dunn J, Burbank K, O’Toole E and Pellman D. Genome-wide genetic analysis of polyploidy in yeast. Nature. 2006; 443:541–547.
22. Vitale I, Senovilla L, Jemaa M, Michaud M, Galluzzi L, Kepp O, Nanty L, Criollo A, Rello-Varona S, Manic G, Metivier D, Vivet S, Tajeddine N, Joza N, Valent A, Castedo M, et al. Multipolar mitosis of tetraploid cells: inhibition by p53 and dependency on Mos. EMBO J. 2010; 29:1272–1284.
23. Maley CC, Galipeau PC, Finley JC, Wongsurawat VJ, Li X, Sanchez CA, Paulson TG, Blount PL, Risques RA, Rabinovitch PS and Reid BJ. Genetic clonal diversity predicts progression to esophageal adenocarcinoma. Nat Genet. 2006; 38:468–473.
24. Olaharski AJ, Sotelo R, Solorza-Luna G, Gonsebatt ME, Guzman P, Mohar A and Eastmond DA. Tetraploidy and chromosomal instability are early events during cervical carcinogenesis. Carcinogenesis. 2006; 27:337–343.
25. Galipeau PC, Cowan DS, Sanchez CA, Barrett MT, Emond MJ, Levine DS, Rabinovitch PS and Reid BJ. 17p (p53) allelic losses, 4N (G2/tetraploid) populations, and progression to aneuploidy in Barrett’s esophagus. Proc Natl Acad Sci U S A. 1996; 93:7081–7084.
26. Dewhurst SM, McGranahan N, Burrell RA, Rowan AJ, Gronroos E, Endesfelder D, Joshi T, Mouradov D, Gibbs P, Ward RL, Hawkins NJ, Szallasi Z, Sieber OM and Swanton C. Tolerance of whole-genome doubling propagates chromosomal instability and accelerates cancer genome evolution. Cancer Discov. 2014; 4:175–185.
27. Lundberg G, Jin Y, Sehic D, Ora I, Versteeg R and Gisselsson D. Intratumour diversity of chromosome copy numbers in neuroblastoma mediated by on-going chromosome loss from a polyploid state. PLoS One. 2013; 8:e59268.
28. Zack TI, Schumacher SE, Carter SL, Cherniack AD, Saksena G, Tabak B, Lawrence MS, Zhang CZ, Wala J, Mermel CH, Sougnez C, Gabriel SB, Hernandez B, Shen H, Laird PW, Getz G, et al. Pan-cancer patterns of somatic copy number alteration. Nat Genet. 2013; 45:1134–1140.
29. Nik-Zainal S, Van Loo P, Wedge DC, Alexandrov LB, Greenman CD, Lau KW, Raine K, Jones D, Marshall J, Ramakrishna M, Shlien A, Cooke SL, Hinton J, Menzies A, Stebbings LA, Leroy C, et al. The life history of 21 breast cancers. Cell. 2012; 149:994–1007.
30. Carter SL, Cibulskis K, Helman E, McKenna A, Shen H, Zack T, Laird PW, Onofrio RC, Winckler W, Weir BA, Beroukhim R, Pellman D, Levine DA, Lander ES, Meyerson M and Getz G. Absolute quantification of somatic DNA alterations in human cancer. Nat Biotechnol. 2012; 30:413–421.
31. Beroukhim R, Mermel CH, Porter D, Wei G, Raychaudhuri S, Donovan J, Barretina J, Boehm JS, Dobson J, Urashima M, Mc Henry KT, Pinchback RM, Ligon AH, Cho YJ, Haery L, Greulich H, et al. The landscape of somatic copy-number alteration across human cancers. Nature. 2010; 463:899–905.
32. Pandit SK, Westendorp B and de Bruin A. Physiological significance of polyploidization in mammalian cells. Trends Cell Biol. 2013; 23:556–566.
33. Duncan AW. Aneuploidy, polyploidy and ploidy reversal in the liver. Semin Cell Dev Biol. 2013; 24:347–356.
34. Fox DT and Duronio RJ. Endoreplication and polyploidy: insights into development and disease. Development. 2013; 140:3–12.
35. Ganem NJ, Cornils H, Chiu SY, O’Rourke KP, Arnaud J, Yimlamai D, Thery M, Camargo FD and Pellman D. Cytokinesis failure triggers hippo tumor suppressor pathway activation. Cell. 2014; 158:833–848.
36. Margolis RL, Lohez OD and Andreassen PR. G1 tetraploidy checkpoint and the suppression of tumorigenesis. J Cell Biochem. 2003; 88:673–683.
37. Andreassen PR, Lohez OD, Lacroix FB and Margolis RL. Tetraploid state induces p53-dependent arrest of nontransformed mammalian cells in G1. Mol Biol Cell. 2001; 12:1315–1328.
38. Vitale I, Galluzzi L, Castedo M and Kroemer G. Mitotic catastrophe: a mechanism for avoiding genomic instability. Nat Rev Mol Cell Biol. 2011; 12:385–392.
39. Galluzzi L, Bravo-San Pedro JM, Vitale I, Aaronson SA, Abrams JM, Adam D, Alnemri ES, Altucci L, Andrews D, Annicchiarico-Petruzzelli M, Baehrecke EH, Bazan NG, Bertrand MJ, Bianchi K, Blagosklonny MV, Blomgren K, et al. Essential versus accessory aspects of cell death: recommendations of the NCCD 2015. Cell Death Differ. 2015; 22:58–73.
40. Kroemer G, Galluzzi L, Vandenabeele P, Abrams J, Alnemri ES, Baehrecke EH, Blagosklonny MV, El-Deiry WS, Golstein P, Green DR, Hengartner M, Knight RA, Kumar S, Lipton SA, Malorni W, Nunez G, et al. Classification of cell death: recommendations of the Nomenclature Committee on Cell Death 2009. Cell Death Differ. 2009; 16:3–11.
41. Castedo M, Coquelle A, Vivet S, Vitale I, Kauffmann A, Dessen P, Pequignot MO, Casares N, Valent A, Mouhamad S, Schmitt E, Modjtahedi N, Vainchenker W, Zitvogel L, Lazar V, Garrido C, et al. Apoptosis regulation in tetraploid cancer cells. EMBO J. 2006; 25:2584–2595.
42. Senovilla L, Vitale I, Martins I, Tailler M, Pailleret C, Michaud M, Galluzzi L, Adjemian S, Kepp O, Niso-Santano M, Shen S, Marino G, Criollo A, Boileve A, Job B, Ladoire S, et al. An immunosurveillance mechanism controls cancer cell ploidy. Science. 2012; 337:1678–1684.
43. Boileve A, Senovilla L, Vitale I, Lissa D, Martins I, Metivier D, van den Brink S, Clevers H, Galluzzi L, Castedo M and Kroemer G. Immunosurveillance against tetraploidization-induced colon tumorigenesis. Cell Cycle. 2013; 12:473–479.
44. Coward J and Harding A. Size Does Matter: Why Polyploid Tumor Cells are Critical Drug Targets in the War on Cancer. Front Oncol. 2014; 4:123.
45. Marxer M, Foucar CE, Man WY, Chen Y, Ma HT and Poon RY. Tetraploidization increases sensitivity to Aurora B kinase inhibition. Cell Cycle. 2012; 11:2567–2577.
46. Rello-Varona S, Vitale I, Kepp O, Senovilla L, Jemaa M, Metivier D, Castedo M and Kroemer G. Preferential killing of tetraploid tumor cells by targeting the mitotic kinesin Eg5. Cell Cycle. 2009; 8:1030–1035.
47. Roh M, van der Meer R and Abdulkadir SA. Tumorigenic polyploid cells contain elevated ROS and ARE selectively targeted by antioxidant treatment. J Cell Physiol. 2012; 227:801–812.
48. Lissa D, Senovilla L, Rello-Varona S, Vitale I, Michaud M, Pietrocola F, Boileve A, Obrist F, Bordenave C, Garcia P, Michels J, Jemaa M, Kepp O, Castedo M and Kroemer G. Resveratrol and aspirin eliminate tetraploid cells for anticancer chemoprevention. Proc Natl Acad Sci U S A. 2014; 111:3020–3025.
49. Donovan P, Cato K, Legaie R, Jayalath R, Olsson G, Hall B, Olson S, Boros S, Reynolds BA and Harding A. Hyperdiploid tumor cells increase phenotypic heterogeneity within Glioblastoma tumors. Mol Biosyst. 2014; 10:741–758.
50. Zeng JY, Sharma S, Zhou YQ, Yao HP, Hu X, Zhang R and Wang MH. Synergistic activities of MET/RON inhibitor BMS-777607 and mTOR inhibitor AZD8055 to polyploid cells derived from pancreatic cancer and cancer stem cells. Mol Cancer Ther. 2014; 13:37–48.
51. Liu LL, Long ZJ, Wang LX, Zheng FM, Fang ZG, Yan M, Xu DF, Chen JJ, Wang SW, Lin DJ and Liu Q. Inhibition of mTOR pathway sensitizes acute myeloid leukemia cells to aurora inhibitors by suppression of glycolytic metabolism. Mol Cancer Res. 2013; 11:1326–1336.
52. Liu X and Winey M. The MPS1 family of protein kinases. Annu Rev Biochem. 2012; 81:561–585.
53. Musacchio A and Salmon ED. The spindle-assembly checkpoint in space and time. Nat Rev Mol Cell Biol. 2007; 8:379–393.
54. Abrieu A, Magnaghi-Jaulin L, Kahana JA, Peter M, Castro A, Vigneron S, Lorca T, Cleveland DW and Labbe JC. Mps1 is a kinetochore-associated kinase essential for the vertebrate mitotic checkpoint. Cell. 2001; 106:83–93.
55. Mills GB, Schmandt R, McGill M, Amendola A, Hill M, Jacobs K, May C, Rodricks AM, Campbell S and Hogg D. Expression of TTK, a novel human protein kinase, is associated with cell proliferation. J Biol Chem. 1992; 267:16000–16006.
56. Kusakabe K, Ide N, Daigo Y, Itoh T, Yamamoto T, Hashizume H, Nozu K, Yoshida H, Tadano G, Tagashira S, Higashino K, Okano Y, Sato Y, Inoue M, Iguchi M, Kanazawa T, et al. Discovery of imidazo[1,2-b]pyridazine derivatives: selective and orally available Mps1 (TTK) kinase inhibitors exhibiting remarkable antiproliferative activity. J Med Chem. 2015; 58:1760–1775.
57. Kaistha BP, Honstein T, Muller V, Bielak S, Sauer M, Kreider R, Fassan M, Scarpa A, Schmees C, Volkmer H, Gress TM and Buchholz M. Key role of dual specificity kinase TTK in proliferation and survival of pancreatic cancer cells. Br J Cancer. 2014; 111:1780–1787.
58. Slee RB, Grimes BR, Bansal R, Gore J, Blackburn C, Brown L, Gasaway R, Jeong J, Victorino J, March KL, Colombo R, Herbert BS and Korc M. Selective inhibition of pancreatic ductal adenocarcinoma cell growth by the mitotic MPS1 kinase inhibitor NMS-P715. Mol Cancer Ther. 2014; 13:307–315.
59. Jemaa M, Galluzzi L, Kepp O, Senovilla L, Brands M, Boemer U, Koppitz M, Lienau P, Prechtl S, Schulze V, Siemeister G, Wengner AM, Mumberg D, Ziegelbauer K, Abrieu A, Castedo M, et al. Characterization of novel MPS1 inhibitors with preclinical anticancer activity. Cell Death Differ. 2013; 20:1532–1545.
60. Colombo R, Caldarelli M, Mennecozzi M, Giorgini ML, Sola F, Cappella P, Perrera C, Depaolini SR, Rusconi L, Cucchi U, Avanzi N, Bertrand JA, Bossi RT, Pesenti E, Galvani A, Isacchi A, et al. Targeting the mitotic checkpoint for cancer therapy with NMS-P715, an inhibitor of MPS1 kinase. Cancer Res. 2010; 70:10255–10264.
61. Janssen A, Kops GJ and Medema RH. Elevating the frequency of chromosome mis-segregation as a strategy to kill tumor cells. Proc Natl Acad Sci U S A. 2009; 106:19108–19113.
62. Jelluma N, Brenkman AB, van den Broek NJ, Cruijsen CW, van Osch MH, Lens SM, Medema RH and Kops GJ. Mps1 phosphorylates Borealin to control Aurora B activity and chromosome alignment. Cell. 2008; 132:233–246.
63. Kops GJ, Foltz DR and Cleveland DW. Lethality to human cancer cells through massive chromosome loss by inhibition of the mitotic checkpoint. Proc Natl Acad Sci U S A. 2004; 101:8699–8704.
64. Santaguida S, Tighe A, D’Alise AM, Taylor SS and Musacchio A. Dissecting the role of MPS1 in chromosome biorientation and the spindle checkpoint through the small molecule inhibitor reversine. J Cell Biol. 2010; 190:73–87.
65. Kepp O, Galluzzi L, Lipinski M, Yuan J and Kroemer G. Cell death assays for drug discovery. Nat Rev Drug Discov. 2011; 10:221–237.
66. Galluzzi L, Aaronson SA, Abrams J, Alnemri ES, Andrews DW, Baehrecke EH, Bazan NG, Blagosklonny MV, Blomgren K, Borner C, Bredesen DE, Brenner C, Castedo M, Cidlowski JA, Ciechanover A, Cohen GM, et al. Guidelines for the use and interpretation of assays for monitoring cell death in higher eukaryotes. Cell Death Differ. 2009; 16:1093–1107.
67. Hewitt L, Tighe A, Santaguida S, White AM, Jones CD, Musacchio A, Green S and Taylor SS. Sustained Mps1 activity is required in mitosis to recruit O-Mad2 to the Mad1-C-Mad2 core complex. J Cell Biol. 2010; 190:25–34.
68. Jelluma N, Dansen TB, Sliedrecht T, Kwiatkowski NP and Kops GJ. Release of Mps1 from kinetochores is crucial for timely anaphase onset. J Cell Biol. 2010; 191:281–290.
69. Jemaa M, Galluzzi L, Kepp O, Castedo M, Rello-Varona S, Vitale I and Kroemer G. Transgenerational cell fate profiling: a method for the graphical presentation of complex cell cycle alterations. Cell Cycle. 2013; 12:183–190.
70. Espeut J, Lara-Gonzalez P, Sassine M, Shiau AK, Desai A and Abrieu A. Natural Loss of Mps1 Kinase in Nematodes Uncovers a Role for Polo-like Kinase 1 in Spindle Checkpoint Initiation. Cell Rep. 2015; 12:58–65.
71. von Schubert C, Cubizolles F, Bracher JM, Sliedrecht T, Kops GJ and Nigg EA. Plk1 and Mps1 Cooperatively Regulate the Spindle Assembly Checkpoint in Human Cells. Cell Rep. 2015; 12:66–78.
72. Jemaa M, Galluzzi L, Kepp O, Boileve A, Lissa D, Senovilla L, Harper F, Pierron G, Berardinelli F, Antoccia A, Castedo M, Vitale I and Kroemer G. Preferential killing of p53-deficient cancer cells by reversine. Cell Cycle. 2012; 11:2149–2158.
73. Jemaa M, Vitale I, Kepp O, Berardinelli F, Galluzzi L, Senovilla L, Marino G, Malik SA, Rello-Varona S, Lissa D, Antoccia A, Tailler M, Schlemmer F, Harper F, Pierron G, Castedo M, et al. Selective killing of p53-deficient cancer cells by SP600125. EMBO Mol Med. 2012; 4:500–514.
74. Degenhardt Y, Greshock J, Laquerre S, Gilmartin AG, Jing J, Richter M, Zhang X, Bleam M, Halsey W, Hughes A, Moy C, Liu-Sullivan N, Powers S, Bachman K, Jackson J, Weber B, et al. Sensitivity of cancer cells to Plk1 inhibitor GSK461364A is associated with loss of p53 function and chromosome instability. Mol Cancer Ther. 2010; 9:2079–2089.
75. Jeganathan K, Malureanu L, Baker DJ, Abraham SC and van Deursen JM. Bub1 mediates cell death in response to chromosome missegregation and acts to suppress spontaneous tumorigenesis. J Cell Biol. 2007; 179:255–267.
76. Fang Y, Liu T, Wang X, Yang YM, Deng H, Kunicki J, Traganos F, Darzynkiewicz Z, Lu L and Dai W. BubR1 is involved in regulation of DNA damage responses. Oncogene. 2006; 25:3598–3605.
77. Kilpinen S, Ojala K and Kallioniemi O. Analysis of kinase gene expression patterns across 5681 human tissue samples reveals functional genomic taxonomy of the kinome. PLoS One. 2010; 5:e15068.
78. Mizukami Y, Kono K, Daigo Y, Takano A, Tsunoda T, Kawaguchi Y, Nakamura Y and Fujii H. Detection of novel cancer-testis antigen-specific T-cell responses in TIL, regional lymph nodes, and PBL in patients with esophageal squamous cell carcinoma. Cancer Sci. 2008; 99:1448–1454.
79. Landi MT, Dracheva T, Rotunno M, Figueroa JD, Liu H, Dasgupta A, Mann FE, Fukuoka J, Hames M, Bergen AW, Murphy SE, Yang P, Pesatori AC, Consonni D, Bertazzi PA, Wacholder S, et al. Gene expression signature of cigarette smoking and its role in lung adenocarcinoma development and survival. PLoS One. 2008; 3:e1651.
80. Chng WJ, Braggio E, Mulligan G, Bryant B, Remstein E, Valdez R, Dogan A and Fonseca R. The centrosome index is a powerful prognostic marker in myeloma and identifies a cohort of patients that might benefit from aurora kinase inhibition. Blood. 2008; 111:1603–1609.
81. Salvatore G, Nappi TC, Salerno P, Jiang Y, Garbi C, Ugolini C, Miccoli P, Basolo F, Castellone MD, Cirafici AM, Melillo RM, Fusco A, Bittner ML and Santoro M. A cell proliferation and chromosomal instability signature in anaplastic thyroid carcinoma. Cancer Res. 2007; 67:10148–10158.
82. Yuan B, Xu Y, Woo JH, Wang Y, Bae YK, Yoon DS, Wersto RP, Tully E, Wilsbach K and Gabrielson E. Increased expression of mitotic checkpoint genes in breast cancer cells with chromosomal instability. Clin Cancer Res. 2006; 12:405–410.
83. Thykjaer T, Workman C, Kruhoffer M, Demtroder K, Wolf H, Andersen LD, Frederiksen CM, Knudsen S and Orntoft TF. Identification of gene expression patterns in superficial and invasive human bladder cancer. Cancer Res. 2001; 61:2492–2499.
84. Ling Y, Zhang X, Bai Y, Li P, Wei C, Song T, Zheng Z, Guan K, Zhang Y, Zhang B, Liu X, Ma RZ, Cao C, Zhong H and Xu Q. Overexpression of Mps1 in colon cancer cells attenuates the spindle assembly checkpoint and increases aneuploidy. Biochem Biophys Res Commun. 2014; 450:1690–1695.
85. Gyorffy B, Bottai G, Lehmann-Che J, Keri G, Orfi L, Iwamoto T, Desmedt C, Bianchini G, Turner NC, de The H, Andre F, Sotiriou C, Hortobagyi GN, Di Leo A, Pusztai L and Santarpia L. TP53 mutation-correlated genes predict the risk of tumor relapse and identify MPS1 as a potential therapeutic kinase in TP53-mutated breast cancers. Mol Oncol. 2014; 8:508–519.
86. Brough R, Frankum JR, Sims D, Mackay A, Mendes-Pereira AM, Bajrami I, Costa-Cabral S, Rafiq R, Ahmad AS, Cerone MA, Natrajan R, Sharpe R, Shiu KK, Wetterskog D, Dedes KJ, Lambros MB, et al. Functional viability profiles of breast cancer. Cancer Discov. 2011; 1:260–273.
87. Tannous BA, Kerami M, Van der Stoop PM, Kwiatkowski N, Wang J, Zhou W, Kessler AF, Lewandrowski G, Hiddingh L, Sol N, Lagerweij T, Wedekind L, Niers JM, Barazas M, Nilsson RJ, Geerts D, et al. Effects of the selective MPS1 inhibitor MPS1-IN-3 on glioblastoma sensitivity to antimitotic drugs. J Natl Cancer Inst. 2013; 105:1322–1331.
88. Daniel J, Coulter J, Woo JH, Wilsbach K and Gabrielson E. High levels of the Mps1 checkpoint protein are protective of aneuploidy in breast cancer cells. Proc Natl Acad Sci U S A. 2011; 108:5384–5389.
89. Maachani UB, Kramp T, Hanson R, Zhao S, Celiku O, Shankavaram U, Colombo R, Caplen NJ, Camphausen K and Tandle A. Targeting MPS1 Enhances Radiosensitization of Human Glioblastoma by Modulating DNA Repair Proteins. Mol Cancer Res. 2015.
90. Kagami Y, Nihira K, Wada S, Ono M, Honda M and Yoshida K. Mps1 phosphorylation of condensin II controls chromosome condensation at the onset of mitosis. J Cell Biol. 2014; 205:781–790.
91. Pradhan A, Singh TR, Ali AM, Wahengbam K and Meetei AR. Monopolar spindle 1 (MPS1) protein-dependent phosphorylation of RecQ-mediated genome instability protein 2 (RMI2) at serine 112 is essential for BLM-Topo III alpha-RMI1-RMI2 (BTR) protein complex function upon spindle assembly checkpoint (SAC) activation during mitosis. J Biol Chem. 2013; 288:33500–33508.
92. Mattison CP, Stumpff J, Wordeman L and Winey M. Mip1 associates with both the Mps1 kinase and actin, and is required for cell cortex stability and anaphase spindle positioning. Cell Cycle. 2011; 10:783–793.
93. Lan W and Cleveland DW. A chemical tool box defines mitotic and interphase roles for Mps1 kinase. J Cell Biol. 2010; 190:21–24.
94. Huang YF, Chang MD and Shieh SY. TTK/hMps1 mediates the p53-dependent postmitotic checkpoint by phosphorylating p53 at Thr18. Mol Cell Biol. 2009; 29:2935–2944.
95. Kasbek C, Yang CH and Fisk HA. Mps1 as a link between centrosomes and genomic instability. Environ Mol Mutagen. 2009; 50:654–665.
96. Yeh YH, Huang YF, Lin TY and Shieh SY. The cell cycle checkpoint kinase CHK2 mediates DNA damage-induced stabilization of TTK/hMps1. Oncogene. 2009; 28:1366–1378.
97. Leng M, Chan DW, Luo H, Zhu C, Qin J and Wang Y. MPS1-dependent mitotic BLM phosphorylation is important for chromosome stability. Proc Natl Acad Sci U S A. 2006; 103:11485–11490.
98. Wei JH, Chou YF, Ou YH, Yeh YH, Tyan SW, Sun TP, Shen CY and Shieh SY. TTK/hMps1 participates in the regulation of DNA damage checkpoint response by phosphorylating CHK2 on threonine 68. J Biol Chem. 2005; 280:7748–7757.
99. Fisk HA, Mattison CP and Winey M. A field guide to the Mps1 family of protein kinases. Cell Cycle. 2004; 3:439–442.
100. Fisk HA, Mattison CP and Winey M. Human Mps1 protein kinase is required for centrosome duplication and normal mitotic progression. Proc Natl Acad Sci U S A. 2003; 100:14875–14880.